Opposite Regulation of CD36 Ubiquitination by Fatty Acids and Insulin: EFFECTS ON FATTY ACID UPTAKE (original) (raw)
Abstract
FAT/CD36 is a membrane scavenger receptor that facilitates long chain fatty acid uptake by muscle. Acute increases in membrane CD36 and fatty acid uptake have been reported in response to insulin and contraction. In this study we have explored protein ubiquitination as one potential mechanism for the regulation of CD36 level. CD36 expressed in Chinese hamster ovary (CHO) or HEK 293 cells was found to be polyubiquitinated via a process involving both lysines 48 and 63 of ubiquitin. Using CHO cells expressing the insulin receptor (CHO/hIR) and CD36, it is shown that addition of insulin (100 nm, 10 and 30 min) significantly reduced CD36 ubiquitination. In contrast, ubiquitination was strongly enhanced by fatty acids (200 μm palmitate or oleate, 2 h). Similarly, endogenous CD36 in C2C12 myotubes was ubiquitinated, and this was enhanced by oleic acid treatment, which also reduced total CD36 protein in cell lysates. Insulin reduced CD36 ubiquitination, increased CD36 protein, and inhibited the opposite effects of fatty acids on both parameters. These changes were paralleled by changes in fatty acid uptake, which could be blocked by the CD36 inhibitor sulfosuccinimidyl oleate. Mutation of the two lysine residues in the carboxyl-terminal tail of CD36 markedly attenuated ubiquitination of the protein expressed in CHO cells and was associated with increased CD36 level and enhanced oleate uptake and incorporation into triglycerides. In conclusion, fatty acids and insulin induce opposite alterations in CD36 ubiquitination, modulating CD36 level and fatty acid uptake. Altered CD36 turnover may contribute to abnormal fatty acid uptake in the insulin-resistant muscle.
Long chain fatty acids (FA)3 are the predominant fuel used by oxidative muscle and heart. FA uptake into muscle was shown to involve, in addition to passive diffusion, protein-facilitated transfer with a major role for the FA translocase FAT/CD36 (1–3). CD36 was identified as a FA receptor based on its binding to inhibitors of FA uptake in isolated adipocytes. The most specific of these inhibitors is membrane-impermeable sulfosuccinimidyl oleic acid (SSO), which forms a covalent bond with free amino groups on the protein (4). Transgenic mice that overexpress CD36 in heart and muscle tissues exhibit increased FA utilization and glucose sparing (5). In contrast, deficiency in CD36 is associated in mice (3, 6) and humans (7) with a defect in FA uptake that is most pronounced in the heart and that is compensated for by increased glucose utilization.
Muscle CD36 content is highly regulated both at chronic and acute levels (8). Up-regulation of protein levels has been shown to associate with fasting (9) and insulin resistance in mice (10) and humans (8). Chronic leptin down-regulates the protein (11), whereas resistin (12) has the opposite effect. Acute up-regulation of muscle CD36 has been documented in response to insulin and to contraction and has been postulated to involve recruitment of the protein from intracellular stores to the plasma membrane (2). This recruitment was shown to involve AMPK because it was reproduced by AMPK agonists. Increased membrane CD36 also rapidly follows activation of the nuclear transcription factor FoxO1 (Forkhead box O transcription factor 1) and contributes to the function of FoxO1 in modulating fuel preference by muscle in response to fasting (13).
Persistently increased CD36 at the muscle membrane has been proposed to play a key role in diabetes etiology by enhancing FA uptake, accumulation of intramyocellular lipid derivatives, and impairing insulin-stimulated GLUT4 translocation (14, 15).
In addition to its major role in facilitating FA uptake by muscle (3, 7), CD36 functions as a receptor for a number of ligands including thrombospondin 1, collagen, and Gram-negative bacteria (16). It also contributes to uptake of oxidized lipoproteins (17) and consequently to macrophage conversion to foam cells (18). Macrophage CD36 is increased by lysosomal and proteasomal inhibitors, and decreased CD36 turnover was reported in macrophages from insulin-resistant ob/ob mice (19).
The present study examined the possibility that one potential mechanism for both acute and long term regulation of CD36 level would involve the ubiquitin pathway. Ubiquitination in eukaryotes targets proteins to degradation by the action of the 26 S proteasomes or of lysosomes (20, 21). Ubiquitin is a 76-residue protein that is joined reversibly to protein substrates by linkage between the α-carboxyl group of ubiquitin and ε-amino groups of lysine on the acceptor proteins. Ubiquitin molecules are generally added to the substrate via an enzyme cascade, in which ubiquitin moieties are linked to previously conjugated ubiquitin molecules until multi-ubiquitin chains are formed, which facilitates rapid degradation of the substrate (22). We explored the possibility that CD36 is ubiquitinated and examined whether changes in protein ubiquitination could mediate regulation of protein levels. We found that CD36 is ubiquitinated at carboxyl-terminal lysines, and the process is regulated by both FA and insulin.
EXPERIMENTAL PROCEDURES
_Materials_—Mouse C2C12 myoblasts and human embryonic kidney (HEK) 293 cells were from the American Type Culture Collection (Manassas, VA). Dulbecco's modified Eagle's medium, fetal bovine serum, calf serum, antibiotics, and other cell culture products were from Invitrogen and Roche Applied Science. [9,10(n)-3H]Oleate was from ICN Biochemicals (Costa Mesa, CA). CD36 antibody was from Cascade Biosciences (Winchester, MA) or Santa Cruz Biotechnology, Inc (Santa Cruz, CA), and antibody for ubiquitin from Invitrogen and that for ran from Santa Cruz. Immobile-P membranes for Westerns were from Millipore. Fatty acid-free bovine serum albumin fraction IV (BSA) was from Sigma (St. Louis, MO). SSO was synthesized as described earlier (23), and purity was determined to be greater than 95% by mass spectrometry.
_Cell Culture_—C2C12 cells were maintained in low glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 200 units/ml penicillin, and 50 μg/ml streptomycin. At 90% confluence, the cells were washed with warm PBS, and myotube differentiation was induced by switching the cells to Dulbecco's modified Eagle's medium complemented with 1% horse serum, 200 units/ml penicillin, and 50 μg/ml streptomycin. Fresh medium was supplied every 24 h. Polynucleated myotubes were obtained within 3 days. Chinese hamster ovary (CHO) cells stably expressing human insulin receptor (CHO/hIR) (24) were maintained in Ham's F-12 medium containing 10% fetal bovine serum. HEK 293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum. All of the cells were cultured in a 5% CO2 environment at 37 °C in 100-mm dishes or 12-well plates.
_Plasmid Construction and Transfection_—The carboxyl-FLAG-tagged human CD36 in pcDNA3 was generated as previously described (25). To generate amino-terminal GFP-tagged-CD36, rat CD36 cDNA was cloned into the pEGFP-C1 construct (Invitrogen) using the XhoI and ApaI sites. Human and rat CD36 predicted protein sequences share 79% identity (85% similarity) (26). For generating the CD36 with mutated carboxyl lysines, the human CD36 cDNA was subcloned into the expression vector pcDNA3 as previously reported (25). Site-directed mutagenesis was used to generate alanine substitutions of lysine residues 469 and 472 using the following oligonucleotides: 5′-TATTGTGCATGCAGATCGGCAACAATAAAAGACTACAAGGACGAC-3′; 5′-GTCGTCCTTGTAGTCTTTTATTGTTGCCGATCTGCATGCACAATA-3′; 5′-TATTGTGCATGCAGATCGAAAACAATAGCAGACTACAAGGACGAC-3′; 5′-GTCGTCCTTGTAGTCTCGTATTGTTTTCGATCTGCATGCACAATA-3′; 5′-TATTGTGCATGCAGATCGGCAACAATAGCAGACTACAAGGACGAC-3′; and 5′-GTCGTCCTTGTAGTCTCGTATTGTTGCCGATCTGCATGCACAATA-3′.
Mutant cDNAs were sequenced to verify the expected mutations and the absence of spurious ones and were transfected using Lipofectamine 2000 (Invitrogen) in 6-well plates. All of the transfections were transient.
_Immunoprecipitation and Western Blot Analysis_—Whole cell extracts were prepared using lysis buffer (50 mm Tris·HCl, pH 7.5, 100 mm NaCl, 1% Triton X-100, 10% glycerol, 1 mm EDTA, 10 mm NaF, 1 mm sodium orthovanadate) containing protease inhibitor mixture (Sigma). The cell lysates were clarified by centrifugation at 10,000 × g (10 min, 4 °C), and the clarified lysates were incubated with primary antibodies (overnight at 4 °C). Immune complexes were captured by adding 10 μl of protein A-Sepharose beads (Santa Cruz) followed by incubation with end-over-end rotation overnight at 4 °C followed by washing five times in lysis buffer. Protein was eluted by boiling (10 min) in 30 μl of SDS sample buffer. All of the samples were separated on a 4–20% polyacrylamide-reduced gradient gel and electroblotted onto Immobilon-P polyvinylidene difluoride membranes. The membranes were blocked in TBST (100 mm NaCl, 10 mm Tris-HCl, pH 7.5, 0.1% Tween 20) with 5% nonfat milk and then incubated with primary antibodies in 2% BSA/TBST overnight at 4 °C or 2 h at room temperature. Incubation with secondary horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG followed, and the proteins were visualized by enhanced chemiluminesence (Pierce, Rockford, IL).
_Biotinylation and Purification of Cell Surface Proteins_—Cell surface proteins were labeled with biotin using EZ-Link® Sulfo-NHS-SS-Biotin (Pierce) and purified as described (27). The amount of CD36 protein on the cell surface and that in total cell lysates were evaluated by Western blots and quantified by densitometry.
_Fatty Acid Uptake and SSO Treatment_—Myotubes were washed three times with 2 ml of Krebs-Ringer-Hepes buffer (KRH), pH 7.4, 0.1% BSA and once with 2 ml of KRH. Uptake was at room temperature for the indicated times (28). The assay began with the addition of 2 ml of transport buffer, KRH containing 5 mm glucose and 40 μm oleate, FA:BSA = 1.1 μCi/ml [3H]oleate. It was stopped by medium aspiration and washing with ice-cold KRH, 0.5% BSA. The cells were lysed in 1 ml of 0.1 n NaOH for 30 min, and the aliquots were taken for scintillation counting and protein quantification using a DC protein assay kit (Bio-Rad). The CD36 inhibitor SSO (400 μm) was added when indicated to cells for 30 min before assay of FA uptake.
_Thin Layer Chromatography_—The myotubes were incubated with fresh medium containing [3H]oleate (1 μCi/ml, 40 μm). The cells were washed at 4 °C (three washes with PBS, 0.5% BSA) and scraped in 500 μl of PBS. The lipids were extracted and separated on silica gel 60A plates (Whatman, Clifton, NJ) using hexane:diethyl ether:acetic acid, 75:25:1. Spots corresponding to major lipid species, identified by standards run simultaneously and visualized by iodine vapors, were scraped and counted.
_Measurement of Free Fatty Acids by Mass Spectrometry_—Cell pellets were collected, and total lipids were extracted using a modified Bligh and Dyer technique with 50 mm LiCl in the aqueous layer in the presence of an internal standard (20:0 FA, 2 nmol/mg protein). Mass spectral analysis of free FAs was performed by electrospray ionization utilizing a Finnigan TSQ Quantum spectrometer (Finnigan MAT, San Jose, CA) as previously described (29).
RESULTS
_CD36 Is Constitutively Ubiquitinated_—GFP-tagged CD36 and FLAG-tagged ubiquitin were co-expressed in CHO/hIR cells. Western blot analysis using an anti-FLAG antibody showed (Fig. 1_A_) significant ubiquitination of CD36 from the smear signal obtained that is usually typical of multi ubiquitinated substrates. To determine the type of ubiquitin linkage involved, co-transfection with FLAG-tagged ubiquitin mutated at lysine 48 or 63 was done. The amount of ubiquitinated CD36 was significantly decreased in the presence of either mutated form, suggesting that both processes were involved. To determine whether ubiquitination involved CD36 rather than a CD36-associated protein, cell lysates were boiled in the presence of SDS to dissociate protein-protein associations, then diluted, and processed for CD36 immunoprecipitation. The Western analysis in Fig. 1_B_ shows that the ubiquitination signal obtained was virtually identical to that observed in Fig. 1_A_, supporting direct ubiquitination of CD36.
FIGURE 1.

Ubiquitination of CD36 in CHO/hIR cells. A, CHO/hIR cells were co-transfected with GFP-CD36 and FLAG-ubiquitin wild type (WT) or mutants K48R, K64R, or K/R (double mutant). The cells were lysed, and clarified lysates were immunoprecipitated (IP) with rabbit anti-GFP antibody. The immunocomplexes were resolved by SDS-PAGE and immunoblotted (IB) with antibody against FLAG. B, CHO/hIR cells were co-transfected with GFP-CD36 and FLAG-ubiquitin WT. The cell pellets were collected and boiled in PBS containing 1% SDS. The lysates were diluted 10-fold with lysis buffer prior to immunoprecipitation (IP) and Western blot analysis as described under A. The data shown are representative of two experiments.
_Insulin and FA Regulate CD36 Ubiquitination_—Insulin deficiency, such as with acute diabetes or fasting, is associated with enhanced protein breakdown in skeletal muscle. However, both conditions increase muscle CD36 protein levels (9, 10). Insulin resistance also increases macrophage CD36 via a decrease in CD36 turnover (19). We examined the effects of insulin on ubiquitination of CD36. CHO/hIR cells transfected with CD36-GFP were incubated with 100 nm insulin for 10 and 30 min. As shown in Fig. 2_A_, a decrease in ubiquitination of exogenous CD36 was observed that was quite apparent at 30 min. Because both fasting and insulin resistance are usually associated with an increase in FA levels, we examined how FA treatment would impact CD36 ubiquitination. CHO/hIR cells were incubated with 200 μm oleic acid for 2 h. As shown in Fig. 2_B_, this was associated with a significant increase in CD36 ubiquitination as compared with no FA treatment. In addition to oleic acid (C18:1), we tested palmitic acid (C16:0) and found it equally effective in enhancing CD36 ubiquitination (data not shown).
FIGURE 2.

Ubiquitination of exogenous CD36 in CHO/hIR cells is modulated by insulin and FA. CHO/hIR cells were transfected with GFP alone or GFP-CD36. A, cells were starved and treated with 100 nm insulin (Ins) for the indicated times. The cells were lysed, and clarified lysates were immunoprecipitated (IP) with rabbit anti-GFP antibody. The immunocomplexes were resolved by SDS-PAGE and immunoblotted (IB) with antibodies recognizing ubiquitin (Ubi) or GFP (GFP alone does not appear on the gel because the molecular weight of unconjugated GFP is 28kD versus >110kDa for GFP-CD36). B, cells were treated with 200 μm oleic acid complexed to BSA (FA:BSA = 1) for 2 h, immunoprecipitated (IP) and immunoblotted (IB) as described for A. Similar results were obtained with palmitic acid (data not shown). The data shown are representative of three experiments.
_FA Induces Ubiquitination and Degradation of Endogenous CD36 in C2C12 Myotubes, and This Is Inhibited by Insulin_—We next examined whether ubiquitination could be demonstrated under conditions that more closely resemble the physiological situation. Confluent cultures of myoblasts of the C2C12 line can be induced to differentiate into multinucleated myotubes that express CD36 and recapitulate many features of the muscle oxidative phenotype (13, 28). We examined ubiquitination of endogenous CD36 in C2C12 myotubes. As shown in Fig. 3_A_, CD36 in myotubes is ubiquitinated. Acute exposure of myotubes to 100 nm insulin resulted in a slight decrease of CD36 ubiquitination as compared with untreated cells. The addition of oleic acid (200 μm, 2 h) strongly increased CD36 ubiquitination as compared with untreated myotubes (Fig. 3_A_). This increase was effectively inhibited if myotubes were preincubated with insulin for 30 min. Levels of CD36 immunoprecipitated from cell lysates showed a trend toward more protein in insulin treated cells. CD36 expression in whole cell lysates from myotubes treated with insulin, FA alone, and FA plus insulin is shown in Fig. 3_B_. Insulin increased the CD36 protein level ∼2-fold, and FA treatment decreased it by ∼50%. Preincubation with insulin blocked the effect of FA to decrease CD36 level. The data show that FA-induced ubiquitination of endogenous CD36 is associated with lower CD36 protein levels and that insulin prevents FA effects on ubiquitination and protein loss.
FIGURE 3.

Ubiquitination of endogenous CD36 in C2C12 myotubes and effects of insulin and FA. A, C2C12 myotubes were treated with 100 nm insulin (Ins) for 30 min and/or 200 μm oleic acid (FA:BSA = 1) for 2 h as indicated. The cells were lysed, and clarified lysates were immunoprecipitated (IP) with rabbit anti-CD36 antibody. The immunocomplexes were resolved by SDS-PAGE and immunoblotted (IB) with ubiquitin antibody. B, whole cell lysates were analyzed for CD36 expression by Western blot (upper panel), Ran is the loading control. Signals were quantified by densitometry (lower panel) and CD36 level in FA-, insulin-, and FA- and insulin-treated cells was related to expression in untreated (basal) cells. Although a representative Western blot is shown, the densitometry data are the averages from three experiments with their standard errors. All of the treatments were significantly different (p < 0.05) from untreated cells.
_Insulin and FA Regulate FA Uptake and Triglyceride Synthesis in C2C12 Myotubes_—Alteration of CD36 ubiquitination by altering the total CD36 level could provide a mechanism for regulation of FA uptake. To examine this we determined the effects of insulin and FA pretreatments on FA uptake in myotubes under the same conditions used for ubiquitination. The cells were preincubated with 100 nm insulin for 30 min before assay of oleate uptake. As shown in Fig. 4, insulin increased FA uptake into myotubes by 200% above control untreated cells. In contrast when myotubes were treated with FA for 2 h, thoroughly washed to remove residual FA, and then assayed for uptake, a 50% decrease in rates was observed. (Fig. 4_A_). Preincubation with insulin before FA addition significantly inhibited the ability of FA treatment to reduce FA uptake. The corresponding changes in FA incorporation into triglycerides (TAG) are shown in Fig. 4_B_. Insulin increased TAG synthesis in skeletal muscle 7-fold above untreated cells. FA incorporation into TAG was not increased in FA-treated and FA- and insulin-treated cells. In an attempt to correct for label dilution in cells treated with FA, we measured intracellular free FA by mass spectrometry and found it to be increased, respectively, by 55 and 58% for cells treated with FA and with FA plus insulin. When a correction for potential label dilution is factored into the data (inset in Fig. 4_B_), cells treated with FA in the absence or presence of insulin exhibited enhanced FA incorporation into TAG above control untreated cells. Fig. 4_C_ shows that FA uptake by myotubes is sensitive to the CD36 covalent inhibitor SSO (400 μm), which reduced rates by 70 and 42% in basal and insulin-treated cells, respectively.
FIGURE 4.

Effects of insulin and FA treatments on FA uptake and incorporation into triglycerides by C2C12 myotubes. C2C12 myotubes were treated with 100 nm insulin (Ins) for 30 min and/or 200 μm oleic acid (FA: BSA = 1) for 2 h as indicated. The controls are untreated cells. A, fatty acid uptake values were derived from time courses (1–4 min) performed at room temperature. The data represent the means ± S.E. from six independent experiments. B, FA incorporation into TAG was measured as described under “Experimental Procedures.” The data represent the means ± S.E. from six experiments. Inset, the FA incorporation data after correction for dilution of the isotope by intracellular free FA measured by mass spectrometry. C, cells were treated with or without SSO prior to fatty acid uptake measurement. The data are the means ± S.E. from two experiments with quadruplicate. *, p < 0.05.
The Proteasome Inhibitor MG132 Prevents CD36 Protein _Loss and Enhances FA Uptake and TAG Synthesis in Skeletal Muscle_—Proteasomes are cellular proteinase complexes that degrade ubiquitinated proteins or mediate the degradation of transmembrane proteins in lysosomes. Inhibition of proteosomal activity with MG 132 (10 μg/ml) resulted in accumulation of ubiquitinated CD36 in HEK 293 cells (Fig. 5_A_) and C2C12 myotubes (Fig. 5_B_). In myotubes, MG132 also reduced the loss of CD36 protein caused by a 2-h incubation with 200 μm oleic acid (Fig. 5_C_), confirming proteosomal mediation of FA-induced degradation. Myotubes treated with MG132 for 4 h also exhibited enhanced oleate uptake that was sensitive to SSO inhibition (Fig. 5_D_), consistent with it being mediated by CD36. Label incorporation into TAG (Fig. 5_E_) by MG132-treated myotubes was enhanced above controls, whereas that into intracellular FA or diacylglycerol was unaltered (data not shown). Thus MG132 mimicked the effect of insulin on uptake.
FIGURE 5.

Effects of MG132 on CD36 ubiquitination, CD36 protein levels and fatty acid uptake. A, HEK 293 cells were transfected with FLAG-CD36 construct. The cells were treated with MG132 (10 μm) for the indicated times. The cells were lysed, and clarified lysates were immunoprecipitated (IP) with FLAG affinity gel. The immunocomplexes were eluted with FLAG peptide, resolved by SDS-PAGE, and immunoblotted (IB) with ubiquitin (Ubi) antibody. B, effect of MG132 (4 h) on CD36 ubiquitination in C2C12 myotubes. C, MG132 (4 h) treatment on FA-induced reduction in CD36 levels. The myotubes were treated with 200 μm oleic acid (FA:BSA = 1) for 2 h with or without preincubation with MG132 (10 μm) for 4 h. Whole cell lysates (WCL) were prepared and analyzed by immunoblotting. The data (A–C) are representative of two experiments with similar results. D, effect of SSO on FA uptake by myotubes. C2C12 myotubes were pretreated with 10 μm MG132 for 4 h and/or SSO for 30 min as indicated. Fatty acid uptake rates are from time courses (1–4 min) conducted in quadruplicate from two independent experiments. The data are the means ± S.E. E, effect of MG132 on FA incorporation into TAG. The data (means ± S.E.) are from three independent experiments. *, p < 0.05.
_Mutation of Carboxyl-terminal Lysine Residues (K469A and K472A) Reduces CD36 Ubiquitination and Enhances FA Uptake_—CD36 topography in the plasma membrane is predicted to be hairpin-like with a large extracellular segment, two intramembrane domains, and two short cytoplasmic termini. The only lysines present in the cytoplasmic domain of CD36 are conserved in the human rat and mouse proteins and are located at residues 469 and 472 in the carboxyl-terminal segment (26). These residues would constitute potential ubiquitination sites. To directly determine this, we generated FLAG-CD36 mutants where lysines 469 and 472 were substituted by alanine. The double mutated (K/A)-CD36 was then expressed in HEK 293 cells and effects on ubiquitination monitored. Fig. 6 shows that ubiquitination of the double mutant (K/A) is almost completely blocked as compared with wild type CD36, demonstrating that the carboxyl lysines are the major ubiquitination sites. Identical data were obtained in CHO/IR cells (data not shown).
FIGURE 6.
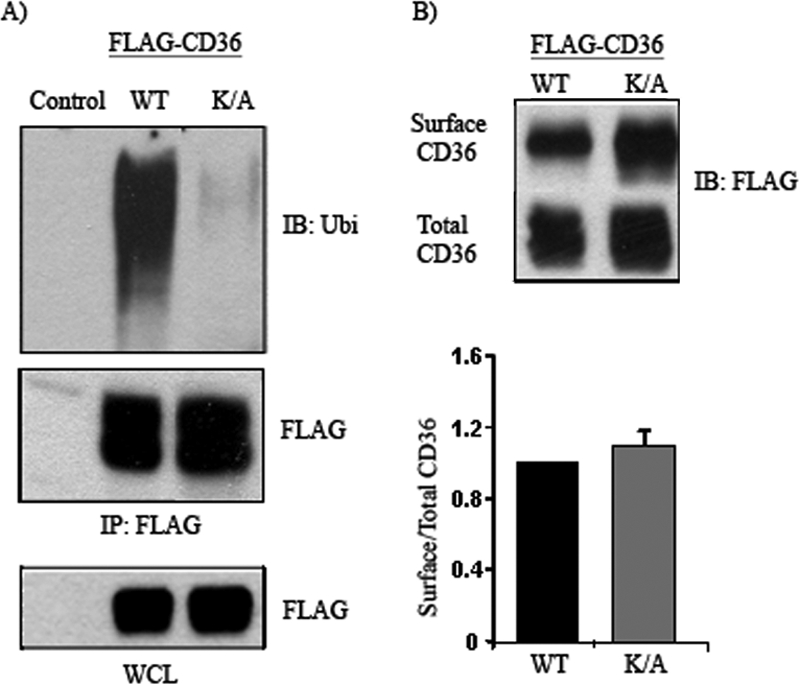
CD36 is ubiquitinated at lysines 469 and 472. A, HEK 293 cells were transfected with FLAG-CD36 wild type (WT) or mutated CD36 with alanine substitutions at lysines 469 and 472, K469A/K472A mutant (K/A). The cells were lysed, and clarified lysates were immunoprecipitated (IP) with FLAG affinity gel. The immunocomplexes were eluted with FLAG peptide, resolved by SDS-PAGE, and immunoblotted (IB) with ubiquitin (Ubi) antibody. B, Cells transfected with WT or K/A FLAG-CD36 were labeled with biotin, and the amounts of CD36 protein on the cell surface and in total cell lysates were evaluated by Western blot and quantified by densitometry. WCL, whole cell lysates.
Fig. 6_B_ shows that the lysine mutant form of CD36 is expressed at higher levels than WT CD36. However, the fraction of total CD36 recovered at the plasma membrane and thus accessible to biotinylation was not significantly changed as shown from the ratio of surface to total CD36. This indicated that ubiquitination does not alter membrane CD36 internalization.
The effect of lysine substitution on FA uptake was examined next. Cells transfected with mutated CD36 where Lys469 and Lys472 were substituted by alanine exhibited consistently ∼50% more CD36 protein as compared with cells expressing the wild type CD36. Expression of ubiquitination-deficient CD36 was associated with a 45% enhancement of FA uptake (Fig. 7). FA incorporation into TAG was increased almost 2-fold, whereas no alterations in label incorporation into FA or diacylglycerol were measured (data not shown). These data show that ubiquitination of the two carboxyl lysines regulates CD36 protein level, and this is reflected in corresponding changes in FA uptake.
FIGURE 7.

Effects of the double CD36 lysine mutant on FA uptake in CHO/hIR cells. CHO/hIR cells were transfected with either FLAG-CD36 wild type (WT) or with CD36 mutated at both lysines 469 and 472 (K/A). The upper panel shows levels of expressed CD36 with Ran as a loading control (Ctrl). The lower panel shows fatty acid uptake rates (means ± S.E.) from two experiments conducted in quadruplicate. **, p < 0.01. IB, immunoblot.
DISCUSSION
This study documented that ubiquitination is a mechanism by which the CD36 protein is acutely regulated in opposite directions by its FA ligand and insulin. Ubiquitination was observed using rat or human CD36 and in three cell types, suggesting that it is a conserved mechanism for regulating CD36 level. Effects on ubiquitination were also shown to be paralleled by changes in uptake and lipid incorporation of the FA. Finally the lysines present in the carboxyl terminus of CD36 were identified as the major ubiquitination sites.
CD36 plays a key role in tissue FA utilization, and appropriate regulation of its level is important especially in muscle which, as a result of its mass and high oxidative needs, exerts a large influence on overall metabolism (3, 30). Mice with muscle-targeted CD36 overexpression (5) exhibit less body fat, lower serum FA and TAG and higher fasting glucose and insulin. Muscle from these mice has enhanced ability for FA oxidation in response to contraction. In contrast, mice null for CD36 have high fasting serum lipids and low glucose and insulin (6). CD36-deficient muscle and heart exhibit a defect in FA uptake and oxidation that is compensated for by a severalfold increase in glucose utilization, changes that are reversed by muscle targeted CD36 rescue (31). In humans, there is strong evidence for the metabolic role of CD36. Common variants in the CD36 gene may impact high density lipoprotein metabolism and susceptibility to the metabolic syndrome (32). Abnormally high CD36 at the sarcolemma was reported in diabetic muscle and proposed to contribute to intramyocellular lipid accumulation (14). CD36 deficiency is associated with an abnormal blood lipid profile and with delayed lipid clearance after glucose or fat loading (7).
Our findings indicate that turnover and level of the CD36 protein can be acutely regulated by its FA ligand. The fact that FA can reduce CD36 content by inducing protein ubiquitination adds another layer to the ability of these molecules to regulate their own utilization (9, 33, 34). For example, FA can modulate activity of proteins involved in their cellular utilization such as the peroxisome proliferator-activated receptors (35, 36) and FoxO1 (9, 37) transcription factors. This study may be the first demonstration of FA modulation of protein ubiquitination, but it is likely that this ability extends, albeit selectively, to proteins other than CD36. We found that FA treatment did not enhance ubiquitination of the epidermal growth factor receptor, EGFR receptor, under basal conditions or in response to EGF (data not shown). In addition, GLUT1 was not detectably ubiquitinated in absence or presence of FA (data not shown). The effects of FA may target proteins important for FA processing or for cellular response to stress (fasting or exercise) and could involve specific E3 ubiquitin ligases or their adaptor proteins. Oleic acid increases expression of the E3 ligases atrogin-1/MAFbx and MuRF-1 in muscle cells (38). FA also enhance expression of TRB3 (37), increased by fasting in adipose tissue (39). TRB3, an adaptor for the ligase COP1, facilitates ubiquitination and degradation of acetyl-CoA carboxylase, leading to enhanced FA oxidation.
Ubiquitination is a major pathway for regulating subcellular localization and turnover of membrane proteins (40) with the most studied being the EGFR receptor (22, 41, 42). Increasing evidence, as recently reviewed (22), now supports the interpretation that ubiquitination is also involved in regulating levels of membrane transporters such as those for bile acids (43), dopamine (44), or the drug transporter P-Glycoprotein (45).
CD36 in muscle cells has been reported to cycle between endosomes and the sarcolemma (46), and this process is regulated by contraction and insulin (8, 47). Insulin increases sarcolemmal CD36 content, which enhances FA uptake and incorporation into TAG. The role of ubiquitination in CD36 cycling remains to be defined. Our data show that ubiquitination does not alter the fraction of CD36 that is at the plasma membrane as shown from comparing the ratio of sarcolemmal to total WT and K/A CD36. Thus ubiquitination does not appear linked to CD36 internalization, similar to findings with a number of other membrane receptors. For example, mutation of the major ubiquitination sites in EGFR receptor did not influence its internalization (47). Similarly, ubiquitination of the β2-adrenergic receptor was not necessary for its internalization but was critical for endosomal sorting to lysosomal degradation (48). In the case of CD36, ubiquitination was linked to degradation because the proteosomal inhibitor MG132 resulted in accumulation of ubiquitinated CD36. CD36 facilitated FA uptake and incorporation into TAG appeared a function of total protein level because they were enhanced in the presence of increased CD36 level whether the protein was present in the ubiquitinated form (MG132 treatment) or as the K/A-mutated form that is not ubiquitinated. Uptake in MG132-treated cells, which accumulate ubiquitinated CD36, was also sensitive to the membrane-impermeable SSO. Together these data suggest that CD36 ubiquitination occurs at the membrane and is independent of CD36 internalization. However, it is likely linked to sorting of endosomal CD36 between degradation pathways and recycling to the membrane. As a result, ubiquitination could acutely regulate the amount of sarcolemmal CD36. Trafficking kinetics of wild type and K/A CD36 could help clarify this point.
In summary, CD36 level and consequently FA uptake are acutely regulated by ubiquitination and degradation of the protein, a process that is modulated in opposite direction by FA and insulin. In vivo, these factors by regulating CD36 turnover would induce rapid changes in FA handling by muscle tissue. Insulin could regulate CD36 ubiquitination via at least two pathways, a direct one involving signaling and an indirect one via effects on FA metabolism. Which predominates would depend on the physiological or cellular context. Chronic FA intake and/or insulin resistance probably associate with abnormal CD36 ubiquitination. CD36 turnover may also integrate influences that are insulin- or FA-independent. Interestingly, the two lysines in the carboxyl terminus of CD36 are located adjacent to cysteines that can be palmitoylated (25) and to serines and threonines that may be subject to phosphorylation.
CD36 ubiquitination could alter other important functions of the protein such as binding/uptake of oxidized low density lipoprotein or signal transduction after TSP1 and other ligands. Because ubiquitination does not seem to alter membrane versus total CD36 distribution, these functions may be impacted in a way similar to FA uptake. However, this will have to be tested directly. More information related to the factors and molecular players involved in regulating CD36 ubiquitination should enhance our understanding of muscle FA metabolism under normal or disease conditions. It will also be important to evaluate other CD36 functions within the context of its ubiquitin-mediated turnover.
Acknowledgments
We thank Dr. Douglas Lublin (Washington University) and Dr. David Orlicky (University of Colorado Health Sciences Center, Aurora, CO), respectively, for FLAG-CD36 and GFP-CD36, Dr. Jeff Pessin (Albert Einstein College of Medicine) for the CHO/hIR, Dr. Kiran Madura (State University of New Jersey, Rutgers) for the FLAG ubiquitin constructs, and Dr. Xianlin Han (Bioorganic Chemistry) for help with the analysis of intracellular free FA.
*
This work was supported, in whole or in part, by National Institutes of Health Grants R01DK 33301 (to N. A.) and 2R01GM42259 (to P. S.), Clinical Nutrition Research Unit Grant DK56351, and NHLBI Cardiovascular System: Function, Regulation, Pharmacology Training Grant T32 HL-007275 (to J. S.). This work was also supported by the Human Center for Nutrition, Washington University (to X. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “_advertisement_” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
3
The abbreviations used are: FA, long chain fatty acid; KRH, Krebs-Ringer-Hepes; SSO, sulfosuccinimidyl oleate; TAG, triglycerides; BSA, bovine serum albumin; CHO, Chinese hamster ovary; CHO/hIR, CHO cells expressing the insulin receptor; HEK, human embryonic kidney; PBS, phosphate-buffered saline; GFP, green fluorescent protein; E3, ubiquitin-protein isopeptide ligase; EGF, epidermal growth factor.
References
- 1.Hajri, T., and Abumrad, N. A. (2002) Annu. Rev. Nutr. 22 383–415 [DOI] [PubMed] [Google Scholar]
- 2.Glatz, J. F., Luiken, J. J., and Bonen, A. (2001) J. Mol. Neurosci. 16 123–132 [DOI] [PubMed] [Google Scholar]
- 3.Coburn, C. T., Knapp, F. F., Jr., Febbraio, M., Beets, A. L., Silverstein, R. L., and Abumrad, N. A. (2000) J. Biol. Chem. 275 32523–32529 [DOI] [PubMed] [Google Scholar]
- 4.Harmon, C. M., and Abumrad, N. A. (1993) J. Membr. Biol. 133 43–49 [DOI] [PubMed] [Google Scholar]
- 5.Ibrahimi, A., Bonen, A., Blinn, W. D., Hajri, T., Li, X., Zhong, K., Cameron, R., and Abumrad, N. A. (1999) J. Biol. Chem. 274 26761–26766 [DOI] [PubMed] [Google Scholar]
- 6.Febbraio, M., Abumrad, N. A., Hajjar, D. P., Sharma, K., Cheng, W., Pearce, S. F., and Silverstein, R. L. (1999) J. Biol. Chem. 274 19055–19062 [DOI] [PubMed] [Google Scholar]
- 7.Yamashita, S., Hirano, K., Kuwasako, T., Janabi, M., Toyama, Y., Ishigami, M., and Sakai, N. (2007) Mol. Cell Biochem. 299 19–22 [DOI] [PubMed] [Google Scholar]
- 8.Bonen, A., Benton, C. R., Campbell, S. E., Chabowski, A., Clarke, D. C., Han, X. X., Glatz, J. F., and Luiken, J. J. (2003) Acta Physiol. Scand. 178 347–356 [DOI] [PubMed] [Google Scholar]
- 9.Nahlé, Z., Hsieh, M., Pietka, T., Coburn, C. C., Grimaldi, P. A., Michael, Q., Zhang, Debopriya Das, and Abumrad, N. A. (February 28, 2008) J. Biol. Chem. 10.1074/jbc.M706478200 [DOI] [PMC free article] [PubMed]
- 10.Greenwalt, D. E., Scheck, S. H., and Rhinehart-Jones, T. (1995) J. Clin. Investig. 96 1382–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinberg, G. R., Dyck, D. J., Calles-Escandon, J., Tandon, N. N., Luiken, J. J., Glatz, J. F., and Bonen, A. (2002) J. Biol. Chem. 277 8854–8860 [DOI] [PubMed] [Google Scholar]
- 12.Palanivel, R., and Sweeney, G. (2005) FEBS Lett. 579 5049–5054 [DOI] [PubMed] [Google Scholar]
- 13.Bastie, C. C., Nahle, Z., McLoughlin, T., Esser, K., Zhang, W., Unterman, T., and Abumrad, N. A. (2005) J. Biol. Chem. 280 14222–14229 [DOI] [PubMed] [Google Scholar]
- 14.Glatz, J. F., Bonen, A., Ouwens, D. M., and Luiken, J. J. (2006) Cardiovasc. Drugs Ther. 20 471–476 [DOI] [PubMed] [Google Scholar]
- 15.Osanai, T., and Okumura, K. (2007) Circ. Res. 100 1106–1108 [DOI] [PubMed] [Google Scholar]
- 16.Febbraio, M., and Silverstein, R. L. (2007) Int. J. Biochem. Cell Biol. 39 2012–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Endemann, G., Stanton, L. W., Madden, K. S., Bryant, C. M., White, R. T., and Protter, A. A. (1993) J. Biol. Chem. 268 11811–11816 [PubMed] [Google Scholar]
- 18.Nicholson, A. C., and Hajjar, D. P. (2004) Vascul. Pharmacol. 41 139–146 [DOI] [PubMed] [Google Scholar]
- 19.Liang, C. P., Han, S., Okamoto, H., Carnemolla, R., Tabas, I., Accili, D., and Tall, A. R. (2004) J. Clin. Investig. 113 764–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciechanover, A. (1994) Cell 79 13–21 [DOI] [PubMed] [Google Scholar]
- 21.Hochstrasser, M. (1996) Annu. Rev. Genet. 30 405–439 [DOI] [PubMed] [Google Scholar]
- 22.Miranda, M., and Sorkin, A. (2007) Mol. Interv. 7 157–167 [DOI] [PubMed] [Google Scholar]
- 23.Harmon, C. M., Luce, P., and Abumrad, N. A. (1992) Biochem. Soc. Trans. 20 811–813 [DOI] [PubMed] [Google Scholar]
- 24.Dong, C., Waters, S. B., Holt, K. H., and Pessin, J. E. (1996) J. Biol. Chem. 271 6328–6332 [DOI] [PubMed] [Google Scholar]
- 25.Tao, N., Wagner, S. J., and Lublin, D. M. (1996) J. Biol. Chem. 271 22315–22320 [DOI] [PubMed] [Google Scholar]
- 26.Abumrad, N. A., el-Maghrabi, M. R., Amri, E. Z., Lopez, E., and Grimaldi, P. A. (1993) J. Biol. Chem. 268 17665–17668 [PubMed] [Google Scholar]
- 27.Su, X., Lodhi, I. J., Saltiel, A. R., and Stahl, P. D. (2006) J. Biol. Chem. 281 27982–27990 [DOI] [PubMed] [Google Scholar]
- 28.Bastie, C. C., Hajri, T., Drover, V. A., Grimaldi, P. A., and Abumrad, N. A. (2004) Diabetes 53 2209–2216 [DOI] [PubMed] [Google Scholar]
- 29.Su, X., Han, X., Yang, J., Mancuso, D. J., Chen, J., Bickel, P. E., and Gross, R. W. (2004) Biochemistry 43 5033–5044 [DOI] [PubMed] [Google Scholar]
- 30.Luiken, J. J., Arumugam, Y., Dyck, D. J., Bell, R. C., Pelsers, M. M., Turcotte, L. P., Tandon, N. N., Glatz, J. F., and Bonen, A. (2001) J. Biol. Chem. 276 40567–40573 [DOI] [PubMed] [Google Scholar]
- 31.Irie, H., Krukenkamp, I. B., Brinkmann, J. F., Gaudette, G. R., Saltman, A. E., Jou, W., Glatz, J. F., Abumrad, N. A., and Ibrahimi, A. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 6819–6824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love-Gregory, L., Sherva, R., Sun, L., Wasson, J., Schappe, T., Doria, A., Rao, D., Hunt, S. C., Klein, S., Neuman, R. J., Permutt, M. A., and Abumrad, N. A. (2008) Hum. Mol. Genet, in press [DOI] [PMC free article] [PubMed]
- 33.Hajri, T., Hall, A. M., Jensen, D. R., Pietka, T. A., Drover, V. A., Tao, H., Eckel, R., and Abumrad, N. A. (2007) Diabetes 56 1872–1880 [DOI] [PubMed] [Google Scholar]
- 34.Yang, J., Sambandam, N., Han, X., Gross, R. W., Courtois, M., Kovacs, A., Febbraio, M., Finck, B. N., and Kelly, D. P. (2007) Circ. Res. 100 1208–1217 [DOI] [PubMed] [Google Scholar]
- 35.Muoio, D. M., and Koves, T. R. (2007) Appl. Physiol. Nutr. Metab. 32 874–883 [DOI] [PubMed] [Google Scholar]
- 36.Grimaldi, P. A. (2001) Curr. Opin. Clin. Nutr. Metab. Care 4 433–437 [DOI] [PubMed] [Google Scholar]
- 37.Martinez, S. M., Tanabe, K., Cras-Méneur, C., Abumrad, N. A., Bernal-Mizrachi, E., and Permutt, M. A. (2008) Diabetes 57 846–859 [DOI] [PubMed] [Google Scholar]
- 38.Zhou, Q., Du, J., Hu, Z., Walsh, K., and Wang, X. H. (2007) Endocrinology 148 5696–5705 [DOI] [PubMed] [Google Scholar]
- 39.Qi, L., Heredia, J. E., Altarejos, J. Y., Screaton, R., Goebel, N., Niessen, S., Macleod, I. X., Liew, C. W., Kulkarni, R. N., Bain, J., Newgard, C., Nelson, M., Evans, R. M., Yates, J., and Montminy, M. (2006) Science 312 1763–1766 [DOI] [PubMed] [Google Scholar]
- 40.Marmor, M. D., and Yarden, Y. (2004) Oncogene 23 2057–2070 [DOI] [PubMed] [Google Scholar]
- 41.Huang, F., Goh, L. K., and Sorkin, A. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 16904–16909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su, X., Kong, C., and Stahl, P. D. (2007) J. Biol. Chem. 282 21278–21284 [DOI] [PubMed] [Google Scholar]
- 43.Xia, X., Roundtree, M., Merikhi, A., Lu, X., Shentu, S., and Lesage, G. (2004) J. Biol. Chem. 279 44931–44937 [DOI] [PubMed] [Google Scholar]
- 44.Miranda, M., Wu, C. C., Sorkina, T., Korstjens, D. R., and Sorkin, A. (2005) J. Biol. Chem. 280 35617–35624 [DOI] [PubMed] [Google Scholar]
- 45.Zhang, Z., Wu, J. Y., Hait, W. N., and Yang, J. M. (2004) Mol. Pharmacol. 66 395–403 [DOI] [PubMed] [Google Scholar]
- 46.Bonen, A., Luiken, J. J., Arumugam, Y., Glatz, J. F., and Tandon, N. N. (2000) J. Biol. Chem. 275 14501–14508 [DOI] [PubMed] [Google Scholar]
- 47.Luiken, J. J., Koonen, D. P., Willems, J., Zorzano, A., Becker, C., Fischer, Y., Tandon, N. N., Van Der Vusse, G. J., Bonen, A., and Glatz, J. F. (2002) Diabetes 51 3113–3119 [DOI] [PubMed] [Google Scholar]
- 48.Shenoy, S. K., McDonald, P. H., Kohout, T. A., and Lefkowitz, R. J. (2001) Science 294 1307–1313 [DOI] [PubMed] [Google Scholar]