Crystal Structure of a Thermally Stable Rhodopsin Mutant (original) (raw)
. Author manuscript; available in PMC: 2008 Oct 5.
Published in final edited form as: J Mol Biol. 2007 Mar 12;372(5):1179–1188. doi: 10.1016/j.jmb.2007.03.007
Abstract
We determined the structure of the rhodopsin mutant N2C/D282C expressed in mammalian cells; the first structure of a recombinantly produced G protein-coupled receptor (GPCR). The mutant was designed to form a disulfide bond between the N-terminus and loop E3 which allows handling of opsin in detergent solution and increases thermal stability of rhodopsin by 10°C. It furthermore allowed us to crystallize a fully deglycosylated rhodopsin (N2C/N15D/D282C). N15 mutations are normally misfolding and cause retinitis pigmentosa in humans. Microcrystallographic techniques and a 5μm x-ray beam were used to collect data along a single needle measuring 5x5x90μm3. The disulfide introduces only minor changes but fixes the N-terminal cap over the β-sheet lid covering the ligand binding site, a likely explanation for the increased stability. This work allows structural investigation of rhodopsin mutants and shows the problems encountered during structure determination of GPCRs and other mammalian membrane proteins.
Introduction
Transmission of signals across the plasma membrane is fundamental for all multicellular organisms. The largest group of membrane proteins involved in this process is the G protein-coupled receptors (GPCRs). Not surprisingly this class of membrane proteins contains many major drug targets 1, including those against cardiovascular and gastrointestinal diseases, central nervous system and immune disorders, and cancer.
Despite the physiological and medical relevance of this superfamily of receptors, to date there exist crystal structures for only one GPCR, bovine rhodopsin. Detailed molecular models have been obtained for rhodopsin from x-ray diffraction data of 3D crystals with P41 2; 3 and P31 symmetry 4. There are a number of reasons that account for the success of rhodopsin in these studies. First, rhodopsin is arguably the best characterized GPCR, as studies of it date back over 100 years and provide a rich biochemical literature. Second, the covalently bound chromophore, 11-cis-retinal, significantly enhances the stability of rhodopsin in the ground state such that a wide range of detergents may be used to solubilize the protein without loss of activity. This allows a greater range of crystallization conditions to be explored and increases the chances of obtaining good crystals suitable for high resolution structure determination. Third, and perhaps most importantly, rhodopsin is easily isolated in large quantities (>100 mg) from a naturally occurring source, bovine retina. These characteristics differentiate rhodopsin from all other GPCRs.
The absence of high-abundance sources is a acute problem because GPCRs are difficult to express in functional form in bacterial systems. GPCRs generally express well in mammalian cell-culture systems, but generation of the milligram quantities required for crystallization trials with these systems is expensive, labor intensive, and the solubilized proteins can be unstable. In addition, post-translational modification, in particular glycosylation, of the recombinant GPCRs from mammalian cells is often highly heterogeneous. As a consequence, only a few examples of GPCRs expressed and purified in functional form in sufficient amounts for crystallization studies have been reported 5; 6.
These same limitations apply as well to recombinant rhodopsin. Thus, rhodopsin has not been successfully expressed in bacterial systems. It has been expressed in mammalian cell-culture systems 7; 8, but the expression levels are low and the purified protein can be highly heterogeneous with respect to its oligosaccharyl chains. A further complication is that the cultured cells do not produce 11-cis-retinal, which must be supplied exogenously before solubilization as the apoprotein form opsin is not stable in detergent solution. Therefore, while it is true that rhodopsin has been crystallized and its structure determined by x-ray diffraction, it is also true that this work has been restricted to the naturally occurring wild-type protein and has not been open to the large number of interesting mechanism- and disease-related mutants.
We present here the crystal structure of a recombinant rhodopsin, isolated from mammalian cells heterologously expressing the protein in culture. The recombinant protein was a mutant form of rhodopsin containing an engineered disulfide bond between Cys residues at positions 2 and 282 in the polypeptide chain. The double mutant N2C/D282C was initially designed to replace the hydrogen bond between Asn 2 and Asp 282 without changing the overall fold of the protein. This disulfide has been shown to increase thermal stability of opsin in detergent solution 9. The mutant protein was expressed transiently in COS cells, combined with retinal chromophore, and then purified for use in crystallization trials. The resulting crystals were too small to be used at standard synchrotron sources but were suitable for the micro-focusing optics of the ID13 beamline at the ESRF in Grenoble. The structure, solved by molecular replacement, is the first for any recombinant GPCR and opens the way for structural investigation not only of rhodopsin mutants but of other GPCRs expressed heterologously in cultured mammalian cells as well.
Results
Preliminary Studies
Initially, three recombinant constructs of rhodopsin were screened for use in crystallization trials: the wild-type protein and two mutants, N2C/D282C and N2C/N15D/D282C. The N2C/D282C double mutant is a thermally stable mutant containing an engineered disulfide between the introduced Cys residues at positions 2 and 282, as described previously 9. The N2C/N15D/D282C triple mutant was made to remove both N-linked glycosylation sites (Asn 2 and Asn 15) found in rhodopsin. Our interest in removing glycosylation sites stemmed from concern over the possible adverse effect of carbohydrate heterogeneity on ability of the protein to crystallize. When rhodopsin is expressed in and purified from transfected cells in culture the protein runs on SDS-PAGE gels as a smear due to heterogeneous glycosylation. Although it is well known that Asn2 can be substituted without effect on the stability or activity of the protein, mutations of Asn15 are not tolerated 10. In fact, mutations that disrupt the Asn15 glycosylation site are found in patients with retinitis pigmentosa, and Asn15 mutants from transfected COS cells cannot fold properly and do not bind the 11-cis-retinal chromophore 11. To test if an Asn15 mutation might be tolerated in the context of the stabilizing N2C/D282C disulfide the N2C/N15D/D282C triple mutant was constructed. N2C/N15D/D282C expresses reasonably well in transfected COS cells (to a level about 50% of that of the wild-type protein and the N2C/D282C double mutant), displays a wild-type absorption spectrum, and has a specific activity for light-dependent activation of transducin that is indistinguishable from that of the N2C/D282C mutant (data not shown). Most importantly, the heterogeneity seen on SDS-PAGE gels is absent in the N2C/N15D/D282C triple mutant, and the protein runs as a single homogeneous band (Figure 1_A_).
Figure 1. Western blot of rhodopsin mutants and three-dimensional crystals of recombinant rhodopsins.

A: The proteins were expressed and purified from transfected COS cells. Primary antibody used in the blot was 1D4. Lane 1: The N2C/D282C mutant has lost the carbohydrate chain on residue 2 but retains that attached to residue 15. Lane 2: The fully deglycosylated mutant N2C/N15D/D282C shows a single homogeneous band. Mutations in position 15 are known to cause autosomal dominant retinitis pigmentosa in humans. Photos from crystals of recombinant rhodopsins were taken under infrared illumination (>850nm). B: Crystals of recombinant wild type rhodopsin. C: Crystals of thermally stabilized rhodopsin N2C/D282C. D: Crystals of stabilized fully deglycosylated rhodopsin N2C/N15D/D282C.
In previous work only the apoprotein or opsin form of the mutant was examined for enhanced stability 9, but it seemed likely that the holoprotein or rhodopsin form possessed increased thermal stability as well. This indeed turned out to be the case as is shown in Figure 2 where the thermal stability of the N2C/D282C mutant from COS cells is compared with that of native rhodopsin from bovine retina. Both proteins were purified in the presence of the detergent n-octyotetraoxyethylene (C8E4), chosen for these studies because it is a relatively harsh detergent used in the final steps of purification of the proteins for crystal trials. The proteins were then incubated for 30 min at varying temperatures and the remaining pigment determined by absorption spectroscopy. The temperature at which 50% of the bound retinal is lost (T50) was ∼10°C higher for the N2C/D282C mutant (47.2°C) than that of the native control (37.3°C). In the presence of 50 mM β-mercaptoethanol the T50 value for the mutant drops to 41.3°C whereas the wild type is unaffected (37.5°C). The higher stability of the reduced mutant, compared to the wild-type, implies that reduction of the disulfide has not been complete under these conditions. Most of the engineered disulfide bonds are however readily reduced and, once reduced, do not longer contributes to the increased stability of the mutant. In contrast, the native disulfide between Cys residues 110 and 187, which is present in both the mutant and the wild-type and is important for stability of the wild type 12, is apparently not reduced under these conditions. In total, these data show that the engineered disulfide significantly increases stability not only of the apoprotein opsin but of the holoprotein rhodopsin as well.
Figure 2. Thermal stability of N2C/D282C rhodopsin in the detergent C8E4 used for crystallization.
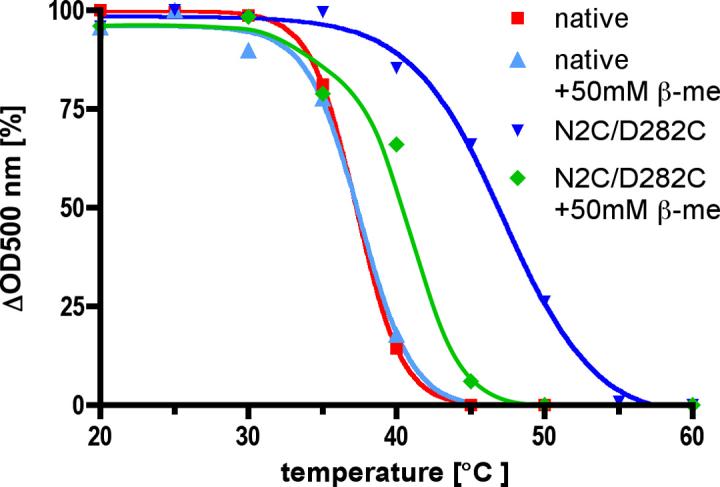
The N2C/D282C mutant was purified by 1D4 immunoaffinity chromatography. Rhodopsin solubilized from retinas (native) was purified by Con A affinity chromatography. In both cases a Sephadex G50 column was used to exchange the detergent from 0.02% (w/v) DDM to 0.2% (w/v) C8E4 immediately before the experiment. The protein solutions were incubated for 30 minutes at various temperatures before an absorption spectrum has been recorded. The amount of bound retinal was determined by the absorption at 500 nm, corrected by the protein absorption at 280 nm. To investigate the effect of reduced disulfide bonds the same experiment was performed in the presence of 50 mM β-mercaptoethanol (β-me). Using the program Prism sigmoidal curves were fitted to the individual data points. The T50 values obtained from these curves (wt: 37.3°C., wt+β-me: 37.5°C., N2C/D282C: 47.2°C., N2C/D282C+β-me: 41.3°C.) show a ∼10°C. increase in stability of the mutant which is mostly lost in the presence of β-mercaptoethanol.
Each of the proteins, wild type, N2C/D282C, and N2C/N15D/D282C, were expressed in transfected COS cells. Typically, the yield following immunoaffinity purification was about 2.5 mg rhodopsin from 50 15-cm plates of COS cells. The final yield from a typical preparation used for crystallization was ∼0.6 mg of rhodopsin with a protein/chromophore ratio (OD280/OD500) of 1.65. All three proteins produced up to 100 μm long needle-like crystals (Figure 1 B to D) under conditions similar to the ones established previously for native rhodopsin isolated from bovine retina 13, but only crystals of wild type and the N2C/D282C mutant were found to diffract. This was surprising given the fact that both wild type and N2C/D282C are considerably more heterogenous as a result of their oligosaccharyl chains than is the N2C/N15D/D282C triple mutant. While both wild-type and N2C/D282C crystals did diffract, the double mutant crystals diffracted to higher resolution and the patterns were more isotropic than for the wild type. For these reasons, further efforts were focused exclusively on the stabilized N2C/D282C mutant.
Data collection and Processing
Due to their small size, diffraction data from rhodopsin crystals required the microdiffractometer at the ESRF, Grenoble 14; 15. In this setup the x-ray beam is guided and focused through a central bore in a co-axial objective, followed by a beam defining aperture (4 μm), a guard aperture (100 μm) and a guard metal capillary. Together with slits in the focusing mirror system this resulted in a clean focused beam spot of 5 μm with minimal tails and a flux of 1011 photons/s. This was essential to reduce beam tail induced radiation damage, necessary to preserve high-quality diffraction from neighboring positions on the microcrystalline needle. To further reduce background from air scattering of the primary beam, a final metal guard tube and the beamstop were placed very close to the crystal. The adjusted beam size matched the width of the crystalline needles to ensure maximal beam intensity on the crystal volume with minimal additional background scattering from the surrounding solvent. The possibility to position the beam with the microdiffractometer setup allowed us to place the 5 μm beam on specific locations on a microcrystalline needle measuring 5×5×90 μm3 (Fig. 3_A_). The diffraction qualities of initial crystals obtained with wild-type, N2C/D282C and N2C/N15D/D282C recombinant rhodopsin were compared. Only crystals obtained with wild-type and N2C/D282C rhodopsin showed promising diffraction, with the crystals containing the opsin stabilizing disulfide bond diffracting more isotropically to higher resolution.
Figure 3. Data collection on a recombinant microcrystal.
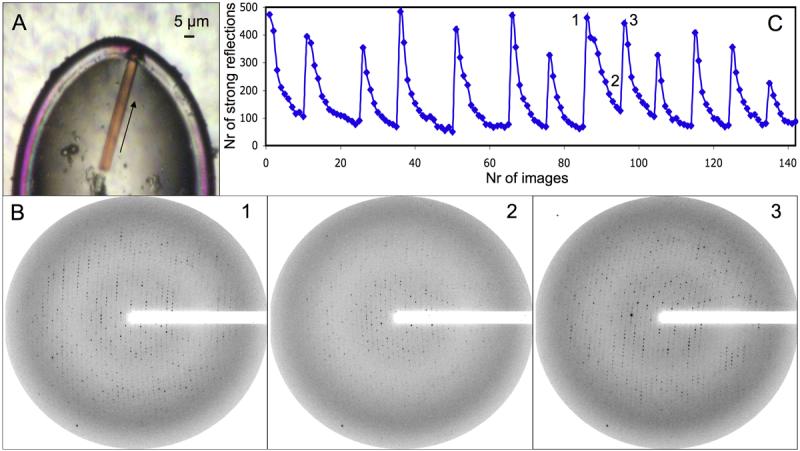
A: Frozen microcrystal used for data collection. The arrow represents the axis along which the data was collected on 15 distinct positions. B: Diffraction pattern obtained after 15 s exposure and 1° rotation. 1: The first diffraction pattern collected at a new position on the needle. 2: The eighth consecutive frame collected at the same position. A strong reduction in diffraction intensity caused by radiation damage is observed. 3: Fully restored diffraction intensity is obtained after the beam had been translated along the needle axis. C: Diffraction intensity represented by the number of strong spots (NSTRONG, XDS) on each frame of the 13 indexed wedges. At each minimum in the number of strong spots the beam was translated to a new position on the needle axis. The positions of diffraction patterns shown in B are marked.
The diffraction limit achieved with the microcrystallographic setup was ∼3.5 Å after 15 s exposure (Fig. 3_B_) in about 10% of the recombinant crystals. This success rate is roughly ten times higher than with crystals previously grown from native rhodopsin isolated from the retina.
The long exposure times needed to produce sufficient diffraction however reduced the resolution to less then 5 Å after about 8 collected frames (Fig. 3_B_). We solved this problem by progressively translating the crystalline needle in the microbeam, successively exposing small sections. This fully restored diffraction after most translations (Fig. 3_B_ and C) and allowed us to collect 15 wedges each containing 5-15 degrees rotation from a single crystal with a length of 90 μm. The data obtained from these wedges varied in quality and two of the fifteen wedges were not good enough to be indexed correctly. The remaining 13 wedges were indexed and showed no significant differences in unit cell (a=b=109.3, c=77.7) and symmetry (P31). The 13 indexed wedges however varied in their overall intensity and quality as apparent from the varying number of strong spots on the first frames (Fig. 3_C_). These quality variations appeared because in some of the wedges we did not fully hit the needle or did not translate the beam far enough from the previous position.
As a guide to which data to use, we visually investigated the frames for radiation damage and then tried different merging combinations to optimize completeness and redundancy without including too many frames with strong radiation damage. The quality of the merging strategies was furthermore judged by the quality of the electron density maps obtained after molecular replacement, which improved even when frames with several times reduced intensity were included. In the case of our data, in which the radiation damaged frames are equally distributed, the gain obtained with higher redundancy and completeness thus made it worth including frames which showed a strong decrease in diffraction. The final merge included data from 10 wedges with 8-10 frames each, yielding a consistent and complete dataset (Table 1) sufficient for structure determination.
Table 1.
Data collection and refinement statistics. Data was collected at ID13, ESRF Grenoble
| Data collection and refinement of N2C/D282C rhodopsin | |
|---|---|
| Data collection | |
| Space group | P31 |
| Cell dimensions | |
| a, b, c (Å) | 109.3, 109.3, 77.7 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 50-3.4 (3.5-3.4) |
| _R_merge | 0.24 (0.703) |
| I / s_I_ | 5.42 (1.79) |
| Completeness (%) | 95.9 (92.3) |
| Redundancy | 3.0 (2.6) |
| Molecular Replacement with Monomer A of 1GZM | |
| R-factor | 0.455 |
| Correlation coefficient | 0.344 |
| Refinement | |
| Resolution (Å) | 50-3.4 |
| No. reflections | 13282 |
| _R_work / _R_free | 0.287/0.325 |
| No. atoms | |
| Protein | 5178 |
| Ligand/ion | 67 |
| _B_-factors | 67 |
| Protein | 69 |
| Ligand/ion | 33 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.014 |
| Bond angles (°) | 1.66 |
Structure determination
The N2C/D282C structure was solved by molecular replacement using the coordinates of monomer A from the 2.65 Å resolution structure of rhodopsin 4 as a search model. The coordinates were modified before use by changing the Asp residues at positions 2 and 15 to Ala and removing the attached carbohydrate chains and the retinal chromophore. The initial rotation and translation searches gave a unique solution with good statistics (Table 1). Phases were improved by solvent flattening, rigid body refinement and density modification, and the resulting map showed continuous density for most of the protein chain. The electron density map shows obvious density for carbohydrate at position 15 and for the retinal chromophore despite the fact that both were omitted from the search model (Fig. 4_A_). Furthermore, no density is visible for carbohydrate adjacent to the mutated residue 2 where the N-glycan would have been in wild-type rhodopsin, as is shown in Figure 4_B_ for the electron density map obtained after refinement. Significantly, there is new density in this map corresponding to the engineered disulfide bond between the introduced Cys residues at positions 2 and 282. These data confirmed that the solution obtained from molecular replacement was correct. The model was completed and manually fitted to the electron density prior to further refinement. After multiple rounds of building and refinement the model converged with an R-factor of 0.287 and a free R-factor of 0.325. The geometry is in close agreement with ideal values (Table 1) and 97.4 % of the residues are in the allowed regions of the Ramachandran plot.
Figure 4. Electron density maps of N2C/D282C rhodopsin.
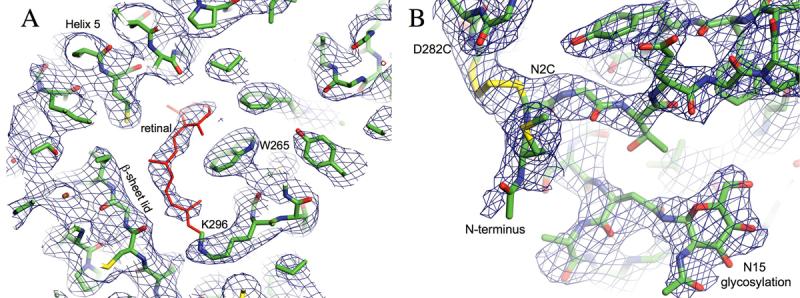
A: Retinal Omit electron density map (0.75 sigma) obtained after molecular replacement and density modification. The retinal (red) was not included but appears clearly in the electron density. B: Disulfide bond and N15 glycosylation. Sigma-A weighted 2Fo-Fc Electron density contoured at sigma 0.9. The disulfide bond between the mutated residues C2 and C282 is well resolved, as is the first residue of the N-glycosylation linked to N15. No density is visible adjacent to the mutated residue 2, which would have been glycosylated in wild-type rhodopsin.
Packing within the Unit Cell
Packing of native bovine rhodopsin in the trigonal crystal form has been discussed previously 13; 16. Briefly, the two rhodopsin dimers in the asymmetric unit are related by a nearly perfect 2-fold non-crystallographic symmetry (NCS) axis and have antiparallel contacts along the entire length of helix 5. These antiparallel dimers are stacked according to the 31 screw symmetry, forming protein columns parallel to the c-axis of the unit cell. The protein columns are arranged around a solvent filled channel along the long axis of the crystalline needles. Crystal contacts mainly involve transmembrane helices 1, 4 and 5, and the packing is thus remarkably hydrophobic. The hydrophilic loops and the termini, including the glycosylation sites, are located in the solvent filled channel, virtually undisturbed by crystal contacts.
Packing of the recombinant N2C/D282C mutant is different, as the antiparallel interface along helix 5 of two monomers in the non-crystallographic dimer is shifted 1.5 helix turns with respect to the 1GZM structure (Fig. 5_A_). The two helices, 5 and 5′, are thus in contact over their whole length and not offset as much as in the 1GZM structure. This leads to a rotation of monomers within the asymmetric unit widening the solvent-filled channel and increasing axes a and b of the unit cell by 5.5 Å. It is unlikely that this difference in dimer arrangement and crystal packing is due to the mutations or altered glycosylation pattern in the protein as the sites of these modifications are far from the dimer interface or location of crystal contacts. A more likely explanation is that the predominance of hydrophobic contacts within this particular crystal form, as a consequence of low specificity, allows for variations in crystal packing. Indeed, variation in unit cell dimensions were noted previously for native rhodopsin in this same crystal form 13.
Figure 5. Comparison of recombinant and native rhodopsin.
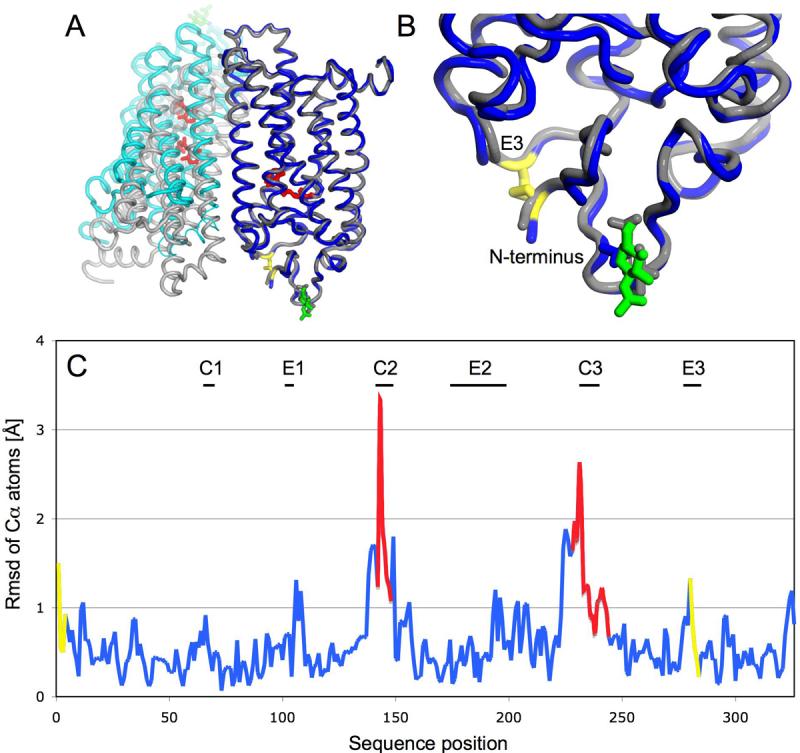
A: Superposition emphasizing overall similarity of the individual monomers and their different dimer packing along helix 5. Blue: Monomer A, Cyan: Monomer B, Grey: native rhodopsin monomers A and B obtained from 1GZM coordinates, Yellow: Designed disulfide bond between C2 and C282, Red:retinal, Green: N15 linked glycosylation B: Enlargement of the extracellular region with the introduced disulfide bond. C: Coordinate differences between monomers A of both structures plotted as the root mean square deviation (rmsd) of Cα atoms against sequence position (blue). Poorly ordered regions with a B-factor higher than 100 are marked red. The designed disulfide includ ing two amino acids on each side is labeled yellow. The largest differences are found in loops C2 and C3 and are due to the inherent flexibility and poor order of these loops. The end of helix 5 before the C3 loop is shifted about 2 Å due to the different dimer packing. The disulfide is introducing small but significant changes in the backbone of the E3 loop and the N-terminus.
Structure of the N2C/D282C Mutant
The refined structure of N2C/D282C contains with 326 of the 348 residues most of the polypeptide chain. NCS restraints were applied throughout the refinement, and both monomers of the asymmetric unit are highly similar as demonstrated by their low average Cα root mean square (rms) deviation of 0.143 Å. Post-translational modifications are observed in both molecules, including acetylation of the amino terminus, N-linked glycosylation of Asn15, and attachment of the 11-cis-retinal through Schiff base linkage to Lys296. Palmitoylation of C322 and C323 could not be observed, probably due to the high flexibility of these modifications and the limited resolution of the data. All seven of the transmembrane helices, the amphipathic helix 8 and most of the loops are well defined, whereas the C2 and C3 loops known to be highly flexible from the wildtype structure 4 are not.
The electron density map shows clear density for the engineered 2-282 disulfide and surrounding region in both molecules of the dimer, although it is better defined in monomer A (Fig. 4_B_). While the N2C mutation removed one glycosylation site, and as noted above there is no density for the carbohydrate at this position, the second oligosaccaryl chain attached to Asn15 is clearly evident in the map with strong density for the first sugar residue and partial density for the second. In addition, other distinctive features of the rhodopsin structure in this region are well defined, notably the N-terminal domain and three extracellular loops. The short E3 loop contains only 8 amino acids, including the Cys282 mutation, and connects transmembrane helices 6 and 7. The N-terminal 36 amino acids form an independent domain making multiple contacts with the E1 and E2 loops as well as the ends of helices 1 and 3 burying 974 Å2 of surface area in the process (Fig. 6). The N-terminal domain appears to function predominantly as a cap for the E2 loop which itself forms a lid for the retinal-binding pocket. In the N2C/D282C mutant this cap is covalently crosslinked to the E3 loop and hence is anchored in the protein structure at two locations on opposite sides of the domain. This fixes the N-terminal cap permanently over the β-sheet lid of the ligand binding pocket (Fig. 6).
Figure 6. The N-terminal Cap domain.
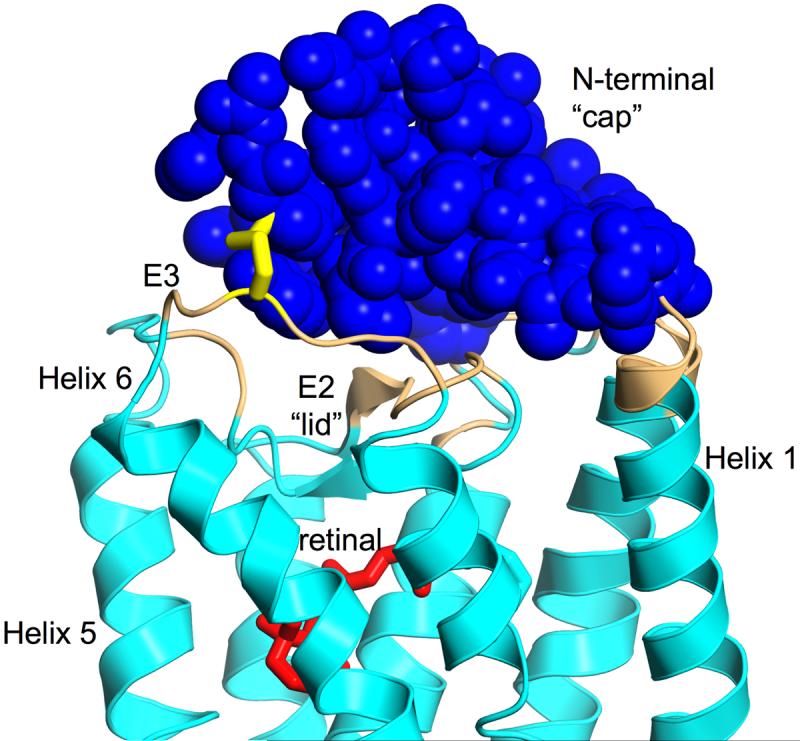
Contacts between the N-terminal cap (blue) and the rest of the receptor (cyan). Atoms of the receptor with a minimum distance of 5 Å to residues 1-33 of the N-terminus are colored beige. The covalently bound retinal (red) is separated from the N-terminal cap by the lid formed from the E2 loop. The designed disulfide between C2 and C282 connecting the N-terminal cap with loop E3 is shown as yellow sticks. The molecule is viewed with the extracellular site facing up.
Comparison with native rhodopsin (1GZM)
The overall Cα rms difference between the refined structure of recombinant N2C/D282C rhodopsin and the model used for molecular replacement (1GZM, monomer A) is 0.745 Å (0.800 Å for 1GZM, monomer A). The rms differences generally track with B factors: lower in helical regions and greater in the cytoplasmic loops. Accordingly the Cα rms difference drops to 0.619 Å if residues with a B-factor higher than 100 are omitted (Fig. 5_C_). In addition, the changed dimer interface and crystal contacts in the mutant result in small differences at the cytoplasmic sides of helix 3 (residues 137-140) and helix 5 (residues 223-232), which are each shifted with respect to 1GZM by 1.5 Å and 2.0 Å. However, this does not effect the overall fold seen in the EM structure of rhodopsin in 2D crystals 16.
The extracellular domain includes the three loops (E1, residues 101-105; E2, 174-199; and E3, 280-285) and N-terminal cap (residues 1-34) and has a much higher degree of order than the cytoplasmic side in both the mutant and native protein. It is also the site of the engineered disulfide bond in the mutant. Although the immediate sites of mutation, positions 2 and 282, have a Cα-Cα distance slightly closer than in the 1GZM structure (4.8 Å versus 5.2 Å), and residues 1 through 5 and the short E3 loop show Cα rms deviations higher than 1 Å with respect to the 1GZM structure (Fig. 5), it is striking that the extracellular domain is overall one of the regions least different from that of the published P31 structure (Cα rms difference of 0.599 Å). Clearly, formation of the engineered disulfide has had a minimal impact on the global conformation of the protein.
Discussion
Structure determination of eukaryotic membrane proteins remains a challenging task despite technological advances. This is especially true for proteins that are not highly abundant in natural sources and thus have to be produced recombinantly. The only eukaryotic integral membrane proteins that have been successfully overexpressed and used for structure determination are the plant aquaporin 0 17, the voltage-gated potassium channel 18, both expressed in the yeast Pichia Pastoris 19; 20 and, very recently, the rat aquaporin 4 expressed in the insect cell baculovirus system 21. Of even greater challenge are eukaryotic membrane proteins for which heterologous expression of functional protein is limited to mammalian cells in culture, as is the case with many of the G protein-coupled receptors 5. Our interest in recombinant rhodopsin stems from the fact that most GPCRs, and certainly most interesting functional mutants of GPCRs, are available only from recombinant sources and often only a very small amount of high quality protein is available, like in our case where we isolated 0.6mg of crystallization competent protein in one preparation.
The use of recombinant protein also enables the exploration of mutants that are designed to improve crystal quality. For example, the N2C/D282C mutant used in this study was selected under the assumption that its enhanced thermal stability might lead to crystals of higher quality for x-ray diffraction studies. Similarly, the triple mutant N2C/N15D/D282C was designed to remove all carbohydrate chains from rhodopsin in the hope that eliminating this source of heterogeneity might also improve crystal quality. While removing carbohydrate proved crucial in crystallization of the mammalian voltage-gated potassium channel 18; 20, it did not help in our case with rhodopsin. Crystals from the N2C/N15D/D282C mutant were similar in appearance to those of N2C/D282C but the diffraction patterns were of lower resolution and were more anisoptropic. We suspect that the reason that removing glycosylation sites did not improve crystal quality was because of the unusual hydrophobic packing found in the rhodopsin P31 crystals where the oligosaccharyl chains extend from the amino-terminal domain into a solvent-filled channel and are not involved in crystal contacts 13; 16. Indeed complete removal of glycosylation seemed to result in decreased diffraction quality. Due to the limited number of crystallization experiments and tested crystals the reason for this is unclear at this stage, it is however possible that the destabilizing removal of the second carbohydrate chain introduced micro-heterogeneities which may have interfered with crystal order. Nevertheless, the engineered N2C/D282C mutant was stabilizing enough to isolate and crystallize a fully deglycosylated protein which causes autosomal dominant retinitis pigmentosa in humans 11. A 10°C thermal stabilization of rhodopsin solubilized in a harsh detergent is striking especially if it is achieved by a single engineered disulfide bond. A possible explanation is that fixing the N-terminal cap microdomain over the binding pocket lid (Fig. 4) has prevented a specific unfolding pathway of the receptor.
Interestingly, recombinant rhodopsin proved to be better than the native protein in terms of the fraction of protein preparations yielding diffraction quality crystals. The yield with recombinant rhodopsin was about 10%, whereas the yield with native rhodopsin isolated from bovine retina was about 1%. While we did not systematically explore the source of this difference it is likely that the greater reproducibility of the recombinant crystals is a result of the enhanced stability found with the thermally stable mutant. It is also possible that the greater reproducibility is a consequence of the recombinant crystals being in general much smaller than those of the native protein and therefore suffer less from long range disorder and mosaicity. This is an interesting consideration because crystal optimization is usually geared toward development of conditions leading to greater size of the crystal. But the larger size may actually be a disadvantage with the P31 rhodopsin crystals. A clear disadvantage of the smaller crystals is the greater beam intensities, and consequent radiation damage, required to observe higher resolution reflections in comparison with crystals of larger volume. The crystal volume exposed by the 5 μm beam contains about 450×450×650 unit cells or 2.6×108 rhodopsin molecules. To obtain sufficient diffraction long exposures were necessary which resulted in deterioration of the diffraction patterns through radiation damage after only a few frames were recorded (Fig. 3 B and C). For this reason, data were collected from multiple positions along the long axis of a single small crystal, a procedure facilitated by the improved data collection strategies employed at the microfocus beamline at ESRF 22; 23. Generally, radiation damage manifests itself in a decrease in overall resolution and an increase in B-factors of the refined structure and, in severe cases, even a change in unit cell size is observed 24. Specific damage is generally observed on side chains of amino acids, frequently involving loss of sulfhydryl groups of cysteins, hydroxyl groups from tyrosines and decarboxylation of aspartate and glutamate 25; 26. In our case, radiation damage was clearly present but the effect was minimized because a different fresh position of the crystal was sampled every few frames. Consequently, the damage was distributed equally over Fourier space and did not accumulate in one region. A similar data collection strategy would be useful on other poorly reproducible, non-isomorphous crystals, which are thin in one or two dimensions, as for example hexagonal plates of the plant light harvesting complex II 27 or any other difficult to crystallize protein.
In conclusion, determination of the structure of the rhodopsin N2C/D282C mutant is an important first step in development of a generally applicable approach to crystallization of heterologously expressed GPCRs. However, important hurdles remain. First and foremost is obtaining sufficient quantities of the proteins from cell culture systems to allow for large-scale crystallization trials. With some effort and straightforward modification of existing expression procedures 7; 28 we have been recently able to produce tens of milligrams of rhodopsin mutants in both COS and HEK-293 cells. This is a quantity of protein that is sufficient for large-scale screening of crystallization conditions. Second is the issue of heterogeneity, primarily with regard to oligosaccharyl chains. While carbohydrate heterogeneity did not present a problem in the current study, it would without doubt severely limit success in broad-based crystallization screens. Two straight-forward approaches to eliminate this problem are mutation of the N-linked consensus sites, as was done after stabilization in our study, and the use of a glycosyl transferase minus cell line, as was done by Reeves et al. 8. Third, is the stability of the receptor. A more stable and thus more detergent tolerant receptor as used here would allow a greater range of crystallization conditions to be explored and thereby increase the chances of obtaining good crystals suitable for structure determination. Finally, the technology for data collection from small crystals needs further development to eliminate the impact of radiation damage on data quality. In particular, it is essential to develop smaller, more focused beams (e.g., 1μm), and the ability to record from a greater number of different and independent positions on a single crystal, ideally collecting in the limit each image from a different position on the crystal. This could prove particularly useful not only for mutants of rhodopsin and other GPCRs but in a broader context for proteomic projects in which expression and optimization are still major bottlenecks.
Materials and Methods
Expression and purification
Expression and initial purification has been performed as described previously 7; 9. Briefly, for recombinant rhodopsins COS-1 cells from 50 plates (15cm) were transiently transfected with DEAE dextran. Cell pellets were solubilized with 1% DDM for 1hour at 4°C. Nuclei were spun out and the supernatant was incubated with 1D4 antibody coupled to CnBr activated sepharose (Amersham Biosciences). After 3-4 hours the column material was washed with PBS pH 7, 0.1% DDM. 11-cis retinal (50 μM) was added to the matrix. After incubation at 4°C overnight the matrix was washed with PBS pH 6, 0.1% DDM followed by 2 mM NaPi pH 6, 0.02% DDM. The purified protein was eluted with the peptide TETSQVAPA (80μM). All procedures after reconstitution were performed under dim red light.
About 2.5 mg of purified rhodopsin was concentrated and detergent partially exchanged using a Sephadex G50 column (Amersham Biosciences), pre-equilibrated with 20 mM Tris pH 8.5, 1 mM EDTA 2 mM MgCl2, 0.2% C8E4 (Bachem). The sample was loaded onto a Mono-Q 5/50 ion exchange column (Amersham Biosciences) in the same buffer. The fractions eluting around 0.05 M NaCl crystallized reproducibly.
Native rhodopsin was purified from retinas as described 13, only that 0.02% DDM was used during ConA chromatography instead of LDAO. The DDM was partially exchanged to C8E4 as described for the recombinant rhodopsin to ensure comparability of the stability measurements.
Stability measurements
Protein after the Sephadex G50 detergent exchange step was diluted to a concentration of 0.05 mg/ml in 20 mM Tris pH 8.5, 1 mM EDTA 2 mM MgCl2, 0.2% C8E4. The thermal denaturation was investigated by 30 minutes incubation in a PRC machine at various temperatures and subsequent recording of UV/VIS spectra. The absorption at 500 nm was corrected by the 280 nm absorption of the polypeptide and plotted against the temperature. Sigmoidal curves were fitted to the data points using the program Prism.
Crystallization setups
Fractions were concentrated to 10mg/ml with a Centricon 30 and Microcon 30 (Millipore) and LDAO (Fluka) was added to a final concentration of 0.05%. Sitting drops (1μl protein +1μl motherliquor) were setup over a range of precipitants. A motherliquor containing 1.2-1.7 M lithium sulfate, 0.1 M Hepes pH 7.5 reproducibly yielded crystals within 2 weeks. Crystals to be harvested were first transferred to a drop of mother liquor containing 20% glycerol and then picked into cryoloops under infrared illumination 13.
Data collection and processing
The possibility to position the 5μm beam on the right part of a difficult to visualize microcrystal was crucial for data collection at the ID13 microfocus beamline. To achieve maximum precision the inline optics were used to align the guard aperture and the beam defining aperture, before the rotation axis was aligned to the microbeam. During alignment, the focused beam was visualized by a removable fluorescent crystal.
A complete dataset was obtained by combining wedges of 8-15° collected from different positions on one crystalline needle. Data were indexed and scaled with XDS 29. Consistent indexing was maintained by re-indexing each wedge under the four indexing regimes of the predicted P31 symmetry. The overlap, the correlation coefficients and the Rmerge between each indexing regime and the best wedge as a reference were used to find the correct solutions. Once a solution was found the wedge was added to the reference and the re-indexing merging cycle repeated until a full dataset was obtained. Consistency was furthermore verified by testing each indexing regime against the full dataset.
Structure determination and refinement
The structure was solved by molecular replacement using MOLREP 30. As a search model the 2.65 Å structure of bovine rhodopsin (1GZM) solved from crystals with trigonal spacegroup was used 4. All heteroatoms including the retinal and the glycosylations were removed from this model prior to molecular replacement. The initial solution was improved using rigid body refinement and solvent flattening. The model was deployed to a simulated annealing prior to alternating rounds of manual model building (programs O 31 and COOT 32) and energy minimization followed by restrained B-factor refinement. Throughout refinement strong NCS restraints were maintained. If not stated otherwise, the CNS suite of programs 33 was used.
Coordinates and structure factors are deposited in the Protein Data Bank under ID code 2j4y. Superposition and rmsd calculation were carried out with the program ProFit (http://www.bioinf.org.uk/software/profit/). Mutual contact surface areas were calculated with the CCP4 program AREAIMOL. Figures were prepared with PYMOL (http://www.pymol.org).
Supplementary Material
01
Acknowledgements
We thank Mathew Higgins for help in the initial crystallization trials and Jade Li for helpful suggestions on molecular replacement and refinement of the structure. For stimulating discussions and critical reading of the manuscript we thank Phil Evans and Chris Tate. We thank the National Institute of Health and Rosalie Crouch at the University of South Carolina for the kind gift of 11-cis retinal. The work was financially supported by a Human Frontier Science Project (HFSP) program grant (RG/0052), a European Commission FP6 specific targeted research project (LSH-2003-1.1.0-1), and by NIH grant EY007965 (DDO).
Abbreviations
GPCR
G protein-coupled receptor
C8E4
n-octyotetraoxyethylene
DDM
β-D-dodecylmaltoside
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Drews J, Ryser S. The role of innovation in drug development. Nat Biotechnol. 1997;15:1318–9. doi: 10.1038/nbt1297-1318. [DOI] [PubMed] [Google Scholar]
- 2.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 3.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–83. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–38. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 5.Sarramegn V, Muller I, Milon A, Talmont F. Recombinant G protein-coupled receptors from expression to renaturation: a challenge towards structure. Cell Mol Life Sci. 2006 doi: 10.1007/s00018-005-5557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tate CG. Overexpression of mammalian integral membrane proteins for structural studies. FEBS Lett. 2001;504:94–8. doi: 10.1016/s0014-5793(01)02711-9. [DOI] [PubMed] [Google Scholar]
- 7.Oprian DD, Molday RS, Kaufman RJ, Khorana HG. Expression of a synthetic bovine rhodopsin gene in monkey kidney cells. Proc Natl Acad Sci U S A. 1987;84:8874–8. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reeves PJ, Callewaert N, Contreras R, Khorana HG. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci U S A. 2002;99:13419–24. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie G, Gross AK, Oprian DD. An opsin mutant with increased thermal stability. Biochemistry. 2003;42:1995–2001. doi: 10.1021/bi020611z. [DOI] [PubMed] [Google Scholar]
- 10.Kaushal S, Ridge KD, Khorana HG. Structure and function in rhodopsin: the role of asparagine-linked glycosylation. Proc Natl Acad Sci U S A. 1994;91:4024–8. doi: 10.1073/pnas.91.9.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kranich H, Bartkowski S, Denton MJ, Krey S, Dickinson P, Duvigneau C, Gal A. Autosomal dominant ‘sector’ retinitis pigmentosa due to a point mutation predicting an Asn-15-Ser substitution of rhodopsin. Hum Mol Genet. 1993;2:813–4. doi: 10.1093/hmg/2.6.813. [DOI] [PubMed] [Google Scholar]
- 12.Davidson FF, Loewen PC, Khorana HG. Structure and function in rhodopsin: replacement by alanine of cysteine residues 110 and 187, components of a conserved disulfide bond in rhodopsin, affects the light-activated metarhodopsin II state. Proc Natl Acad Sci U S A. 1994;91:4029–33. doi: 10.1073/pnas.91.9.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards PC, Li J, Burghammer M, McDowell JH, Villa C, Hargrave PA, Schertler GF. Crystals of native and modified bovine rhodopsins and their heavy atom derivatives. J Mol Biol. 2004;343:1439–50. doi: 10.1016/j.jmb.2004.08.089. [DOI] [PubMed] [Google Scholar]
- 14.Perrakis A, Cipriani F, Castagna JC, Claustre L, Burghammer M, Riekel C, Cusack S. Protein microcrystals and the design of a microdiffractometer: current experience and plans at EMBL and ESRF/ID13. Acta Crystallogr D Biol Crystallogr. 1999;55:1765–70. doi: 10.1107/s0907444999009348. [DOI] [PubMed] [Google Scholar]
- 15.Cusack S, Belrhali H, Bram A, Burghammer M, Perrakis A, Riekel C. Small is beautiful: protein micro-crystallography. Nat Struct Biol. 1998;5(Suppl):634–7. doi: 10.1038/1325. [DOI] [PubMed] [Google Scholar]
- 16.Schertler GF. Structure of rhodopsin and the metarhodopsin I photointermediate. Curr Opin Struct Biol. 2005;15:408–15. doi: 10.1016/j.sbi.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 17.Tornroth-Horsefield S, Wang Y, Hedfalk K, Johanson U, Karlsson M, Tajkhorshid E, Neutze R, Kjellbom P. Structural mechanism of plant aquaporin gating. Nature. 2006;439:688–94. doi: 10.1038/nature04316. [DOI] [PubMed] [Google Scholar]
- 18.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 19.Karlsson M, Fotiadis D, Sjovall S, Johansson I, Hedfalk K, Engel A, Kjellbom P. Reconstitution of water channel function of an aquaporin overexpressed and purified from Pichia pastoris. FEBS Lett. 2003;537:68–72. doi: 10.1016/s0014-5793(03)00082-6. [DOI] [PubMed] [Google Scholar]
- 20.Parcej DN, Eckhardt-Strelau L. Structural characterisation of neuronal voltage-sensitive K+ channels heterologously expressed in Pichia pastoris. J Mol Biol. 2003;333:103–16. doi: 10.1016/j.jmb.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 21.Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, Mizoguchi A, Fujiyoshi Y. Implications of the aquaporin-4 structure on array formation and cell adhesion. J Mol Biol. 2006;355:628–39. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- 22.Riekel C. Recent developments in micro-diffraction on protein crystals. J Synchrotron Radiat. 2004;11:4–6. doi: 10.1107/s090904950302541x. [DOI] [PubMed] [Google Scholar]
- 23.Riekel C, Burghammer M, Schertler G. Protein crystallography microdiffraction. Curr Opin Struct Biol. 2005;15:556–62. doi: 10.1016/j.sbi.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 24.Ravelli RB, Theveneau P, McSweeney S, Caffrey M. Unit-cell volume change as a metric of radiation damage in crystals of macromolecules. J Synchrotron Radiat. 2002;9:355–60. doi: 10.1107/s0909049502014541. [DOI] [PubMed] [Google Scholar]
- 25.Burmeister WP. Structural changes in a cryo-cooled protein crystal owing to radiation damage. Acta Crystallogr D Biol Crystallogr. 2000;56:328–41. doi: 10.1107/s0907444999016261. [DOI] [PubMed] [Google Scholar]
- 26.Garman EF, Owen RL. Cryocooling and radiation damage in macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2006;62:32–47. doi: 10.1107/S0907444905034207. [DOI] [PubMed] [Google Scholar]
- 27.Standfuss J, Terwisscha van Scheltinga AC, Lamborghini M, Kuhlbrandt W. Mechanisms of photoprotection and nonphotochemical quenching in pea light-harvesting complex at 2.5 A resolution. Embo J. 2005;24:919–28. doi: 10.1038/sj.emboj.7600585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reeves PJ, Kim JM, Khorana HG. Structure and function in rhodopsin: a tetracycline-inducible system in stable mammalian cell lines for high-level expression of opsin mutants. Proc Natl Acad Sci U S A. 2002;99:13413–8. doi: 10.1073/pnas.212519199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- 30.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 1997;30:1022–1025. [Google Scholar]
- 31.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Cryst. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite formacromolecular structure determination. Acta Cryst. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01