Phosphorylation of Sodium Channel Nav1.8 by p38 Mitogen-Activated Protein Kinase Increases Current Density in Dorsal Root Ganglion Neurons (original) (raw)
Abstract
The sensory neuron-specific sodium channel Nav1.8 and p38 mitogen-activated protein kinase are potential therapeutic targets within nociceptive dorsal root ganglion (DRG) neurons in inflammatory, and possibly neuropathic, pain. Nav1.8 channels within nociceptive DRG neurons contribute most of the inward current underlying the depolarizing phase of action potentials. Nerve injury and inflammation of peripheral tissues cause p38 activation in DRG neurons, a process that may contribute to nociceptive neuron hyperexcitability, which is associated with pain. However, how substrates of activated p38 contribute to DRG neuron hyperexcitability is currently not well understood. We report here, for the first time, that Nav1.8 and p38 are colocalized in DRG neurons, that Nav1.8 within DRG neurons is a substrate for p38, and that direct phosphorylation of the Nav1.8 channel by p38 regulates its function in these neurons. We show that direct phosphorylation of Nav1.8 at two p38 phospho-acceptor serine residues on the L1 loop (S551 and S556) causes an increase in Nav1.8 current density that is not accompanied by changes in gating properties of the channel. Our study suggests a mechanism by which activated p38 contributes to inflammatory, and possibly neuropathic, pain through a p38-mediated increase of Nav1.8 current density.
Keywords: channel modulation, SB203580, kinase inhibitor, inflammation, nociception, anisomycin, stress
Introduction
Although the molecular pathophysiology of inflammatory and neuropathic pain is not fully understood, it is known that tissue injury and nerve injury cause the release of pronociceptive cytokines and growth factors, and alter ion conductances, leading to sensitization and hyperexcitability of nociceptive neurons. For example, tumor necrosis factor α (TNF-α), a major pronociceptive cytokine (McMahon et al., 2005; Myers et al., 2006), can modulate nociceptor firing directly via TNF-α receptors or indirectly by inducing the production of other pronociceptive factors including neurotrophic growth factor (NGF) (McMahon et al., 2005; Pezet and McMahon, 2006). Pronociceptive cytokines activate downstream signaling pathways including p38 mitogen-activated protein kinase (MAPK), a process that has been implicated in inducing hyperexcitability of injured dorsal root ganglion (DRG) neurons (Schafers et al., 2003; Obata et al., 2004a). In fact, acute application of TNF-α to these neurons in culture increases the tetrodotoxin-resistant (TTX-R) current density in a p38-dependent manner (Jin and Gereau, 2006). These results suggest that Nav1.8 current density may be regulated by activated p38, but do not answer the question of whether the Nav1.8 channel is a substrate for direct phosphorylation by the activated p38 MAPK enzyme, or whether phosphorylation of the channel is necessary for the increase in current density.
The voltage-gated sodium channel Nav1.8 produces a slowly inactivating TTX-R current and is preferentially expressed in DRG and trigeminal ganglion neurons (Akopian et al., 1996; Sangameswaran et al., 1996), most of which are nociceptive (Djouhri et al., 2003), and thus is a likely target for injury-induced p38 MAPK modulation. Indeed, Nav1.8-null mice show a delayed response to carrageenan-evoked (Akopian et al., 1999) and attenuated NGF-evoked (Kerr et al., 2001) thermal hyperalgesia, and reduced visceral pain responses to capsaicin or acetylcholine (Laird et al., 2002). Acute application of inflammatory mediators on cultured DRG neurons increases the TTX-R current attributable to Nav1.8 (Gold et al., 1996; Zhang et al., 2002). Additionally, knockdown of Nav1.8 (Khasar et al., 1998; Gold et al., 2003; Joshi et al., 2006; Dong et al., 2007) and an isoform-specific small molecule blocker of this channel (Jarvis et al., 2007) have been shown to ameliorate both inflammatory and neuropathic pain symptoms. Because Nav1.8 contributes most of the inward current underlying the depolarizing phase of action potentials (Renganathan et al., 2001; Blair and Bean, 2002) and significantly influences neuronal excitability (Rush et al., 2006), acute as well as long-term regulation of this channel could significantly impact the excitability of nociceptive neurons after tissue or nerve injury.
We show here, for the first time, direct evidence that sodium channel Nav1.8 colocalizes with, and is a substrate for, p38 within DRG neurons. Activation of p38 increases Nav1.8 current density in DRG neurons without changing the activation or steady-state inactivation properties of the channel. We demonstrate that the p38-mediated increase in Nav1.8 current density in DRG neurons is dependent on direct phosphorylation on serine residues S551 and S556. Our findings indicate that p38 directly modulates Nav1.8 in vivo and provides a potentially rapid mechanism that can regulate nociceptive neuron excitability after injury.
Materials and Methods
Reagents.
Monoclonal pan-sodium channel antibody (K58/35) was purchased from Sigma (St. Louis, MO) and polyclonal anti-Nav1.8 antibody (lot AN#1) was purchased from Alomone Labs (Jerusalem, Israel). The specificity of the Nav1.8 antibody was tested on Western blots, immunoprecipitation (IP) assay, and immunofluorescence assays (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Rabbit anti-IgG antibody (Vector Laboratories, Burlingame, CA) was used as a control for immunoprecipitation. The rabbit polyclonal anti-phospho p38 MAPK (9211S) was purchased from Cell Signaling Technology (Danvers, MA) and the mouse monoclonal anti-peripherin antibody was purchased from Abcam (Cambridge, MA). Secondary antibodies for Western blot analysis, anti-rabbit IgG and anti-mouse IgG coupled to HRP, were purchased from DakoCytomation (Carpinteria, CA); secondary antibodies used in the immunostaining experiments, goat anti-rabbit IgG F(ab′)2 fragment Alexa Fluor 546, goat anti-rabbit IgG F(ab′)2 fragment Alexa Fluor 488, and goat anti-mouse IgG-Alexa 488 were purchased from Invitrogen (Carlsbad, CA), and goat anti-rabbit IgG-Cy3 was purchased from GE Healthcare Bio-Sciences (Piscataway, NJ). Recombinant activated p38α MAPK was purchased from the Roche Protein Expression group (Roche, Indianapolis, IN). Anisomycin; 4-(4-fluorophenyl)-2-(4-methylsulfonylphenyl)-5-(4-pyridyl)-1_H_-imidazole, HCl (SB203580); and 4-ethyl-2(_p_-methoxyphenyl)-5-(4′-pyridyl)-1_H_-imidazole, 2HCl (SB202474) were obtained from Calbiochem (La Jolla, CA). The [γ-32P]ATP was purchased from GE Healthcare Bio-Sciences.
Animals.
Adult male Sprague Dawley rats (250–300 g) were used according to Veterans Administration Connecticut Healthcare System Institutional Animal Care and Use Committee guidelines. For some experiments, rats were deeply anesthetized with ketamine/xylazine (80/10 mg/kg, i.p.) and complete Freund's adjuvant (CFA) (100 μl) was injected into the plantar surface of the right hindpaw. Two days later, rats were deeply anesthetized and perfused as described below for immunocytochemical investigation.
Plasmids.
The plasmid carrying full-length rat Nav1.8 insert (GenBank accession no. U53833), pRK-Nav1.8, was a gift from Dr. John Wood (University College London, London, UK). DNA inserts encoding the N and C termini, and the loops joining domains I and II (L1), domains II and III (L2), and domains III and IV (L3) were amplified by PCR using the Nav1.8 plasmid as a template and were subcloned into the _Nde_I and _Bam_H1 sites of pET15B. The plasmids are designated pET-Nav1.8-N (amino acids 1–125), pET-Nav1.8-L1 (amino acids 400–659), pET-Nav1.8-L2 (amino acids 890–1151), pET-Nav1.8-L3 (amino acids 1420–1472), and pET-Nav1.8-C (amino acids 1724–1956). The truncated derivative of pET-Nav1.8-L1 was designated as pET-Nav1.8-L1trunc (amino acids 400–516) and was produced by inserting a stop codon in pET-Nav1.8-L1. Amino acid substitutions and the internal deletion in L1 (see Figs. 5, 6) were accomplished using QuikChange XL site-directed mutagenesis reagents (Stratagene, La Jolla, CA). All constructs were confirmed by sequencing the inserts (Howard Hughes Medical Institute/Keck Biotechnology Resource Laboratory at Yale University).
Figure 5.
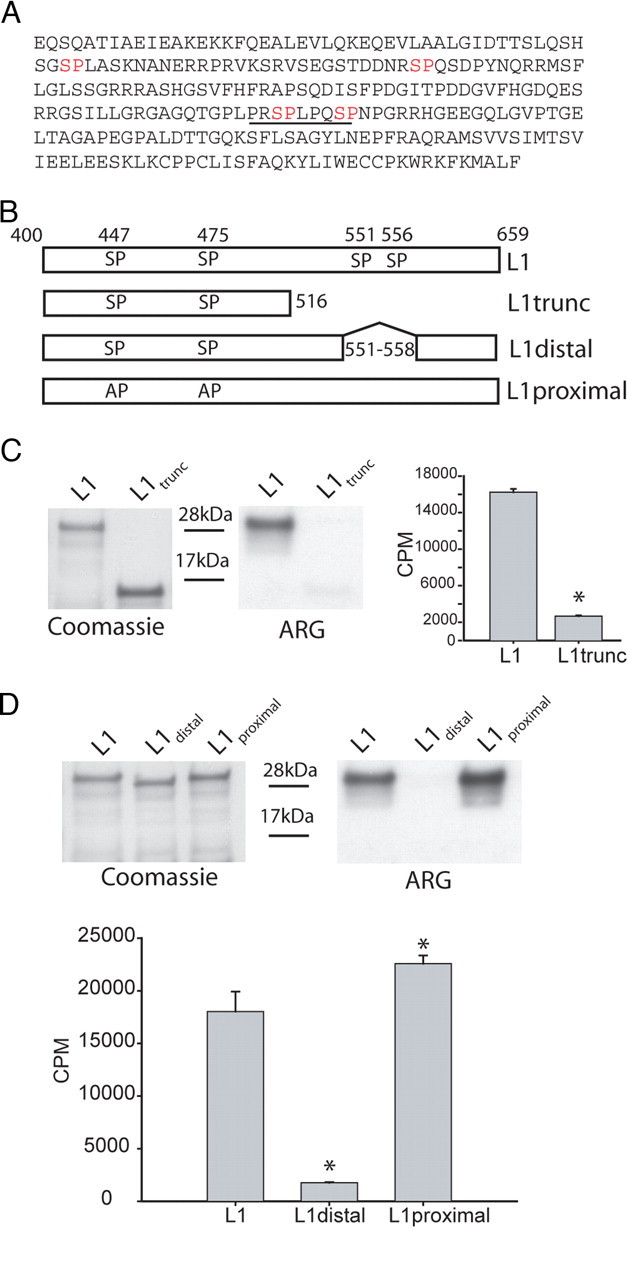
Identification of the p38 phosphorylation sites in L1 of Nav1.8. Deletion analysis and site-directed mutagenesis were used to identify the specific proline-directed serine residues required for p38 phosphorylation of L1. A, Amino acid sequence of the L1 region linking domains I and II shows four potential p38 phosphorylation sites (i.e., proline-directed serine or SP dipeptide; red lettering). B, Schematic diagram illustrating L1 (amino acids 400–659), L1-trunc (amino acids 400–516), L1-proximal (site-specific mutagenesis of S447 and S475 to alanine), and L1-distal (deletion of amino acids 551–558) that were expressed in bacteria as 6×His-tagged fusion proteins and used as substrates for p38 kinase assays. C, Autoradiogram illustrating that L1-trunc (deleting amino acids 517–659 in L1) is a poor substrate for p38 phosphorylation compared with L1 (right ARG). The relative level of each fusion protein used in the kinase assay is shown by Coomassie blue staining (left gel). Incorporation of 32P is determined as Cerenkov counts after p38 phosphorylation of L1 and L1-trunc (*p < 0.01; n = 4; mean ± SD). D, Autoradiogram illustrating that deleting amino acids 551–558 in L1 (L1-distal) disrupt p38 phosphorylation compared with L1 and mutation of the proximal proline-directed serine residues at 447 and 475 (right ARG). The relative level of each fusion protein in the kinase assay is shown by Coomassie blue staining (left gel). Incorporation of 32P is determined as Cerenkov counts after p38 phosphorylation of L1, L1-proximal, and L1-distal (*p < 0.01; n = 4; mean ± SD).
Figure 6.
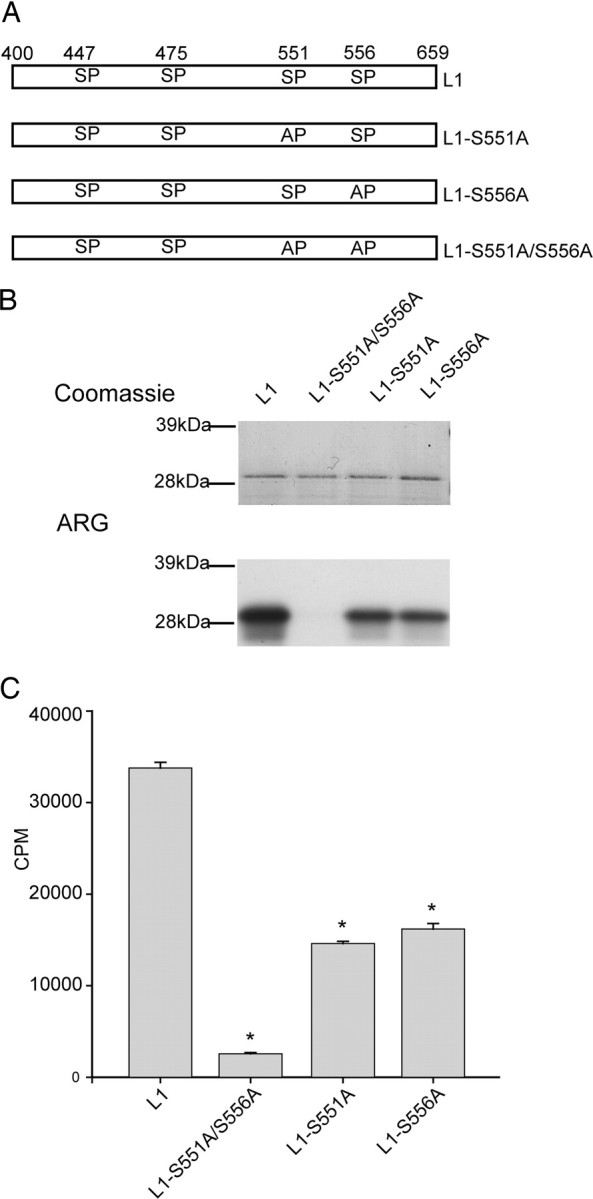
S551 and S556 contribute equally to the p38 phosphorylation of L1 of Nav1.8. Site-directed mutagenesis of S551 and S556 (single and double mutants) was used to examine their relative contribution to p38 phosphorylation of L1. A, Schematic diagram illustrating L1 (amino acids 400–659), L1-S551A (serine 551 mutated to alanine), L1-S556A (serine 556 mutated to alanine), and L1-S551A/S556A (serine 551 and serine 556 mutated to alanines) that were expressed in bacteria as 6×His-tagged fusion proteins and used as substrates for p38. B, Autoradiogram illustrating that S551 and S556 both contribute to the p38 phosphorylation of L1 (bottom ARG). The relative levels of each fusion protein used in the kinase assay visualized by Coomassie blue (top gel). C, Cerenkov counts of p38 phosphorylation demonstrate that single- and double-mutant substrates are significantly less phosphorylated compared with WT (*p < 0.01; n = 4; mean ± SD).
DRG culture.
Adult Sprague Dawley rats (1–2 months of age) were deeply anesthetized with ketamine/xylazine (80/10 mg/kg, i.p.) and decapitated, and L4 and L5 DRG were quickly removed and desheathed in sterile complete saline solution (CSS) (in mm: 137 NaCl, 5.3 KCl, 1 MgCl2, 25 d-sorbitol, 3 CaCl2, and 10 HEPES, pH 7.2 adjusted with NaOH), as described previously (Rizzo et al., 1994). The tissue was then enzymatically digested at 37°C for 25 min with collagenase A (1 mg/ml; Roche) in CSS and for 25 min with collagenase D (1 mg/ml; Roche) and papain (30 U/ml; Worthington, Lakewood, NJ) in CSS at 37°C. Treated tissues were gently centrifuged (100 × g for 3 min), and the pellets were triturated in DRG media (1:1 DMEM/F12, 10% FCS, 100 U/ml penicillin, and 0.1 mg/ml streptomycin) containing 1.5 mg/ml BSA (fraction V; Sigma) and 1.5 mg/ml trypsin inhibitor (Sigma). Cells were then plated on poly-l-ornithine–laminin-coated glass coverslips, flooded with DRG media after 1 h, and incubated at 37°C in a humidified 95% air–5% CO2 incubator.
Immunohistochemistry.
Rats injected with CFA in the right hindpaw to induce inflammation were killed 2 d later to study the effect on inflammation-induced activation of p38. Rats were deeply anesthetized with ketamine/xylazine (80/10 mg/kg, i.p.) and perfused with 4% paraformaldehyde in 0.14 m Sorensen's phosphate buffer, pH 7.4. L4 and L5 DRGs were harvested, rinsed in PBS, and cryoprotected overnight in 30% sucrose in PBS. Cryosections were incubated with polyclonal antibody against phosphorylated p38 (1:1000; Cell Signaling Technology) and monoclonal antibody against peripherin (1:1000). Secondary antibodies were goat anti-rabbit Cy3 and goat anti-mouse-Alexa Fluor 488 (1:1000).
For Nav1.8 and p38 colocalization, anisomycin-treated DRG cultures and DRG tissue sections were incubated sequentially in the following: (1) blocking solution (PBS with 5% normal goat serum, 1% bovine serum albumin, 0.1% Triton X-100, and 0.02% sodium azide) for 30 min, (2) anti-phosphorylated p38, (3) PBS wash, (4) goat anti-rabbit IgG F(ab′)2 fragment Alexa Fluor 488 or 546, (5) PBS wash, (6) anti-Nav1.8, (7) PBS wash, (8) goat anti-rabbit IgG F(ab′)2 fragment Alexa Fluor 546 or 488, and (9) PBS wash. Control experiments in which the second primary antibody was omitted, with sequential first and second secondary antibody incubations, only exhibited the fluorochrome of the first secondary antibody, confirming the specificity of the immunostaining signal.
Sections were examined with a Nikon (Tokyo, Japan) PCM 2000 confocal microscope or Nikon E800 light microscope. Images were collected with SimplePCI (C-Imaging Systems, Cranberry Township, PA) or Meta-Vue (Molecular Devices, Downington, PA) software, and were arranged with Adobe (San Jose, CA) Photoshop.
Nav1.8 channel phosphorylation by p38.
Using a protocol for immunoprecipitation of Nav1.8 channels from DRG neurons (Choi et al., 2006), lumbar (L2–L6) DRGs from two young adult male Sprague Dawley rats (250–275 g) were homogenized in 0.5 ml of buffer (200 mm NaCl, 20 mm Tris, pH 7.4), 1% Triton X-100, and Complete protease inhibitor mixture containing 1 mm EDTA (Roche). Triton X-100-soluble proteins were collected after centrifugation for 20 min at 15,000 × g at 4°C and precleared using protein A-agarose (Upstate, Lake Placid, NY). The cleared DRG lysate was incubated with 2 μg of Nav1.8-specific polyclonal antibody (Alomone Labs) for 2–3 h at 4°C before addition of protein A-agarose for an additional 1.5 h at 4°C. The beads were washed extensively in binding buffer and incubated with activated p38 as described below.
Purification of 6×His-tagged fusion proteins.
Purification of 6×His-tagged fusion proteins was described previously (Choi et al., 2006). Briefly, BL21-Rossetta cells (Stratagene) were transformed with constructs encoding 6×His fusion proteins of each of the major cytoplasmic regions of Nav1.8 (N terminus, L1, L2, L3, and C terminus) and grown in the presence of carbenicillin (100 μg/ml) and chloramphenicol (50 μg/ml). Large-scale growths were inoculated using an overnight starter culture (1:100) and grown to an OD595 of 0.6 at 37°C in 2XYT media. The cells were chilled on ice to <10°C and 1 mm isopropyl β-d-thiogalactoside was added to the culture, which was then incubated with shaking for 4 h at 30°C to induce recombinant protein expression. The bacterial pellets were collected by centrifugation at 6000 × g for 10 min, washed in ice-cold PBS, and frozen at −80°C.
Frozen bacterial pellets were resuspended in 20 ml of ice-cold lysis buffer (10 mm Tris, pH 8, 0.1 mm EDTA, 300 mm NaCl) supplemented with protease inhibitor mixture minus EDTA (Roche) and lysed using a Microfluidics (Newton, MA) device. Lysed cells were incubated on ice for 45 min in 1% Tween 20, and then sonicated and centrifuged at 10,000 × g for 20 min. The supernatant was diluted with 5–10 vol of 50 mm phosphate buffer, pH 8.0 plus 300 mm NaCl, and applied to nitrilotriacetic acid-agarose (Invitrogen). Bound proteins were washed in 5 column vol of phosphate buffer plus 5 mm imidazole and finally in 10 column vol of 25 mm imidazole. Enriched 6×His-tagged proteins were eluted using 250 mm imidazole in phosphate buffer. Positive fractions were pooled and concentrated using Amicon Ultra4 5000 molecular weight cutoff concentrators (Millipore, Bedford, MA). The proteins were diluted two to three times in phosphate buffer and reconcentrated in Amicon Ultra4 to reduce the imidazole in the sample before being stored in phosphate buffer/10% glycerol at −80°C. Purified fusion proteins were quantified using Bradford assay (BSA as a standard) and equal protein levels were visually inspected using Coomassie blue staining. The molecular weights of the purified proteins are consistent with the predicted size: N terminus, 14 kDa; L1, 27 kDa, L2, 27 kDa; L3, 6 kDa; C terminus, 24 kDa.
In vitro p38 kinase assays.
Phosphorylation assays using immunoprecipitation (IP) samples or 6×His fusion proteins were performed in 200 and 50 μl reactions, respectively, in 25 mm HEPES, pH 7.4, 2 mm DTT, 10 mm MgCl2, 100 μm ATP, and 5 μCi of [γ-32P]ATP. Activated p38 (final concentration, 1 ng/μl) was added to each reaction tube (30°C) and allowed to proceed for 2–3 min. For the IP samples, the kinase reactions were performed as described above in the presence or absence of the active p38-specific inhibitor (SB203580), and were terminated with 5 vol (1 ml) of PBS containing 20 mm EDTA. Protein A beads were again collected by centrifugation and washed three times in PBS plus EDTA to further reduce the background. Proteins were eluted from the beads using 2× lithium dodecyl sulfate–PAGE sample buffer for gel analysis. The presence of the sodium channel was verified by Western analysis using the pan-sodium channel antibody, and the phosphorylated channels were detected via autoradiography. Phosphorylation of bacterially expressed fusion proteins was performed as described above for fusion proteins immobilized on beads, or on purified samples as described previously using Cerenkov counting in a scintillation counter (Wittmack et al., 2005). Similar results were obtained for all kinase assays on soluble or bead-immobilized proteins using different preparations of IP samples and 6×His fusion proteins.
Western blotting and autoradiography.
The IP proteins were separated on NUPAGE Bis-Tris 4–12% gels (Invitrogen) using a 3-(_N_-morpholino)propanesulfonic acid-based buffer system; the fusion protein samples were processed on 12% gels using MES as the running buffer (Invitrogen). Western blotting for native Nav1.8 samples was performed as described previously (Choi et al., 2006). Briefly, proteins were transferred to nitrocellulose for 2 h at 30 V using the XCell II system (Invitrogen) and blocked overnight in 5% nonfat milk. The membrane was washed in TBST (Tris-buffered saline plus 0.2% Tween 20) five times for 10 min each after primary and secondary antibody incubations. Chemiluminescent detection was performed using Lightening Plus (PerkinElmer, Boston, MA) and detected on Hyperfilm ECL (Amersham). Gels for autoradiography were first stained by Coomassie blue and destained to compare loading levels of fusion proteins tested in the kinase assays.
Electroporation of Nav1.8 channels into mouse Nav1.8−/− DRG neurons.
DRG neurons from Nav1.8-null mice (Nav1.8−/− on C57BL/6 genetic background) (Akopian et al., 1999) were transfected with Nav1.8 or its mutant derivatives S551A, S556A, and the double mutant S551–556A using the Nucleofector system (Amaxa, Gaithersburg, MD) as described previously (Choi et al., 2006). Briefly, adult Nav1.8-null mice were deeply anesthetized and decapitated, and 10 pairs of DRG were quickly removed and desheathed in sterile calcium- and magnesium-free HBSS, pH 7.2. The tissue was treated at 37°C for 50 min with dispase (5 mg/ml; Roche), collagenase A (2 mg/ml; Roche), and DNase I (0.1 mg/ml; Roche) in HBSS, and triturated in culture media (DMEM supplemented with GlutaMax I, 10% FCS, 2 mm l-glutamine, 16.5 mm NaHCO3, 6 g/L glucose, 100 U/ml penicillin, and 0.1 mg/ml streptomycin). Cells were centrifuged for 5 min at 80 × g, and the pellet was resuspended in 100 μl of rat neuron Nucleofector solution (VPG-1003; Amaxa) to a final concentration of 104 cells/μl (neurons and non-neuronal cells). Wild-type (WT) Nav1.8 or mutant plasmids (10 μg) and 2 μg of EGFP (enhanced green fluorescent protein) DNA (Clontech, Palo Alto, CA) were mixed with DRG cell suspension. Cells were electroporated using the preset program O-003, allowed to recover in Ca2+-free DRG media for 10 min at 37°C in a humidified 95% air–5% CO2 incubator, and plated on poly-l-ornithine–laminin-coated glass coverslips. The cells were flooded with DRG medium after 1 h and returned to the incubator. Using these methods, we routinely obtain transfection efficiencies of 15–20%.
Electrophysiological recordings.
Whole-cell patch-clamp recordings of sodium currents were performed 24–34 h after plating of native rat DRG neurons, or 24–52 h after plating of transfected Nav1.8−/− mouse DRG neurons. Almost all of the transfected mouse DRG neurons (99%) that expressed green fluorescent protein (GFP) also expressed slowly inactivating TTX-R sodium currents. Because slowly inactivating TTX-R currents are not observed in untransfected Nav1.8−/− neurons or Nav1.8−/− neurons transfected with GFP alone (Cummins et al., 2001; Herzog et al., 2003; Rush et al., 2005a; Choi et al., 2006), this confirmed that most of the transfected cells that displayed green fluorescence also had been cotransfected successfully with the recombinant Nav1.8 channel constructs.
Conventional whole-cell patch-clamp recordings were made from small DRG neurons (≤25 μm diameter), using Axopatch 200B amplifiers (Molecular Devices). For currents >20 nA, we used a 50 MΩ feedback resistor (β of 0.1), which can pass up to 200 nA. Micropipettes (0.6–0.9 MΩ) were pulled from capillary glass (PG10165-4; WPI, Sarasota, FL) with a Flaming Brown P80 micropipette puller (Sutter Instruments, Novato, CA). The pipette solution contained the following (in mm): 140 CsF, 1 EGTA, 10 NaCl, and 10 HEPES, pH 7.3 (osmolarity was adjusted to 310 mOsm/L with sucrose, and pH was adjusted by CsOH). The Na+ bath solution contained the following (in mm): 70 NaCl, 70 choline-Cl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 glucose, and 10 HEPES, pH 7.3 (osmolarity was adjusted to 320 mOsm/L with sucrose, and pH was adjusted by NaOH). To isolate TTX-R currents, 20 mm TEA-Cl, 0.1 mm CdCl2, and 300 nm TTX were included in the bath solution to inhibit endogenous K+, Ca2+, and TTX-sensitive Na+ currents, respectively. The fluoride-based pipette solution was used in these studies to facilitate the separation of Nav1.8 slowly inactivating TTX-R current from the Nav1.9 persistent TTX-R current (Cummins et al., 1999; Rush et al., 2005b; Choi et al., 2006). It is important to note that activation and steady-state inactivation properties of Nav1.8 are not altered in fluoride-based or chloride-based pipette solutions (Saab et al., 2003; Coste et al., 2004). Pipette potential was zeroed before seal formation and voltages were not corrected for liquid junction potential. Whole-cell Na+ currents were filtered at 5 kHz and acquired at 50 kHz, using Clampex 8.2 software (Molecular Devices). Capacity transients were cancelled and series resistance (1.1 ± 0.1 MΩ; n = 231) was compensated (90–95%) in all experiments. All experiments were performed at room temperature (21–25°C).
Electrophysiological protocols and data analysis.
For voltage-clamp studies, wild-type DRG neurons were held at −70 mV, and recording was started 10 min after establishing whole-cell configuration to allow currents to stabilize, and to minimize the contamination of residual persistent TTX-R Nav1.9 currents (Cummins et al., 1999; Choi et al., 2006). Cells with a leakage current that was >500 pA were discarded. Leakage current was digitally subtracted on-line using hyperpolarizing control pulses, applied after the test pulse (P/N subtraction). Data were analyzed using Clampfit 8.2 software (Molecular Devices) and Origin 7.5 (Microcal Software, Northampton, MA).
A stock solution of anisomycin (Sigma) was prepared in DMSO and added to the recording chamber to produce a final concentration of 10 μg/ml anisomycin in 0.1% DMSO. SB203580 and SB202474 were dissolved in distilled water and used at a final concentration of 10 μm. All chemicals for p38 activation and inactivation were purchased from Calbiochem. To activate p38, cultured native DRG neurons were treated with 10 μg/ml anisomycin for 30 min. Medium was then replaced with the bath solution without the drug, and electrophysiological recordings were performed during a 30 min interval after the withdrawal of anisomycin. In experiments using the p38 inhibitor SB203580 or the inactive analog SB202474, cells were treated with 10 μm of the drug for 60 min preceding the recordings. In the experiments that included anisomycin and p38 inhibitors, cells were treated for 30 min with SB203580 or SB202474 and then 30 min with anisomycin in the continued presence of the inhibitor before starting the recording of sodium currents. Maximal peak currents were measured in native and transfected DRG neurons under control conditions or after the various treatments. We have reported the peak current density to compensate for variations in cell sizes as reflected in their measured capacitance.
Statistics.
The F statistic associated with ANOVA was used to test the null hypothesis of no differences between experimental conditions, and a post hoc Tukey's test was conducted to examine significance at an α level of 0.05 in multigroup comparisons. Student's t test was used to examine significance between two groups. Descriptive data are presented as the mean ± SD for the biochemistry, and mean ± SE for the electrophysiology, experiments. The ANOVA and post hoc analyses and Student's t test were conducted using SPSS 12.0 or Origin 7.5 (Microcal).
Results
Sodium channel Nav1.8 and p38 MAPK colocalize in DRG neurons
Nav1.8 is the main contributor to the inward sodium current in DRG neurons (Renganathan et al., 2001; Blair and Bean, 2002); thus, acute or chronic modulation of this channel could significantly influence DRG neuron excitability. The p38 MAPK has been shown to be activated in DRG neurons after inflammation or nerve injury (Ji et al., 2002; Obata et al., 2004a,b,c). Because previous studies have reported that the dominant nodal sodium channel Nav1.6 is a substrate of p38 (Wittmack et al., 2005) and that the TTX-R current in DRG neurons is modulated by p38 (Jin and Gereau, 2006), we hypothesized that Nav1.8 channels are substrates for activated p38.
We investigated whether Nav1.8 and activated (phosphorylated) p38 are colocalized in DRG neurons. We first examined the activation of p38 in DRG neurons after CFA-induced inflammation of rat hindpaw (Fig. 1). Activated p38 was detected in ipsilateral small diameter, peripherin-positive, DRG neurons (Fig. 1A, right, yellow), and in medium-sized, peripherin-negative, DRG neurons (Fig. 1A, right, red). Peripherin is used in these studies as a marker for unmyelinated nociceptor primary afferents (Goldstein et al., 1991; Potrebic et al., 2003), which have been shown to express Nav1.8 (Rush et al., 2005b; Stirling et al., 2005). We then asked whether Nav1.8 and phospho-p38 are colocalized in DRG neurons. Nav1.8-specific immunolabeling (green) colocalized (yellow) with immunolabeling of activated p38 (red) in adult rat small- and medium-diameter DRG neurons (Fig. 1B). The colocalization of the two proteins suggests that p38 could contribute to the regulation of Nav1.8 current density and/or gating properties when activated in nociceptive neurons after stress or injury.
Figure 1.
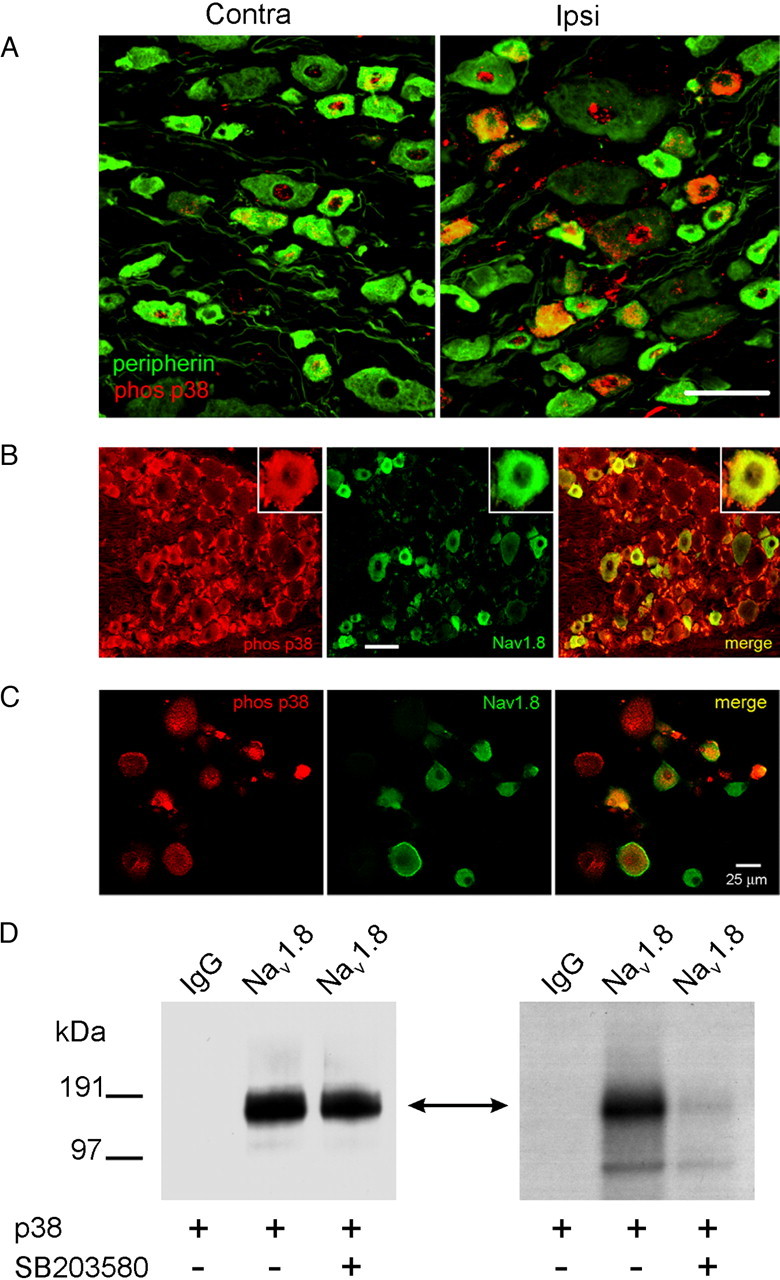
Native Nav1.8 sodium channels are colocalized and phosphorylated by p38 MAPK. A, CFA injection induces activated p38 in DRG neurons. Two days after CFA injection into the hindpaw, activated (phosphorylated) p38 immunolabeling (red) is exhibited predominantly in ipsilateral (right) compared with contralateral (left) DRG neurons. Peripherin (green) labels small-diameter neurons that project unmyelinated fibers. Colocalization of phosphorylated p38 and peripherin appears yellow. Scale bar, 50 μm. B, Nav1.8 (green) and phospho-p38 (red) immunolabeling in DRG neurons. The merged image demonstrates colocalization of Nav1.8 and phospho-p38 (yellow) in a subset of small- to medium-size DRG neurons. Scale bar, 50 μm. C, Phosphorylated p38 and Nav1.8 colocalization in anisomycin-treated DRG neurons. Phosphorylated p38 (red) and Nav1.8 (green) colocalization (yellow) is displayed in some DRG neurons, whereas other neurons exhibit only phosphorylated p38 or Nav1.8 immunolabeling. D, Native Nav1.8 is phosphorylated by activated p38. Lane 1 of the Western (left panel) and the ARG (right panel) shows that no protein product was immunoprecipitated nor phosphorylated by the control IgG antibody. Lanes 2 and 3 of the Western blot demonstrate that equal levels of immunoprecipitated Nav1.8 channel were used in the kinase assay. Lanes 2 and 3 of the ARG demonstrate that p38 phosphorylation of the Nav1.8 immunoprecipitated product (double-headed arrow) is only observed in the absence of the specific p38 inhibitor (SB203580; 10 μm).
To determine the extent of colocalization of Nav1.8 and phospho-p38 under the experimental conditions that we used in this study, cultured DRG neurons that had been treated with anisomycin for 30 min were labeled with phosphorylated p38 and Nav1.8 antibodies (Fig. 1C). Almost 62% of neurons in culture were immunolabeled by the Nav1.8 antibody (74 of 120 neurons), with approximately one-half of these neurons also displaying phosphorylated p38 immunolabeling (38 of 74). Thus, the effect of activation of p38 on Nav1.8 TTX-R current is expected to be diluted by inclusion in the analysis of current density measurements from a subpopulation of neurons that do not produce phosphorylated p38.
Native Nav1.8 sodium channel is phosphorylated by p38 MAP kinase
To determine whether full-length Nav1.8 channel is a substrate for p38 MAP kinase, native Nav1.8 channels from DRG were immunoprecipitated using a Nav1.8-specific polyclonal antibody, and the IP complex was incubated with activated p38 (Fig. 1D). A control IgG antibody was used to validate the specificity of the IP reaction and kinase assay. Pan-sodium channel antibody cross-reacted with a protein of the expected molecular weight of Nav1.8 in the Nav1.8-specific IP protein complex (Fig. 1D, left). The absence of a protein cross-reacting with the pan-sodium channel antibody in the control IP sample supports the specificity of the anti-Nav1.8 channel antibody used in this assay (Fig. 1D, left; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Control and Nav1.8 IP complexes were incubated with recombinant activated p38 and [γ-32P]ATP in the presence (10 μm, final concentration) or absence of an active p38-specific inhibitor (SB203580). Equivalent samples of the Nav1.8 IP complex were used (confirmed by similar immunolabeled bands on the Western blot) in the kinase assay. The autoradiogram (ARG) in Figure 1D, right, is consistent with native Nav1.8 being a substrate for p38. Including the specific p38 inhibitor (SB203580) in the reaction mixture prevented Nav1.8 phosphorylation, further supporting the conclusion that phosphorylation is mediated by p38 and not by other kinases that might have coimmunoprecipitated with the channel.
Anisomycin, a p38 activator, modulates Nav1.8 sodium channel current density in DRG neurons
To investigate whether p38 modulates Nav1.8 currents, we treated DRG neurons with anisomycin, a cell-permeable antibiotic routinely applied to activate p38 (Cano and Mahadevan, 1995; Ogawa et al., 2004; Wittmack et al., 2005). Native DRG neurons in culture for 24 h were treated with anisomycin (10 μg/ml) or vehicle (0.1% DMSO) for 30 min before Nav1.8 TTX-R sodium currents were measured (Fig. 2). As shown in Figure 2A, peak current density of Nav1.8 (667.3 ± 74.4 pA/pF; n = 16) was significantly increased after treatment of DRG neurons with anisomycin (893.4 ± 66.8 pA/pF; n = 16; p < 0.05). However, the time constants for activation and inactivation kinetics of Nav1.8 in treated DRG neurons obtained by fitting the data with a monoexponential function were not changed at any voltage [for example, τ was 1.64 ± 0.14 ms (activation) and 19.38 ± 2.24 ms (inactivation) at −10 mV for vehicle-treated neurons (n = 12); τ was 1.70 ± 0.24 ms (activation) and 18.58 ± 1.90 ms (inactivation) at −10 mV for anisomycin-treated neurons (n = 9)]. Therefore, the apparent increase in late current at the end of the 40 ms pulse reflects an increase in the peak current amplitude and not a change in kinetics of inactivation.
Figure 2.
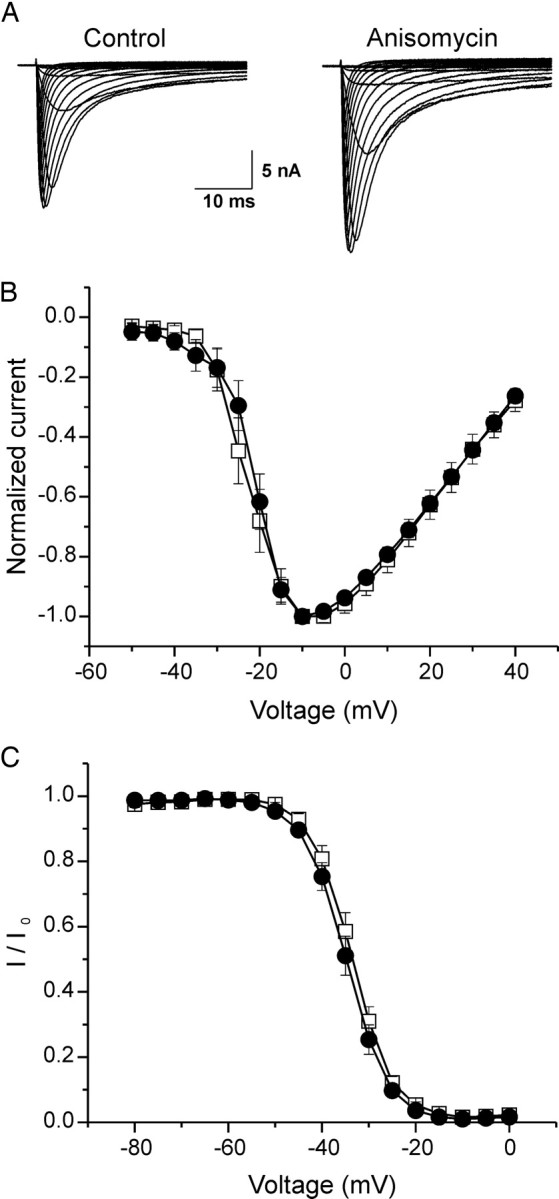
Anisomycin increases peak current density of Nav1.8 in native rat DRG neurons. A, Representative families of current traces that were recorded in the presence of 0.3 μm TTX are shown, in which cells were depolarized to a variety of potentials (−50 to +40 mV) from a holding potential of −70 mV to elicit Nav1.8 sodium current. Cells were treated with vehicle (DMSO; control) or anisomycin (10 μg/ml) for 30 min before recording. B, Mean normalized I–V curves for Nav1.8 currents pretreated with vehicle (□; _V_1/2 = −20.28 ± 0.26 mV; k = 5.28 ± 0.18; n = 12) or anisomycin (●; _V_1/2 = −19.42 ± 0.46 mV; k = 5.04 ± 0.32; n = 9) for 30 min revealed an identical voltage dependence of activation. C, Best-fit curves of steady-state fast inactivation were generated by Boltzmann distribution equation: vehicle (□; n = 9; _V_1/2 = −33.54 ± 0.65 mV; k = 4.86 ± 0.25); anisomycin (●; n = 8; _V_1/2 = −35.16 ± 0.73 mV; k = 5.10 ± 0.27). Error bars indicate SE.
We also examined the effects of anisomycin treatment on voltage dependence of activation and steady-state fast inactivation of Nav1.8 (Fig. 2B,C). Nav1.8 TTX-R currents have been shown to undergo steady-state fast inactivation and slow inactivation depending on the stimulus duration (Ogata and Tatebayashi, 1993; Rush et al., 1998; Blair and Bean, 2003; Choi et al., 2007); fast inactivation occurs when the stimulus is applied in short pulses of tens of milliseconds, whereas slow inactivation occurs when a stimulus is applied for longer duration (seconds), or when a train of pulses is applied. Because we have used shorter prepulses (500 ms), the remaining amplitude indicates available current from steady-state fast-inactivated channels.
The voltage-dependent activation of the channel (_V_1/2 = −20.28 ± 0.26 mV; slope k = 5.28 ± 0.18 mV; n = 12) (Fig. 2B) under control conditions (vehicle) were not significantly different after treatment of DRG neurons with anisomycin: (_V_1/2 = −19.42 ± 0.46 mV; k = 5.04 ± 0.32 mV; n = 9) (Fig. 2B). For steady-state fast inactivation, we determined the channel availability after 500 ms prepulses at various potentials (Fig. 2C). The _V_1/2 of voltage dependence of fast inactivation was −33.54 ± 0.65 mV (n = 9) using vehicle alone, and −35.16 ± 0.73 mV (n = 8) after anisomycin treatment. Compared with vehicle alone, the voltage-dependent activation and steady-state fast inactivation of Nav1.8 current after anisomycin treatment of DRG neurons were not significantly different.
The p38 inhibitor SB203580 prevents anisomycin-induced increase in Nav1.8 current density in DRG neurons
To determine whether the increase in Nav1.8 current density after anisomycin treatment is caused specifically by p38 activity, DRG neurons were preincubated with the p38-specific inhibitor SB203580, or the inactive analog SB202474 as a negative control, for 30 min before anisomycin treatment (Fig. 3A). Sixty minute treatment of overnight cultures of DRG neurons with SB202474 or SB203580 did not significantly change the current density of native Nav1.8 (SB202474, 649.9 ± 94.8 pA/pF, n = 32; SB203580, 626.8 ± 78.9 pA/pF, n = 31), suggesting that p38 is not activated before anisomycin treatment of these cultures. Pretreatment of sister cultures with the inactive analog SB202474 for 30 min followed by a 30 min cotreatment with anisomycin increased the current density of Nav1.8 (826.5 ± 77.0 pA/pF; n = 30), similar to that for Nav1.8 current density observed with anisomycin treatment alone (911.8 ± 57.0 pA/pF; n = 30). In contrast, pretreatment of DRG neurons with SB203580 for 30 min followed by cotreatment with anisomycin (Fig. 3B) blocked the increase in Nav1.8 current density (586.6 ± 78.8 pA/pF; n = 30; p < 0.05). Together with the data in Figure 1, these observations favor the hypothesis that anisomycin-induced increase of native Nav1.8 current density results from p38 phosphorylation of the channel.
Figure 3.

The specific p38 MAPK inhibitor SB203580 blocks anisomycin-mediated increase in Nav1.8 current density. A, Representative families of current traces that were recorded in the presence of 0.3 μm TTX are shown, in which cells were depolarized to a variety of potentials (−50 to +40 mV) from a holding potential of −70 mV to elicit Nav1.8 sodium current. Cells were treated with anisomycin (10 μg/ml) for 30 min, the specific p38 MAP kinase inhibitor SB203580 (10 μm) for 30 min followed by cotreatment with anisomycin for 30 min, or the inactive analog SB202474 (10 μm) for 30 min followed by cotreatment with anisomycin for 30 min. B, The increase in peak current density of Nav1.8 induced by anisomycin treatment is blocked by treatment with SB203580 but not by the inactive analog SB202474. *p < 0.05. Error bars indicate SE.
Nav1.8 L1, the cytoplasmic loop joining domains I and II, is phosphorylated by p38
The schematic diagram in Figure 4A illustrates the structural topology common to all eukaryotic sodium channels (Catterall, 2000). Potential p38 phosphorylation sites (i.e., proline-directed serine, SP dipeptide) are present within several major cytoplasmic regions of Nav1.8 sodium channel (N and C termini, L1 and L2). These channel regions were produced and purified as 6×His-tagged fusion proteins as described in Materials and Methods. Purified fusion proteins (shown in Fig. 4B, top) were incubated with recombinant activated p38 in the presence of [γ-32P]ATP to determine whether they are substrates for this enzyme. The autoradiogram in Figure 4B of the reaction products shows that L1 is the only p38 substrate. Quantification of the phosphorylation products by Cerenkov counting (Fig. 4C) shows ∼10-fold more 32P incorporation into L1 (19,972 ± 661 cpm) compared with the other channel fragments: N terminus (2126 ± 149 cpm), L2 (2369 ± 278 cpm), L3 (2694 ± 510 cpm), and C terminus (490 ± 190 cpm); the counts from the substrates other than L1 are considered background levels because L3 carries no putative p38 phosphorylation sites.
Figure 4.
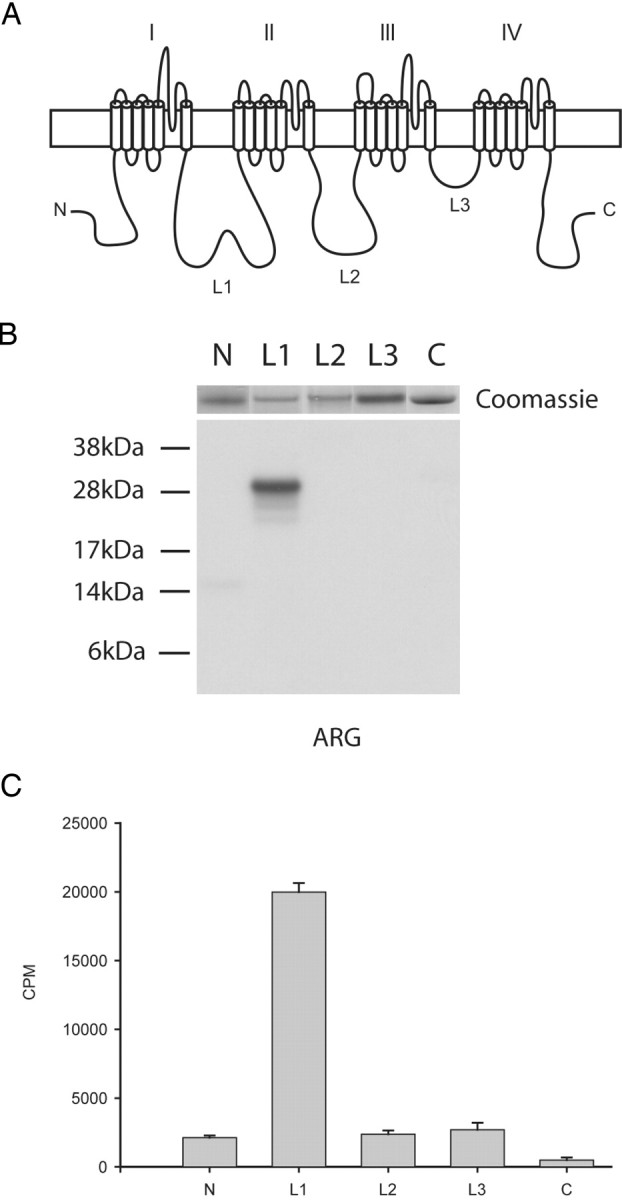
Nav1.8 L1 is phosphorylated by p38 MAPK. A, Schematic diagram illustrating the cytoplasmic fragments (N, 14 kDa; L1, 27 kDa; L2, 27 kDa; L3, 6 kDa; C terminus, 24 kDa) that were expressed in bacteria as 6×His-tagged fusion proteins and tested as substrates for p38. B, Autoradiogram illustrating that the L1 fusion protein was the only 32P-labeled substrate phosphorylated by p38 (see Materials and Methods). The relative levels of each fusion protein present in the kinase assay can be seen in the top panel as stained by Coomassie blue. C, Quantitative measurements of phosphorylation as assessed using the P81 filter assay and Cerenkov counting (n = 4; mean ± SD).
L1 is phosphorylated by p38 at two serine residues
A survey of the L1 primary sequence revealed that there are four putative p38 phospho-acceptor sites (SP dipeptide) (Fig. 5A). To identify the phospho-acceptor amino acid(s) in L1 (amino acids 400–659), we first constructed a truncation mutant in L1 by inserting a stop codon after residue 516 to separate the SP dipeptides in L1 into proximal and distal groups (Fig. 5B). The new construct, termed L1-trunc, spanned amino acids 400–516 of L1. Although L1-trunc contains consensus p38 phosphorylation sites at S447 and S475, this fusion protein was a poor substrate for activated p38, because no radioactive signal could be detected on the autoradiogram compared with full-length L1 (Fig. 5C); 32P incorporation was measured by Cerenkov counts and confirmed the data from autoradiography: L1 (16,235 ± 358 cpm) compared with L1-trunc (2669 ± 91 cpm) (Fig. 5C, histogram).
To test whether L1 is phosphorylated by p38 at the distal SP dipeptides, S551 and S556, a new fusion protein designated L1-distal was constructed, which deletes amino acids 551–558 (Fig. 5B). Although Coomassie blue staining of gels suggests equal loadings of the fusion proteins (Fig. 5D, top left), the autoradiogram (Fig. 5D, ARG, top right) and Cerenkov counting [L1 (18,030 ± 1891 cpm); L1-distal (1759 ± 63 cpm)] (Fig. 5D, histogram) demonstrate that deleting amino acids 551–558 renders L1 a poor p38 substrate.
The deletion analysis described above strongly suggests that the distal S551 and S556 are the primary p38 phosphorylation site(s) in L1. However, to eliminate the possibility that truncation of L1 or deletion of 551–558 residues could remove the docking site for p38 or alter conformation of L1 to inhibit the phosphorylation of proximal SP sites, we substituted S447 and S475 by alanine in full-length L1 (Fig. 5B) and tested the new fusion protein (L1-proximal) as a substrate for activated p38 (Fig. 5D). Coomassie blue staining (Fig. 5D, left) shows that equivalent levels of L1, L1-proximal, and L1-distal fusion proteins were used in the kinase assay. The S447A–S475A substitutions in L1-proximal did not impair phosphorylation of this protein as shown by the autoradiogram (Fig. 5D, right) and by Cerenkov counts (Fig. 5D, histogram) (L1, 18,030 ± 1891 cpm; L1-proximal, 22,575 ± 782 cpm). The difference in phosphorylation observed between L1-proximal and L1 is possibly caused by small differences in the level of proteins that were used as substrates, or by L1-proximal being a better substrate for p38 in our assays.
p38 phosphorylates L1 at serine 551 and 556
The L1-distal protein is missing an 8 aa sequence containing two SP dipeptides between amino acid positions 551 and 557. The fact that the L1-distal fusion protein was not phosphorylated implicates S551 and S556 as the p38 phosphorylation site(s). To determine the relative contribution of these sites to the p38 phosphorylation of L1, S551 and S556 were substituted by alanine both individually and collectively (Fig. 6A). Comparison of wild-type L1 (33,772 ± 617 cpm) to individually substituted L1/S551A (14,601 ± 235 cpm), S556A (16,176 ± 604 cpm), as well as the L1/S551A–S556A double mutant (2552 ± 139 cpm), shows that both S551 and S556 contribute equally to the total phosphorylation of L1, whereas the double mutant becomes a poor substrate for p38 (Fig. 6B,C). The Coomassie blue staining profile shows that equal amounts of L1 and its mutant derivatives were used in the kinase assay (Fig. 6B, top), supporting the conclusion that S551 and S556 contribute equally to the total p38 phosphorylation of L1. Together, our biochemical data strongly suggest that activated p38 phosphorylates Nav1.8 on two specific serine residues, S551 and S556, in L1.
S551A or S556A substitution disrupts p38 modulation of Nav1.8 current density within DRG neurons
To test the effects of S551A and S556A substitutions, we examined p38-mediated modulation of the current density of Nav1.8**/**S551A–S556A in Nav1.8−/− DRG neurons. Nav1.8−/− knock-out mice do not produce any Nav1.8 current (Akopian et al., 1999; Cummins et al., 1999), and their DRG neurons therefore provide an optimal neuronal background in which it is possible to test modulation of Nav1.8 channels in their native environment.
It is not unreasonable to suspect that transfection of DRG neurons might invoke a stress response including p38 activation. Therefore, we first examined the current density of Nav1.8 after anisomycin treatment of Nav1.8−/− mouse DRG neurons transfected with wild-type channels. The current density of WT Nav1.8 was not significantly different with (780.3 ± 119.5 pA/pF; n = 25) or without (670.5 ± 96.7 pA/pF; n = 23) treatment with anisomycin.
One explanation for the lack of a robust anisomycin effect would be that p38 was already activated in transfected DRG neurons. We tested this hypothesis by investigating the presence of activated p38 in Nav1.8−/− neurons in culture for 24 h, with or without treatment with anisomycin. Figure 7A, top panels, shows a weak immunofluorescence signal in untransfected cells using an antibody that is specific for activated p38 (left), whereas an enhanced signal is observed in DRG neurons that were treated with anisomycin (right). This result is in agreement with the functional data using rat DRG cultures (Figs. 2, 3). However, transfection of Nav1.8−/− DRG neurons by electroporation (Fig. 7A, bottom panels) induces activation of p38 without treatment with anisomycin (left). Activated p38 is present in cells with or without green fluorescence, indicating that transfection per se was sufficient to activate p38 under these experimental conditions. Thus, the activation of p38 by electroporation of DRG neurons might explain the lack of an effect of anisomycin treatment on the peak Nav1.8 current density in transfected neurons. Indeed, pretreatment of transfected DRG neurons by the p38 specific inhibitor SB203580 significantly reduced the current density of WT Nav1.8 in Nav1.8−/− mouse DRG neurons, compared with the inactive analog SB202474 (Fig. 7B) (SB203580, 485.9 ± 71.2 pA/pF, n = 36; vs SB202474, 710.3 ± 80.7 pA/pF, n = 36; p < 0.05). Therefore, in contrast to primary cultured rat DRG neurons, which required anisomycin treatment to activate p38 (Figs. 2, 3), electroporation appears to be a potent activator of p38 in transfected Nav1.8−/− DRG neurons.
Figure 7.

Phosphorylation of S551 and S556 in L1 of Nav1.8 by p38 mediates the increase in current density. A, Top panels, Untransfected Nav1.8−/− DRG neurons in culture for 24 h do not exhibit activated (phosphorylated) p38 immunolabeling, but an enhanced immunolabeling signal (red) from activated p38 is displayed by DRG neurons after the treatment with anisomycin. A, Bottom panels, transfected Nav1.8−/− DRG neurons in culture for 24 h exhibit activated p38 immunolabeling (red) both with and without anisomycin treatment. Scale bar, 50 μm. B, The current density of WT Nav1.8 in transfected Nav1.8−/− DRG neurons was significantly (*p < 0.05) smaller after treatment with SB203580 compared with SB202474. C, Peak current density of Nav1.8/S551–556A did not show significant reduction with SB203580 treatment compared with SB202474. Error bars indicate SE.
To determine the contribution of p38 phosphorylation at S551 and S556 of Nav1.8 to the increase in current density, both serine residues were substituted with alanine (i.e., S551A–S556A) and the mutant channel Nav1.8**/S551A–S556A was transfected into Nav1.8−/− mouse DRG neurons. We compared the current density for the Nav1.8/S551A–S556A channels in the presence of either the active (SB203580) or inactive (SB202474) inhibitor of p38, and found that, unlike WT Nav1.8 (Fig. 7B), there was no significant difference (p > 0.05) (Fig. 7C) in the current density of Nav1.8/S551A–S556A: SB203580 (416.3 ± 60.0 pA/pF; n = 26) compared with SB202474 (457.3 ± 51.3 pA/pF; n = 23). We also examined the effects of p38 specific inhibitor treatment on activation and steady-state inactivation of Nav1.8/S551A–S556A. The _V_1/2 for voltage-dependent activation of Nav1.8/S551A–S556A was −19.95 ± 1.58 mV (n = 18) in SB202474-treated, and −20.61 ± 1.80 mV (n = 17) in SB203580-treated, transfected DRG neurons. For channel availability, the _V_1/2 of voltage dependence of steady-state inactivation of Nav1.8/S551A–S556A was −36.76 ± 0.94 mV (n = 15) in SB202474-treated, and −37.52 ± 0.89 mV (n = 12) in SB203580-treated, transfected DRG neurons. Neither the voltage-dependent activation nor steady-state inactivation of Nav1.8/**S551A–S556A channels after SB203580 or SB202474 treatments were significantly different.
In separate experiments, we tested the individual roles of S551 and S556 phosphorylation in p38-mediated modulation of Nav1.8 in transfected Nav1.8−/− DRG neurons. Similar to the experiments described above, we compared the current densities for the Nav1.8**/S551A and Nav1.8/S556A channels in transfected Nav1.8−/− DRG neurons after inhibiting p38. The peak current densities for Nav1.8/S551A after treatment with the inactive analog SB202474 (238 ± 75 pA/pF; n = 20), or the active inhibitor SB203580 (201 ± 43 pA/pF; n = 18), are not significantly different (p > 0.05). Similarly, peak current densities for Nav1.8/S556A after treatment with the inactive analog SB202474 (477 ± 124 pA/pF; n = 21), or the active inhibitor SB203580 (464 ± 94 pA/pF; n = 24), are not significantly different (p > 0.05). The voltage-dependent activation and steady-state inactivation of Nav1.8/**S551A and Nav1.8/S556A channels after SB203580 or SB202474 treatments were not significantly different (data not shown). These results show that a single substitution at either S551A or S556A is sufficient to prevent the p38-mediated increase in Nav1.8 current density in DRG neurons.
Discussion
Sodium channel Nav1.8 is a key contributor to excitability and repetitive firing of normal DRG neurons (Renganathan et al., 2001; Blair and Bean, 2002), and to hyperexcitability of these neurons in an inherited painful neuropathy (Rush et al., 2006). We show here that Nav1.8 is modulated by p38 MAPK phosphorylation of S551 and S556 in L1 of the channel, leading to an increase in the current density, but without altering gating properties of the channel. Our functional data demonstrate that, within DRG neurons, the native cell background for Nav1.8 channels, substituting S551 or S556 to the non-phospho-acceptor amino acid alanine abrogates p38 regulation of the channel. Together, these data suggest that nociceptive neurons in which Nav1.8 and p38 are coexpressed may produce a more robust response to stimulation on activation of p38, for example after exposure to proinflammatory agents (Sommer and Kress, 2004).
A relatively brief treatment with the protein synthesis inhibitor anisomycin (30 min) is sufficient to activate p38 and cause a change in the current density of sodium channels (Wittmack et al., 2005; Jin and Gereau, 2006; this study). It is conceivable that inhibition of protein synthesis, independent of p38 activation, might contribute to the increase in the Nav1.8 current density that we report here. However, a direct effect of activated p38 on Nav1.8 current density, independent of inhibition of protein synthesis, is supported by the following observations: (1) available data show that the anisomycin effect on Nav1.8 (Jin and Gereau, 2006; this study) and Nav1.6 (Wittmack et al., 2005) is blocked by the coapplication of the p38-specific inhibitor SB203580; (2) transfection of DRG neurons by electroporation causes an increase in the Nav1.8 current density that is independent of anisomycin treatment but is blocked by the p38-specific inhibitor SB203580 (Fig. 7); (3) the increase in Nav1.8 current density that is caused by transfection of DRG neurons is blocked by the specific substitution of the p38 phospho-acceptor sites S551A or S556A. In addition, consistent with our findings, the anisomycin effect on gap junction intracellular communication is caused by the phosphorylation of connexin 43 by activated p38 and is independent of the inhibition of protein synthesis (Ogawa et al., 2004). Together, these data strongly suggest that the increase in Nav1.8 current density is caused by a direct effect of p38 on the channel.
What is the mechanism underlying the increase in Nav1.8 currents after p38 activation? Although p38 signaling is typically associated with long-term changes in cell function because of its role in regulating gene expression, our data favor the hypothesis that p38 acutely regulates Nav1.8 channel density at a posttranscriptional level. In support of this idea, previous reports have shown that CFA-induced inflammation produced no change in the level of Nav1.8 mRNA in DRG neurons (Okuse et al., 1997), but there is an increase in the number of Nav1.8-positive myelinated and unmyelinated axons in sciatic nerve (Coggeshall et al., 2004). These data favor the proposal that Nav1.8 protein translation, turnover, and translocation to axons and nerve endings might be regulated by inflammatory mediators via signaling pathways that involve p38. By analogy to TRPV1 (Ji et al., 2002), phosphorylation of Nav1.8 by p38 might lead to transport of the channels to the periphery under inflammatory conditions. These findings are consistent with a localized mechanism for acute inflammatory pain that depends on the elevated Nav1.8 channel protein levels along axons and in the nerve terminals in inflamed skin, which could enhance the successful initiation of action potentials and their propagation centrally.
Acute modulation of Nav1.8 current density could be mediated by binding of channel partners or by direct phosphorylation of the channel. To date, an increase in Nav1.8 current density has been shown to occur after binding of annexin II (p11) to the N terminus (Okuse et al., 2002), calmodulin binding to the C terminus (Choi et al., 2006), and protein kinase A (PKA)/protein kinase C (PKC) phosphorylation within L1 (Fitzgerald et al., 1999; Vijayaragavan et al., 2004) of the channel. The pathways leading to elevated levels of Nav1.8 at the plasma membrane by the binding to p11, calmodulin, or after PKA/PKC phosphorylation are not well understood, but could encompass regulation of insertion or retention of these channels at the plasma membrane. In our study, the p38-mediated increase in Nav1.8 current density was relatively rapid (<30 min of treatment with anisomycin), in agreement with a similar finding using acute application of TNF-α to DRG neurons in culture (Jin and Gereau, 2006). However, the potential for a p38-mediated alteration in single channel conductance causing the increase in current density cannot be discounted. Alternative mechanisms that might account for phosphorylation-mediated increase in Nav1.8 current density include the following: (1) stabilizing membrane-bound channels [for example, by inhibiting removal of the channel from the plasma membrane (Liu et al., 2005)] or (2) regulating the insertion of a pool of “silent” channels into the plasma membrane.
We demonstrate in this study that the TTX-R Nav1.8 channel is phosphorylated by p38 at two serine residues at positions S551 and S556 in L1 of the channel, and that substituting S551 or S556 individually to alanine residues abrogates the effect of p38 on rat Nav1.8 current density. Our data thus show, for the first time, that the phosphorylation of two serine residues within this proline-rich segment of L1 is necessary to mediate the effect of p38 on the channel function. Importantly, we show that preventing phosphorylation of either site is sufficient to block this effect. PKA/PKC phosphorylation of Nav1.8 occurs at multiple serine residues within L1 (S463, S487, S499, S510, and S536) (Fitzgerald et al., 1999; Vijayaragavan et al., 2004). Similarly, PKA and PKC phosphorylation at several serine residues within L1 have previously been shown to cooperate in regulating Nav1.2 and Nav1.7 (Cantrell and Catterall, 2001; Chahine et al., 2005). The voltage dependence of activation and steady-state inactivation of Nav1.8 were not altered by p38 phosphorylation of the channel, unlike the PKA-mediated increase in current density of Nav1.8 in COS cells, which was associated with a shift in voltage dependence of activation and slowing in the inactivation at hyperpolarized potentials (Fitzgerald et al., 1999). These data strongly suggest that p38 modulates Nav1.8 channels in a manner distinct from that of PKA.
The p38-mediated increase in Nav1.8 current density (this study) and decrease in Nav1.6 current density (Wittmack et al., 2005) parallel the effect of PKA and PKC modulation of TTX-R compared with TTX-S channels (Chahine et al., 2005), supporting the hypothesis that different isoforms of sodium channels differ in their functional response to phosphorylation in L1. Additionally, our in vitro data demonstrate that there is no preferential order of phosphorylation of S551 and S556, although it is conceivable that alanine substitution itself disrupts the structure of L1 to obscure such a mechanism. Whether the charge differential within a small region of the channel alters the local conformation of L1, or whether the phosphorylation state of S551 or S556 promotes or prevents the binding of accessory proteins, remains to be determined.
Modulation of TTX-R currents, specifically the current produced by Nav1.8 channels, by activated MAPKs (Jin and Gereau, 2006; this study) is likely to contribute to inflammatory, and possibly neuropathic, pain. We have shown in this study that a subset of small diameter DRG neurons, which are known to produce Nav1.8 (Fjell et al., 1999; Amaya et al., 2000; Sleeper et al., 2000; Rush et al., 2005b), also produce activated p38 MAPK. Given the central role of Nav1.8 in action potential firing (Renganathan et al., 2001; Blair and Bean, 2002), the increase in the slowly inactivating TTX-R current density, which is mediated by direct phosphorylation of Nav1.8 channels by p38 MAPK, can increase DRG neuron excitability under pathological conditions. Our results thus add to the evidence for p38 MAPK as a therapeutic target in chronic pain.
Footnotes
This work was supported in part by grants from the National Multiple Sclerosis Society and the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America and the United Spinal Association with Yale University. We thank Dr. Theodore R. Cummins for helpful discussions, and Rachael Blackman, Larry Macala, and Bart Toftness for technical assistance.
References
- Akopian et al., 1996.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- Akopian et al., 1999.Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Amaya et al., 2000.Amaya F, Decosterd I, Samad TA, Plumpton C, Tate S, Mannion RJ, Costigan M, Woolf CJ. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- Blair and Bean, 2002.Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22:10277–10290. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair and Bean, 2003.Blair NT, Bean BP. Role of tetrodotoxin-resistant Na+ current slow inactivation in adaptation of action potential firing in small-diameter dorsal root ganglion neurons. J Neurosci. 2003;23:10338–10350. doi: 10.1523/JNEUROSCI.23-32-10338.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano and Mahadevan, 1995.Cano E, Mahadevan LC. Parallel signal processing among mammalian MAPKs. Trends Biochem Sci. 1995;20:117–122. doi: 10.1016/s0968-0004(00)88978-1. [DOI] [PubMed] [Google Scholar]
- Cantrell and Catterall, 2001.Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Catterall, 2000.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Chahine et al., 2005.Chahine M, Ziane R, Vijayaragavan K, Okamura Y. Regulation of Na v channels in sensory neurons. Trends Pharmacol Sci. 2005;26:496–502. doi: 10.1016/j.tips.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Choi et al., 2006.Choi J, Hudmon A, Waxman S, Dib-Hajj S. Calmodulin regulates current density and frequency-dependent inhibition of sodium channel Nav1.8 in DRG neurons. J Neurophysiol. 2006;96:97–108. doi: 10.1152/jn.00854.2005. [DOI] [PubMed] [Google Scholar]
- Choi et al., 2007.Choi JS, Dib-Hajj SD, Waxman S. Differential slow inactivation and use-dependent inhibition of Nav1.8 channels contribute to distinct firing properties in IB4+ and IB4− DRG neurons. J Neurophysiol. 2007;97:1258–1265. doi: 10.1152/jn.01033.2006. [DOI] [PubMed] [Google Scholar]
- Coggeshall et al., 2004.Coggeshall RE, Tate S, Carlton SM. Differential expression of tetrodotoxin-resistant sodium channels Nav1.8 and Nav1.9 in normal and inflamed rats. Neurosci Lett. 2004;355:45–48. doi: 10.1016/j.neulet.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Coste et al., 2004.Coste B, Osorio N, Padilla F, Crest M, Delmas P. Gating and modulation of presumptive Nav1.9 channels in enteric and spinal sensory neurons. Mol Cell Neurosci. 2004;26:123–134. doi: 10.1016/j.mcn.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Cummins et al., 1999.Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci. 1999;19(RC43):1–6. doi: 10.1523/JNEUROSCI.19-24-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins et al., 2001.Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib-Hajj SD, Waxman SG. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci. 2001;21:5952–5961. doi: 10.1523/JNEUROSCI.21-16-05952.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouhri et al., 2003.Djouhri L, Fang X, Okuse K, Wood JN, Berry CM, Lawson S. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol (Lond) 2003;550:739–752. doi: 10.1113/jphysiol.2003.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong et al., 2007.Dong XW, Goregoaker S, Engler H, Zhou X, Mark L, Crona J, Terry R, Hunter J, Priestley T. Small interfering RNA-mediated selective knockdown of Nav1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience. 2007;146:812–821. doi: 10.1016/j.neuroscience.2007.01.054. [DOI] [PubMed] [Google Scholar]
- Fitzgerald et al., 1999.Fitzgerald EM, Okuse K, Wood JN, Dolphin AC, Moss SJ. cAMP-dependent phosphorylation of the tetrodotoxin-resistant voltage-dependent sodium channel SNS. J Physiol (Lond) 1999;516:433–446. doi: 10.1111/j.1469-7793.1999.0433v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell et al., 1999.Fjell J, Cummins TR, Dib-Hajj SD, Fried K, Black JA, Waxman SG. Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Mol Brain Res. 1999;67:267–282. doi: 10.1016/s0169-328x(99)00070-4. [DOI] [PubMed] [Google Scholar]
- Gold et al., 1996.Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold et al., 2003.Gold MS, Weinreich D, Kim CS, Wang R, Treanor J, Porreca F, Lai J. Redistribution of Nav1.8 in uninjured axons enables neuropathic pain. J Neurosci. 2003;23:158–166. doi: 10.1523/JNEUROSCI.23-01-00158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein et al., 1991.Goldstein ME, House SB, Gainer H. NF-L and peripherin immunoreactivities define distinct classes of rat sensory ganglion cells. J Neurosci Res. 1991;30:92–104. doi: 10.1002/jnr.490300111. [DOI] [PubMed] [Google Scholar]
- Herzog et al., 2003.Herzog RI, Cummins TR, Ghassemi F, Dib-Hajj SD, Waxman SG. Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J Physiol (Lond) 2003;551:741–750. doi: 10.1113/jphysiol.2003.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis et al., 2007.Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roeloffs R, Zhong C, Mikusa JP, Hernandez G, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci USA. 2007;104:8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji et al., 2002.Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Jin and Gereau, 2006.Jin X, Gereau RW. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi et al., 2006.Joshi SK, Mikusa JP, Hernandez G, Baker S, Shieh CC, Neelands T, Zhang XF, Niforatos W, Kage K, Han P, Krafte D, Faltynek C, Sullivan JP, Jarvis MF, Honore P. Involvement of the TTX-resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain. 2006;123:75–82. doi: 10.1016/j.pain.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Kerr et al., 2001.Kerr BJ, Souslova V, McMahon SB, Wood JN. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. NeuroReport. 2001;12:3077–3080. doi: 10.1097/00001756-200110080-00019. [DOI] [PubMed] [Google Scholar]
- Khasar et al., 1998.Khasar SG, Gold MS, Levine JD. A tetrodotoxin-resistant sodium current mediates inflammatory pain in the rat. Neurosci Lett. 1998;256:17–20. doi: 10.1016/s0304-3940(98)00738-1. [DOI] [PubMed] [Google Scholar]
- Laird et al., 2002.Laird JM, Souslova V, Wood JN, Cervero F. Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. J Neurosci. 2002;22:8352–8356. doi: 10.1523/JNEUROSCI.22-19-08352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu et al., 2005.Liu C, Cummins TR, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD. CAP-1A is a novel linker that binds clathrin and the voltage-gated sodium channel Nav1.8. Mol Cell Neurosci. 2005;28:636–649. doi: 10.1016/j.mcn.2004.11.007. [DOI] [PubMed] [Google Scholar]
- McMahon et al., 2005.McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp Neurol. 2005;192:444–462. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Myers et al., 2006.Myers RR, Campana WM, Shubayev VI. The role of neuroinflammation in neuropathic pain: mechanisms and therapeutic targets. Drug Discov Today. 2006;11:8–20. doi: 10.1016/S1359-6446(05)03637-8. [DOI] [PubMed] [Google Scholar]
- Obata et al., 2004a.Obata K, Yamanaka H, Dai Y, Mizushima T, Fukuoka T, Tokunaga A, Noguchi K. Differential activation of MAPK in injured and uninjured DRG neurons following chronic constriction injury of the sciatic nerve in rats. Eur J Neurosci. 2004a;20:2881–2895. doi: 10.1111/j.1460-9568.2004.03754.x. [DOI] [PubMed] [Google Scholar]
- Obata et al., 2004b.Obata K, Yamanaka H, Dai Y, Mizushima T, Fukuoka T, Tokunaga A, Noguchi K. Activation of extracellular signal-regulated protein kinase in the dorsal root ganglion following inflammation near the nerve cell body. Neuroscience. 2004b;126:1011–1021. doi: 10.1016/j.neuroscience.2004.04.036. [DOI] [PubMed] [Google Scholar]
- Obata et al., 2004c.Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004c;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata and Tatebayashi, 1993.Ogata N, Tatebayashi H. Kinetic analysis of two types of Na+ channels in rat dorsal root ganglia. J Physiol (Lond) 1993;466:9–37. [PMC free article] [PubMed] [Google Scholar]
- Ogawa et al., 2004.Ogawa T, Hayashi T, Kyoizumi S, Kusunoki Y, Nakachi K, MacPhee DG, Trosko JE, Kataoka K, Yorioka N. Anisomycin downregulates gap-junctional intercellular communication via the p38 MAP-kinase pathway. J Cell Sci. 2004;117:2087–2096. doi: 10.1242/jcs.01056. [DOI] [PubMed] [Google Scholar]
- Okuse et al., 1997.Okuse K, Chaplan SR, McMahon SB, Luo ZD, Calcutt NA, Scott BP, Akopian AN, Wood JN. Regulation of expression of the sensory neuron-specific sodium channel SNS in inflammatory and neuropathic pain. Mol Cell Neurosci. 1997;10:196–207. doi: 10.1006/mcne.1997.0657. [DOI] [PubMed] [Google Scholar]
- Okuse et al., 2002.Okuse K, Malik-Hall M, Baker MD, Poon WY, Kong H, Chao MV, Wood JN. Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature. 2002;417:653–656. doi: 10.1038/nature00781. [DOI] [PubMed] [Google Scholar]
- Pezet and McMahon, 2006.Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Potrebic et al., 2003.Potrebic S, Ahn AH, Skinner K, Fields HL, Basbaum AI. Peptidergic nociceptors of both trigeminal and dorsal root ganglia express serotonin 1D receptors: implications for the selective antimigraine action of triptans. J Neurosci. 2003;23:10988–10997. doi: 10.1523/JNEUROSCI.23-34-10988.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renganathan et al., 2001.Renganathan M, Cummins TR, Waxman SG. Contribution of Nav1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86:629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- Rizzo et al., 1994.Rizzo MA, Kocsis JD, Waxman SG. Slow sodium conductances of dorsal root ganglion neurons: intraneuronal homogeneity and interneuronal heterogeneity. J Neurophysiol. 1994;72:2796–2815. doi: 10.1152/jn.1994.72.6.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush et al., 1998.Rush AM, Brau ME, Elliott AA, Elliott JR. Electrophysiological properties of sodium current subtypes in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 1998;511:771–789. doi: 10.1111/j.1469-7793.1998.771bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush et al., 2005a.Rush AM, Dib-Hajj SD, Waxman SG. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurons. J Physiol (Lond) 2005a;564:803–815. doi: 10.1113/jphysiol.2005.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush et al., 2005b.Rush AM, Craner MJ, Kageyama T, Dib-Hajj SD, Waxman SG, Ranscht B. Contactin regulates the current density and axonal expression of tetrodotoxin-resistant but not tetrodotoxin-sensitive sodium channels in DRG neurons. Eur J Neurosci. 2005b;22:39–49. doi: 10.1111/j.1460-9568.2005.04186.x. [DOI] [PubMed] [Google Scholar]
- Rush et al., 2006.Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci USA. 2006;103:8245–8250. doi: 10.1073/pnas.0602813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab et al., 2003.Saab CY, Cummins TR, Waxman SG. GTP(gammaS) increases Nav1.8 current in small-diameter dorsal root ganglia neurons. Exp Brain Res. 2003;152:415–419. doi: 10.1007/s00221-003-1565-7. [DOI] [PubMed] [Google Scholar]
- Sangameswaran et al., 1996.Sangameswaran L, Delgado SG, Fish LM, Koch BD, Jakeman LB, Stewart GR, Sze P, Hunter JC, Eglen RM, Herman RC. Structure and function of a novel voltage-gated, tetrodoxtoxin-resistant sodium channel specific to sensory neurons. J Biol Chem. 1996;271:5953–5956. doi: 10.1074/jbc.271.11.5953. [DOI] [PubMed] [Google Scholar]
- Schafers et al., 2003.Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeper et al., 2000.Sleeper AA, Cummins TR, Dib-Hajj SD, Hormuzdiar W, Tyrrell L, Waxman SG, Black JA. Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J Neurosci. 2000;20:7279–7289. doi: 10.1523/JNEUROSCI.20-19-07279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer and Kress, 2004.Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–187. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Stirling et al., 2005.Stirling LC, Forlani G, Baker MD, Wood JN, Matthews EA, Dickenson AH, Nassar MA. Nociceptor-specific gene deletion using heterozygous NaV1.8-Cre recombinase mice. Pain. 2005;113:27–36. doi: 10.1016/j.pain.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Vijayaragavan et al., 2004.Vijayaragavan K, Boutjdir M, Chahine M. Modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by protein kinase A and protein kinase C. J Neurophysiol. 2004;91:1556–1569. doi: 10.1152/jn.00676.2003. [DOI] [PubMed] [Google Scholar]
- Wittmack et al., 2005.Wittmack EK, Rush AM, Hudmon A, Waxman SG, Dib-Hajj SD. Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J Neurosci. 2005;25:6621–6630. doi: 10.1523/JNEUROSCI.0541-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al., 2002.Zhang YH, Vasko MR, Nicol GD. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. J Physiol (Lond) 2002;544:385–402. doi: 10.1113/jphysiol.2002.024265. [DOI] [PMC free article] [PubMed] [Google Scholar]