Dichotomous Dopaminergic Control of Striatal Synaptic Plasticity (original) (raw)
. Author manuscript; available in PMC: 2010 Mar 8.
Published in final edited form as: Science. 2008 Aug 8;321(5890):848–851. doi: 10.1126/science.1160575
Abstract
Dopamine (DA) is important to a wide range of striatal functions, including associative learning and habit formation. At synapses between cortical pyramidal neurons and principal striatal medium spiny neurons (MSNs), postsynaptic D1 and D2 DA receptors are postulated to be necessary for the induction of long-term potentiation and depression, respectively. Because these receptors are restricted to two distinct MSN populations, this postulate demands that synaptic plasticity be unidirectional in each cell type. Using brain slices from DA receptor transgenic mice, we show that this is not the case. Rather, DA plays complementary roles in these two types of MSN to ensure that synaptic plasticity is bidirectional and Hebbian. In models of Parkinson's disease, this system is thrown out of balance, leading to unidirectional changes in plasticity that could underlie network pathology and symptoms.
The striatal release of DA is intimately linked to associative learning and habit formation (1, 2). This role is thought to be mediated by controlling corticostriatal synaptic plasticity (3-5). However, efforts to characterize how this control is exerted have met with only modest success. Principal MSNs are heterogeneous in their expression of DA receptors (6), falling into one of two equally-sized, morphologically similar groups. One group expresses predominantly D1 DA receptors (D1 MSNs) whereas the other expresses D2 DA receptors (D2 MSNs). These two receptors appear to modulate long-term changes in glutamatergic synaptic plasticity in MSNs in different ways. D1 DA receptor signaling promotes long-term potentiation (LTP) (3, 7) whereas D2 DA receptor signaling promotes long-term depression (LTD) (8, 9). In animal models of Parkinson's disease (PD), where striatal DA levels are very low, both forms of synaptic plasticity in MSNs appear to be lost, suggesting that DA receptor signaling is necessary for their induction (7, 8).
This simple picture poses an obvious conceptual puzzle. If DA is necessary for the induction of synaptic plasticity and D1 and D2 receptors are expressed by different MSNs, then synaptic plasticity must be unidirectional in each population. However, a high percentage of MSNs display both forms of plasticity (3, 7, 9).
In an attempt to resolve this paradox, we re-examined glutamatergic synaptic plasticity in brain slices from transgenic mice in which the expression of D1 or D2 receptors was reported by co-expression of green fluorescent protein (GFP). GFP expression in these mice faithfully reports MSN phenotype, allowing D1 and D2 receptor expressing MSNs to be reliably sampled (10, 11). We induced plasticity by pairing afferent stimulation with postsynaptic spikes in short bursts that were repeated at a theta frequency (5 Hz). At most synapses, a Hebbian form of spike-timing dependent plasticity (STDP) is induced by this protocol (12). That is, when presynaptic activity precedes postsynaptic spiking, LTP is induced, whereas reversing the order induces LTD (12-14). To induce STDP, glutamatergic afferent fibers were stimulated with a small pipette close (∼100 μm) to the soma of an identified MSN that was driven to spike by current injected through a somatic perforated membrane patch (Fig. 1A to C) (12, 13, 15).
Fig. 1.

D2 MSNs displayed bidirectional Hebbian STDP dependent upon D2 and A2a receptors. (A) Schematic illustration of the recording/stimulation configuration. (B) The theta-burst pairing protocols for induction of LTP and (C) LTD. (B and C) Scale bars: 40 mV × 200 ms. (D) LTP induced in by a positive timing pairing. Plots show EPSP amplitude and input resistance as a function of time. The dashed line shows the average EPSP amplitude before induction. The induction was performed at the vertical bar. Filled symbol shows the averages of 12 trials (± s.e.m.). The averaged EPSP traces before and after induction are showed at the top. Scale bars: 2 mV × 100 ms. (E) LTD induced by a negative timing pairing. Plots and EPSP traces as in (D). Scale bars: 2 mV × 100 ms. (F) In the presence of D2 receptor antagonist sulpiride (10 μM), negative timing pairing failed to alter EPSP amplitude. But co-application of A2a adenosine receptor agonist CGS 21680 (100 nM) and sulpiride led to LTP (n = 6; P < 0.05, Wilcoxon). G) LTP induction (n = 11; P < 0.01, Wilcoxon) was disrupted by the NMDA receptor antagonists APV (50 μM) and MK-801 (20 μM) or the A2a receptor antagonist SCH58261 (100 nM). (H) In the presence of D2 receptor agonist quinpirole (10 μM, n = 6), the application of the positive timing protocol leads to induction of LTD. Application of quinpirole and CGS21680 (100 nM, n = 6) together restored LTP with a positive timing pairing. (I) Schematic illustration shows that activation of A2a and NMDA receptors leads to LTP and activation of D2 and mGluR5 receptors and CaV1.3 channels leads to LTD. Moreover, A2a and D2 receptor activation opposes each other in inducing plasticity. Glu, glutamate; EC, endocannabinoid.
In D2 MSNs (fig. S1), repeated pairing of a synaptic stimulation with a postsynaptic spike 5 ms later resulted in LTP of the synaptic response (Fig. 1D). In contrast, preceding synaptic stimulation (−10 ms) with a short burst of postsynaptic spikes induced LTD (Fig. 1E). There were no lasting alterations in synaptic strength with unpaired presynaptic or postsynaptic activity (fig. S1).
Previous studies of striatal LTD induced by conventional plasticity protocols have underscored the importance of D2 receptors (7, 8, 16). In D2 MSNs, timing-dependent LTD was disrupted by antagonizing D2 receptors with sulpiride (control n = 5; sulpiride n = 5; P < 0.05, Mann-Whitney rank sum test), suggesting a similar involvement of D2 receptors (Fig. 1F). Moreover, LTD was disrupted by antagonizing CB1 endocannabinoid (fig. S2) or mGluR5 glutamate receptors (fig. S3). The combination of presynaptic activity and activation of terminal CB1 receptors leads to a lasting reduction in glutamate release probability underlying conventional LTD (17). Indicating a presynaptic expression mechanism, LTD was accompanied by increased trial-to-trial variation in excitatory postsynaptic potential (EPSP) amplitude (fig. S4).
The determinants of LTP at glutamatergic synapses of MSNs are less well characterized. D1 receptors are thought to be important, but they are not expressed by D2 MSNs. Moreover, blocking D1 receptors with SCH23390 had no effect on timing-dependent LTP in D2 MSNs (fig. S5). Adenosine A2a receptors, which couple to the same second messenger cascades as D1 receptors, are robustly and selectively expressed by D2 MSNs (18). Antagonizing these receptors – not D1 receptors – disrupted the induction of LTP in D2 MSNs (control n = 11; SCH58261 n = 7; P < 0.001, Mann-Whitney test) (Fig. 1G). Blocking NMDA receptors also prevented the induction of timing-dependent LTP, as with conventional LTP (7) (APV and MK-801 n = 5; P < 0.01, Mann-Whitney test) (Fig. 1G). Furthermore, the variation in EPSP amplitude was unchanged after LTP induction, suggested that the expression of plasticity was postsynaptic (fig. S4).
Bidirectional STDP depends upon opponent processes controlling LTD and LTP (13, 19, 20). The net change in synaptic strength is hypothesized to reflect the interaction between cellular processes sensitive to the timing of pre- and postsynaptic activity (e.g., NMDA receptors, L-type Ca2+ channels) and G-protein coupled receptor (GPCR) regulated intracellular signaling cascades. By altering the balance between GPCR cascades controlling plasticity, the timing dependence of STDP can be diminished (19, 20). We therefore elevated D2 receptor stimulation by bath application of quinpirole during pairing of presynaptic stimulation with a trailing (+5 ms) postsynaptic spike, a protocol that normally induces LTP. This resulted in a robust LTD (n = 6; P < 0.05, Wilcoxon signed rank test) (Fig. 1H, I). Boosting A2a receptor signaling by bath application of CGS21680 restored LTP (n = 6; P < 0.05, Wilcoxon test) (Fig. 1H, I). Conversely, bath application of the A2a receptor agonist in the presence of a D2 receptor antagonist led to the induction of LTP, even when postsynaptic spiking preceded presynaptic stimulation (Fig. 1F).
When the same STDP protocols were applied to D1 MSNs (fig. S1), a different picture emerged. Pairing presynaptic activity with a trailing postsynaptic spike (+5 ms) induced a robust LTP (Fig. 2A). LTP was dependent upon NMDA receptors (control n = 5; APV and MK-801 n = 4; P < 0.05, Mann-Whitney test) (Fig. 2B) and appeared to be postsynaptically expressed (fig. S4). However, when presynaptic activity followed postsynaptic spiking (−10 ms), EPSP amplitude did not change (Fig. 2C). The absence of LTD in D1 MSNs was consistent with recent work using a conventional plasticity protocol and has been attributed to the failure to generate endocannabinoids postsynaptically during induction, rather than the absence of presynaptic CB1 receptors (8), an inference consistent with the inability of AM-251 to affect the response (control n = 6; AM-251 n = 5; P > 0.05, Mann-Whitney test) (Fig. 2D). We reasoned that this failure could be due to the activation of the GPCR signaling responsible for LTP induction. To test this hypothesis, D1 receptors – commonly believed to be necessary for LTP induction in MSNs (3, 7) – were blocked by bath application of SCH23390 and the protocol repeated. In the absence of D1 receptor activity, pairing postsynaptic spiking with a trailing presynaptic volley led to a robust LTD (n = 6; P < 0.05, Wilcoxon test) (Fig. 2D) that was blocked by an mGluR5 antagonist (fig. S3). However, antagonizing D2 receptors did not disrupt LTD induction in D1 MSNs (fig. S6), suggesting that D2 receptor sensitive interneuronal signaling was not engaged by local, minimal stimulation (16).
Fig. 2.

D1 MSNs displayed bidirectional Hebbian STDP dependent upon D1 receptors. (A) LTP induction by a positive timing pairing protocol. EPSP amplitude and input resistance of the recorded cell were plotted as a function of time. The averaged EPSP traces before and after induction are shown at the top. Scale bars: 2 mV × 100 ms. (B) LTP induction (n = 10; P < 0.01, Wilcoxon test) was blocked by APV (50 μM) and MK-801 (20 μM). (C) LTD was not induced in D1 neurons with a negative pairing. Plots and EPSP traces are from a single cell as in (A). Scale bars: 2 mV × 100 ms. (D) In the presence of D1 receptor antagonist SCH23390 (3 μM), a negative timing pairing revealed LTD. But in the presence of CB1 receptor antagonist AM-251 (2 μM), negative pairing failed to alter EPSP amplitude. (E) LTP induced by a positive timing pairing was blocked by SCH23390, revealing LTD. LTD induced in the presence of SCH23390 was disrupted by AM-251. (F) Schematic drawing shows that activation of D1 and NMDA receptors evokes LTP and activation of mGluR5 receptor and CaV1.3 channels evokes LTD. Moreover, D1 and mGluR5 receptor activation opposes each other in inducing plasticity.
To determine whether attenuating D1 receptor signaling altered the timing dependence of plasticity, the effect of the positive timing protocol that normally induced a robust LTP (Fig. 2E) was re-examined in the presence of a D1 receptor antagonist. This not only prevented LTP induction, it led to the induction of LTD (SCH23390 n = 6; P < 0.05, Wilcoxon test) that was dependent upon endocannabinoid CB1 receptors (SCH23390 n = 6; SCH23390 and AM-251 n = 5; P < 0.01, Mann-Whitney test) (Fig. 2E, F), establishing a mechanistic parallel to LTD in D2 MSNs. Antagonizing downstream presynaptic CB1 receptors alone did not alter LTP induction (fig. S7).
These experiments show that while DA makes STDP in striatal MSNs bidirectional and Hebbian (12), it is not necessary for the induction of synaptic plasticity. This stands in contrast to previous work asserting that DA is essential for plasticity and that striatal DA depletion in animal models of PD eliminates both LTD and LTP at MSN glutamatergic synapses (7, 8). To revisit this issue, two well-established mouse models of PD were examined: in one, DA neurons were lesioned by injecting the toxin 6-hydroxydopamine (6-OHDA) into the medial forebrain bundle (Fig. 3A) and in the other DA was depleted by disrupting the monoamine vesicular transporter with reserpine (15). In both models, striatal DA levels were dramatically reduced and STDP altered in the same way. In D2 MSNs, LTP was induced not only by the usual pairing protocol (6-OHDA n = 6; P < 0.05, Wilcoxon test) (Fig. 3B) (reserpine n = 6; P < 0.05, Wilcoxon test) (Fig. 3C), but also when spiking preceded synaptic stimulation, a protocol that normally induced LTD (n = 10; P < 0.05, Wilcoxon test) (Fig. 3D). As in normal tissue, this LTP was sensitive to A2a receptor block (reserpine n = 6; SCH58261 n = 4; P < 0.05, Mann-Whitney test) (Fig. 3C). Re-establishing D2 receptor activity with exogenous quinpirole rescued LTD (n = 6; P < 0.05, Wilcoxon test) (Fig. 3D). In contrast, LTP was not induced in D1 MSNs by pairings in which synaptic activity preceded postsynaptic spiking, rather, this protocol induced a robust LTD (6-OHDA n = 6; P < 0.05, Wilcoxon test) (Fig. 3E) (reserpine n = 6; P < 0.05, Wilcoxon test) (Fig. 3F) that was sensitive to CB1 receptor block (reserpine n = 6; AM-251 n = 4; P < 0.01, Mann-Whitney test) (Fig. 3F). Reestablishing D1 receptor activity with exogenous SKF81297 rescued LTP with this protocol (n = 6; P < 0.05, Wilcoxon test) (Fig. 3F).
Fig. 3.
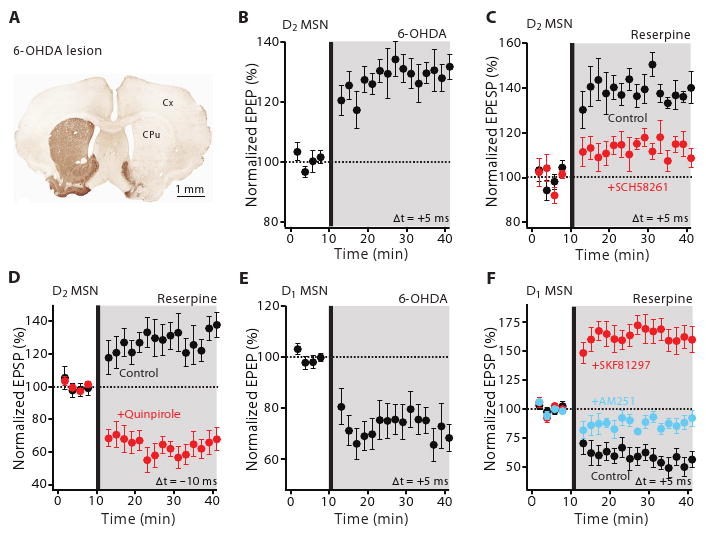
Bidirectional Hebbian STDP is disrupted in MSNs from parkinsonian mice. (A) Light microscopic image of a coronal section showing the loss of immunoreactivity for TH following unilateral 6-OHDA lesioning. Cx, cortex; CPu, caudate putamen. (B) LTP was induced from lesioned D2 mice and (C) reserpine treated mice following a positive timing protocol. Plot of average EPSP amplitude as a function of time. In (C), timing-dependent LTP induced in reserpine treated animals was blocked by SCH58261 (100 nM). (D) LTP also was induced with a negative timing protocol that would normally induce LTD. In contrast, perfusion of quinpirole (10 μM) restored LTD. (E) timing-dependent LTD was evoked in D1 MSNs from lesioned D1 mice and (F) reserpine-treated mice. In (F), D1 receptor agonist SKF81297 (3 μM) restored LTP following a positive timing protocol. The LTD induced in reserpine-treated mice was disrupted by AM-251 (2 μM).
Our studies demonstrate that DA is critical for the induction of bidirectional, timing-dependent (Hebbian) plasticity at glutamatergic synapses formed on striatal MSNs. This finding is consistent with a recent study (14), but conflicts with another (21). The discrepancy between studies could be attributable to heterogeneity in glutamatergic synapses (22) or the engagement of striatal interneurons capable of modulating the induction of plasticity (7, 16). Although the role of these factors is important to sort out, our goal in using focal stimulation near synaptic sites was to isolate the direct actions of DA on the induction of plasticity at glutamatergic synapses of largely cortical origin. This strategy revealed that the type of DA receptor present at the postsynaptic site governed the actions of DA. Furthermore, the signaling determinants of STDP resolved in this way were very similar to those reported for conventional plasticity in MSNs (7-9), suggesting they engaged the same cellular machinery. This insight and the recognition that DA does not act alone in regulating the induction of plasticity, but in concert with glutamate and adenosine, resolves the conflict posed by the apparent obligatory roles of D1 and D2 receptors in the induction of plasticity and their segregation in different MSN classes. It also shows how the activity of DA neurons might serve to reshape the striatal network during associative learning (4). In the absence of behaviorally significant stimuli, DA neurons spike autonomously to maintain striatal DA concentrations at levels sufficient to keep high-affinity D2 DA receptors active, but not low affinity D1 DA receptors (23, 24) – in principle enabling bidirectional, Hebbian plasticity in D2 MSNs, but not in D1 MSNs where the low level of D1 receptor activity should permit only LTD. However, when behaviorally significant stimuli drive phasic spiking of mesencephalic DA neurons, striatal DA levels rise transiently and activate D1 DA receptors (23); this should enable the induction of Hebbian LTP in D1 MSNs. This stark dichotomy provides a cellular foundation for the view that the networks anchored by these two MSNs regulate distinct aspects of behavior and learning (25, 26).
The existence of opponent processes regulating the induction of plasticity also has implications for disease states where DA signalling is abnormal. In both hyper-dopaminergic states, like drug abuse or schizophrenia (27), and hypodopaminergic states, like PD, the imposition of Hebbian rules on the sculpting of synaptic strength by experience will be disrupted, leading to inappropriate associations. This regulatory balance was absent in PD models. In this hypo-dopaminergic state, plasticity at glutamatergic synapses was still present, but it had lost its bidirectionality and dependence upon the temporal structure of pre- and postsynaptic activity. This might help to explain why striatal learning in PD patients is dysfunctional rather than simply absent (28). The cellular specificity of this imbalance also helps to explain the contrasting effects of DA depletion on the synaptic connectivity of D1 and D2 MSNs (10, 29) and the longer term adaptations in network activity thought to underlie the motor symptoms of PD (30).
Materials and Methods
Slice preparation
Parasagittal corticostriatal slices were obtained from 19–26 days old BAC D1/BAC D2 enhanced GFP transgenic mice (S1), in accord with the procedures approved by the Northwestern University Animal Care and Use Committee. The mice were deeply anesthetized intraperitoneally (i.p.) with a mixture of ketamine (50 mg kg−1) and xylazine (4.5 mg kg−1) and perfused transcardially with 5–10 ml of ice-cold artificial CSF (aCSF) comprising (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, 26 NaHCO3 and 14 glucose, continuously bubbled with carbogen (95% O2 and 5% CO2). The brain was quickly removed, blocked in parasagittal plane, glued to the stage of a VT1000S slicer, and immersed in the ice-cold aCSF. Sections through the striatum were cut at a thickness of 275 μm and then transferred to a holding chamber where they were completely submerged in aCSF and was maintained at 34°C for 30 min. Slices were then kept in the holding chamber at the room temperature (22–23°C) for another 20 min before recording.
Electrophysiology
Individual slices were transferred to a recording chamber and were continuously perfused (2–3 ml min−1) with aCSF for the duration of the experiment. A 40× water immersion objective was used to examine the slice with standard infrared differential interference contrast (IR-DIC) video microscopy and a digital CCD camera. Experiments were performed in the dorsal striatum (both medial and lateral regions) at room temperature.
Patch pipettes were pulled from thick-walled borosilicate glass (1.5 mm outer diameter) on a Sutter P-97 puller and fire-polished before use. Pipette resistance was typically 3–4 MΩ when filled with recording solution. The internal recording solution comprised (in mM): 126 KMeSO4, 14 KCl, 3 MgCl2, 0.5 CaCl2, 5 EGTA, 10 HEPES; pH was adjusted to 7.25 with NaOH and osmolarity to 275–280 mOsm l−1. Electrical access was achieved through the perforated-patch method using amphotericin B (S2, S3). A stock solution of amphotericin B (60 mg ml−1) in dimethyl sulfoxide (DMSO) was prepared and diluted in the recording solution immediately before use yielding a final concentration of 180 μg ml−1. Capacitance current was continuously monitored during perforation by applying a 5–10 mV pulse from a holding potential of −70 mV. The membrane potentials were routinely corrected for liquid junction potential. The perforated-patch recordings were made using a MultiClamp 700A amplifier interfaced to a PC running pClamp8 or 9. The signal was filtered at 2 kHz and digitized at 10 kHz with a Digidata 1322A. The amplifier bridge circuit was adjusted to compensate for serial resistance and continuously monitored during recordings. Electrode capacitance was also compensated. Data were excluded when the input resistance (Ri) changed >20 % over the time course of the experiment, which is often accompanied by an increase of serial resistance, indicating a deterioration of patch configuration. EPSPs were evoked by focal extracellular stimulation with a small theta glass electrode positioned ∼100 μm from the recording electrode. Stimulation intensity (0.2 ms, 5–30 μA) was adjusted to evoke baseline single component EPSPs. Backpropagating APs were evoked by direct somatic current injection (5 ms, 1–1.5 nA). Long-lasting synaptic plasticity was induced using protocols consisting of subthreshold synaptic stimulation paired with bAPs at theta frequency (5 Hz). These protocols consisted of 10–15 trains of five bursts repeated at 0.1 Hz, with each burst composed of three bAPs preceded with three EPSPs at 50 Hz (positive timing, +5ms) or three bAPs followed by one EPSP (negative timing, −10 ms). The time interval Δt was defined as the time between the onset of the EPSP and the AP closest in time to the EPSP. To ensure induction of consistent synaptic plasticity, postsynaptic neurons were depolarized to −70 mV from their resting membrane potentials (typically, −85 mV) during their induction (S4). GABAA were blocked by the bath application of bicuculline methiodide (10 μM) or gabazine (10 μM). In preliminary experiments, GABAB was also blocked by CGP55845 (2 μM).
Single cell RT-PCR analysis
Acutely dissociated, individual MSNs were patched in the cell-attached mode and then aspirated into an electrode. Electrodes contained 1.64 μl diethyl carbonate-treated water, 0.2 μl 10× RT buffer, 0.08 μl MgCl2 (25 mM) and 0.08 μl RNaseOUT (40 U μl−1). After aspiration of the neuron, the electrode tip was broken in a sterile eppendorf tube containing 1.4 μl diethyl carbonate-treated water, 0.5 μl MgCl2 (25 mM), 1.0 μl mixed dNTPs (10 mM), 0.7 μl RNaseOUT (40 U μl−1), 0.7 μl BSA (143 μg μl−1), 0.7 μl oligo-dT (0.5 μg μl−1) and 1.0 μl Random Hexamer (50 ng μl−1), and the contents were ejected. The cell mixture was heated to 65°C for 5 min to denature the nucleic acids and then incubated on ice for 1 min. Single-strand cDNA was synthesized from the cellular mRNA by adding 1.0 μl 10× RT buffer, 1.5 μl MgCl2 (25 mM), 1.0 μl DTT (0.1M), 0.5 μl RNase Out (40 U μl−1) and 1.0 μl Superscript III Reverse Transcriptase (200 U μl−1) and then incubating the mixture at 42°C for 90 min. The reaction was terminated by heating the mixture to 85°C for 5 min. The RNA strand in the RNA-DNA hybrid was then removed by adding 1 μl of RNase H (2 U ml−1) and incubating at 37° C for 20 min.
The single-cell cDNAs of interest were then amplified by PCR using specific primers that were developed from GenBank sequences with OLIGO. Standard methods were employed for PCR product detection. Primers for substance P (SP), enkephalin (ENK), D1 and D2 DA receptors have been described before (S5).
Two-photon imaging
BAC D1 or D2 neurons in 275 μm thick slices were identified by somatic eGFP two-photon excited fluorescence using an Ultima Laser Scanning Microscope system. A Dodt contrast was used to provide a bright-field transmission image in registration with the fluorescent images. The green eGFP signals (490–560 nm) were acquired from BAC D1 and BAC D2 neurons using an 810 nm excitation wavelength. The display software was a custom-written shareware package.
DA depletion
The acute DA depletions were produced by administering reserpine (5 mg kg−1, i.p.) for 5 successive days (S3,S6). Animals were killed for experiments 2–3 h after the final injection. Unilateral lesions of nigrostriatal DA neurons were achieved by injecting 6-OHDA into the medial forebrain bundle (S3, S7). Mice were anethetized with i.p. injection of a mixture of ketamine and xylazine and placed on a stereotactic frame. Animals also received an injection of desipramine (15 mg kg−1, i.p.), a norepinephrine reuptake inhibitor, 20–30 min before the infusion of 6-OHDA. 6-OHDA was dissolved at a concentration of 2.5 μg μl−1 in 0.02 % ascorbic acid and injected in final dosage of 2.5 μg. Injections were performed using calibrated glass micropipettes at the following coordinates: AP: 0.7 mm; ML 1.1 mm; DV: 4.8 mm. 6-OHDA was ejected at a rate of 0.05 ml min−1. The micropipette was left in situ for another 30 min after the injection to maximize tissue retention of 6-OHDA.
Limb-use asymmetry test
The degree of damage to nigrostriatal DA neurons was routinely assessed with a forelimb-use asymmetry test (S8). Forelimb use during explorative activity was analyzed by observing mice in a transparent cylinder (8 cm diameter and 12 cm height) for 5 min immediately after animals were placed in the cylinder. The extent of forelimb-use asymmetry displayed by the animal during exploration of the cylinder wall was recorded. The percentages of use of the non-impaired and impaired forelimbs relative to the total number of limb-use movements were obtained.
Tyrosine hydroxylase immunoreactivity
The degree of DA depletion was verified in a subset of mice using TH staining (3). Seven days after 6-OHDA injection, mice were anesthetized (135 mg kg−1 sodium pentobarbital, i.p.) and perfused transcardially with 3% paraformaldehyde in 0.1 M phosphate buffer, pH 7.3. Tissue blocks containing the striatum were cut on a vibrating microtome at a thickness of 50 μm and were incubated with a TH antibody (MAB318, 1:1000) in phosphate-buffered saline (PBS) containing 10% normal goat serum (NGS) and 0.1% Triton X-100 (Tx) for 24 h at 4°C. After washes in PBS, the sections were incubated for 2 h at room temperature with biotinylated donkey anti-mouse IgGs diluted 1:200 in PBS-Tx containing 1% NGS. The sections were then washed and reacted with avidin-biotin peroxidase complex at room temperature for 2 h. Bound peroxidase enzyme activity was revealed using Tris-buffered saline (pH 7.3) containing 0.025 % 3–3-diaminobenzidine tetrahydrochloride (DAB), 0.05% nickel chloride and 0.003 % hydrogen peroxide. No specific immunostaining for respective molecules was observed (data not shown) as sections were incubated with the omission of each of the primary antibodies. Images were captured with a digital camera mounted on fixed-stage, upright microscope. Analysis of immunostaining was performed on scans of each section using ImageJ. The dorsal and ventral striatum ipsilateral and contralateral to the lesion and rostral to the decussation of the anterior commissure was measured. Mean optical density (OD) values were corrected for background staining. TH immunostaining in the striatum ipsilateral to the injection was expressed as a percentage of TH immunostaining in the contralateral striatum.
Coefficient of variance (CV) analysis
If the synaptic transmission is presumed a bionomial distribution, changes in release probability or number of release sites are associated with a change in the CV of the synaptic responses. In contrast, postsynaptic changes should have little effect on CV (S9). Means and CVs were calculated from 50–60 EPSPs immediately before induction and 15–25 min after the end of induction.
Data analysis and statistics methods
Data analysis was done with Igor Pro 6 and Clampfit 9. Both EPSP slope and amplitude were calculated from 50–60 sweeps immediately before the start of induction and 20–30 min after the end of induction. These measures yielded qualitatively similar results. As a consequence, the less derived measure – EPSP amplitude – was used in the presentation. The change in EPSP amplitude was calculated by averaging EPSP values after the induction and normalized to the average baseline EPSP amplitude. Compiled data were expressed as mean ± s.e.m. Statistical tests were performed using Excel and SigmaStat. Non-matched samples were analyzed with the non-parametric Mann-Whitney rank sum test. Matched samples were analyzed with Wilcoxon signed rank test. The threshold for significance was P < 0.05.
Chemicals and reagents
Reagents were obtained as follow: bicuculline methiodide, amphotericin B, MK-801, SKF81927, SCH23390, quinpirole and sulpiride (Sigma); D-APV, SR95531 (gabazine), CGP55845, CGS21680, SCH58261, AM-251 and MPEP (Tocris Cookson).
Supplementary Material
Acknowledgments
This work was supported by NIH grant NS 34696 and a grant from the Picower Foundation. We thank Q. Ruan, K. Saporito and S. Ulrich for their technical assistance; E. Ilijic for generating TH staining picture; M. Day for two-photon image; N. Spruston, R. Malenka and the members of the Surmeier lab for helpful discussion; and N. Spruston and A. Contractor for their comments on the manuscript.
Footnotes
References and Notes
- 1.Graybiel AM, Aosaki T, Flaherty AW, Kimura M. Science. 1994;265:1826. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- 2.Yin HH, Knowlton BJ. Nat Rev Neurosci. 2006;7:464. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds JN, Hyland BI, Wickens JR. Nature. 2001;413:67. doi: 10.1038/35092560. [DOI] [PubMed] [Google Scholar]
- 4.Schultz W. J Neurophysiol. 1998;80:1. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Surmeier DJ, Ding J, Day M, Wang Z, Shen W. Trends Neurosci. 2007;30:228. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 6.Gerfen CR, et al. Science. 1990;250:1429. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 7.Calabresi P, Picconi B, Tozzi A, Di Filippo M. Trends Neurosci. 2007;30:211. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 8.Kreitzer AC, Malenka RC. Nature. 2007;445:643. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- 9.Gerdeman GL, Ronesi J, Lovinger DM. Nat Neurosci. 2002;5:446. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- 10.Day M, et al. Nat Neurosci. 2006;9:251. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- 11.Shen W, et al. Nat Neurosci. 2007;10:1458. doi: 10.1038/nn1972. [DOI] [PubMed] [Google Scholar]
- 12.Sjostrom PJ, Rancz EA, Roth A, Hausser M. Physiol Rev. 2008;88:769. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- 13.Nevian T, Sakmann B. J Neurosci. 2006;26:11001. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pawlak V, Kerr JN. J Neurosci. 2008;28:2435. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.See Supporting Online Materials available on Science Online.
- 16.Wang Z, et al. Neuron. 2006;50:443. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Adermark L, Lovinger DM. J Neurosci. 2007;27:6781. doi: 10.1523/JNEUROSCI.0280-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Trends Neurosci. 2006;29:647. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Seol GH, et al. Neuron. 2007;55:919. doi: 10.1016/j.neuron.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tzounopoulos T, Rubio ME, Keen JE, Trussell LO. Neuron. 2007;54:291. doi: 10.1016/j.neuron.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fino E, Glowinski J, Venance L. J Neurosci. 2005;25:11279. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolam JP, Hanley JJ, Booth PA, Bevan MD. J Anat. 2000;196:527. doi: 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonon F. J Neurosci. 1997;17:5972. doi: 10.1523/JNEUROSCI.17-15-05972.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richfield EK, Penney JB, Young AB. Neuroscience. 1989;30:767. doi: 10.1016/0306-4522(89)90168-1. [DOI] [PubMed] [Google Scholar]
- 25.Middleton FA, Strick PL. Brain Res Brain Res Rev. 2000;31:236. doi: 10.1016/s0165-0173(99)00040-5. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura K, Hikosaka O. J Neurosci. 2006;26:5360. doi: 10.1523/JNEUROSCI.4853-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montague PR, Hyman SE, Cohen JD. Nature. 2004;431:760. doi: 10.1038/nature03015. [DOI] [PubMed] [Google Scholar]
- 28.Dujardin K, Laurent B. Curr Opin Neurol. 2003;16 2:S11. doi: 10.1097/00019052-200312002-00003. [DOI] [PubMed] [Google Scholar]
- 29.Mallet N, Ballion B, Le Moine C, Gonon F. J Neurosci. 2006;26:3875. doi: 10.1523/JNEUROSCI.4439-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ. Trends Neurosci. 2002;25:525. doi: 10.1016/s0166-2236(02)02235-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.