Alzheimer Disease With Amygdala Lewy Bodies: A Distinct Form of α-Synucleinopathy (original) (raw)
. Author manuscript; available in PMC: 2017 Nov 29.
Abstract
Lewy bodies (LBs) are α-synuclein-immunoreactive neuronal inclusions with a predilection for specific cortical and subcortical regions, including the amygdala. In this study, the presence of LBs was assessed in 347 cases of Alzheimer disease (AD). In 87 cases, LB pathology was diagnostic of brainstem (n = 3), transitional (n = 32), or diffuse (n = 52) Lewy body disease (LBD). The remaining 260 cases of AD were screened for amygdala LBs (AD/ALB) and 62 (24%) cases were found. If AD/LBD cases are included, LBs were detected in 149 (43%) cases of AD. The presence α-synuclein pathology was assessed in multiple brain regions of the 62 cases of AD/ALB and 57 randomly selected cases of AD, and only sparse α-synuclein pathology was detected in both. The burden of α-synuclein pathology in brainstem nuclei, amygdala, and neocortex was significant lower in AD/ALB than in AD/LBD. In comparison to AD/LBD, AD/ALB did not differ in age at death, disease duration, male-to-female ratio, brain weight, Braak neurofibrillary tangle stage, average senile plaque density, or apolipoprotein E ∈4 allele frequency. The results suggest that AD/ALB is pathologically different from AD/LBD, suggesting that it is a neuropathologically distinct and isolated α-synucleinopathy.
Keywords: Alzheimer disease, α-synuclein, Amygdala, Immunohistochemistry, Lewy bodies
INTRODUCTION
Lewy body disease (LBD) is the most common pathologic substrate for the clinical syndromes of Parkinson disease (PD) and dementia with Lewy bodies (DLB) (1). It is characterized by α-synuclein-immunoreactive Lewy bodies (LBs) and Lewy neurites as well as neuronal loss and gliosis in select subcortical nuclei. LBD is considered to be the second most common neurodegenerative process after Alzheimer disease (AD). Most cases of LBD are accompanied by varying degrees of Alzheimer-type pathology, including neurofibrillary tangles (NFTs) and senile plaques (SPs). In some cases, the Alzheimer-type pathology is insufficient to warrant a diagnosis of AD and is more consistent with pathologic aging (2), but in other cases, SPs and NFTs are abundant and consistent with a diagnosis of concurrent AD (3). The latter is sometimes referred to as the Lewy body variant of AD (4).
Several recent studies using α-synuclein immunohistochemistry have drawn attention to the presence of LBs in sporadic AD (5–12). Seminal studies by Schmidt and coworkers drew attention to the pleomorphic nature of the lesions in the amygdala and the frequent co-occurrence of Lewy-like lesions in neurons that also had neurofibrillary pathology (13). Although it might be argued that this form of α-synuclein pathology is distinct from that in LBD, there is no significant difference in morphology from that seen in LBD. It is clear from these reports that the amygdala is particularly vulnerable to α-synuclein pathology, particularly in familial AD (14) and Down syndrome (15). The amygdala is also one of the regions most severely affected in DLB with severity of pathology correlating with visual hallucinations, one of the cardinal clinical features of DLB (16–18).
The amygdala has long been known to be vulnerable to Alzheimer-type pathology (19), and it has been described as one of earliest locations to develop Alzheimer pathology in Down syndrome (20). The amygdala is thought to be second only to periallocortex in NFT severity in AD (21). Neuronal loss in the amygdala ranges from 35% to 70% in AD (22). In addition, Tsuchiya et al reported that neuronal loss in the corticomedial was more severe than in the basolateral region of the amygdala with the distribution of neuronal loss in the amygdala paralleling that of NFTs (23). In contrast, the distribution of SPs did not follow that of neuronal loss (23). Severity of amygdala pathology correlates with disease duration in AD (24), and amygdala pathology has been associated with emotional and memory disturbances (25, 26).
In the present study, we screened cases of AD for α-synuclein pathology in the amygdala and identified a distinct form of α-synuclein pathology with LBs or pleomorphic neuronal inclusions and a variable number of neurites relatively confined to the amygdala, which we operationally term Alzheimer disease with amygdala LBs (AD/ALB). Cases of AD/ALB were compared with AD with varying degrees of Lewy body pathology—AD with diffuse LBD (AD/DLBD) and AD with transitional LBD (AD/TLBD)—that were collected during the same time period with respect to density and distribution of Alzheimer and α-synuclein pathology as well as demographic, genetic, and clinical features.
MATERIALS AND METHODS
From 1998 to 2001, there were 347 consecutive cases of AD accessioned in the Mayo Clinic Jacksonville brain bank. Of this total, 242 cases (70%) were derived from State of Florida Alzheimer Disease Initiative, 75 cases (22%) from Mayo Clinic Jacksonville Memory Disorder Clinic, and the remaining 30 cases were from a variety of referral sources, including 15 cases (4%) from the Einstein Aging Study, 12 cases (3%) from the Mayo Clinic Movement Disorder Clinic, and 3 cases (1%) from referring pathologists. The State of Florida Alzheimer Disease Initiative is a state-supported resource to provide autopsy confirmation of dementing conditions for individuals who are seen at least once in one of 13 state-sponsored memory disorders clinics (27). It is not a standardized, prospective, or longitudinal study, and the extent of accompanying clinical information is variable from case to case depending on the referral center. Cases from the Einstein Aging Study, which is a prospective, longitudinal study, had consistent antemortem longitudinal clinical information.
The study cohort consisted of 152 men and 195 women with an age range of 57 to 102 years. All cases met pathologic criteria for AD by Khachaturian criteria (28) as well as more stringent NIA-Reagan criteria (3). They all had a Braak NFT stage of IV or greater with a median score of VI, indicative of advanced AD in almost all cases (29). Most of the cases (n = 278) were white, with a few Hispanic (n = 9) and black (n = 9). In some cases (n = 55), race was not stated.
All cases underwent a uniform neuropathologic evaluation with standardized tissue sampling and quantitative assessment of SP and NFT densities with thioflavin-S fluorescent microscopy. The SPs were assessed at ×100 and NFTs at ×400. The analysis included 5 neocortical sections and the entorhinal cortex as well as the basal nucleus of Meynert (nbM), 4 sectors of the hippocampus; and basolateral and corticomedial zones of the amygdala. Given that it is impossible to precisely define subnuclei of the amygdala without special methods (e.g. cholinesterase histochemistry) and because the anatomic boundaries are often severely distorted in AD, in the present study “corticomedial” is used to refer to the medial part of the amygdala near the cortical transitional zone and “basolateral” to refer to the region of the ventral and lateral region of the amygdala. The corticomedial region roughly corresponds to the corticomedial complex of the amygdala, which is composed of central, medial, and cortical nuclei together (30).
Of these 347 cases of AD, 87 cases had neuronal loss and gliosis with readily apparent LBs in the nbM, SN, LC, and dorsal motor nucleus of the vagus (DMN) with hematoxylin and eosin stains. For these 87 cases, multiple cortical sections as well as the sections of the basal forebrain and brainstem were also immunostained. Based on the density and distribution of LBs, the type of LBD was classified as brainstem-predominant (BLBD), transitional (TLBD), or diffuse (DLBD) (AD/BLBD: n = 3; AD/TLBD: n = 32; AD/DLBD: n = 52). For the purposes of this study, AD cases with varying degrees cerebrovascular pathology or argyrophilic grains were included. The 3 cases of AD/BLBD were not included in comparative evaluations, but had typical Alzheimer-type dementia without significant psychiatric or extrapyramidal signs. The remaining 260 AD cases were operationally considered to be “pure” with respect to overt LB pathology.
For these 260 cases, a section of the basal forebrain at the level of the infundibulum, which includes the hypothalamus, substantia innominata, lentiform nucleus, and amygdala, was screened with α-synuclein immunohistochemistry. When LBs were detected in the amygdala, additional sections of brainstem (including midbrain, pons, and medulla) as well as hippocampus with entorhinal cortex (ERC), anterior cingulate gyrus (Cing), middle frontal gyrus (MF), superior temporal gyrus (ST), and inferior parietal gyrus (IP) were immunostained for α-synuclein. In 62 cases, α-synuclein pathology was detected in the amygdala, which constitutes the cohort of AD/ALB, which were studied in more detail.
Of the other remaining AD cases without amygdala LBs (n = 198), 57 cases were randomly selected, and brainstem regions of these cases were immunostained to detect inconspicuous or incidental α-synuclein pathology not associated with overt neuronal loss and gliosis or readily apparent LBs. In this group of 57 cases, we found 6 cases with sparse α-synuclein pathology, mostly in the form of pale bodies and neuritic pathology, which were not visible with routine histologic methods. Figure 1 shows an algorithm for the classification of cases and the overall study design.
FIGURE 1.
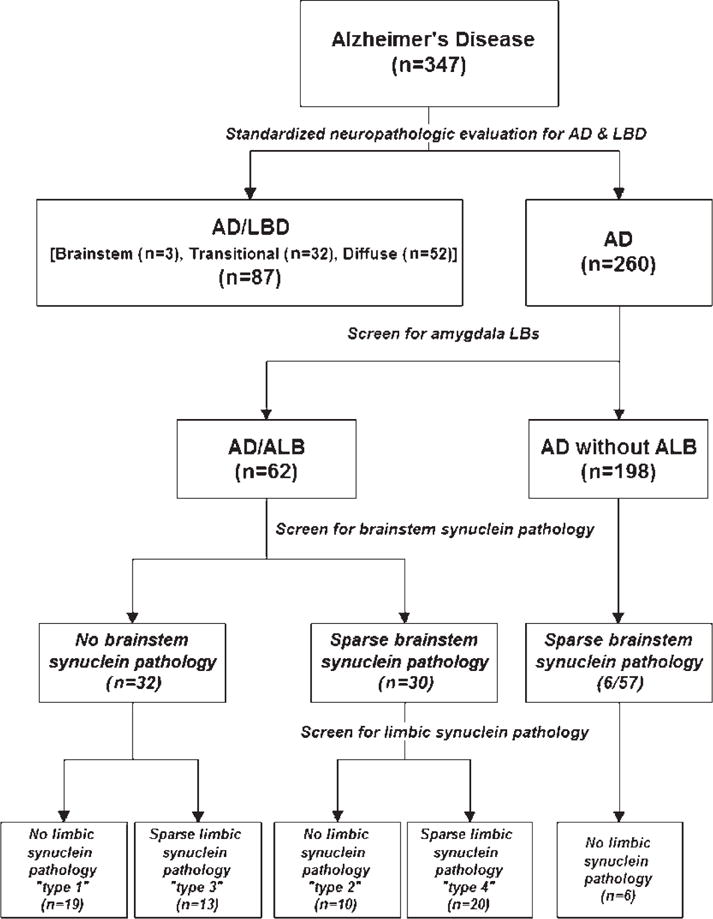
A flow chart of the study design and classification of cases stemming from standardized neuropathologic evaluation to diagnose Alzheimer disease (AD) or AD with concurrent Lewy body disease, followed by screening of the amygdala section for α-synuclein pathology in all AD cases. For those cases with any amygdala α-synuclein pathology, brainstem sections and limbic areas (entorhinal and cingulate gyrus) are screened for additional α-synuclein pathology. A randomly selected series of AD cases without amygdala α-synuclein pathology are processed in the same manner.
Immunohistochemistry
For all regions of interest (amygdala, nbM, MF, IP, Cing, ERC, SN, LC, and DMN), 5-μm-thick paraffin sections were immunostained using a previously characterized rabbit polyclonal antibody to α-synuclein (NACP; 1:3000 [31]). The deparaffinized and rehydrated sections were pretreated with 95% formic acid for 30 minutes and then steamed in distilled water for 30 minutes. Immunohistochemistry was performed with a DAKO Autostainer (DAKO, Carpinteria, CA) using 3, 3-diaminobenzidine (DAB) as the chromogen. After immunostaining, the sections were counterstained with hematoxylin.
Results with the polyclonal antibody were compared with immunostaining with a commercial monoclonal antibody to α-synuclein (LB509; Zymed, South San Francisco, CA; 1:100) using various pretreatments. The sections were pretreated with 95% formic acid (FA) for 30 minutes or with proteinase K (Sigma, St. Louis, MO; 10 mg/mL, 1:100) for 10 minutes at 37°C. Sections were also processed according to the procedure advocated by Hamilton (11) with Sigma protease XXIV (Sigma; 0.1 mg/mL) for 10 minutes at room temperature. Protease treatment often revealed more neuritic pathology, but at the cost of tissue digestion and loss of histologic integrity. In no case did we detect differences in α-synuclein pathology with LB509, nor with the various pretreatments with the polyclonal antibody or steam heat and formic acid, using the DAKO Envision-Plus signal amplification method.
To characterize the nature of amygdala α-synuclein pathology, a subset of cases was studied with double immunolabeling immunohistochemistry. In particular, sections were double stained for tau and α-synuclein using immunohistochemistry and immunofluorescence, as described previously (32). The antibodies used were as follows: NACP, CP13 (mouse monoclonal IgG1 specific to tau phosphorylated at residue 202, 1:500; from Peter Davies, Albert Einstein College of Medicine, New York), and MC1 (monoclonal mouse IgG1 to a conformational epitope in tau in neurofibrillary lesions, 1:50; from Peter Davies, Albert Einstein College of Medicine).
Several cases were also studied with single and double immunoelectron microscopy using rabbit polyclonal antibody to α-synuclein (NACP) and mouse monoclonal antibody to tau (CP13) as previously described (33). Small pieces (1 mm3) of amygdala from formalin-fixed autopsy brain were postfixed in 4% paraformaldehyde in 100 mM phosphate buffer; dehydrated in 30%, 50%, 70%, and 90% ethanol; and embedded in LR White resin (Polysciences, Warrington, PA). Ultrathin sections collected on Formvar-coated nickel grids were incubated in primary antibodies overnight at 4°C followed by species-specific secondary antibodies conjugated with colloidal gold particles of 2 sizes (5 nm and 18 nm).
Evaluation of Alzheimer-Type Pathology
As part of the standardized neuropathologic evaluation of all AD cases, SPs and NFTs are counted with thioflavin-S fluorescent microscopy. The average SP density was determined at a ×100 magnification from the following cortical regions: MF, ST, IP as well as the primary motor and visual cortices. The average NFT density was determined in the same regions at ×400 magnification. The density of SPs and NFTs were also assessed in corticomedial and basolateral regions of the amygdala. Specific amygdala nuclei are not readily detected with routine histologic methods, particularly with fluorescent histochemistry. Nevertheless, consistent separation of nonoverlapping fields from basolateral and corticomedial regions of the amygdala was possible. A Braak NFT stage was assigned based on the distribution of NFTs observed with thioflavin-S fluorescent microscopy (29).
Determination of the α-Synuclein Score
Four areas (nbM, SN, LC, and DMN) were selected for semiquantitative evaluation of α-synuclein pathology. The α-synuclein score was assigned as follows: 0 = absent, 1 = mild, 2 = moderate, and 3 = severe. In this assessment, not only perikaryal LBs, but also intraaxonal LBs, cortical-type LBs, pleomorphic LBs, and Lewy neurites were taken into consideration.
Determination of Lewy Body Counts
Six anatomic regions, including amygdala, ERC, Cing, ST, MF, and IP, were used in the diagnostic assessment of LBD with α-synuclein immunohistochemistry. After examining the entire region of interest under low magnification, the microscopic field with the highest density of LBs was identified and the number of LBs was counted at ×200 magnification. For this evaluation, we did not take Lewy neurites into consideration and each LB had to have a visible nucleus to be counted.
APOE Genotype and Tau Haplotype Analysis
Genomic DNA extracted from frozen brain tissue was used for determining the tau haplotype and APOE genotypes according to methods described in previous reports (34, 35). APOE genotyping was available on 306 AD cases (175 women and 131 men; age range, 57–102 years; mean age, 80.4 years). The tau haplotype was available on 305 AD cases (174 women and 131 men; age range, 57–102 years; mean age, 80.4 years).
Distribution of Lewy Bodies in Alzheimer Disease With Amygdala Lewy Bodies
The distribution of LBs in AD/ALB was assessed with Parkinson disease staging scheme proposed by Braak and coworkers (36). In this evaluation, the distribution of α-synuclein-immunoreactive lesions was considered more important than the density and all types of neuronal and neuritic lesions were taken into consideration.
Clinical Features of Alzheimer Disease With Amygdala Lewy Bodies
For all cases in which detailed clinical notes were available, the medical records were evaluated to determine the presence (at any time during the disease course) of extrapyramidal symptoms (rigidity, bradykinesia, and tremor) and neuropsychiatric symptoms (hallucinations, delusions, depression, and anxiety). A comparable number of cases of AD without ALB were studied in a similar way, blinded to pathologic diagnosis.
Statistical Methods
Data were analyzed with SigmaStat 3.0 (Systat Software, Inc., Point Richmond, CA) and the significance level was set at p < 0.05. For the comparison of AD, AD/ALB, AD/TLBD, and AD/DLBD with respect to age at death, male-to-female ratio, brain weight, Braak NFT stage, disease duration, and average density of SPs and NFTs, we used Kruskal-Wallis analysis of variance (ANOVA) on ranks. For the comparison of cases with respect to tau H1 haplotype, APOE ∈4 carrier frequency, and clinical differences between AD and AD/ALB, either the Fisher exact or chi-squared test was performed as appropriate. AD/BLBD cases were not included in this analysis as a result of their small number. The differences between α-synuclein scores and LBs counts in the groups were examined with Kruskal-Wallis ANOVA on ranks. If there was a significant difference, Dunn’s method was performed for pairwise comparisons. Spearman’s rank order correlation was used to evaluate correlations between LBs in the amygdala with α-synuclein score, cortical LB counts, average density of SPs, and NFTs in the amygdala, age at death, disease duration, and clinical features.
RESULTS
Frequency of Alzheimer Disease With Amygdala Lewy Bodies
Of the 347 cases of AD accessioned in the study period, 87 cases (25%) were readily recognized as AD/LBD based on the presence of LBs in vulnerable brainstem nuclei. These cases were classified after immunostaining of cortical and subcortical brain regions with α-synuclein immunohistochemistry as AD/DLBD (n = 52), AD/TLBD (n = 32), and AD/BLBD (n = 3). After screening the remaining 260 cases, α-synuclein pathology was detected in the amygdala of 62 of the remaining 260 AD cases (24%). When AD/ALB and AD/LBD cases are combined, the overall frequency of amygdala LBs in AD was 43%. To determine if LBs might be detected in AD cases without ALB, a series of 57 cases were selected at random and screened for α-synuclein pathology as mentioned previously. We found inconspicuous α-synuclein pathology in 6 cases (11%). It must be emphasized that these latter cases did not have overt neuronal loss or LBs in vulnerable brainstem nuclei, except the locus ceruleus, and most of the α-synuclein pathology was in sparse neurites.
Morphology of α-Synuclein Pathology in Alzheimer Disease With Amygdala Lewy Bodies
The AD/ALB cases all had α-synuclein-positive neuronal lesions that resembled LBs or more pleomorphic cytoplasmic inclusions. The lesions had a range of morphologies, including small dense cytoplasmic spots (Fig. 2A), globose-shaped lesions (Fig. 2B) and, rarely, hyaline Lewy-like inclusions (Fig. 2C). A similar range of amygdala inclusions is present in LBD. Although not all lesions resembled classic LBs, we have operationally defined such cases as having AD/ALB. The number of amygdala LBs varied widely between cases (from one to more than 50), but all cases had neuronal cytoplasmic inclusions that were distinguished from α-synuclein-immunoreactive dystrophic neurites that are sometimes present in SPs. The highest density of amygdala LBs was in the corticomedial region of the amygdala, which is composed of the central, medial, and cortical nuclei (30). A few dystrophic neurites were sometimes detected in AD/ALB, but they were far fewer than in AD/LBD cases.
FIGURE 2.
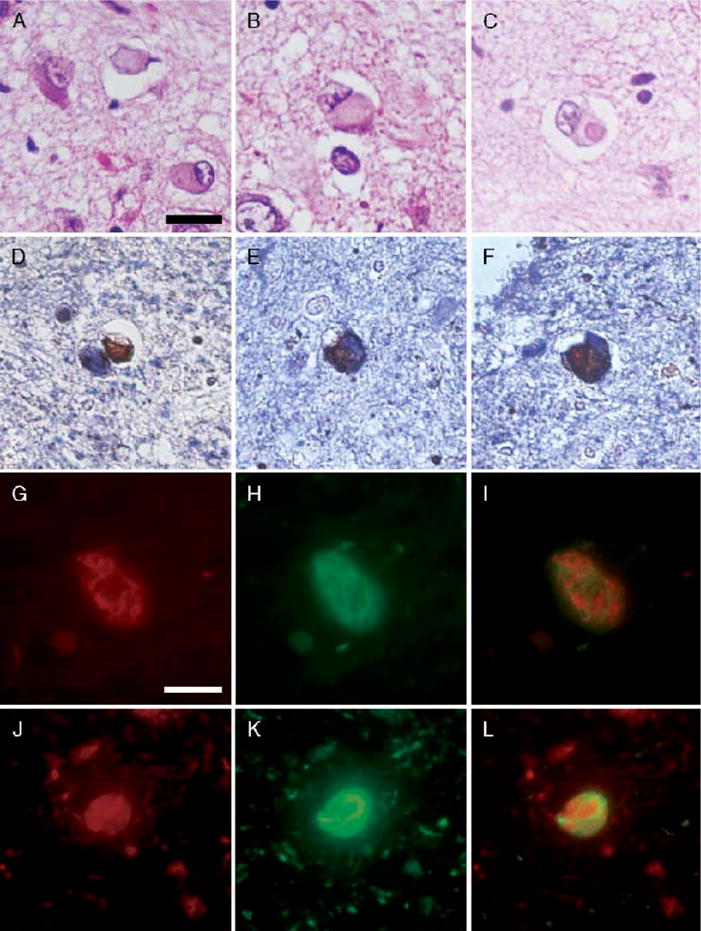
(A–C) Range of morphologies of amygdala Lewy bodies (LBs) with hematoxylin and eosin. Only exceptionally do amygdala LBs have dense hyaline appearance (C); more often, they are pleomorphic lesions indistinguishable from neurofibrillary tangles (NFTs) (A, B). Double immunostaining with α-synuclein (brown) and phospho-tau (blue) shows a range of morphologies as well. The α-synuclein-positive lesions may be separate from tau-positive NFTs (D) or variably intermingled within the same neuron (E, F). Double immunofluorescence for α-synuclein (red) and tau (green). In the merged images (I, L), colocalization appears yellow. The neuron in (G–I) shows intermingling of 2 distinct types of filaments within the same neuron, whereas the neuron in (J–L) shows an α-synuclein-immunoreactive, Lewy-like inclusion surrounded by a tau-immunoreactive NFT. Scale bars = (A–F) 25 μm; (D–I) 10 μm.
On a subset of AD/ALB cases, double immunohistochemistry and immunofluorescent microscopy was performed for α-synuclein and tau to characterize the lesions. Although the α-synuclein-positive Lewy-like inclusions were often found in neurons with tau-immunoreactive neurofibrillary pathology, a few LBs were found in neurons with no tau-immunoreactivity (Fig. 2D). When tau and α-synuclein were colocalized, they were either intermingled (Fig. 2E, G–I) or discrete lesions within the same neuron (Fig. 2F, J–L). The discrete inclusions most often appeared to have a central α-synuclein inclusion surrounded by tau-immunoreactive neurofibrillary pathology.
Several cases were also studied with double immunolabeling electron microscopy. The amygdala neurons showed intermingling of 2 morphologically distinct types of filaments: paired helical filaments or straight filaments that were immunolabeled for tau and granule-coated filaments immunolabeled for α-synuclein (Fig. 3). In some neurons, there was separation of the two 2 of filamentous aggregates by other cytoplasmic structures. The α-synuclein-immunoreactive neuronal lesions were morphologically similar to cortical-type LBs.
FIGURE 3.
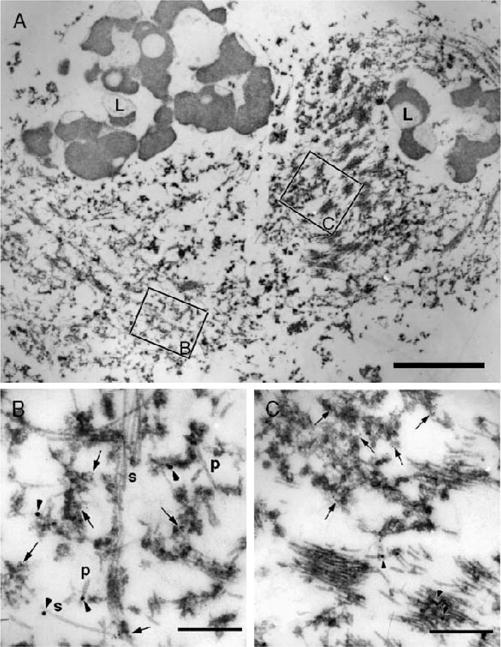
Immunoelectron microscopy with double labeling for phospho-tau (18-nm gold particles) and α-synuclein (5-nm gold particles) in a case of Alzheimer disease with amygdala Lewy bodies. (A) At low magnification, a neuron with separate filamentous aggregates (right) is distinguished from the neuron with intermingled filaments (left). L, lipofuscin. Boxed areas are enlarged at the same magnification in (B) and (C). (B) In the intermingled area, tau filaments (arrowheads) have the morphology of paired helical filaments (P) or straight filaments (S). α-synuclein is localized to granulofilamentous aggregates, arrows). (C) Tightly packed tau filaments (arrowheads) are separate from α-synuclein-positive granulofilamentous aggregates (arrows). The α-synuclein filaments are thinner than tau filaments. Scale bars = (A) 1 μm; (B, C) 300 nm.
Demographic and Pathologic Measures in Alzheimer Disease With Amygdala Lewy Bodies
The demographics and pathologic characteristics of the 62 AD/ALB cases, as well as 198 AD, 52 AD/DLBD, 32 AD/TLBD, and 3 AD/BLBD cases, are summarized in Table 1. The 6 AD cases with inconspicuous α-synuclein pathology are not separated from the rest of the AD group. The age at death, male-to-female ratio, average Braak NFT stage, brain weight, and disease duration did not differ between the groups. The AD/DLBD group had a lower average density of NFTs than the AD, AD/ALB, and AD/TLBD groups, although there were no differences between the groups for average SP density. The AD/BLBD group was not included in this comparison as a result of the small sample size. The APOE ∈4 carrier and the tau H1 haplotype frequencies did not differ between the groups.
TABLE 1.
Clinical and Pathologic Characteristics of Case Material
| AD | AD/ALB | AD/DLBD | AD/TLBD | AD/BLBD | |
|---|---|---|---|---|---|
| (n = 198) | (n = 62) | (n = 52) | (n = 32) | (n = 3) | |
| Age at death (years) | 80 ± 8 | 82 ± 8 | 80 ± 7 | 82 ± 8 | 87 ± 5 |
| Sex ratio (M:F) | 92:106 | 26:36 | 25:27 | 7:25 | 2:1 |
| Braak NFT stage | 5.4 ± 0.7 | 5.6 ± 0.5 | 5.1 ± 0.7 | 5.5 ± 0.6 | 5.3 ± 1.2 |
| Brain weight (g) | 1100 ± 150 | 1110 ± 220 | 1080 ± 173 | 1000 ± 128 | 1040 ± 42 |
| Disease duration (years) | 9.5 ± 4.9 | 10 ± 3.0 | 8.8 ± 5.6 | NA | NA |
| Average NFT count (×400) | 5.5 ± 0.3 | 5.4 ± 0.5 | 3.5 ± 0.6* | 6.5 ± 1.0 | 2.7 ± 1.2 |
| Average SP count (×100) | 41 ± 6 | 42 ± 76 | 40 ± 7 | 42 ± 6 | 41 ± 1 |
| _APOE_∈ carrier frequency (%) | 33 | 39 | 41 | 33 | 50 |
| _MAPT_H1 frequency (%) | 80 | 80 | 80 | 76 | 75 |
Paucity of α-Synuclein Pathology in Subcortical Nuclei in Alzheimer Disease With Amygdala Lewy Bodies
We investigated the presence and severity of α-synuclein pathology in subcortical nuclei, including nbM, SN, LC, and DMV. A score was assessed semiquantitatively in each region based on the overall density of α-synuclein pathology, including LBs and neurites (Fig. 4). The highest α-synuclein score was in the DMN in all groups (AD/ALB 0.5 ± 0.1; AD/TLBD 2.4 ± 0.1; AD/DLBD 2.9 ± 0.05; mean ± standard error of the mean). The α-synuclein score in DMN, LC, SN, and nbM was significantly less in AD/ALB than in all of the other groups. The α-synuclein score was also significantly less in AD/TLBD than in AD/DLBD.
FIGURE 4.
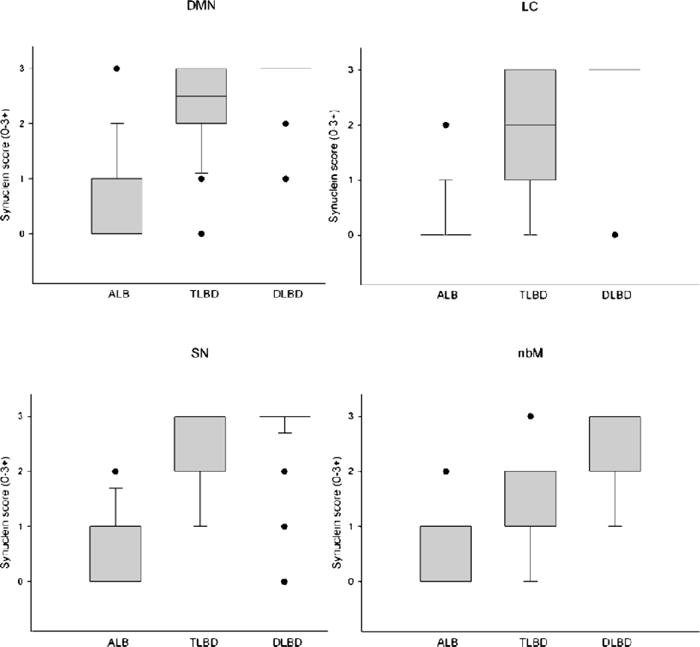
Comparison of α-synuclein score in subcortical regions vulnerable to α-synuclein pathology (DMN, LC, SN, and nbM) for AD/ALB, AD/TLBD, and AD/DLBD. The boxes show median and 25th and 75th percentiles with whisker plots showing 10th and 90th percentiles. The outliers are shown as filled circles. (Median scores for DMN, LC, SN, and nbM are, respectively: AD/ALB 0, 0, 0, 0; AD/TLBD 2.4, 1.7, 2.3, 1.4; and AD/DLBD 2.9, 2.9, 2.8, 2.1.) The α-synuclein scores in DMN, LC, SN and nbM are significantly less in AD/ALB than in AD/TLBD and AD/DLBD (*, p < 0.05). The α-synuclein scores in DMN, LC, SN, and nbM are significantly less in AD/TLBD than in AD/DLBD (#, p < 0.05). DMN, dorsal motor nucleus; nbM, nucleus of Meynert; AD/ALB, Alzheimer disease with amygdala Lewy bodies; AD/TLBD, Alzheimer disease with transitional Lewy body disease; AD/DLBD, Alzheimer disease with diffuse Lewy body disease.
Paucity of Lewy Bodies in Cortical Areas in Alzheimer Disease With Amygdala Lewy Bodies
The density of cortical and amygdala LBs was determined with α-synuclein immunohistochemistry in AD/ALB, AD/TLBD, and AD/DLBD (Fig. 5). The highest LB density was in the amygdala for all groups and the lowest LB density was in the inferior parietal cortex. The LB density in the amygdala was significantly less in AD/ALB than in the other groups (AD/ALB 18 ± 2; AD/TLBD 29 ± 3; AD/DLBD 46 ± 4). The LB density in the amygdala tended to be greater in AD/DLBD than in AD/TLBD. Only ST had rare LBs in AD/ALB. The LB density in neocortical regions (MF and IP) was significantly greater in AD/DLBD than in both AD/TLBD and AD/ALB. The LB density in limbic cortices (ERC and Cing) and the ST gyrus was significantly greater in AD/DLBD than in both AD/TLBD and AD/ALB and significantly greater in AD/TLBD than in AD/ALB.
FIGURE 5.
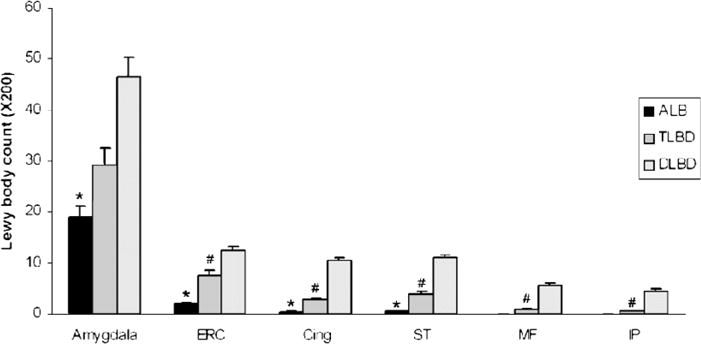
Comparison of Lewy body (LB) density in amygdala and cortical regions for AD/ALB, AD/TLBD and AD/DLBD. Bar charts show mean and error bars show standard errors of the mean. The LB density in the amygdala was significantly less (*) in AD/ALB than in AD/TLBD and AD/DLBD. The LB density in the limbic cortices (ERC and Cing) and ST was significantly less (*) in AD/ALB than in AD/TLBD and AD/DLBD. The LB density in the limbic cortices and neocortical regions (ST, MF, and IP) was significantly less (#) in AD/TLBD than AD/DLBD. AD/ALB, Alzheimer disease with amygdala Lewy bodies; AD/TLBD, Alzheimer disease with transitional Lewy body disease; AD/DLBD, Alzheimer disease with diffuse Lewy body disease; ERC, entorhinal cortex; Cing, anterior cingulate gyrus; ST, superior temporal gyrus; MF, middle frontal gyrus; IP, inferior parietal gyrus.
Neuropathologic Features of Alzheimer Disease With Amygdala Lewy Bodies
Distribution of Lewy Bodies in Alzheimer Disease With Amygdala Lewy Bodies
In this study of 62 cases of AD/ALB, α-synuclein immunostaining of multiple brain regions showed sparse α-synuclein pathology in SN (n = 22), LC (n = 13), DMN (n = 20), Cing (n = 13), ERC (n = 33), and ST (n = 14). Most AD/ALB cases except type 1 had sparse α-synuclein pathology in either brainstem or limbic areas. This α-synuclein pathology was not apparent with hematoxylin and eosin-stained sections (Fig. 6A, B). In contrast, in AD/DLBD, LBs and neuronal loss were readily detected in vulnerable brainstem nuclei with both hematoxylin and eosin stain and α-synuclein immunostaining (Fig. 6C, D).
FIGURE 6.
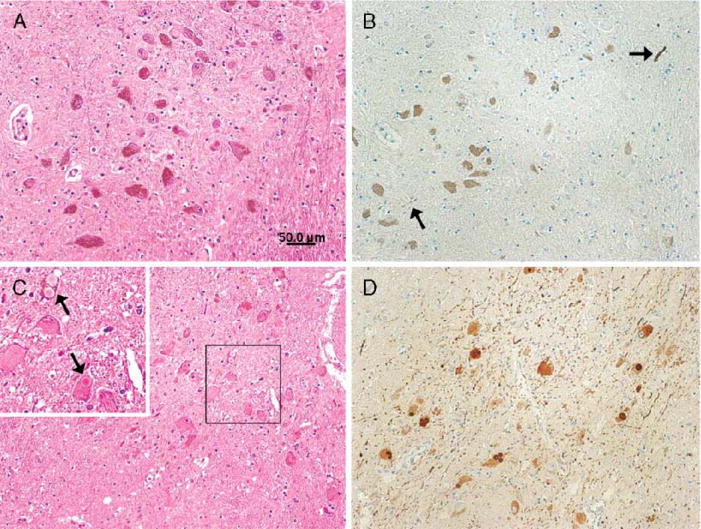
(A) Neuronal population is well preserved in the substantia nigra with hematoxylin and eosin stain. (B) A few Lewy neurites (arrows) are found in the substantia nigra with α-synuclein immunostaining. (C) Neuronal loss with Lewy bodies (LBs) is found in the substantia nigra. Higher magnification (upper left corner, arrows) shows brainstem type LBs clearly. (D) Large amounts of LBs and Lewy neurites are found in the substantia nigra with α-synuclein immunostaining. (A, B) Alzheimer disease with amygdala Lewy bodies case, (C, D) Alzheimer disease with diffuse Lewy body disease case. Scale bars = (A–D) 50 μm.
The AD/ALB cases could be subdivided into 4 types based on the distribution of α-synuclein pathology in brainstem and limbic cortices (type 1−/−, type 2+/−, type 3−/+, and type 4+/+) (Table 2). Without exception, both type 1 and type 3 AD/ALB did not fit the Parkinson disease staging scheme proposed by Braak and coworkers because neither type had brainstem LBs (36). The distribution of α-synuclein pathology in type 2 cases is summarized in Table 3A and that of type 4 in Table 3B. Most type 2 and type 4 AD/ALB cases also failed to fit the Parkinson disease staging scheme (type 2: 80%; type 4: 85%) as a result of the fact that some anatomic regions in brainstem and basal forebrain were unaffected. If all AD/ALB cases are combined, 92% (57 of 62) of AD/ALB did not fit the Parkinson disease staging scheme. In comparison, only 6% (3 of 52) of AD/DLBD and 19% (6 of 32) of AD/TLBD did not fit the Parkinson disease staging scheme, with the brainstem region most often unaffected being the LC. These results are not entirely unexpected given that neuronal loss is severe in the LC even in AD cases without LBD. The present results demonstrate that the distribution of α-synuclein pathology in AD/ALB is different from that in LBD. Type 4 had a significantly greater amygdala LB density than type 1 (Table 2; Fig. 7), but there were no differences in amygdala LB densities between the other groups. Average SP and NFT densities did not differ between the 4 subtypes of AD/ALB.
TABLE 2.
Summary of Lewy Body and Alzheimer-type Pathology in Alzheimer Disease With Amygdala With Lewy Bodies
| α-Synuclein Pathology | Amygdala LBs | Cortical SP | Cortical NFT | ||
|---|---|---|---|---|---|
| Brainstem | Limbic Cortex | ||||
| Type 1 (n = 19) | – | – | 10 ± 1 | 43 ± 0.8 | 5.8 ± 0.6 |
| Type 2 (n = 10) | + | – | 15 ± 2 | 41 ± 1 | 4.8 ± 0.5 |
| Type 3 (n = 13) | – | + | 18 ± 2 | 39 ± 1 | 5.2 ± 0.4 |
| Type 4 (n = 20) | + | + | 30 ± 3* | 43 ± 0.6 | 5.4 ± 0.5 |
TABLE 3.
Distribution of α-Synuclein Pathology
| Cing | ERC | Amygdala | nbM | SN | LC | DMN | PD Stage |
|---|---|---|---|---|---|---|---|
| a. Distribution of α-synuclein pathology in type 2 AD/ALB | |||||||
| − | − | + | + | + | + | + | 4 |
| − | − | ++ | + | + | + | + | 4 |
| − | − | + | − | + | NA | − | 4* |
| − | − | + | − | + | + | NA | 4* |
| − | − | + | + | − | − | + | 4* |
| − | − | + | − | − | − | + | 4* |
| − | − | ++ | − | + | − | − | 4* |
| − | − | ++ | − | − | − | + | 4* |
| − | − | ++ | − | + | − | NA | 4* |
| − | − | ++ | + | + | − | + | 4* |
| b. Distributions of α-synuclein pathology in type 4 AD/ALB | |||||||
| − | + | ++ | + | + | + | + | 4 |
| − | + | ++ | + | + | + | + | 4 |
| − | + | ++ | NA | + | + | + | 4 |
| − | + | + | + | − | + | + | 4* |
| − | + | ++ | − | + | − | − | 4* |
| − | + | ++ | + | + | − | − | 4* |
| − | + | ++ | − | − | − | + | 4* |
| − | + | ++ | + | − | + | + | 4* |
| − | + | ++ | NA | + | − | + | 4* |
| − | + | ++ | − | + | − | NA | 4* |
| NA | + | ++ | + | − | + | + | 4* |
| + | + | + | − | + | − | + | 5* |
| + | + | + | + | + | − | + | 5* |
| + | + | + | − | + | + | ++ | 5* |
| + | + | + | + | + | − | NA | 5* |
| + | + | ++ | − | + | − | − | 5* |
| + | + | ++ | + | − | + | − | 5* |
| + | + | ++ | + | + | − | + | 5* |
| + | + | ++ | − | − | + | + | 5* |
| + | + | ++ | − | + | + | + | 5* |
FIGURE 7.
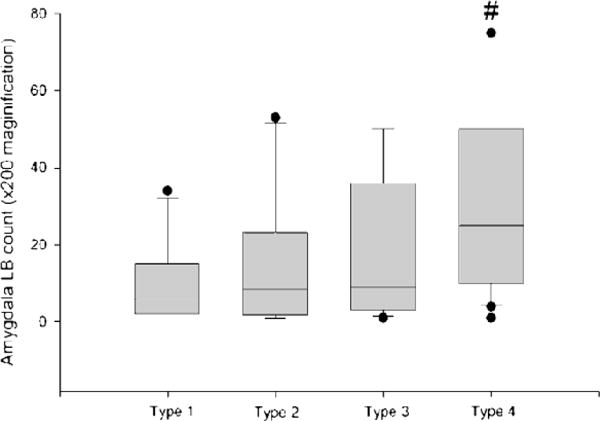
Average amygdala Lewy body counts in 4 subtypes of Alzheimer disease with amygdala Lewy bodies. The boxes show median and 25th and 75th percentiles with whisker plots showing 10th and 90th percentiles. The outliers are shown as filled circles. Type 1 cases have no α-synuclein pathology in the brainstem and limbic cortex. Type 2 cases have α-synuclein pathology only in the brainstem. Type 3 cases have α-synuclein pathology only in the limbic cortex. Type 4 cases have α-synuclein pathology in the brainstem and limbic cortex. #, Type 4 significantly different from type 1. LB, Lewy body.
Correlation Between Amygdala Lewy Bodies, α-Synuclein, and Alzheimer Disease Pathology in Alzheimer Disease With Amygdala Lewy Bodies
The relationship between amygdala LB density and various pathologic measures was assessed with Spearman rank order correlation analysis for all 62 cases of AD/ALB (Table 4). The amygdala LB density in AD/ALB correlated with the density of NFTs, but not SP, in the amygdala, particularly the density of NFTs in the corticomedial zone of the amygdala, which is also the region that consistently shows the highest density of LBs. There was no correlation of amygdala LBs with cortical SPs or NFTs. There was a correlation between amygdala LB density and LBs in ERC, but not for LBs in cingulate gyrus or for α-synuclein scores in brainstem nuclei. These results suggest that amygdala LBs in AD/ALB are more closely linked to concurrent Alzheimer pathology in the amygdala than to brainstem α-synuclein pathology.
TABLE 4.
Correlations Between Amygdala LBs and Pathologic Parameters in Alzheimer Disease With Amygdala Lewy Bodies
| α- Synuclein Score | LBs | Senile Plaques | Neurofibrillary Tangles | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DMN | LC | SN | ERC | Cing | Ctx | Med Amyg | Lat Amyg | Ctx | Med Amyg | Lat Amyg |
| r = 0.24 | r = 0.23 | r = 0.09 | r = 0.41 | r = 0.12 | r = 0.18 | r = 0.14 | r = −0.02 | r = 0.15 | r = 0.44 | r = 0.36 |
| NS | NS | NS | p < 0.01 | NS | NS | NS | NS | NS | p < 0.001 | p < 0.01 |
Clinical Features of Alzheimer Disease With Amygdala Lewy Bodies
We investigated clinical features in available medical records that might be predicted to correlate with amygdala LBs such as psychiatric symptoms, and we also determined the frequency of extrapyramidal signs that might reflect pathology in brainstem nuclei vulnerable to LBs. A series of AD cases matched for age and sex, but without amygdala LBs, was included for comparison. The results are summarized in Table 5. There was no significant difference between AD/ALB and AD for extrapyramidal signs or neuropsychiatric symptoms. The initial presentation of AD/ALB was cognitive impairment (n = 24), depression (n = 8), aggression (n = 1), and delusion (n = 1). Given the lack of standardized data collection and the large amount of missing data, these results need to be interpreted with caution.
TABLE 5.
Clinical Features of AD/ALB Compared With AD
| AD/ALB (n = 62) | AD (n = 52) | Fisher Exact Test | |
|---|---|---|---|
| Extrapyramidal signs | |||
| Rigidity | 5/16 | 12/40 | NS |
| Bradykinesia | 3/13 | 7/37 | NS |
| Tremor | 4/22 | 3/38 | NS |
| Neuropsychiatric symptoms | |||
| Hallucinations | 10/24 | 9/30 | NS |
| Delusions | 15/23 | 12/21 | NS |
| Depression | 17/23 | 15/26 | NS |
| Anxiety | 11/15 | 9/10 | NS |
DISCUSSION
Recognition of Alzheimer Disease With Amygdala Lewy Bodies as a Distinct Type of α-Synucleinopathy
In this study, we describe a type of α-synuclein pathology that is characteristic of advanced AD and unlikely related to that which occurs in the setting of PD and DLB. It is characterized by α-synuclein-immunoreactive neuronal lesions (which resemble cortical-type LBs at the light and electron microscopic level) that are relatively restricted to the amygdala. We have operationally defined this process as AD/ALB. Whether it is related to LBD remains to be further investigated. Among 260 cases of AD that were free of LBD, we found 62 cases with AD/ALB (24%). Although some of these cases had sparse α-synuclein pathology in brainstem or limbic areas, the density and distribution of this pathology was not consistent with a diagnosis of LBD. Most previous studies of α-synuclein pathology in AD have failed to draw attention to this form of LBD, which explains some of the discrepancy between reported frequency of LBs in AD, which range from less than 15% to over 60% (5, 6, 9–12).
The terminology used for LBD in the setting of AD is varied and includes senile dementia of Lewy body type (37), Lewy body variant of AD (LBV) (4), and the common variant of DLBD (38). It seems unlikely that any of these published studies specifically addressed AD/ALB because a diagnosis of LBD would not be immediately suggested in these cases given that they have virtually no LBs and minimal or no neuronal loss in subcortical nuclei that are vulnerable to LBD such as the nbM, SN, and LC.
Frequency of α-Synuclein Pathology in Alzheimer Disease
In this study of 347 cases of AD, α-synuclein pathology was detected in 62 cases of AD/ALB and 149 cases of AD/LBD. In a random screen of 57 AD cases without amygdala LBs, approximately 10% had inconspicuous α-synuclein pathology (mostly scattered Lewy neurites) in brainstem nuclei. It remains to be seen if these cases represent so-called incidental LBD (39). Extrapolation of these findings to the entire cohort of AD cases without amygdala LBs would yield another 20 cases with at least some α-synuclein pathology for a total of 169 cases (49%). Thus, some degree of α-synuclein pathology can be expected in approximately half of AD cases. Whether it has any clinical relevance is questionable for those cases in which it appears to be characterized by sparse, mostly neuritic, α-synuclein pathology in brainstem nuclei or LBs relatively restricted to the amygdala (i.e. AD/ALB).
The frequency of α-synuclein pathology in AD has been reported to range from 20% to 60% (10, 11). The results of the present study are within this range. Arai et al found 13 of 27 (48%) sporadic AD cases had α-synuclein pathology and reported that α-synuclein pathology was seen most frequently in the amygdala (9). Hamilton et al examined 145 AD cases and found 60% with α-synuclein pathology and that the amygdala was involved in all cases (11). In these studies, cases with overt LBD were often included, making it difficult to determine the frequency of AD/ALB from published studies. One thing that is clear from these studies of AD is that the amygdala, not brainstem monoaminergic nuclei, is the most vulnerable region for α-synuclein pathology in AD.
One of the few published studies that is comparable to the present report is that of Jellinger et al who reported that 52% of AD cases (57 of 110) had LB pathology in one or more regions of the brain (10). In that study, 20% (20 of 101) of the AD cases had LBs relatively confined to the amygdala. This result is consistent with the present report, in which the frequency of AD/ALB was 24%.
As noted previously, our findings suggest that up to 50% of AD cases have some degree of α-synuclein pathology, ranging from inconspicuous brainstem neurites to widespread neocortical LBs. Similarly, the frequency of LBs has been reported to be over 60% of familial AD (14) and 50% in Down syndrome with AD pathology (15). These findings are in striking contrast to the frequency of α-synuclein pathology in tauopathies such as progressive supranuclear palsy (PSP) and Pick disease. Recently, we reported that the frequency of α-synuclein pathology in PSP (40) and Pick disease to be 11% and 19%, respectively. These findings suggest an interaction between α-synuclein and AD pathology rather than between α-synuclein and tau. Nevertheless, the present results indicate a stronger correlation, at least in the amygdala, between LBs and NFTs than between LBs and SPs.
Comparison of Alzheimer Disease With Amygdala Lewy Bodies and Alzheimer Disease With Lewy Body Disease
We compared α-synuclein pathology between AD/ALB and AD/LBD to determine if AD/ALB was different from AD/DLBD and AD/TLBD. As a result, both the α-synuclein score in brainstem nuclei and density of LBs in several cortical regions and the amygdala were significantly lower in AD/ALB than in AD/DLBD and AD/TLBD. These results support the notion that AD/ALB is distinct from the more widely recognized AD/LBD subtypes.
The Distribution of Lewy Bodies in Alzheimer Disease With Amygdala Lewy Bodies
In the present study, the immunohistochemical distribution of α-synuclein pathology was investigated in AD/ALB. We found sparse and inconsistent α-synuclein pathology in brainstem and limbic regions in some of the cases. Only the type that had α-synuclein pathology in both brainstem and limbic regions (type 4) was different from the rest and possibly an early stage of AD/LBD (Table 2). Interestingly, the amygdala LB density was also higher in type 4 AD/ALB than in the other types. When all types of AD/ALB are considered together, in most cases (92%), the distribution of LBs in AD/ALB did not fit the Parkinson disease staging scheme recently proposed by Braak and coworkers (36). From these findings, one might propose that α-synucleinopathy in AD/ALB starts in the amygdala and spreads to the entorhinal cortex and brainstem regions, or both, as the disease progresses. The so-called “spread” of pathology in AD/ALB is distinctly different from that described in Parkinson disease, in which α-synucleinopathy is proposed to start in the medulla (36). Thus, AD/ALB appears to show a spread from upper neuraxis to lower regions (“top-down”), whereas Parkinson disease is associated with spread from lower neuraxis to higher regions (“bottom-up”).
In light of this hypothesis, it is of note that a previous study by Marui et al investigated the progression of LB pathology in the cerebrum of DLB and proposed that LB pathology began in the amygdala with subsequent spread to the limbic cortex and higher-order neocortices (17). Unfortunately, these investigators did not include examination of brainstem regions. Recently, this same group reported on AD with amygdala LBs that did not meet the proposed Parkinson disease staging scheme of Braak and coworkers (41). Because their AD cases did not include AD/LBD cases, all AD cases were thought to be AD/ALB cases. In another recent study, Saito et al speculated that α-synucleinopathy pathology associated with AD may begin in the amygdala, although the study was based on a small sample size and was thus underpowered to make conclusive statements (7). Nevertheless, our results support the speculation that AD/ALB is a distinct form of α-synucleinopathy that is seen in advanced AD.
As an aside, we noted that even among the AD/LBD cases included in this study, several did not fit the Parkinson disease staging scheme. This was most often the result of lack of α-synuclein pathology in the LC. A possible explanation for this is the fact that the LC is vulnerable to NFTs and neuronal loss in AD. When neuronal loss is severe, it may preclude detection of LBs. We have noted a similar phenomenon in the SN in PSP with concurrent LBs (40). When the neuronal loss in the SN was very severe, LB density was low or absent in this nucleus.
Correlations Between Lewy Bodies and Alzheimer Disease Pathology in Alzheimer Disease With Amygdala Lewy Bodies
Giasson et al pointed out that tau and α-synuclein may reciprocally promote aggregation in vitro (42). In transgenic models, there have been several reports of a synergistic relationship between both proteins (43). Some studies in autopsied brains also suggest an interaction between LBs and AD pathology (5, 9, 12, 13, 17, 44, 45). It remains to be seen, however, if there is a direct relationship tau and α-synuclein in AD/ALB. Given evidence that LBs frequently occur in the same neurons that contain NFTs (9, 12, 44, 45), this seems possible. Moreover, in the present study, we noted that amygdala LBs correlated with amygdala NFTs but not with LBs or SPs. The strongest correlation was for NFTs in the corticomedial region, which is the amygdala region most vulnerable to both LBs and NFTs. Moreover, some investigators have speculated that AD-related synuclein pathology associated with neurofibrillary lesions in the amygdala is a “secondary” α-synucleinopathy (46).
Arguing against a direct interaction between tau and α-synuclein at the cellular level is evidence from cases with LBs in the setting of a 3R tauopathy (Pick disease) (unpublished data) and a 4R tauopathy (PSP) (40). These studies suggest that LBs in 3R and 4R tauopathies represent an independent disease process that is unrelated to tau pathology. The missing link in this correlation is amyloid, which is present in AD, but not in PSP or Pick disease. This might suggest that it is amyloid rather than tau pathology that leads to the increased frequency of α-synuclein pathology in AD compared with the tauopathies. Of note in this regard is the recent report by Mikolaenko et al who showed that the frequency of α-synuclein pathology increased as the density of neuritic plaques increased (5). In addition, the study by Pletnikova et al concluded that α-amyloid enhanced the development of cortical α-synuclein pathology in PD (47). Support for this hypothesis also comes from studies of transgenic mice that develop enhanced α-synuclein (or tau) pathology when they are engineered to also deposit β-amyloid (48, 49).
Although we could not find any correlation between LBs and SPs in the present study of AD/ALB, it might be related to the fact that the SP counts used in the present study may not be sensitive to amyloid burden. Future studies that more rigorously measure amyloid content with biochemical methods or image analytic methods will be necessary to explore this issue.
APOE and Tau Haplotype in Alzheimer Disease With Amygdala Lewy Bodies
Recently, Tsuang et al reported that the APOE ∈4 allele was associated with the presence of concomitant LBs in AD (50). They referred to their cases as LBV of AD and did not specifically analyze cases that would fall under the category of AD/ALB. Therefore, the present report is the first to investigate the genetic influence, or lack thereof, of tau haplotype and APOE ∈4 allele frequencies in AD/ALB. In support of the idea that AD/ALB is similar to AD, we could find no genetic differences between AD/ALB and AD.
Clinical Features of Alzheimer Disease With Amygdala Lewy Bodies
A number of clinicopathologic studies have compared clinical features between AD and AD/LBD. Lopez et al reported that AD/LBD develop extrapyramidal signs and diurnal hypersomnia earlier and have shorter time to institutionalization than AD, but there were no differences in cognitive or functional decline or survival (51). Heyman et al compared AD and LBV of AD and found LBV of AD to have a greater frequency of extrapyramidal signs (52). Serby et al demonstrated that the co-occurrence of AD and LBD is associated with a greater degree of dementia (53); however, the clinical features of LBD were masked by concurrent AD pathology (54). Because severe cognitive dysfunction and extrapyramidal signs often appear at late stages of AD, it is thought to be difficult to evaluate the clinical features of AD/LBD. In fact, recent neuropathologic criteria have adopted this approach to the diagnosis of DLB with high-likelihood diagnoses only possible in cases with minimal or no Alzheimer-type pathology in the setting of TLBD or DLBD (55).
Given that all the cases in the present study had moderate to marked Alzheimer pathology, it is perhaps not surprising that we were unable to find clinical differences between AD and AD/ALB. We were also unable to find any demographic differences between AD/ALB and AD with respect to age at death, male-to-female ratio, and disease duration. A serious limitation of the present study, however, is the retrospective nature of the cohort under investigation, most of which were derived from the State of Florida Alzheimer Disease Initiative, in which a standardized clinical protocol is not used.
SUMMARY
In conclusion, the present study indicates that AD/ALB is a distinct form of α-synucleinopathy that occurs in the setting of advanced AD with no significant demographic or genetic differences between AD and AD/ALB. Although we were unable to find any clinical differences between AD and AD/ALB, the results need to be interpreted with caution given the retrospective nature of our study design. Future prospective studies will be needed to determine the clinical significance, if any, of AD/ALB.
Acknowledgments
The authors thank Virginia Phillips and Linda Rousseau for their histologic support and Natalie Thomas for genetic studies.
This study was supported by NIH grants P50-AG16574, P01-AG17216, P01-AG14449, P01-AG03949, P50-AG25711, and P50-NS40256; the Mayo Foundation; and State of Florida Alzheimer Disease Initiative.
References
- 1.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology. 1996;47:1113–24. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 2.Dickson DW, Crystal HA, Mattiace LA, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–89. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 3.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–97. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Hansen L, Salmon D, Galasko D, et al. The Lewy body variant of Alzheimer’s disease: a clinical and pathologic entity. Neurology. 1990;40:1–8. doi: 10.1212/wnl.40.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Mikolaenko I, Pletnikova O, Kawas CH, et al. Alpha-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: evidence from the Baltimore Longitudinal Study of Aging (BLSA) J Neuropathol Exp Neurol. 2005;64:156–62. doi: 10.1093/jnen/64.2.156. [DOI] [PubMed] [Google Scholar]
- 6.Parkkinen L, Soininen H, Alafuzoff I. Regional distribution of alpha-synuclein pathology in unimpaired aging and Alzheimer disease. J Neuropathol Exp Neurol. 2003;62:363–67. doi: 10.1093/jnen/62.4.363. [DOI] [PubMed] [Google Scholar]
- 7.Saito Y, Kawashima A, Ruberu NN, et al. Accumulation of phosphorylated alpha-synuclein in aging human brain. J Neuropathol Exp Neurol. 2003;62:644–54. doi: 10.1093/jnen/62.6.644. [DOI] [PubMed] [Google Scholar]
- 8.Dickson DW. Alpha-synuclein and the Lewy body disorders. Curr Opin Neurol. 2001;14:423–32. doi: 10.1097/00019052-200108000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Arai Y, Yamazaki M, Mori O, et al. Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation. Brain Res. 2001;888:287–96. doi: 10.1016/s0006-8993(00)03082-1. [DOI] [PubMed] [Google Scholar]
- 10.Jellinger KA. Alpha-synuclein pathology in Parkinson’s and Alzheimer’s disease brain: incidence and topographic distribution—a pilot study. Acta Neuropathol (Berl) 2003;106:191–201. doi: 10.1007/s00401-003-0725-y. [DOI] [PubMed] [Google Scholar]
- 11.Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–84. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marui W, Iseki E, Ueda K, et al. Occurrence of human alpha-synuclein immunoreactive neurons with neurofibrillary tangle formation in the limbic areas of patients with Alzheimer’s disease. J Neurol Sci. 2000;174:81–84. doi: 10.1016/s0022-510x(99)00327-5. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt ML, Martin JA, Lee VM, Trojanowski JQ. Convergence of Lewy bodies and neurofibrillary tangles in amygdala neurons of Alzheimer’s disease and Lewy body disorders. Acta Neuropathol (Berl) 1996;91:475–81. doi: 10.1007/s004010050454. [DOI] [PubMed] [Google Scholar]
- 14.Lippa CF, Fujiwara H, Mann DM, et al. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–70. doi: 10.1016/s0002-9440(10)65722-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lippa CF, Schmidt ML, Lee VM, et al. Antibodies to alpha-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann Neurol. 1999;45:353–57. doi: 10.1002/1531-8249(199903)45:3<353::aid-ana11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 16.Rezaie P, Cairns NJ, Chadwick A, et al. Lewy bodies are located preferentially in limbic areas in diffuse Lewy body disease. Neurosci Lett. 1996;212:111–14. doi: 10.1016/0304-3940(96)12775-0. [DOI] [PubMed] [Google Scholar]
- 17.Marui W, Iseki E, Nakai T, et al. Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci. 2002;195:153–59. doi: 10.1016/s0022-510x(02)00006-0. [DOI] [PubMed] [Google Scholar]
- 18.Harding AJ, Broe GA, Halliday GM. Visual hallucinations in Lewy body disease relate to Lewy bodies in the temporal lobe. Brain. 2002;125:391–403. doi: 10.1093/brain/awf033. [DOI] [PubMed] [Google Scholar]
- 19.Hopper MW, Vogel FS. The limbic system in Alzheimer’s disease. A neuropathologic investigation. Am J Pathol. 1976;85:1–20. [PMC free article] [PubMed] [Google Scholar]
- 20.Mann DM, Yates PO, Marcyniuk B, et al. The topography of plaques and tangles in Down’s syndrome patients of different ages. Neuropathol Appl Neurobiol. 1986;12:447–57. doi: 10.1111/j.1365-2990.1986.tb00053.x. [DOI] [PubMed] [Google Scholar]
- 21.Arnold SE, Hyman BT, Flory J, et al. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1:103–16. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 22.Vereecken TH, Vogels OJ, Nieuwenhuys R. Neuron loss and shrinkage in the amygdala in Alzheimer’s disease. Neurobiol Aging. 1994;15:45–54. doi: 10.1016/0197-4580(94)90143-0. [DOI] [PubMed] [Google Scholar]
- 23.Tsuchiya K, Kosaka K. Neuropathological study of the amygdala in presenile Alzheimer’s disease. J Neurol Sci. 1990;100:165–73. doi: 10.1016/0022-510x(90)90029-m. [DOI] [PubMed] [Google Scholar]
- 24.Arriagada PV, Growdon JH, Hedley-Whyte ET, et al. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–69. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 25.Zald DH. The human amygdala and the emotional evaluation of sensory stimuli. Brain Res Brain Res Rev. 2003;41:88–123. doi: 10.1016/s0165-0173(02)00248-5. [DOI] [PubMed] [Google Scholar]
- 26.Rolls E. Neurophysiology and functions of the primate amygdala. In: Aggleton JP, editor. The Amygdala. New York: Wiley-Liss; 1992. pp. 143–65. [Google Scholar]
- 27.Barker WW, Luis CA, Kashuba A, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002;16:203–12. doi: 10.1097/00002093-200210000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Khachaturian ZS. Diagnosis of Alzheimer’s disease. Arch Neurol. 1985;42:1097–105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 29.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 30.Gloor P. The amygdala system. In: Gloor P, editor. The Temporal Lobe and Limbic System. New York: Oxford University Press; 1997. pp. 591–721. [Google Scholar]
- 31.Gwinn-Hardy K, Mehta ND, Farrer M, et al. Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol (Berl) 2000;99:663–72. doi: 10.1007/s004010051177. [DOI] [PubMed] [Google Scholar]
- 32.Uchikado H, Delledonne A, Uitti R, Dickson DW. Coexistence of PSP and MSA: A case report and review of the literature. Acta Neuropathol (Berl) 2006;111:186–92. doi: 10.1007/s00401-005-0022-z. [DOI] [PubMed] [Google Scholar]
- 33.Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am J Pathol. 2003;162:213–18. doi: 10.1016/S0002-9440(10)63812-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crook R, Hardy J, Duff K. Single-day apolipoprotein E genotyping. J Neurosci Methods. 1994;53:125–27. doi: 10.1016/0165-0270(94)90168-6. [DOI] [PubMed] [Google Scholar]
- 35.Baker M, Litvan I, Houlden H, et al. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:7111–15. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- 36.Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 37.Perry RH, Irving D, Blessed G, et al. Senile dementia of Lewy body type. A clinically and neuropathologically distinct form of Lewy body dementia in the elderly. J Neurol Sci. 1990;95:119–39. doi: 10.1016/0022-510x(90)90236-g. [DOI] [PubMed] [Google Scholar]
- 38.Kosaka K. Diffuse Lewy body diseases in Japan. J Neurol. 1990;237:197–204. doi: 10.1007/BF00314594. [DOI] [PubMed] [Google Scholar]
- 39.Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745–52. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uchikado H, DelleDonne A, Ahmed Z, Dickson DW. Lewy bodies in progressive supranuclear palsy represent an independent disease process. J Neuropathol Exp Neurol. 2006;65:387–95. doi: 10.1097/01.jnen.0000218449.17073.43. [DOI] [PubMed] [Google Scholar]
- 41.Marui W, Iseki E, Kato M, et al. Pathological entity of dementia with Lewy bodies and its differentiation from Alzheimer’s disease. Acta Neuropathol (Berl) 2004;108:121–28. doi: 10.1007/s00401-004-0869-4. [DOI] [PubMed] [Google Scholar]
- 42.Giasson BI, Forman MS, Higuchi M, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–40. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 43.Geddes JW. Alpha-synuclein: A potent inducer of tau pathology. Exp Neurol. 2005;192:244–50. doi: 10.1016/j.expneurol.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 44.Iseki E, Marui W, Kosaka K, et al. Frequent coexistence of Lewy bodies and neurofibrillary tangles in the same neurons of patients with diffuse Lewy body disease. Neurosci Lett. 1999;265:9–12. doi: 10.1016/s0304-3940(99)00178-0. [DOI] [PubMed] [Google Scholar]
- 45.Iseki E, Takayama N, Marui W, et al. Relationship in the formation process between neurofibrillary tangles and Lewy bodies in the hippocampus of dementia with Lewy bodies brains. J Neurol Sci. 2002;195:85–91. doi: 10.1016/s0022-510x(01)00689-x. [DOI] [PubMed] [Google Scholar]
- 46.Saito Y, Ruberu NN, Sawabe M, et al. Lewy body-related alpha-synucleinopathy in aging. J Neuropathol Exp Neurol. 2004;63:742–49. doi: 10.1093/jnen/63.7.742. [DOI] [PubMed] [Google Scholar]
- 47.Pletnikova O, West N, Lee MK, et al. Abeta deposition is associated with enhanced cortical alpha-synuclein lesions in Lewy body diseases. Neurobiol Aging. 2005;26:1183–92. doi: 10.1016/j.neurobiolaging.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 48.Masliah E, Rockenstein E, Veinbergs I, et al. Beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98:12245–50. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lewis J, Dickson DW, Lin WL, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–91. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 50.Tsuang DW, Wilson RK, Lopez OL, et al. Genetic association between the APOE*4 allele and Lewy bodies in Alzheimer disease. Neurology. 2005;64:509–13. doi: 10.1212/01.WNL.0000150892.81839.D1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopez OL, Wisniewski S, Hamilton RL, et al. Predictors of progression in patients with AD and Lewy bodies. Neurology. 2000;54:1774–79. doi: 10.1212/wnl.54.9.1774. [DOI] [PubMed] [Google Scholar]
- 52.Heyman A, Fillenbaum GG, Gearing M, et al. Comparison of Lewy body variant of Alzheimer’s disease with pure Alzheimer’s disease: Consortium to establish a registry for Alzheimer’s disease, part XIX. Neurology. 1999;52:1839–44. doi: 10.1212/wnl.52.9.1839. [DOI] [PubMed] [Google Scholar]
- 53.Serby M, Brickman AM, Haroutunian V, et al. Cognitive burden and excess Lewy-body pathology in the Lewy-body variant of Alzheimer disease. Am J Geriatr Psychiatry. 2003;11:371–74. [PubMed] [Google Scholar]
- 54.Merdes AR, Hansen LA, Jeste DV, et al. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology. 2003;60:1586–90. doi: 10.1212/01.wnl.0000065889.42856.f2. [DOI] [PubMed] [Google Scholar]
- 55.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]