Barrett’s esophagus: Incidence, etiology, pathophysiology, prevention and treatment (original) (raw)
. 2007 Dec;3(6):1035–1145.
Abstract
Barrett’s esophagus is a metaplastic alteration of the normal esophageal epithelium that is detected on endoscopic examination and pathologically confirmed by the presence of intestinal metaplasia on biopsy. Its major significance is as a predisposing factor for esophageal adenocarcinoma, which carries a high mortality rate and a rapidly growing incidence in the United States. Detection of Barrett’s esophagus allows for endoscopic surveillance in order to detect the potential development of dysplasia and early cancer before symptoms develop, and thereby significantly increases treatment options and may lower mortality from esophageal adenocarcinoma. Much current work in the field is aimed at reducing the risk of progression from Barrett’s esophagus to cancer, and in the identification of biomarkers that may predict progression towards cancer. Barrett’s esophagus is present in 10%–20% of patients with gastroesophageal reflux disease (GERD) and has also been detected in patients who deny classic GERD symptoms and are undergoing endoscopy for other indications. We used an evidence-based approach to describe treatment options for patients with Barrett’s esophagus.
Keywords: Barrett’s esophagus, esophageal adenocarcinoma, evidence-based approach, endoscopic surveillance
Introduction
Esophageal adenocarcinoma carries a grave prognosis, with a relative 3-year survival rate of only 20% in the United States from 1995–1998 (Polednak 2003). According to data from the Surveillance, Epidemiology, and End Results program (SEER) database, the incidence of esophageal carcinoma is rising more rapidly than any other form of cancer, with a six-fold increase from 1975 to 2001 (Devesa et al 1998; Brown and Devesa 2002; Pera et al 2005; Pohl and Welch 2005). As a predisposing condition to esophageal adenocarcinoma, gastro-esophageal reflux disease (GERD) is one of the most common medical conditions in the US, causing symptoms in up to 40% of individuals living in Western populations every month and 7% per week (Gallup Organization 1988; Moayyedi and Axon 2005).
The link between GERD and esophageal adenocarcinoma is Barrett’s esophagus (BE), a condition characterized by metaplastic changes in the esophageal epithelium. In this review, we summarize the current knowledge regarding the epidemiology, pathophysiology, and treatment of patients with this diagnosis.
Methods
We performed a review of the literature published in English from 1970 to 2006, using PUBMED/MEDLINE to obtain references for topics addressed herein. Abstracts corresponding to potentially relevant titles were reviewed, and relevant articles were retrieved to evaluate data and content.
For the prevention and treatment sections, an evidence-based approach to the literature was used, employing a standard scoring system (Dent et al 1999). Evidence was placed in one of five categories. All references were examined independently by the two authors, who assigned a categorical rating to each evidence-based treatment statement. If there was a difference of opinion, a consensus was reached. Rating categories were defined as follows:
- Evidence rated Category A was obtained from randomized clinical trials.
- Evidence rated Category B was from cohort or case-control studies.
- Evidence rated Category C was based on case reports, or flawed clinical trials.
- Evidence rated Category D was limited to the clinical experience of the supervising author.
- Where evidence was insufficient to form an opinion, it was rated Category E.
Incidence
Original retrospective data suggested an incidence of BE amongst patients with reflux symptoms between 8% and 20%, but this estimate may have been influenced by the presence of selection bias (Cameron 1997). For example, in two recent prospective studies, patients presenting for colonoscopy who agreed to upper endoscopy for study purposes were examined. In the first study, approximately 8% of subjects who reported any history of heartburn had endoscopic findings of Barrett’s esophagus, compared to 6% of those who did not report such GERD symptoms (Rex et al 2003). In the second study, a high overall rate of BE was seen, but again absence of reflux symptoms did not dramatically lower the risk of finding metaplasia; 20% of patients with symptoms, compared to 15% of asymptomatic patients, were found to have BE (Ward et al 2006). In this study, males were twice as likely as females to have BE (22% vs 11%), consistent with prior studies and the 2:1 ratio found by recent meta-analysis of the gender ratio for Barrett’s esophagus (Cook et al 2005). Taken together, these newer studies confirm an 8%–20% rate of BE amongst patients with reflux symptoms, similar to the prior retrospective work. In addition, they suggest an equally high rate of BE in the general population without GERD symptoms.
Our own work in patients presenting for screening sigmoidoscopy at a Veteran’s Hospital, who agreed to upper endoscopy for study purposes, found that 25% of patients without significant reflux symptoms had BE detected (Gerson et al 2002). The higher rate of BE in this study could have been influenced by the predominantly male population in the study, the fact that patients screened were all at least 50 years of age, and the inclusion of patients who reported having GERD symptoms once a month or less. However, the majority (51%) of the patients denied the presence of GERD symptoms in their lifetimes.
Using a different approach to examine the incidence of BE in the general population, one study prospectively evaluated unselected autopsy materials from patients at the Mayo Clinic. Only about 1% of 733 deceased patients evaluated by autopsy were found to have Barrett’s esophagus; when adjusted for age and gender, this may suggest a much lower incidence of BE in the general population of 0.4% (Cameron et al 1990; Cameron 1997). Despite this low percentage, 5 out of the 7 autopsy cases of BE did not have previously known disease, and the authors were able to demonstrate that the rate of BE determined by autopsy is much greater than the rate of known BE cases in the local community.
Cases of BE can be further divided by the length of BE segment. Short-segment disease is generally defined as intestinal metaplasia of the distal esophagus that is less than 3 cm in length, while long-segment BE refers to segments measuring 3 cm or greater. Interestingly, short-segment disease appears to be at least 3 times more common than long-segment disease (Hirota et al 1999; Csendes et al 2003; Hanna et al 2006), and longer segment length has been correlated with greater acid exposure (Fass et al 2001). However, once BE develops its length does not generally change, so that short-segment BE normally remains short even in the context of ongoing esophageal exposure to acid (Cameron and Lomboy 1992). The rate of dysplasia has been directly correlated with segment length (Hirota et al 1999; Csendes et al 2003). Because both long and short segment disease are associated with development of dysplasia and adenocarcinoma (Sharma et al 1997a), both forms of BE are treated similarly with regards to endoscopic surveillance and treatment.
Misdiagnosis of BE can occur for a variety of reasons. The diagnosis of BE is dependent upon the identification of any length of distal esophageal columnar-lined tissue containing goblet cells in biopsy specimens. If biopsies are obtained in the setting of a normal esophagogastric junction (EGJ) and demonstrate intestinal metaplasia, then the patient has intestinal metaplasia of the GE junction (SIM-EGJ), a condition found in 10%–15% of patients undergoing upper endoscopy for any indication (Hirota et al 1999) that is more common in patients infected with Helicobacter pylori. SIM-EGJ is not an indication for entry into an endoscopic surveillance program. The second reason for misdiagnosis is based on the landmark used for determining the location of the EGJ. Based on the Prague C and M Criteria for Barrett’s esophagus, the upper end of the gastric folds is used in order to define the location of the EGJ. (Armstrong 2004) It is important to identify the EGJ correctly since most cases of BE are of short length. Correct identification of the gastric folds requires that air must be properly deflated during endoscopy and other factors, such as respiratory movements and cardiac pulsations, be taken into consideration (Amano et al 2006).
Etiology and pathophysiology
GERD is accepted as the primary etiologic factor for BE, which is in turn the major predisposing condition for esophageal adenocarcinoma. From a pathophysiologic perspective, BE is thought to be the result of esophageal epithelial response to injury. Acid-induced injury to the native squamous cell epithelium of the esophagus leads to epithelial repair; eventually, but only in some cases, columnar epithelium can replace the native epithelium (Spechler 2002), offering greater tolerance to low pH, but also a tendency towards dysplastic change predisposing to esophageal adenocarcinoma. A summary of this model of progression from normal esophagus to BE to esophageal cancer is shown in Figure 1, including endoscopic and histologic appearance of the esophageal mucosa during this progression.
Figure 1.
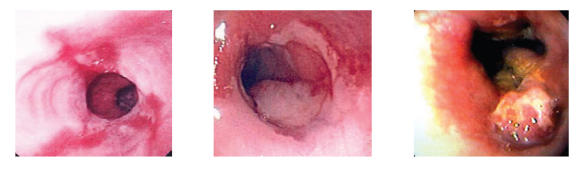
Progression of disease, demonstrating changes observed as esophagitis (A) undergoes metaplasia, leading to salmon-colored mucosal changes in the distal esophagus characteristic of Barrett’s esophagus (B) Dysplasia develops (C).
In a canine model of acid reflux disease, dogs with induced GERD were studied for regeneration of injured esophageal epithelium; the majority (7/10) of the dogs developed columnar epithelium in place of the normal squamous epithelium (Li et al 1994, Gillen et al 1988). Of note, in the canine model, experiments demonstrated that metaplastic changes in esophageal epithelium, as opposed to proximal migration of columnar cells from the gastric cardia, were responsible for the changes observed (Gillen et al 1988). Indeed, the replacement of the normal epithelium of the distal esophagus with columnar epithelium is considered a metaplastic process, and the characteristic columnar histology of the altered tissue is described as specialized intestinal metaplasia (as it typically also contains intestinal crypts and goblet cells).
Interestingly, while duration of GERD symptoms is clearly a risk factor both for development of BE (Eisen et al 1997; Lieberman et al 1997) and greater length of BE segment (Fass et al 2001), the extent of BE does not typically expand over time; that is, the length of the Barrett’s segment of distal esophagus is established over a relatively short period of time (months), and changes little over subsequent years (Cameron and Lomboy 1992). Furthermore, use of proton pump inhibitors (PPIs) to prevent acid reflux does not result in significant reduction in the length of existent Barrett’s esophagus (Sharma et al 1997b).
Additional factors that appear to be risk factors for the presence of BE include obesity, the presence of hiatal hernia, and interestingly, the absence of Heliobacter pylori infection. Speculation is that all of these factors contribute to BE by increasing the risk and severity of acid reflux. Hiatal hernia distorts the anatomy the normally protects against reflux by reducing pressure at the lower esophageal sphincter, creating an acidic hernia sac between the diaphragm and the esophagus, and decreasing the efficacy of peristalsis (Gordon et al 2004). One recent study shows that of 50 patients with GERD who developed BE, 63% had the finding of a hiatal hernia (Westhoff et al 2005), and another study demonstrated that longer length of hiatal hernia correlated with longer segment of BE (Dickman et al 2005).
GERD, BE and esophageal adenocarcinoma have all been associated with the presence of obesity. The relationship between GERD and obesity is thought to be in part due to increased gastroesophageal sphincter gradient (Mercer et al 1987), intra-abdominal pressure (El-Serag et al 2006b), and increased incidence of hiatal hernia in obesity (Pandolfino et al 2006). A recent retrospective case-control study showed a strong direct relationship between mean visceral adipose tissue and BE when comparing patients with and without BE who had undergone both endoscopy and an abdominal CT scan at a large Veterans’ Hospital (El-Serag et al 2005). A similar correlation between body mass index (BMI) and BE was found in another study, with an adjusted odds ratio of 1.35 for each five-point increase in BMI (Stein et al 2005).
Heliobacter pylori, in contrast to obesity and hiatal hernia, may affect the risk of BE by physiologic rather than anatomic means; H. pylori can decrease gastric acidity through activity of urease (Sharma and Vakil 2003). The fact that H. pylori may be protective against BE is a contrast to its well established status as a risk factor for peptic ulcer disease (PUD) and gastritis, and indeed eradication of H. pylori for PUD may increase risk of BE. One strain of H. pylori containing the virulence factor cytotoxin-associated gene (cagA) may be particularly protective. In patients with the cagA+ strains of H. pylori, one study of 153 patients who had undergone endoscopy found that patients with BE and particularly those with BE and dysplasia or cancer were much less likely to be infected than controls; 42% control patients were positive for this strain of H. pylori, as opposed to 13% of patients with BE and 0% of patients with BE and dysplasia and cancer (Vicari et al 1998). In a similar study of 251 patients undergoing endoscopy, cagA+ H. pylori was present in 44% of 25 controls, 36% of 36 patients with GERD, 20% of 10 patients with short-segment BE, and 0% of 18 patients with long-segment BE (Vaezi et al 2000), resulting in an odds ratio of 0.27 for BE patients infected with H. pylori compared to patients with reflux but no BE. In addition to this inverse correlation with BE, studies have also shown a protective effect of H. pylori on the development of esophageal adenocarcinoma (de Martel et al 2005). Until the association between BE and its complications with negative H. pylori status is further clarified, it may be prudent to avoid H. pylori eradication in patients whose predominant problem is attributed to GERD rather than gastritis or PUD.
The risk factors for BE highlight again the role of reflux in its pathogenesis. Given the response to injury model of BE, reflux-associated erosive esophagitis (EE) is considered a likely intermediate step on the path towards development of metaplasia. However, to date, no clear data exist to validate that all patients with BE have had prior erosive disease. Recently, a study of 172 Veterans with reflux symptoms demonstrated that 12% of patients with EE on endoscopy (but no evidence of BE) were found to have BE on repeat endoscopy 8–16 weeks later, after completing PPI therapy for EE (Hanna et al 2006). The finding of BE development in some of these patients may suggest that BE was present but not detected in some patients with EE on initial evaluation, due to the clinical appearance of erosive disease masking the presence of metaplasia. In some cases, intestinal metaplasia can be detected if biopsies are obtained in the setting of erosive disease, however in other cases it may be missed due to sampling error because of the limited visibility. However, BE should become apparent due to healing of esophagitis after PPI therapy. Alternatively, BE may have developed in these patients during treatment with PPI, with columnar epithelium developing as part of the healing process. Thus it is not clear whether this study addresses a temporal progression from EE to BE, or a limit of current detection of BE in the setting of active EE.
The relationships among GERD, BE, and esophageal adenocarcinoma are clearly established. Duration and severity of GERD symptoms increases risk not only for BE, but also for esophageal adenocarcinoma; in fact, patients with severe and prolonged symptoms of GERD have an odds ratio of 43.5 for development of esophageal adenocarcinoma compared with patient who did not report any recurrent GERD symptoms (Lagergren et al 1999). However, the same study also showed that 40% of patients with esophageal adenocarcinoma denied having had GERD symptoms, perhaps reflecting the significant portion of BE patients who do not report symptoms of reflux.
Prevention and screening
Treatment of GERD patients with PPIs to prevent development of BE
If acid-induced epithelial injury leads to Barrett’s metaplasia, it follows that acid suppression may prevent such metaplasia. Indeed, amongst patients diagnosed with BE, those who had been treated pharmacologically for acid suppression prior to diagnosis had significantly shorter length of disease. Patients who had not received such treatment had an average BE segment length of 4.8 cm, as opposed to 3.2 cm for those treated with PPI therapy prior to diagnosis (p < 0.001) (El-Serag et al 2004).
Recommendation
No data exist to demonstrate prevention of BE using PPI therapy. However, such treatment is theoretically compelling, and there are the data to suggest shorter segments of BE amongst PPI users. Therefore usage of PPIs in patients with reflux is recommended for control of reflux symptoms and may be associated with the increasing prevalence of short-segment BE. Evidence rating: Category B.
Use of non-steroidal anti-inflammatory drugs (NSAIDs) to prevent BE
There has been interest for some time regarding the usage of non-steroidal anti-inflammatory agents (NSAIDs) and COX-2 inhibitors in order to reduce esophageal inflammation and proneoplastic stimuli (Kaur et al 2002; Altorki et al 2004). Such agents are speculated to prevent either development of metaplasia or progression of metaplasia to dysplasia and cancer. A recent case-control study of patients in Ireland demonstrated that patients who reported use of aspirin or other NSAIDs were less likely to develop BE, with an odds ratio of 0.53 and 0.40 respectively, a difference which was statistically significant but potentially subject to selection or recall bias (Anderson et al 2006). This result is consistent with work in animals showing reduced incidence of esophageal columnar-lined epithelium in a rat model of acid reflux, when those rats were treated with a COX-2 inhibitor (Oyama et al 2005). Furthermore, data from case-control studies and a prospective cohort study suggest that aspirin and other NSAIDs reduce the risk of development of esophageal cancer (Farrow et al 1998; Jolly et al 2002). A prospective study of the duration, frequency and recency of NSAID usage and the risk of esophageal carcinoma in the Seattle BE cohort of 350 patients revealed that the 5-year cumulative incidence of esophageal adenocarcinoma was 14% for never users, 10% for former users, and 7% for current NSAID users. Compared to never users, current NSAID users had less aneuploidy (n = 35, hazard ratio of 0.25) and tetraploidy (n = 45, HR = 0.44) (Vaughan et al 2006).
Recommendation
Case control studies suggest a protective effect of NSAIDs for the development of BE and the progression to dysplasia and/or carcinoma. The potential benefit from usage of NSAIDs in patients with BE must be weighed against their significant potential for adverse effects (including esophageal injury). Evidence rating: Category B.
Screening for development of BE in GERD patients
The association between longstanding GERD symptoms and development of BE has historically been strong (Eisen et al 1997), with one study showing an odds ratio of 3.0 and 6.4, respectively, for development of BE in patients having GERD symptoms for 1–5 years versus greater than 10 years (Lieberman et al 1997). Males with long-standing reflux symptoms appear to be at the greatest risk for the development of BE (Gerson et al 2001). Based on the prior literature, endoscopic screening for BE in reflux patients who report >1 year of GERD symptoms is recommended.
There is now a growing body of evidence that there is an equally high rate of BE amongst patients who do not report significant or longstanding GERD symptoms (but who likely have asymptomatic reflux). The significance of this “asymptomatic” cohort is substantiated by the fact that a significant portion of patients who develop esophageal adenocarcinoma have no history of GERD symptoms; in one key study, 76 out of 189 patients (40%) with this form of cancer had no history of symptomatic GERD (Lagergren et al 1999). This is similar to the 43% of patients with no history of GERD symptoms found to have esophageal carcinoma in a 1984 study, in which all 26 patients evaluated had BE with dysplasia surrounding the cancer at time of diagnosis (Smith et al 1984). Remarkably, in a systematic review of the literature, only about 5% of patients with esophageal adenocarcinoma had a known history of BE prior to diagnosis of cancer (Dulai et al 2002), reflecting serious limitations to our current ability to catch premalignant BE by screening. As discussed in the section on treatment, premalignant diagnosis of BE substantially improves survival of subsequent cancer, as cancer is caught early due to surveillance measures.
Recommendation
Patients with longstanding (>1 year) GERD symptoms should be screened for development of BE, dysplasia, and early esophageal adenocarcinoma. Additionally, all patients found to have erosive esophagitis should have follow-up endoscopy after treatment both to determine clearance of esophagitis and absence of BE. If the development of esophageal cancer can be prevented by endoscopic detection of BE, then future studies should focus on the identification of who should undergo endoscopic screening regardless of the presence of classic reflux symptoms. As molecular markers for risk of malignant transformation are refined (see below), it may become clinically beneficial and cost-effective to screen a larger segment of the general population for BE. Currently, however, insufficient data exist to recommend screening in the general population (Gerson and Triadafilopoulos 2002), since the risk of developing esophageal cancer remains remains low overall. Evidence rating: Category B.
Treatment
Once Barrett’s esophagus has developed, treatment is primarily directed at prevention of progression to esophageal adenocarcinoma, as well as at control of GERD symptoms. Cancer prevention is currently achieved primarily by monitoring for progression to dysplasia, and then consideration of action to remove the dysplastic tissue before it progresses to malignancy. Treatment to reduce acidity of stomach content is employed not to treat the BE itself, but to treat GERD symptoms; its unclear role in prevention of cancer is also discussed below.
Role of PPI therapy in patients with BE
PPI use does not typically result in reduction of BE length once it has formed (Sharma et al 1997b), although very aggressive acid suppression therapy may yield slight reduction in length of metaplastic tissue (Peters et al 1999). PPI therapy may also lead to islands within segments of Barrett’s that have macroscopic appearance of squamous epithelium (Sharma et al 1998). These islands are of unclear significance, and about a third of them have the pathological appearance of specialized intestinal metaplasia despite reversal of macroscopic appearance (Sharma et al 1998).
A problem currently under debate in the treatment of acid reflux is that of inadequate pH neutralization. Available data demonstrate that even twice-daily PPI administration allows for periods of significant nocturnal gastric acidity with pH < 4.0 in the majority of patients, despite good control of symptoms in most (Katz et al 1998). In the same study, half of all Barrett’s patients also had abnormal percentage of the time that the esophageal pH readings were <4.0 despite the PPI therapy.
Our work with various PPIs confirms the frequent occurrence of inadequate pH control. We have previously shown that patients with BE are less likely to achieve adequate esophageal pH control on PPIs compared to patients with GERD alone (50% vs 58%), and the degree of acid reflux in treated BE patients was more pathologic compared to the GERD cohort (Gerson et al 2004a). second, control of reflux symptoms on PPIs does not indicate adequate control of acid reflux into the esophagus: 62% of BE patients treated with esomeprazole had pathologic esophageal acidity, particularly at night, despite control of symptoms (Yeh et al 2003). Finally, our study of BE patients using three different PPIs showed that intragastric pH was <4.0 fully 46% of the time for patients taking omeprazole, 71% of the time for patients on lansoprazole, and 51% of the time for patients on rabeprazole, correlating with a high rate of pathologic esophageal pH (Gerson et al 2005).
Experimental evidence based on metaplastic tissue collected from BE patients and studied in tissue culture demonstrates that pulsatile exposure to low pH leads to hyperproliferation relative to growth at neutral pH, whereas continuous exposure to acidic fluid actually suppresses proliferation (Fitzgerald et al 1996). While pulsatile exposure of esophageal tissue to acidic fluid is characteristic of GERD, the effect of more limited (nocturnal) pulsatile acid exposure in patients with specialized intestinal metaplasia who manifest inadequate acid suppression on PPIs is not known. One study of 39 patients with BE found that after six months on PPIs, biopsy specimens showed a decrease in the expression of a proliferation marker (PCNA) and and increase in expression of a differentiation marker (villin) in patients who had good control of esophageal pH but not in those found to have persistent acid reflux (Ouatu-Lascar et al 1999). This suggests that control of acid reflux may, indeed, interfere with development of dysplasia. Additionally, while clinical data are limited, results from an observations trial at a Veterans’ hospital has supported the idea that use of PPIs after BE diagnosis may significantly lower risk of progressing to dysplasia, finding a hazard ratio of only 0.25 compared with those who did not receive PPIs (El-Serag et al 2004).
Another interesting finding is that patients with limited control of acid reflux on ranitidine showed a significant increase in an index of proliferation of metaplastic tissue biopsies taken over two years of treatment, whereas patients with much tighter control on omeprazole showed no change in proliferation (Peters et al 2000). This was not a controlled study, and thus it is unclear how changes in proliferation on ranitidine compare to the natural course of the disease towards dysplasia.
Recommendation
Limited clinical data suggests that PPI might prevent progression of metaplastic tissue towards dysplasia and/or cancer. Use of H2 blockers is associated with inferior control of reflux symptoms and intra-esophageal pH, and is not currently recommended in BE. Evidence rating: Category B.
Surgical treatment of reflux in BE patients
Currently available evidence suggests that surgery targeted at reducing reflux (such as fundoplication) may not significantly reduce risk of progression from BE to esophageal cancer. A 2003 meta-analysis of 34 studies in patients with BE involving surgery to reduce reflux (4678 patient years) compared with medical therapy (4906 patient years) found no statistically significant difference in rates of progression to cancer (Corey et al 2003). These rates were 3.8 cancers per 1000 patient years in the surgery group vs. 5.3 cancers per 1000 patient years in the medical group (p = 0.3), with the rate of cancer in the medical group dropping to 4.2 per 1000 patient years when only considering studies from 1996–2001 (likely reflecting improvement in pharmacotherapy). One limitation of these data is that they are derived from nonrandomized cohort studies and allow for significant selection bias, such that patients with more significant symptoms may have been more likely to choose surgery. Long-term follow-up from a randomized controlled trial of patients with GERD also concluded that surgical intervention (open Nissen fundoplication) was not significantly better at preventing esophageal carcinoma than medical treatment, but the study was insufficiently powered to detect modest differences (Spechler et al 2001).
Recommendation
Surgical intervention should not be employed to prevent reflux in an effort to reduce risk of esophageal adenocarcinoma in patients with BE, as it does not significantly alter risk of malignant progression. Evidence rating: Category B.
Surveillance of BE for progression to dysplasia
While the metaplastic changes characteristic of Barrett’s esophagus represent a step towards development of cancer, the risk of progression to esophageal adenocarcinoma in patients with BE is only about 0.5% per year (Shaheen et al 2000). Given this relatively low risk and the limits of currently available treatments, ablation or removal of BE is not currently advised unless there are additional risk factors for malignant transformation, such as the finding of dysplastic change on pathology. Patients shown to have high-grade dysplasia appear to have a variable subsequent 5-year risk of 16%–60% for the development of malignancy (Weston et al 2000; Reid et al 2000b; Schnell et al 2001). A prospective study of patients at a Veterans Affairs Hospital showed that only 16% of 75 patients with high-grade dysplasia developed cancer within a mean surveillance period of 7.3 years (Schnell et al 2001); of note, patients were excluded in this study if they developed cancer within the first year of discovering high-grade dysplasia, as it was considered likely that such patients had cancer at time of initial diagnosis of high-grade dysplasia that was missed due to sampling error. The Seattle BE cohort experience of 76 patients with high-grade dysplasia found a 59% 5-year risk of cancer (Reid et al 2000b); when the 27 patients with incident HGD were analyzed, the incidence of cancer was 31% (Reid et al 2000b). Both of these studies lacked external pathologic confirmation of high-grade dysplasia, likely overestimating the diagnosis of high-grade dysplasia. Even amongst pathologists highly experienced in gastrointestinal disease approximately 15% of the pathological specimens diagnosed as high-grade dysplasia may not be read as such by a second pathologist (inter-observer disagreement) (Reid et al 1988; Montgomery et al 2001). This fact highlights the need for obtaining confirmation from a second pathologist experienced in this area prior to conclusive diagnosis of high-grade dysplasia.
The diagnosis of low-grade dysplasia is of more limited utility than that of high-grade dysplasia, both because it is associated with a great deal of diagnostic imprecision (Skacel et al 2000; Montgomery et al 2001) and because its association with esophageal cancer is weaker (Skacel et al 2000; Weston et al 2001). It is, however, associated with a significant risk of progression to high-grade dysplasia (Skacel et al 2000; Weston et al 2001), which is increased when multiple pathologists agree with the diagnosis of low-grade dysplasia (Skacel et al 2000). Studies have demonstrated that up to 30% of patients with low grade dysplasia will show regression to normal tissue (Miros et al 1991; Sharma et al 1997a).
Rational for surveillance, however, is not based on risk of development of cancer alone; rather, it is based on the observation that patients undergoing a surveillance regimen have significantly greater survival rates when cancer is detected. In one study, 86% of 16 patients with esophageal adenocarcinoma found by surveillance of known Barrett’s were alive two years after cancer diagnosis, as opposed to 43% of 54 patients who presented initially with cancer without prior known Barrett’s or surveillance (van Sandick et al 1998). In a similar study, 73% of cancer patients detected by surveillance, as opposed to 12% of those discovered without surveillance were alive 2 years after cancer diagnosis (Corley et al 2002). While lead-time bias certainly accounts for some of this difference, actual survival is almost certainly prolonged, as nodal involvement is much less common in the surveillance patients (van Sandick et al 1998) and additional data demonstrate that patients diagnosed by rigorous surveillance are much more likely to have resectable esophageal tumors than those initially presenting with cancer (Fitzgerald et al 2001). A decision analysis demonstrated that screening and surveillance of BE is cost effective when patients found to have esophageal cancer who are not surgical candidates are offered endoscopic therapy (Gerson et al 2004b).
Recommendation
We currently advise utilization of a surveillance approach proposed by the American College of Gastroenterology in their Practice Guidelines for Barrett’s Esophagus updated in 2002 (Sampliner 2002):
- 1. No Dysplasia on two EGDs with biopsy → 3 year follow-up endoscopy
- 2. Low-Grade Dysplasia on endoscopy → 1 year follow-up until no dysplasia
- 3. High-Grade Dysplasia without cancer, confirmed by experienced pathologist → 3 month follow-up or intervention Evidence rating: Category B.
Treatment of patients with high-grade dysplasia
A number of options are available to patients who have developed high-grade dysplasia. Some overlap exists between treatment options for high-grade dysplasia and those for esophageal adenocarcinoma, as well as for selected patients with lower-grade dysplastic changes. The three major approaches to high-grade dysplasia are observation with endoscopic surveillance, endoscopic ablation, and surgical intervention (principally esophagectomy).
The goal of observation with frequent surveillance is early detection of progression to cancer (Sampliner 2002). Reasons to manage a high grade dysplasia patient with observation include the highly variable risk (18%–60%) of progression to esophageal cancer, the very high morbidity and mortality of esophagectomy (up to 15% in low volume centers), and the risk of masking progression to cancer through ablation of the superficial esophageal epithelium. As markers of progression towards malignancy (see below) become more established, patients under observation will be able to decide when to opt for more aggressive intervention based on molecular findings which correlate with cancer risk more accurately than dysplasia alone. Observation also allows patients to select more advanced treatments as they become available. The risk of observation, or course, is the inherent possibility of disease progression, the fact that intervention is less likely to succeed with more advanced disease (cancer), and the possibility that progression will be missed on surveillance (ie, sampling error). Prior studies have suggested that up to 30% of patients with high grade dysplasia harbor esophageal cancer, but this figure was mainly derived from patients who were not enrolled in an intensive endoscopic surveillance program (Edwards et al 1996; Heitmiller et al 1996; Incarbone et al 2002; Tseng et al 2003). In order to minimize the risk of missing progression to cancer, the recommended biopsy protocol in intensive, and involves four-quadrant biopsies at 1 cm intervals, plus biopsies of any mucosal irregularities, every 3 months (Reid et al 2000a).
Endoscopic ablation, including argon plasma coagulation and photodynamic therapy, are better tolerated than esophagectomy and offer the potential of complete obliteration of dysplastic epithelium (Johnston 2005). Major concerns with endoscopic ablation include the risk of stricture formation (Overholt et al 1999) and, importantly, the risk of subsquamous metaplasia (Barham et al 1997). In the latter case, normal squamous epithelium grows to replace the ablated metaplastic/dysplastic epithelium, but islands of metaplastic cells survive underneath the normal-appearing epithelium. These can grow and progress to cancer without being visible on subsequent endoscopy, allowing for the much-feared possibility that advanced cancer can develop underneath the squamous epithelium.
While ablative techniques now offer an alternative, esophagectomy remains the definitive treatment in surgically fit patients who have advanced high-grade dysplasia, especially when it is multifocal or associated with other mucosal irregularities, or in those who have progressed to early cancer. It offers the most reliable method of preventing progression to advanced or invasive cancer (Spechler 2002). However, it is an especially high-risk procedure, especially at low-volume institutions, where it carries a mortality rate as high as 15%, and an association with complications in two-thirds of patients; at tertiary care facilities with considerable experience in esophagectomy, the mortality risk drops to 3%–5% and the complication rate drops to just over half (Swisher et al 2000; van Lanschot et al 2001).
In patients with early esophageal cancer who are not operative candidates, endoscopic mucosal resection, photo-dynamic therapy, and/or laser therapy have been shown to be reasonable treatment options associated with remission rates between 45% and 75%. (Overholt et al 1999; Ell et al 2000; Van Laethem et al 2001; Wolfsen et al 2002). Local recurrence or metachronous cancer has been reported in up to 30% of patients but can be treated with endoscopic therapy with similar prognostic results.
Recommendations
Patients with high-grade dysplasia without multifocal features or mucosal irregularity can be followed using an intensive biopsy protocol (4-quadrant biopsies every 1 cm with 3 month follow-up), or may opt for more aggressive intervention. Surgical candidates with high-grade dysplasia with concerning features, or early adenocarcinoma, should strongly consider esophagectomy as possible cure, balancing the risk of aggressive cancer with the considerable risk of surgery. Endoscopic ablation should be offered to patients who are not surgical candidates or who have serious reservations about surgery. Evidence rating: Category B.
Future possibilities in the treatment of patients with Barrett’s esophagus
Screening for Barrett’s esophagus in appropriately selected individuals is currently recommended because pathologic findings can lead to surveillance methods that ultimately reduce the risk of mortality from cancer. An ideal screening tool would allow for simultaneous treatment of patients found to have risk-associated pathology, such as polyp removal during screening colonoscopy to prevent progression to colon cancer. At this point, no such option exists for Barrett’s esophagus, as no endoscopic treatment has been shown to safely eliminate or reduce the risk of progression to cancer. However, as techniques for endoscopic ablation become more advanced, it may become advantageous to eliminate Barrett’s mucosa early on, when only low-grade dysplasia or even metaplasia alone is present. Such an approach may not only reduce the risk of malignancy, but also eliminate the need for costly and tedious long-term surveillance.
Current techniques being used or explored for endoscopic ablation include photodynamic therapy, laser therapy, multipolar electrocoagulation, argon plasma coagulation, endoscopic mucosal resection, radiofrequency ablation, and cryotherapy (Johnston 2005). Preliminary work with radio-frequency ablation, as an example, suggests that it may be capable of full-thickness epithelial ablation without injury to the submucosa or stricture formation (Ganz et al 2004; Johnston 2005). If further data demonstrate this technique is indeed capable ablating Barrett’s mucosa while minimizing the risk of hidden subsquamous metaplasia and of complications such as strictures, it may allow for relatively safe elimination of metaplastic epithelium in patients with non-dysplastic Barrett’s esophagus; clinical trials in such patients are reportedly underway (Ganz et al 2004). Until such data exist, use of ablative technology in patients without dysplasia is not indicated or appropriate outside of the realm of research.
Another area where progress is being made is that of biomarkers of transformation risk. Identification of dysplasia or even high-grade dysplasia offers limited ability to risk-stratify, as illustrated by the need for continued surveillance. Additional markers of transformation risk would thus be valuable tools in determining appropriate treatment. As an example, of 322 Barrett’s patients studied by a combination of histology and flow cytometric analysis for abnormal chromosomal number (aneuploidy or 4N), 247 had baseline histology that was either negative, indefinite, or showed low-grade dysplasia (Reid et al 2000b). Of this subset, 215 patient had neither aneuploidy nor 4N on flow cytometry; these patients had a 5-year cumulative cancer incidence of 0%, as opposed to 28% of the 32 patients from the same group who were positive for either aneuploidy or 4N. Such numbers would clearly have implications for surveillance need. To date, a number of biomarkers have been identified that can predict increased risk of progression of Barrett’s metaplasia to cancer by either flow cytometric analysis or gene chip technology (Reid et al 2003; Helm et al 2005; El-Serag et al 2006a); use of such markers may soon play a more routine role in surveillance of BE patients.
Recommendation
- Ablation of non-dysplastic mucosa should occur in a clinical research setting and cannot currently be recommended for all BE patients. Evidence rating: Category E.
- Use of biomarkers is compelling but not yet standardized. Flow cytometry is commercially available at the University of Washington and can be used to risk stratify patients with non-dysplastic BE as well as patients with high grade dysplasia. Evidence rating: Category B.
References
- Altorki NK, Subbaramaiah K, Dannenberg AJ. Semin Oncol. 2004;31:30–6. doi: 10.1053/j.seminoncol.2004.03.043. [DOI] [PubMed] [Google Scholar]
- Amano Y, Ishimura N, Furuta K, et al. Gastrointest Endosc. 2006;64:206–11. doi: 10.1016/j.gie.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Anderson LA, Johnston BT, Watson RG, et al. Cancer Res. 2006;66:4975–82. doi: 10.1158/0008-5472.CAN-05-4253. [DOI] [PubMed] [Google Scholar]
- Armstrong D. Aliment Pharmacol Ther. 2004;20:40–7. doi: 10.1111/j.1365-2036.2004.02132.x. [DOI] [PubMed] [Google Scholar]
- Barham CP, Jones RL, Biddlestone LR, Hardwick RH, Shepherd NA, Barr H. Gut. 1997;41:281–4. doi: 10.1136/gut.41.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LM, Devesa SS. Surg Oncol Clin N Am. 2002;11:235–56. doi: 10.1016/s1055-3207(02)00002-9. [DOI] [PubMed] [Google Scholar]
- Cameron AJ. Gastroenterol Clin North Am. 1997;26:487–94. doi: 10.1016/s0889-8553(05)70308-3. [DOI] [PubMed] [Google Scholar]
- Cameron AJ, Lomboy CT. Gastroenterology. 1992;103:1241–5. doi: 10.1016/0016-5085(92)91510-b. [DOI] [PubMed] [Google Scholar]
- Cameron AJ, Zinsmeister AR, Ballard DJ, Carney JA. Gastroenterology. 1990;99:918–22. doi: 10.1016/0016-5085(90)90607-3. [DOI] [PubMed] [Google Scholar]
- Cook MB, Wild CP, Forman D. Am J Epidemiol. 2005;162:1050–61. doi: 10.1093/aje/kwi325. [DOI] [PubMed] [Google Scholar]
- Corey KE, Schmitz SM, Shaheen NJ. Am J Gastroenterol. 2003;98:2390–4. doi: 10.1111/j.1572-0241.2003.08702.x. [DOI] [PubMed] [Google Scholar]
- Corley DA, Levin TR, Habel LA, Weiss NS, Buffler PA. Gastroenterology. 2002;122:633–40. doi: 10.1053/gast.2002.31879. [DOI] [PubMed] [Google Scholar]
- Csendes A, Smok G, Burdiles P, Korn O, Gradiz M, Rojas J, Recio M. Dis Esophagus. 2003;16:24–8. doi: 10.1046/j.1442-2050.2003.00284.x. [DOI] [PubMed] [Google Scholar]
- de Martel C, Llosa AE, Farr SM, Friedman GD, Vogelman JH, Orentreich N, Corley DA, Parsonnet J. J Infect Dis. 2005;191:761–7. doi: 10.1086/427659. [DOI] [PubMed] [Google Scholar]
- Dent J, Brun J, Fendrick A, Fennerty M, Janssens J, Kahrilas P, Lauritsen K, Reynolds J, Shaw M, Talley N. Gut. 1999;44(Suppl 2):S1–16. doi: 10.1136/gut.44.2008.s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devesa SS, Blot WJ, Fraumeni JF., Jr Cancer. 1998;83:2049–53. [PubMed] [Google Scholar]
- Dickman R, Green C, Chey WD, Jones MP, Eisen GM, Ramirez F, Briseno M, Garewal HS, Fass R. Gastrointest Endosc. 2005;62:675–81. doi: 10.1016/j.gie.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Dulai GS, Guha S, Kahn KL, Gornbein J, Weinstein WM. Gastroenterology. 2002;122:26–33. doi: 10.1053/gast.2002.30297. [DOI] [PubMed] [Google Scholar]
- Edwards MJ, Gable DR, Lentsch AB, Richardson JD. Ann Surg. 1996;223:585–9. doi: 10.1097/00000658-199605000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen GM, Sandler RS, Murray S, Gottfried M. Am J Gastroenterol. 1997;92:27–31. [PubMed] [Google Scholar]
- Ell C, May A, Gossner L, Pech O, Gunter E, Mayer G, Henrich R, Vieth M, Muller H, Seitz G, Stolte M. Gastroenterology. 2000;118:670–7. doi: 10.1016/s0016-5085(00)70136-3. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, Aguirre T, Kuebeler M, Sampliner RE. Aliment Pharmacol Ther. 2004;19:1255–60. doi: 10.1111/j.1365-2036.2004.02006.x. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, Kvapil P, Hacken-Bitar J, Kramer JR. Am J Gastroenterol. 2005;100:2151–6. doi: 10.1111/j.1572-0241.2005.00251.x. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, Nurgalieva Z, Souza RF, Shaw C, Darlington G. Gastrointest Endosc. 2006a;64:17–26. doi: 10.1016/j.gie.2005.08.041. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, Tran T, Richardson P, Ergun G. Scand J Gastroenterol. 2006b;41:887–91. doi: 10.1080/00365520500535402. [DOI] [PubMed] [Google Scholar]
- Farrow DC, Vaughan TL, Hansten PD, Stanford JL, Risch HA, Gammon MD, Chow WH, Dubrow R, Ahsan H, Mayne ST, Schoenberg JB, West AB, Rotterdam H, Fraumeni JF, Jr, Blot WJ. Cancer Epidemiol Biomarkers Prev. 1998;7:97–102. [PubMed] [Google Scholar]
- Fass R, Hell RW, Garewal HS, Martinez P, Pulliam G, Wendel C, Sampliner RE. Gut. 2001;48:310–3. doi: 10.1136/gut.48.3.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald RC, Omary MB, Triadafilopoulos G. J Clin Invest. 1996;98:2120–8. doi: 10.1172/JCI119018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald RC, Saeed IT, Khoo D, Farthing MJ, Burnham WR. Dig Dis Sci. 2001;46:1892–8. doi: 10.1023/a:1010678913481. [DOI] [PubMed] [Google Scholar]
- Ganz RA, Utley DS, Stern RA, Jackson J, Batts KP, Termin P. Gastrointest Endosc. 2004;60:1002–10. doi: 10.1016/s0016-5107(04)02220-5. [DOI] [PubMed] [Google Scholar]
- Gerson LB, Boparai V, Ullah N, Triadafilopoulos G. Aliment Pharmacol Ther. 2004a;20:637–43. doi: 10.1111/j.1365-2036.2004.02127.x. [DOI] [PubMed] [Google Scholar]
- Gerson LB, Edson R, Lavori PW, Triadafilopoulos G. Am J Gastroenterol. 2001;96:2005–12. doi: 10.1111/j.1572-0241.2001.03933.x. [DOI] [PubMed] [Google Scholar]
- Gerson LB, Groeneveld PW, Triadafilopoulos G. Clin Gastroenterol Hepatol. 2004b;2:868–79. doi: 10.1016/s1542-3565(04)00394-5. [DOI] [PubMed] [Google Scholar]
- Gerson LB, Shetler K, Triadafilopoulos G. Gastroenterology. 2002;123:461–7. doi: 10.1053/gast.2002.34748. [DOI] [PubMed] [Google Scholar]
- Gerson LB, Shetler K, Triadafilopoulos G. Dig Liver Dis. 2005;37:651–8. doi: 10.1016/j.dld.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Gerson LB, Triadafilopoulos G. Am J Med. 2002;113:499–505. doi: 10.1016/s0002-9343(02)01234-2. [DOI] [PubMed] [Google Scholar]
- Gillen P, Keeling P, Byrne PJ, West AB, Hennessy TP. Br J Surg. 1988;75:113–5. doi: 10.1002/bjs.1800750208. [DOI] [PubMed] [Google Scholar]
- Gordon C, Kang JY, Neild PJ, Maxwell JD. Aliment Pharmacol Ther. 2004;20:719–32. doi: 10.1111/j.1365-2036.2004.02149.x. [DOI] [PubMed] [Google Scholar]
- Hanna S, Rastogi A, Weston AP, Totta F, Schmitz R, Mathur S, McGregor D, Cherian R, Sharma P. Am J Gastroenterol. 2006;101:1416–20. doi: 10.1111/j.1572-0241.2006.00631.x. [DOI] [PubMed] [Google Scholar]
- Heartburn across America: A Gallup Organization national survey. Princeton NJ: Gallup Organization; 1988. [Google Scholar]
- Heitmiller RF, Redmond M, Hamilton SR. Ann Surg. 1996;224:66–71. doi: 10.1097/00000658-199607000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm J, Enkemann SA, Coppola D, Barthel JS, Kelley ST, Yeatman TJ. Clin Cancer Res. 2005;11:2478–85. doi: 10.1158/1078-0432.CCR-04-1280. [DOI] [PubMed] [Google Scholar]
- Hirota WK, Loughney TM, Lazas DJ, Maydonovitch CL, Rholl V, Wong RK. Gastroenterology. 1999;116:277–85. doi: 10.1016/s0016-5085(99)70123-x. [DOI] [PubMed] [Google Scholar]
- Incarbone R, Bonavina L, Saino G, Bona D, Paracchia A. Surg Endosc. 2002;16:264–6. doi: 10.1007/s00464-001-8161-3. [DOI] [PubMed] [Google Scholar]
- Johnston MH. Nat Clin Pract Gastroenterol Hepatol. 2005;2:323–30. doi: 10.1038/ncpgasthep0214. [DOI] [PubMed] [Google Scholar]
- Jolly K, Cheng KK, Langman MJ. Drugs. 2002;62:945–56. doi: 10.2165/00003495-200262060-00006. [DOI] [PubMed] [Google Scholar]
- Katz PO, Anderson C, Khoury R, Castell DO. Aliment Pharmacol Ther. 1998;12:1231–4. doi: 10.1046/j.1365-2036.1998.00419.x. [DOI] [PubMed] [Google Scholar]
- Kaur BS, Khamnehei N, Iravani M, Namburu SS, Lin O, Triadafilopoulos G. Gastroenterology. 2002;123:60–7. doi: 10.1053/gast.2002.34244. [DOI] [PubMed] [Google Scholar]
- Lagergren J, Bergstrom R, Lindgren A, Nyren O. N Engl J Med. 1999;340:825–31. doi: 10.1056/NEJM199903183401101. [DOI] [PubMed] [Google Scholar]
- Li H, Walsh TN, O’Dowd G, Gillen P, Byrne PJ, Hennessy TP. Surgery. 1994;115:176–81. [PubMed] [Google Scholar]
- Lieberman DA, Oehlke M, Helfand M. Am J Gastroenterol. 1997;92:1293–7. [PubMed] [Google Scholar]
- Mercer CD, Wren SF, DaCosta LR, Beck IT. J Med. 1987;18:135–46. [PubMed] [Google Scholar]
- Miros M, Kerlin P, Walker N. Gut. 1991;32:1441–6. doi: 10.1136/gut.32.12.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moayyedi P, Axon AT. Aliment Pharmacol Ther. 2005;22(Suppl 1):11–9. doi: 10.1111/j.1365-2036.2005.02605.x. [DOI] [PubMed] [Google Scholar]
- Montgomery E, Bronner MP, Goldblum JR, Greenson JK, Haber MM, Hart J, Lamps LW, Lauwers GY, Lazenby AJ, Lewin DN, Robert ME, Toledano AY, Shyr Y, Washington K. Hum Pathol. 2001;32:368–78. doi: 10.1053/hupa.2001.23510. [DOI] [PubMed] [Google Scholar]
- Ouatu-Lascar R, Fitzgerald RC, Triadafilopoulos G. Gastroenterology. 1999;117:327–35. doi: 10.1053/gast.1999.0029900327. [DOI] [PubMed] [Google Scholar]
- Overholt BF, Panjehpour M, Haydek JM. Gastrointest Endosc. 1999;49:1–7. doi: 10.1016/s0016-5107(99)70437-2. [DOI] [PubMed] [Google Scholar]
- Oyama K, Fujimura T, Ninomiya I, Miyashita T, Kinami S, Fushida S, Ohta T, Koichi M. Carcinogenesis. 2005;26:565–70. doi: 10.1093/carcin/bgh340. [DOI] [PubMed] [Google Scholar]
- Pandolfino JE, El-Serag HB, Zhang Q, Shah N, Ghosh SK, Kahrilas PJ. Gastroenterology. 2006;130:639–49. doi: 10.1053/j.gastro.2005.12.016. [DOI] [PubMed] [Google Scholar]
- Pera M, Manterola C, Vidal O, Grande L. J Surg Oncol. 2005;92:151–9. doi: 10.1002/jso.20357. [DOI] [PubMed] [Google Scholar]
- Peters FT, Ganesh S, Kuipers EJ, Sluiter WJ, Karrenbeld A, de Jager-Krikken A, Klinkenberg-Knol EC, Lamers CB, Kleibeuker JH. Scand J Gastroenterol. 2000;35:1238–44. doi: 10.1080/003655200453557. [DOI] [PubMed] [Google Scholar]
- Peters FT, Ganesh S, Kuipers EJ, Sluiter WJ, Klinkenberg-Knol EC, Lamers CB, Kleibeuker JH. Gut. 1999;45:489–94. doi: 10.1136/gut.45.4.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl H, Welch HG. J Natl Cancer Inst. 2005;97:142–6. doi: 10.1093/jnci/dji024. [DOI] [PubMed] [Google Scholar]
- Polednak AP. Int J Cancer. 2003;105:98–100. doi: 10.1002/ijc.11029. [DOI] [PubMed] [Google Scholar]
- Reid BJ, Blount PL, Feng Z, Levine DS. Am J Gastroenterol. 2000a;95:3089–96. doi: 10.1111/j.1572-0241.2000.03182.x. [DOI] [PubMed] [Google Scholar]
- Reid BJ, Blount PL, Rabinovitch PS. Gastrointest Endosc Clin N Am. 2003;13:369–97. doi: 10.1016/s1052-5157(03)00006-0. [DOI] [PubMed] [Google Scholar]
- Reid BJ, Haggitt RC, Rubin CE, Roth G, Surawicz CM, Van Belle G, Lewin K, Weinstein WM, Antonioli DA, Goldman H, et al. Hum Pathol. 1988;19:166–78. doi: 10.1016/s0046-8177(88)80344-7. [DOI] [PubMed] [Google Scholar]
- Reid BJ, Levine DS, Longton G, Blount PL, Rabinovitch PS. Am J Gastroenterol. 2000b;95:1669–76. doi: 10.1111/j.1572-0241.2000.02196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex DK, Cummings OW, Shaw M, Cumings MD, Wong RK, Vasudeva RS, Dunne D, Rahmani EY, Helper DJ. Gastroenterology. 2003;125:1670–7. doi: 10.1053/j.gastro.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Sampliner RE. Am J Gastroenterol. 2002;97:1888–95. doi: 10.1111/j.1572-0241.2002.05910.x. [DOI] [PubMed] [Google Scholar]
- Schnell TG, Sontag SJ, Chejfec G, Aranha G, Metz A, O’Connell S, Seidel UJ, Sonnenberg A. Gastroenterology. 2001;120:1607–19. doi: 10.1053/gast.2001.25065. [DOI] [PubMed] [Google Scholar]
- Shaheen NJ, Crosby MA, Bozymski EM, Sandler RS. Gastroenterology. 2000;119:333–8. doi: 10.1053/gast.2000.9302. [DOI] [PubMed] [Google Scholar]
- Sharma P, Morales TG, Bhattacharyya A, Garewal HS, Sampliner RE. Am J Gastroenterol. 1997a;92:2012–6. [PubMed] [Google Scholar]
- Sharma P, Morales TG, Bhattacharyya A, Garewal HS, Sampliner RE. Am J Gastroenterol. 1998;93:332–5. doi: 10.1111/j.1572-0241.1998.00332.x. [DOI] [PubMed] [Google Scholar]
- Sharma P, Sampliner RE, Camargo E. Am J Gastroenterol. 1997b;92:582–5. [PubMed] [Google Scholar]
- Sharma P, Vakil N. Aliment Pharmacol Ther. 2003;17:297–305. doi: 10.1046/j.1365-2036.2003.01428.x. [DOI] [PubMed] [Google Scholar]
- Skacel M, Petras RE, Gramlich TL, Sigel JE, Richter JE, Goldblum JR. Am J Gastroenterol. 2000;95:3383–7. doi: 10.1111/j.1572-0241.2000.03348.x. [DOI] [PubMed] [Google Scholar]
- Smith RR, Hamilton SR, Boitnott JK, Rogers EL. Am J Surg Pathol. 1984;8:563–73. doi: 10.1097/00000478-198408000-00001. [DOI] [PubMed] [Google Scholar]
- Spechler SJ. N Engl J Med. 2002;346:836–42. doi: 10.1056/NEJMcp012118. [DOI] [PubMed] [Google Scholar]
- Spechler SJ. Am J Gastroenterol. 2005;100:927–35. doi: 10.1111/j.1572-0241.2005.41201.x. [DOI] [PubMed] [Google Scholar]
- Spechler SJ, Lee E, Ahnen D, Goyal RK, Hirano I, Ramirez F, Raufman JP, Sampliner R, Schnell T, Sontag S, Vlahcevic ZR, Young R, Williford W. Jama. 2001;285:2331–8. doi: 10.1001/jama.285.18.2331. [DOI] [PubMed] [Google Scholar]
- Stein DJ, El-Serag HB, Kuczynski J, Kramer JR, Sampliner RE. Aliment Pharmacol Ther. 2005;22:1005–10. doi: 10.1111/j.1365-2036.2005.02674.x. [DOI] [PubMed] [Google Scholar]
- Swisher SG, Deford L, Merriman KW, Walsh GL, Smythe R, Vaporicyan A, Ajani JA, Brown T, Komaki R, Roth JA, Putnam JB. J Thorac Cardiovasc Surg. 2000;119:1126–32. doi: 10.1067/mtc.2000.105644. [DOI] [PubMed] [Google Scholar]
- Tseng EE, Wu TT, Yeo CJ, Heitmiller RF. J Gastrointest Surg. 2003;7:164–70. doi: 10.1016/s1091-255x(02)00153-1. [DOI] [PubMed] [Google Scholar]
- Vaezi MF, Falk GW, Peek RM, Vicari JJ, Goldblum JR, Perez-Perez GI, Rice TW, Blaser MJ, Richter JE. Am J Gastroenterol. 2000;95:2206–11. doi: 10.1111/j.1572-0241.2000.02305.x. [DOI] [PubMed] [Google Scholar]
- van Lanschot JJ, Hulscher JB, Buskens CJ, Tilanus HW, ten Kate FJ, Obertop H. Cancer. 2001;91:1574–8. doi: 10.1002/1097-0142(20010415)91:8<1574::aid-cncr1168>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Van Laethem JL, Jagodzinski R, Peny MO, Cremer M, Deviere J. Endoscopy. 2002;33:257–61. doi: 10.1055/s-2001-12803. [DOI] [PubMed] [Google Scholar]
- van Sandick JW, van Lanschot JJ, Kuiken BW, Tytgat GN, Offerhaus GJ, Obertop H. Gut. 1998;43:216–22. doi: 10.1136/gut.43.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan TL, Dong LM, Blount PL, Ayub K, Odze RD, Sanchez CA, Rabinovitch PS, Reid BJ. Lancet Oncol. 2006;7:7–8. doi: 10.1016/S1470-2045(05)70431-9. [DOI] [PubMed] [Google Scholar]
- Vicari JJ, Peek RM, Falk GW, Goldblum JR, Easley KA, Schnell J, Perez-Perez GI, Halter SA, Rice TW, Blaser MJ, Richter JE. Gastroenterology. 1998;115:50–7. doi: 10.1016/s0016-5085(98)70364-6. [DOI] [PubMed] [Google Scholar]
- Ward EM, Wolfsen HC, Achem SR, Loeb DS, Krishna M, Hemminger LL, DeVault KR. Am J Gastroenterol. 2006;101:12–7. doi: 10.1111/j.1572-0241.2006.00379.x. [DOI] [PubMed] [Google Scholar]
- Westhoff B, Brotze S, Weston A, McElhinney C, Cherian R, Mayo MS, Smith HJ, Sharma P. Gastrointest Endosc. 2005;61:226–31. doi: 10.1016/s0016-5107(04)02589-1. [DOI] [PubMed] [Google Scholar]
- Weston AP, Sharma P, Topalovski M, Richards R, Cherian R, Dixon A. Am J Gastroenterol. 2000;95:1888–93. doi: 10.1111/j.1572-0241.2000.02234.x. [DOI] [PubMed] [Google Scholar]
- Weston AP, Banerjee SK, Sharma P, Tran TM, Richards R, Cherian R. Am J Gastroenterol. 2001;96:1355–62. doi: 10.1111/j.1572-0241.2001.03851.x. [DOI] [PubMed] [Google Scholar]
- Wolfsen HC, Woodward TA, Raimondo M. Mayo Clin Proc. 2002;77:1176–81. doi: 10.4065/77.11.1176. [DOI] [PubMed] [Google Scholar]
- Yeh RW, Gerson LB, Triadafilopoulos G. Dis Esophagus. 2003;16:193–8. doi: 10.1046/j.1442-2050.2003.00327.x. [DOI] [PubMed] [Google Scholar]