A Novel Delta Opioid Receptor Antagonist, SoRI-9409, Produces a Selective and Long-Lasting Decrease in Ethanol Consumption in Heavy-Drinking Rats (original) (raw)
. Author manuscript; available in PMC: 2014 Jan 12.
Abstract
Background
Naltrexone, a compound with high affinity for the μ opioid receptor (MOP-R) reduces alcohol consumption. SoRI-9409 is a derivative of naltrexone that has highest affinity at δ opioid receptors (DOP-Rs). We have investigated the effects of SoRI-9409 on ethanol consumption to determine the consequences of altering the naltrexone compound to a form with increased efficacy at DOP-Rs.
Methods
Effects of the opioid receptor antagonists, SoRI-9409 (0–30 mg/kg, IP), naltrexone (0–30 mg/kg, IP), or naltrindole (0–10 mg/kg, IP) on ethanol consumption was measured in high- and low-ethanol–consuming rats with two different drinking paradigms. SoRI-9409-, naltrexone-, and naltrindole-mediated inhibition of DOP-R–stimulated [35S]GTPγS binding was measured in brain membranes prepared from high-ethanol–consuming rats. The effects of SoRI-9409 on morphine-mediated analgesia, conditioned place preference, and anxiety were also examined.
Results
In high- but not low-ethanol–consuming animals, SoRI-9409 is threefold more effective and selective at reducing ethanol consumption when compared with naltrexone or naltrindole for up to 24 hours. SoRI-9409 administered daily for 28 days continuously reduced ethanol consumption, and when the administration of SoRI-9409 was terminated, the amount of ethanol consumed remained lower compared with vehicle-treated animals. Furthermore, SoRI-9409 inhibits DOP-R–stimulated [35S]GTPγS binding in brain membranes of high-ethanol–consuming rats.
Conclusions
SoRI-9409 causes selective and long-lasting reductions of ethanol consumption. This suggests that compounds that have high affinity for DOP-Rs such as SoRI-9409 might be promising candidates for development as a novel therapeutic for the treatment of alcoholism.
Keywords: Alcoholism, ethanol, naltrexone, opioid receptor antagonist, rats, SoRI-9409
Despite its devastating impact on society, there are still few effective medications currently available for the treatment of alcoholism. There have been three medications approved by the U.S. Food and Drug Administration for alcoholism: disulfiram (Antabuse), acamprosate (Campral), and naltrexone (ReVia, Vivitrol). The non-selective opioid antagonist, naltrexone, is reported to effectively reduce ethanol consumption in animals (1,2) and in humans (3,4) and has the most consistent effect in reducing alcohol consumption in combination with behavioral therapy (5). However, not all patients respond to naltrexone, which has been partly explained by genetic variations in the opioid receptors (6). Furthermore, adverse effects such as nausea, vomiting, depression, and dysphoria have been reported (3,7-9). Elevated liver enzymes were reported in patients receiving 6–7 times the usual dose of naltrexone (9,10) but not at the usual oral doses or in depot formulations (11). The aim of our study is to further understand the mechanism of action of naltrexone that will facilitate the design of treatments that have greater efficacy with reduced side effects for alcohol use disorders.
Naltrexone has high affinity at the μ opioid peptide receptor (MOP-R) and moderate activity at the δ opioid peptide receptor (DOP-R) and κ opioid peptide receptor (KOP-R) (12,13). There is a large body of evidence that suggests that the MOP-R plays a significant role in ethanol-mediated reward behavior (1) (see Table 1). In contrast, the role of both the DOP-R and KOP-R is less clear. DOP-R knock-out mice consume more ethanol than their wild-type litter-mates (14). However, in contrast, the administration of DOP-R antagonists in rodents have been reported to decrease ethanol consumption and seeking (see Table 1), although some studies have reported DOP-R antagonists do not effect ethanol intake (1,15). DOP-R antagonists increase the aversive properties of alcohol (16), and administration of an enkephalinase inhibitor, which potentiates the actions of enkephalins, has been reported to increase ethanol intake in alcohol-preferring rats (17). Similar differences have been observed in studies investigating the role of the KOP-R in ethanol consumption (see Table 1).
Table 1.
The Role of Opioid Receptors in Ethanol Consumption
| Opioid Receptor | Effects on Ethanol Consumption | |||||
|---|---|---|---|---|---|---|
| Effects in Knock-Out Mice | Effects of Opioid Ligands in Rats | |||||
| Mice | Refs | Antagonists | Refs | Agonists | Refs | |
| MOP-R | ↓ | 35-37 | ↓ | 1, 22, 26, 39-41 | ↑ | 46 |
| DOP-R | ↑ | 14 | ↓ or no change | 1, 15, 17, 21, 22, 40-43 | Unknown | |
| KOP-R | ↓ | 38 | ↑ or no change | 44, 45 | ↑ or ↓ | 45, 47 |
Therefore, an important step in the development of new opioid receptor antagonists for the treatment of alcohol use disorders would be to identify inhibitors or activators of subtypes of opioid receptors that have increased selectivity for inhibiting ethanol-mediated behaviors. Fusion of the chlorinated-phenylpyridine ring onto the naltrexone structure to form SoRI-9409 results in approximately 20-fold higher binding affinity at the DOP-R (Ki = 2.2 ± .2 nmol/L), approximately 20-fold reduced binding affinity for the MOP-R (Ki = 51 ± 8 nmol/L), with similar affinity at the KOP-R (12). We have investigated the effects of SoRI-9409 in both high- and low ethanol–consuming rats to determine the consequences of altering the naltrexone compound to a form with increased affinity at the DOP-R.
Methods and Materials
Drugs and Chemicals
(5’-(4-Chlorophenyl)-17-(cyclopropylmethyl)-6,7-didehydro-3,14-dihydroxy-4,5-α-epoxypyrido-[2’,3’:6,7]morphinan), SoRI-9409, was synthesized at the Southern Research Institute (Birmingham, Alabama). Ethanol (190 proof) was purchased from the University of California San Francisco storehouse (San Francisco, California). [35S]-guanosine 5’-(γ-thio)triphosphate ([35S]-GTPγS) (250 μCi; 9.25 MBq) was supplied from Perkin-Elmer (Boston, Massachusetts). Naltrexone, naltrindole, [D-Pen2,D-Pen5]-Enkephalin (DPDPE), (+)-4-[(αR)-α-((2S,5R)-4-Allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC80), (2-methyl-4a (α)-(3-hydroxyphenyl)-1,2,3,4,4a,5,12, 12a(α)-octahydro-quinolino[2,3,3,g] isoquinoline) dihydrobromide (TAN67), guanosine 5′-[γ-thio]triphosphate tetralithium salt (GTPγS) and guanosine 5′-diphosphate sodium salt (GDP), 2-hydroxy-ethylpiperazine-_N_-2-ethane sulphonic acid (HEPES), DL-dithiothreitol, tricine, dimethyl sulfoxide (DMSO), magnesium chloride (MgCl2) ethylenediaminetetraacetic acid (EDTA), and saponin were purchased from Sigma-Aldrich (St. Louis, Missouri). Sodium chloride and glacial acetic acid were purchased from Fisher Scientific (Fairlawn, New Jersey). Complete mini protease inhibitor cocktail tablets were purchased from Roche (Indianapolis, Indiana), Wheatgerm Agglutinin SPA Beads were purchased from Amersham (Little Chalfont, England) Biosciences.
Animals and Housing
Adult male Long-Evans rats (175–200 g) were purchased from Harlan (Indianapolis, Indiana) and housed in a temperature-controlled environment (22 ± 2°C) with a 12-hour light/dark cycle. Rats were given at least 1 week to acclimatize with food and water available ad libitum before all experimentation. Protocols related to the experiment were approved by the Institutional Animal Care and Use Committee and are in accordance with the National Institute on Drug Abuse, National Institutes of Health, and the Guide for the Care and Use of Laboratory Animals.
Two-Bottle-Choice Drinking Paradigms
All fluids were presented in 100-mL graduated glass cylinders with stainless steel drinking spouts inserted through two grommets in front of the cage. The placement of the ethanol (or sucrose) bottle was alternated before the start of each drinking session to control for side preferences. Bottles were weighed 30 min and 24 hours after the fluids were presented, and measurements were taken to the nearest gram. The weight of each rat was measured daily to calculate the gram/kilogram of ethanol intake (g/kg). Preference for ethanol was calculated as the amount of ethanol consumed as a percentage of the total fluid consumption: % Preference = grams of ethanol/(grams of ethanol + grams of water) × 100%. Sucrose intake and preference was calculated in a similar manner. A minimum of 4 days between drug administrations was given to allow for the drugs to wash out.
Intermittent-Access to 20% Ethanol
The intermittent access 20% ethanol two-bottle-choice drinking paradigm does not require sucrose fading (18-20). Rats given intermittent access to 20% (v/v) ethanol have been reported to consume large amounts of ethanol (18-20). In brief, ethanolnaive rats (n = 12) were given access to bottles of ethanol (20% v/v) and water for 24-hour-long sessions on alternate days (three 24-hour sessions each week) with water only available on days between ethanol exposures. No sucrose fading was needed, and water was always available ad libitum. Drug administrations began after the rats had maintained stable baseline drinking levels (4.3 ± .6 g/kg/24 hours; 18 ethanol exposures) of the 20% v/v ethanol solution for 6 weeks.
Continuous-Access to 10% Ethanol or 5% Sucrose
After the acclimatization period, rats (n = 12) were given access to a bottle containing a solution of 10% (v/v) ethanol and 10% (w/v) sucrose and a separate water bottle. Over the next 12 days, the sucrose concentration was gradually decreased (i.e., from 10% to 5%, 2%, and 0% sucrose) until rats had continuous access to one bottle of 10% v/v ethanol and one bottle of water. Rats given continuous access to 10% (v/v) ethanol have been reported to consume low to moderate amounts of ethanol (18,19). Drug administrations began after the rats had maintained stable baseline drinking levels for 6 weeks (2.1 ± .2 g/kg/24 hours after 8 weeks of ethanol consumption including the sucrose fading period). A separate group of rats (n = 10) were given continuous daily access to a bottle containing a solution of 5% (v/v) sucrose and a separate water bottle. Drug administrations began after the rats had maintained stable baseline drinking levels for 2 weeks.
Drug Treatments
Groups of rats (n = 12) maintained at a stable level of ethanol consumption under each paradigm for at least 6 weeks received an IP injection of each dose of SoRI-9409 (0, 5, 15, 30 mg/kg), naltrexone (0, 5, 15, 30 mg/kg), or naltrindole (0, 1, 5, 10 mg/kg). All injections (1 mL/kg IP) were freshly prepared and given 30 min before access to bottles of ethanol (10% or 20% v/v) or sucrose (5% v/v) and water solutions. SoRI-9409 was dissolved in 2% dimethyl sulfoxide (DMSO) in distilled water with a drop of glacial acetic acid added to keep the drug in solution (pH 5.3), and naltrexone and naltrindole were dissolved in saline and distilled water, respectively. To examine the effects of the multiple administrations of SoRI-9409 on ethanol consumption in drinking rats, SoRI-9409 (5 mg/kg IP; n = 8) or vehicle (1 mL/kg IP; n = 8) was administered daily for 5 consecutive days (three ethanol exposures) to long-term drinking rats with the intermittent-access 20% ethanol two-bottle paradigm. Rats continued to drink with the same drinking paradigm after cessation of daily administration of either SoRI-9409 or vehicle, facilitating observation of post-treatment drinking levels. To examine the effect of the administration of SoRI-9409 on initial ethanol intake and escalation of ethanol consumption over a longer period of time, SoRI-9409 (5 mg/kg IP; n = 16) or vehicle (1 mL/kg IP; n = 15) was administered daily for 28 consecutive days (12 ethanol exposures) to naive rats given access to intermittent 20% ethanol. After 4 weeks, the daily administration of either SoRI-9409 or vehicle was terminated and the rats continued to drink with the same drinking paradigm for a further 28 days.
[35S]GTPγS Binding in Rat Membranes
After decapitation, brains were removed and the following brain regions were dissected and quickly frozen with liquid nitrogen and stored at 80°C until used: the striatum, brainstem, cerebellum, hippocampus, hypothalamus, cerebral cortex, and midbrain. Rat brain regions were quickly thawed and suspended in a homogenization buffer (50 mmol/L Tris-hydrogen chloride, 1 mmol/L EDTA, 3 mmol/L MgCl2, and two mammalian protease inhibitor tablets/50 mL; pH 7.4; 1 g brain tissue/20 mL buffer). The tissue from each brain region was individually homogenized on ice (900 rpm; 15 strokes) and centrifuged (1000 × g, 10 min, 4°C followed by 20,000 × g, 15 min, 4°C) before resuspension in assay buffer (100 mmol/L HEPES.sodium hydroxide, 8 mmol/L MgCl2, 4 mmol/L EDTA, 10 μg/mL saponin, and two mammalian protease inhibitor tablets/50 mL; pH 7.5) before snap-frozen in liquid nitrogen and stored at −80°C until required. Binding assays (n = 3, each in triplicate) were performed in 96-well plates on ice with each reaction containing [35S]GTPγS (50 pmol/L), cell membrane (10 μg protein), GDP (30 μmol/L), and SPA beads (.5 mg) with assay buffer (as previous) and the opioid ligands. Single drug dose-response curves (.1 nmol/L–100 μmol/L) of [35S]GTPγS-stimulated binding were performed with the DOP-R ligands SNC80, DPDPE, and TAN67 in each rat brain region. Inhibition of SNC80-, TAN67-, and DPDPE (10 μmol/L)-stimulated [35S]GTPγS binding in rat striatal membranes prepared from long-term ethanol drinking rats was performed in the presence of varying concentrations of SoRI-9409, naltrexone, and naltrindole (.1 nmol/L–100 μmol/L). Assay plates were shaken for 45 min at 25°C, and centrifuged (1500 rpm, 5 min, 25°C) before [35S]GTPγS-stimulated binding was assessed with the NXT TOPCOUNTER. [35S]GTPγS-stimulated binding is expressed as a percentage increase in [35S]GTPγS-binding relative to binding in unstimulated samples: % stimulation = (ligand count – no treatment)/(no treatment) × 100%.
Statistics
Statistical analyses were performed with GraphPad Prism (GraphPad, San Diego, California). Behavioral data was analyzed with one-way analysis of variance with repeated measures with the Newman-Keuls post hoc analysis and the Student t test where appropriate with an overall significance criterion of p < .05. Data from in vitro functional binding assays were analyzed by non-linear regression with a sigmoidal curve with variable slope to determine EC50 and IC50 values.
Results
Effects of SoRI-9409 on Ethanol and Sucrose Intake
The effects of SoRI-9409, naltrexone, and naltrindole were examined in high-ethanol–consuming rats with the intermittent-access voluntary 20% ethanol two-bottle choice drinking paradigm (18,19). In rats that had maintained a stable level of ethanol consumption (4.3 ± .6 g/kg/24 hours, mean ± SEM) for at least 6 weeks (18 drinking sessions), SoRI-9409 decreased ethanol consumption for at least 24 hours. There was an overall main effect on ethanol consumption in high-ethanol–consuming rats after 24 hours of ethanol access [_F_(3,11) = 24.6, _p_ < .0001; Figure 1A]. Post hoc analysis revealed that SoRI-9409 significantly reduced ethanol consumption at all doses compared with vehicle (5 mg/kg, p < .05; 15 and 30 mg/kg, _p_ < .001). Water consumption was not significantly affected by SoRI-9409 treatment after 24 hours compared with vehicle treatment [_F_(3,11) = .9, _p_ > .05; Table 2]. SoRI-9409 produced an overall effect on the preference for ethanol [_F_(3,11) = 11.7, _p_ <.0001; Table 2].
Figure 1.
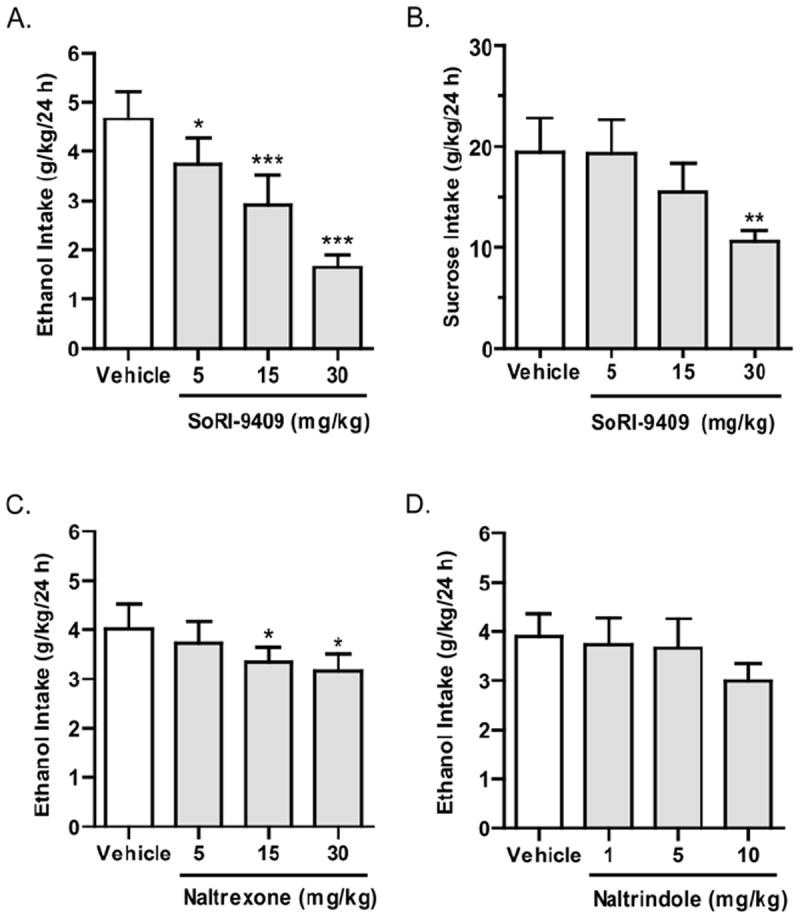
SoRI-9409 selectively reduces ethanol intake in high-ethanol–consuming rats. (A) SoRI-9409 (5, 15, and 30 mg/kg IP, n = 12) significantly and dose-dependently decreases ethanol consumption in rats, using the intermittent access to 20% ethanol drinking paradigm. (B) SoRI-9409 (5 and 15 mg/kg IP, n = 10) does not significantly affect sucrose consumption, using the two-bottle-choice 5% sucrose drinking paradigm. (C) Naltrexone (15 and 30 mg/kg IP, n = 12) significantly decreases ethanol consumption and (D). Naltrindole (1, 5, and 10 mg/kg IP, n = 12) does not significantly decrease ethanol consumption, using the intermittent access to 20% ethanol drinking paradigm. The values are expressed as mean ± SEM ethanol or sucrose consumed (g/kg/24 hours) (repeated measures analysis of variance followed by Newman-Keuls post hoc test *p < .05; **p < .01; ***p < .001; compared with vehicle).
Table 2.
The Effects of SoRI-9409, Naltrexone, and Naltrindole Administration on Water Intake and Ethanol Preference After 24 Hours of Access to Ethanol and Water
| Drug Dose | Intermittent Access to 20% Ethanol | Continuous Access to 10% Ethanol | ||
|---|---|---|---|---|
| Water Intake (mL/24 hours) | Preference for Ethanol (%) | Water Intake (mL/24 hours) | Preference for Ethanol (%) | |
| SoRI-9409 (mg/kg) | ||||
| Vehicle | 11.8 ± 2.0 | 56.9 ± 6.6 | 19.7 ± 2.5 | 41.1 ± 5.5 |
| 5 | 13.7 ± 2.6 | 49.9 ± 7.3 | 18.9 ± 2.1 | 39.5 ± 8.6 |
| 15 | 12.6 ± 1.7 | 42.1 ± 7.4a | 17.6 ± 1.0 | 32.4 ± 7.3b |
| 30 | 11.1 ± .9 | 32.8 ± 4.9a | 21.7 ± 1.6 | 24.0 ± 6.2b |
| Naltrexone (mg/kg) | ||||
| Vehicle | 13.7 ± 2.3 | 53.0 ± 6.8 | 24.0 ± 2.2 | 29.8 ± 4.0 |
| 5 | 11.8 ± 2.2 | 55.2 ± 7.1 | 16.9 ± 1.8b | 32.7 ± 5.9 |
| 15 | 11.3 ± 1.8 | 54.1 ± 6.4 | 17.2 ± 2.1b | 33.8 ± 7.3 |
| 30 | 9.6 ± 1.1b | 54.4 ± 4.7 | 15.5 ± 1.1a | 37.0 ± 4.9 |
| Naltrindole (mg/kg) | ||||
| Vehicle | 13.7 ± 2.2 | 52.7 ± 5.9 | 19.6 ± 1.5 | 39.0 ± 4.4 |
| 1 | 13.0 ± 2.4 | 53.4 ± 7.9 | 18.6 ± 1.6 | 38.2 ± 3.7 |
| 5 | 11.4 ± 2.3 | 55.1 ± 8.3 | 19.6 ± 1.7 | 31.7 ± 4.0 |
| 10 | 12.5 ± 1.9 | 48.8 ± 6.1 | 19.2 ± 1.3 | 31.0 ± 3.3 |
The effects of SoRI-9409 on sucrose intake were measured with continuous access voluntary 5% sucrose two-bottle choice drinking paradigm. SoRI-9409 did not significantly affect sucrose consumption at doses of 5 or 15 mg/kg after 24 hours of sucrose access (p > .05), compared with vehicle (Figure 1B). However, SoRI-9409 at a dose of 30 mg/kg caused a reduction in sucrose intake (p < .01) compared with vehicle [_F_(3,9) = 6.9, _p_ < .01]. No significant differences were found between vehicles and baseline levels for either sucrose or water consumption (_p_ >.05).
Naltrexone produced an overall main effect on ethanol consumption in high-ethanol–consuming rats after 24 hours of ethanol access [_F_(3,11) = 4.5, _p_ < .01; Figure 1C]. Post hoc analysis revealed naltrexone to significantly reduce ethanol consumption at doses of 15 and 30 mg/kg after 24 hours of ethanol access (p < .05). Water consumption was significantly affected by naltrexone treatment after 24 hours compared with vehicle treatment [_F_(3,11) = 3.5, _p_ < .05; Table 2]. Naltrexone produced no significant overall effect on the preference for ethanol [_F_(3,11) = .2, _p_ > .05; Table 2]. Naltrindole did not significantly affect ethanol consumption in high-ethanol–consuming rats at all doses tested after 24 hours of ethanol access [_F_(3,11) = 2.4, _p_ > .05; Figure 1D]. Naltrindole did not significantly affect water consumption [F(3,11) = .7, p >. 05] or preference for ethanol [F(3,11) = .4, p > .05] compared with vehicle-treated rats (Table 2).
Higher Doses of SoRI-9409 Reduce Ethanol Intake in Rats Consuming Lower Amounts of Ethanol
In rats that had maintained a stable level of continuous ethanol consumption (2.2 ± .2 g/kg/24 h, mean ± SEM) for at least 6 weeks (42 drinking sessions after a sucrose fade), SoRI-9409 at 15- and 30-mg/kg doses but not the 5-mg/kg dose decreased ethanol consumption for at least 24 h. There was an overall main effect of SoRI-9409 on ethanol consumption in low-ethanol–consuming rats after 24 hours of ethanol access compared with vehicle-treated rats [_F_(3,9) = 4.3, _p_ < .05; Figure 2A]. Post hoc analysis revealed SoRI-9409 to significantly reduce ethanol consumption at doses of 15 and 30 mg/kg compared with vehicle (p < .05). Water consumption was not significantly affected by SoRI-9409 treatment after 24 hours compared with vehicle treatment [_F_(3,9) = .8, _p_ > .05; Table 2]. SoRI-9409 produced an overall effect on the preference for ethanol [_F_(3,9) = 2.9, _p_ < .05; Table 2]. Naltrexone did not produce any significant effect on ethanol consumption compared with vehicle-treated rats after 24 hours of access to ethanol [_F_(3,10) = .5, _p_ > .05; Figure 2B], although a reduction in ethanol consumption was observed after a limited access period (1 hour; data not shown). Water consumption was significantly affected by naltrexone after 24 hours compared with vehicle treatment [_F_(3,10) = 4.7, _p_ < .01; Table 2]. Naltrexone produced no significant overall effect on the preference for ethanol [_F_(3,10) = .4, _p_ > .05; Table 2]. Naltrindole produced an overall main effect on ethanol consumption in low-ethanol–consuming rats after 24 hours of ethanol access compared with vehicle-treated rats [_F_(3,11) = 3.5, _p_ < .05; Figure 2C]. Post hoc analysis revealed naltrindole to significantly reduce ethanol consumption at doses of 5 and 10 mg/kg compared with vehicle (p < .05). Water consumption was not significantly affected by naltrindole treatment after 24 hours compared with vehicle treatment [_F_(3,11) = .2, _p_ > .05; F = .8, p > .05; Table 2]. Naltrindole produced no significant effects on the preference for ethanol [_F_(3,11) = 2.4, _p_ > .05; Table 2]. Vehicles did not produce any significant effects on ethanol or water consumption after 24 hours of ethanol access, compared with baseline consumption levels (p >.05).
Figure 2.
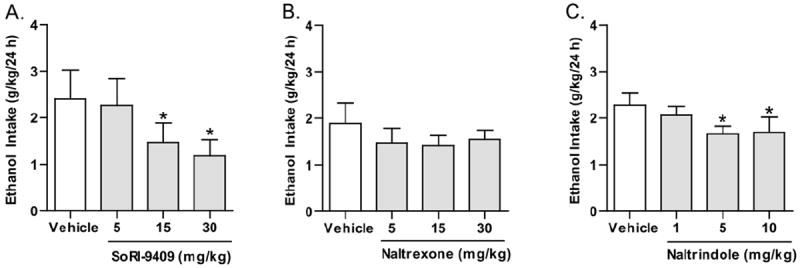
SoRI-9409 reduces ethanol intake, using the continuous access 10% ethanol two-bottle-choice drinking paradigm. (A) SoRI-9409 (5, 15, and 30 mg/kg IP) significantly and dose-dependently decreases ethanol consumption in rats after 24 hours. (B) Naltrexone (5, 15, and 30 mg/kg IP) does not significantly decrease ethanol consumption. (C) Naltrindole (1, 5, and 10 mg/kg IP) significantly decreases ethanol consumption. The values are expressed as mean ± SEM ethanol consumed (g/kg/24 hours) (repeated measures analysis of variance followed by Newman-Keuls post hoc test). *p < .05; compared with vehicle, n = 12.
Multiple Administrations of SoRI-9409 Reduce Ethanol Intake in Long-Term Heavy-Drinking Rats
Administration of SoRI-9409 (5 mg/kg IP) for 5 days to long-term ethanol-drinking rats with the intermittent-access voluntary 20% ethanol two-bottle choice drinking paradigm significantly reduced ethanol intake when compared with baseline drinking levels (Figure 3). Pre-treatment baseline levels of ethanol intake were not significantly different between groups of rats given SoRI-9409 or vehicle (p > .05). Daily administration of vehicle to long-term drinking rats for 5 days did not affect ethanol intake. When daily administrations of SoRI-9409 (or vehicle) were ceased, rats did not display an increased level of ethanol intake compared with pre-treatment baseline drinking levels (Figure 3).
Figure 3.
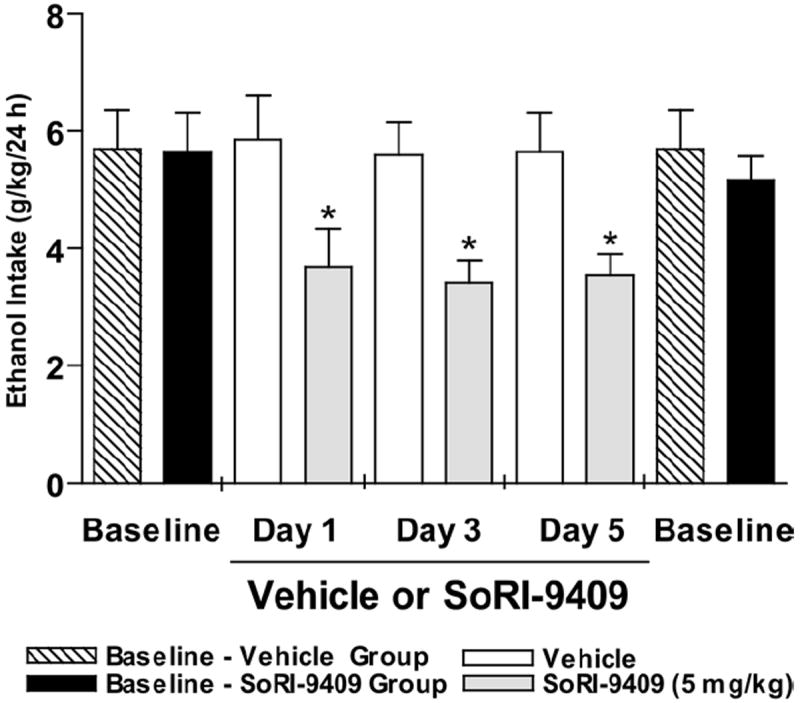
Multiple dosing of SoRI-9409 reduces ethanol intake in high-ethanol–consuming rats. Daily administration of SoRI-9409 (5 mg/kg IP) for 5 days (three drinking sessions) to long-term drinking rats significantly reduced ethanol intake in rats after 24 hours of ethanol access. Daily administration of vehicle to long-term drinking rats for 5 days did not affect ethanol intake (p > .05). When daily administrations of SoRI-9409 (or vehicle) were ceased, rats returned to baseline levels of ethanol intake. The values are expressed as mean ± SEM ethanol consumed (g/kg/24 hours) (Student t test). *p < .05; compared with baseline levels of ethanol intake, n = 8.
SoRI-9409 Inhibits DOP-R–Mediated [35S]GTPγS-stimulated Binding in Long-Term Ethanol-Treated Rats
The DOP-R agonists, SNC80, DPDPE, and TAN67, produced a dose-dependent stimulation of [35S]GTPγS binding in striatal rat membranes in animals consuming high amounts of ethanol for 6 months (Figure 4A; Table 3). SNC80, DPDPE, and TAN67 produced moderate stimulation [35S]GTPγS-binding in the rat cortex and in the spinal cord (Table 3) and weak stimulation in the brainstem, hippocampus, hypothalamus, cerebellum, and midbrain (all EC50s ≥ 10 μmol/L; data not shown). The SNC80-stimulated (10 μmol/L) [35S]GTPγS-binding in the striatal rat membranes was potently inhibited by SoRI-9409 and naltrindole but not by naltrexone (Figure 4B, Table 4). The DPDPE-stimulated (10 μmol/L) [35S]GTPγS-binding in the striatal rat membranes was potently inhibited by SoRI-9409, naltrindole, and naltrexone (Figure 4C, Table 4). The TAN67-stimulated (10 μmol/L) [35S]GTPγS-binding in the striatal rat membranes was potently inhibited by SoRI-9409, in contrast to no binding inhibition produced by either naltrindole or naltrexone (Figure 4D, Table 4).
Figure 4.
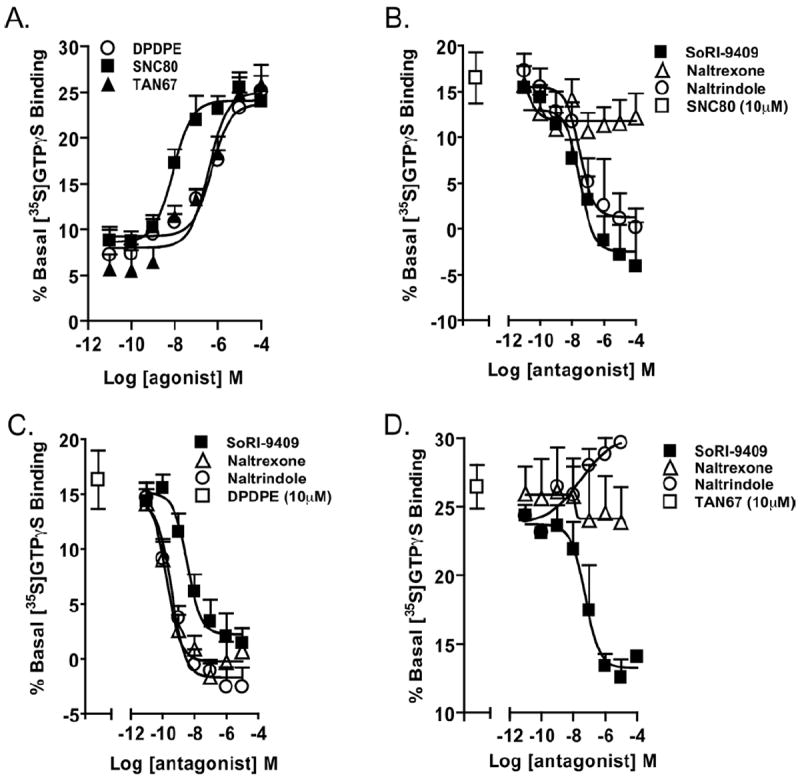
SoRI-9409 inhibits δ opioid receptor (DOP-R)-stimulated [35S]GTPγS binding in the striatum of long-term heavy drinking rats. (A) The DOP-R ligands SNC80, DPDPE, and TAN67 significantly stimulate [35S]GTPγS binding in the striatum of long-term heavy drinking rats. (B) SoRI-9409 and naltrindole but not naltrexone inhibit SNC80-stimulated [35S]GTPγS DOP-R striatal binding. (C) SoRI-9409, naltrindole, and naltrexone inhibit DPDPE-stimulated [35S]GTPγS striatal binding. (D) SoRI-9409 but not naltrindole or naltrexone inhibits TAN67-stimulated [35S]GTPγS DOP-R striatal binding. The values are expressed as mean ± SEM percentage increase in basal [35S]GTPγS binding.
Table 3.
SNC80-, TAN67-, and DPDPE-Mediated [35S]GTPγS-Binding Stimulation in the Striatum, Cerebral Cortex, and Spinal Cord Membranes of Long-Term Ethanol-Consuming Rats
| Compound | Mean (± SEM) EC50 for DOP-R Stimulation | ||
|---|---|---|---|
| Striatum | Cerebral Cortex | Spinal Cord | |
| SNC80 | 8.6 ± .2 nmol/L | 155 ± 6.3 nmol/L | 703 ± 43 nmol/L |
| DPDPE | 514 ± 22 nmol/L | 116 ± 5.1 nmol/L | 4.0 ± .5 μmol/L |
| TAN67 | 372 ± 20 nmol/L | 700 ± 26 nmol/L | 3.7 ± 2.2 μmol/L |
Table 4.
Inhibition of SNC80-, TAN67-, and DPDPE-Mediated (10 μmol/L) [35S]GTPγS-Binding by SoRI-9409, Naltrindole, and Naltrexone in Striatal Rat Membranes of Long-Term Ethanol-Consuming Rats
| Compound | Mean (± SEM) IC50 for Striatal DOP-R Inhibition | ||
|---|---|---|---|
| SNC80 Inhibition | DPDPE Inhibition | TAN67 Inhibition | |
| SoRI-9409 | 31 ± 3.2 nmol/L | 3.5 ± .3 nmol/L | 56 ± 8.2 nmol/L |
| Naltrindole | 31 ± 3.9 nmol/L | .4 ± .02 nmol/L | > 10 μmol/L |
| Naltrexone | > 10 μmol/L | .2 ± .02 nmol/L | > 10 μmol/L |
Long-Term Treatment with SoRI-9409 Reduces Escalation of Ethanol Intake and Induces a Long-Lasting Reduction in Ethanol Intake When the Drug Is No Longer Present
Administration of SoRI-9409 (5 mg/kg IP) for 28 days significantly reduced the escalation and maintenance of ethanol intake compared with vehicle in high-ethanol–consuming rats with the intermittent access 20% ethanol two-bottle-choice drinking paradigm (exposures 2–4, 11: p < .05; exposures 7, 8, 10: _p_ < .01; exposures 5, 6, 9, 12: _p_ < .001; Figure 5). Initial drinking levels in rats administered SoRI-9409 after the first ethanol exposure were not significantly different from vehicle-treated rats (_p_ > .05; Figure 5). The SoRI-9409 reduced the escalation of ethanol intake and over time continued to significantly reduce ethanol intake for up to 4 weeks compared with the vehicle-treated group. When the SoRI-9409 treatment was terminated after 28 days (12 ethanol exposures), the reduced drinking levels were maintained when compared with post-vehicle treatment baseline drinking levels (exposures 17, 19, 20, 22, 23: p < .05; exposures 14–16, 21, 24: _p_ < .01; exposures 13, 18: _p_ < .001; Figure 5). After cessation of SoRI-9409 administration (exposure 13), ethanol intake initially increased compared with the last ethanol exposure during SoRI-9409 treatment (exposure 12; _p_ < .01; Figure 5). However, ethanol intake was not significantly different after subsequent ethanol exposures (exposures 14,15), compared with the last ethanol exposure during SoRI-9409 treatment (exposure 12; _p_ > .05; Figure 5). Cessation of SoRI-9409 treatment did not result in a rapid escalation of ethanol consumption, compared with the profiles of initial escalations in drinking observed in untreated or vehicle-treated rats. The SoRI-9409 treatment reduced drinking for up to 4 weeks after the treatment period was terminated. SoRI-9409 did not decrease water consumption (p > .05) or on alternate water-only days at all time-points (p > .05) and after cessation of daily administered SoRI-9409, there were no further significant decreases in water intake (p > .05) for a further 28 days.
Figure 5.
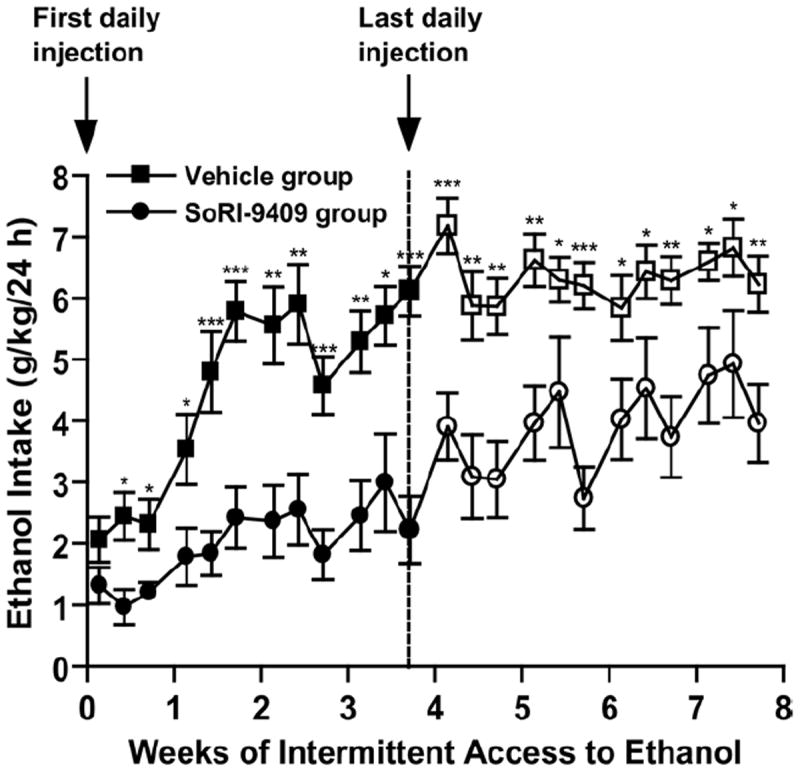
Long-term treatment with SoRI-9409 reduces escalation of ethanol intake and induces a long-lasting reduction in ethanol intake when the drug is no longer present. Rats were administered either SoRI-9409 (5 mg/kg IP n = 16, closed circles) or vehicle (n = 15, closed squares) on each of 28 consecutive days, 30 min before the drinking session. SoRI-9409 significantly reduced ethanol consumption after 24 hours of ethanol access at each drinking session. The administration of either SoRI-9409 (open circles) or vehicle (open squares) was terminated after 28 days (12 ethanol exposures). Ethanol consumption in the SoRI-9409-treated but not the vehicle-treated group remained lower for a further 28 days. The values are expressed as mean ± SEM ethanol consumed (g/kg/24 hours) (Student t test). *p < .05; **p < .01; ***p < .001.
Discussion
SoRI-9409 is a novel opioid receptor antagonist that has greater affinity at DOP-Rs and significantly reduced affinity at MOP-Rs compared with naltrexone. We show that SoRI-9409 administration in high-ethanol–consuming animals is threefold more effective at reducing ethanol consumption compared with either naltrexone or naltrindole. Furthermore, both acute and chronic administration of SoRI-9409 in high-ethanol–consuming animals significantly reduced ethanol intake in rats after 24 hours of ethanol access when compared with vehicle-treated animals. The increased efficacy of SoRI-9409 in decreasing ethanol consumption might be attributed, at least in part, to the specific selectivity of SoRI-9409 for both the DOP-R and MOP-R. The affinity of SoRI-9409 at the DOP-R and the MOP-R seems to lie between that reported for naltrexone and naltrindole (12).
SoRI-9409 did not affect sucrose intake at the lowest doses that effectively reduce ethanol intake (5 and 15 mg/kg), nor did it reduce water intake. This supports previous studies showing that DOP-R antagonists selectively reduce ethanol intake (21,22). In contrast, naltrexone reduced both ethanol and water intake, as shown by other investigators (23-25); however, this has not been found in all studies (22,26).
SoRI-9409 did not reduce morphine-mediated analgesia or induce analgesia and hyperalgesia (Figures 1A and 1B in Supplement 1). In contrast, naltrexone attenuated morphine-mediated analgesia in rats, further evidence suggesting that SoRI-9409 is specifically inhibiting the DOP-R (Figure 1A in Supplement 1). We also show that SoRI-9409 does not produce conditioned place preference or aversion (Figure 1C in Supplement 1). Previous studies have suggested that activation of the MOP-R and DOP-R might regulate anxiety and mood states (27,28) and that DOP-R antagonists have been shown to be anxiogenic in rats (29). We show that SoRI-9409 is not anxiolytic or anxiogenic (Figure 1D in Supplement 1). Taken together this suggests that the specific affinity of SoRI-9409 for the DOP-R and MOP-R might represent a more optimal blockade of these receptors to produce a selective and long-lasting reduction in ethanol consumption without affecting general fluid intake, morphine-mediated analgesia or alter anxiety and mood states.
SoRI-9409 and naltrexone but not naltrindole selectively reduced ethanol intake after 30 min (Figures 2A-2C in Supplement 1). In contrast, SoRI-9409 but not naltrexone or naltrindole reduced ethanol intake after 24 hours. We have previously shown that lower doses of naltrexone (.3–1 mg/kg, IP) reduce ethanol intake after 30 min but not after 24 hours of ethanol access (19). This suggests that SoRI-9409 might be longer-acting than naltrexone in the rat. The fusion of the chlorinated-phenylpyridine ring onto the naltrexone structure to form SoRI-9409 (12) might preclude metabolic transformations such as those that occur with opioid ligands possessing a carbonyl or hydroxy function at the 6-position (30). We did a preliminary investigation into the pharmacokinetics of SoRI-9409 (see Methods in Supplement 1). The elimination half-life of SoRI-9409 (IP) was 2.5 hours with the drug still detectable in plasma at 12 hours after dosing (see Results in Supplement 1). The plasma half-life of SoRI-9409 seems to be longer than that reported for naltrexone (.5–1.4 hours) in rats (31,32); however, it is not known whether this will be the case in humans. The sustained levels of SoRI-9409 detected in plasma up to 12 hours after dosing in the present study might represent significant enterohepatic cycling of this compound to contribute to its long-lasting effects in the rat, but this requires further investigation.
To determine whether SoRI-9409 would also inhibit ethanol intake after long-term treatment in high-ethanol–consuming rats, we acutely administered SoRI-9409 daily to rats for 28 days. SoRI-9409 selectively reduced the onset and escalation of ethanol intake and continued to selectively reduce ethanol intake by > 50% for up to 28 days compared with saline-treated rats. When the administration of SoRI-9409 was terminated after 28 days, the amount of ethanol consumed daily was maintained at approximately one-half of the baseline drinking levels produced in post-vehicle-treated animals, for the next 28 days. Cessation of SoRI-9409 treatment did not result in any significant escalation in ethanol consumption, compared with the profile of rapid escalation in drinking observed in untreated or vehicle-treated rats. Multiple administrations of SoRI-9409 thereby produce long-lasting and permanent effects on the modulation of ethanol consumption. This suggests that activation of DOP-Rs directly modulates ethanol intake during the escalation and maintenance of drinking in high-ethanol–consuming animals. Furthermore, in striatal membranes, prepared from the brains of rats consuming high amounts of ethanol, SoRI-9409 inhibits SNC80-, DPDPE-, and TAN67-stimulated DOP-Rs [35S]GTPγS-binding. This suggests that the DOP-R in the rat striatum can be modulated by SoRI-9409. The dorsal striatum contains major components of the homeostatic pathway controlling ethanol consumption (33,34), the potent inhibition in the rat striatum produced by SoRI-9409 suggests that, in the rat, inhibition of the DOP-R could modulate reward pathways in the striatum; however, this remains to be investigated.
Taken together, our studies suggest that compounds that have higher affinity for DOP-Rs and reduced affinity for MOP-Rs, such as SoRI-9409, might be promising candidates for development as novel therapeutics for the treatment of alcohol use disorders.
Supplementary Material
supplemental
Acknowledgments
This work was supported by funding from the State of California for Medical Research through University of California San Francisco to SEB and Department of Defense Grant W81XWH-07-1-0075 and W81XWH-08-1-0016 to SEB and by the National Institute on Drug Abuse Grant DA008883 to SA. We thank Joan Holgate, Kevin Hur, Mohammad Naeemuddin, Brian Medina, and Tiffany Ho for excellent technical support. We thank the staff of TetraQ at the University of Queensland for technical assistance in conducting the preliminary pharmacokinetic study of SoRI-9409. The experiments contained herein comply with the current laws of the United States of America. All procedures were pre-approved by the Gallo Center Institutional Animal Care and Use Committee and were in accordance with National Institutes of Health guidelines for the Humane Care and Use of Laboratory Animals.
Footnotes
Supplementary material cited in this article is available online.
The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Stromberg MF, Casale M, Volpicelli L, Volpicelli JR, O’Brien CP. A comparison of the effects of the opioid antagonists naltrexone, naltrindole, and beta-funaltrexamine on ethanol consumption in the rat. Alcohol. 1998;15:281–289. doi: 10.1016/s0741-8329(97)00131-6. [DOI] [PubMed] [Google Scholar]
- 2.Altshuler HL, Phillips PE, Feinhandler DA. Alteration of ethanol self-administration by naltrexone. Life Sci. 1980;26:679–688. doi: 10.1016/0024-3205(80)90257-x. [DOI] [PubMed] [Google Scholar]
- 3.Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- 4.O’Malley SS, Krishnan-Sarin S, Farren C, Sinha R, Kreek MJ. Naltrexone decreases craving and alcohol self-administration in alcohol-dependent subjects and activates the hypothalamo-pituitary-adreno-cortical axis. Psychopharmacology (Berl) 2002;160:19–29. doi: 10.1007/s002130100919. [DOI] [PubMed] [Google Scholar]
- 5.Anton RF, O’Malley SS, Ciraulo DA, Cisler RA, Couper D, Donovan DM, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence: The COMBINE study: A randomized controlled trial. JAMA. 2006;295:2003–2017. doi: 10.1001/jama.295.17.2003. [DOI] [PubMed] [Google Scholar]
- 6.Oslin DW, Berrettini WH, O’Brien CP. Targeting treatments for alcohol dependence: The pharmacogenetics of naltrexone. Addict Biol. 2006;11:397–403. doi: 10.1111/j.1369-1600.2006.00036.x. [DOI] [PubMed] [Google Scholar]
- 7.Hollister LE, Johnson K, Boukhabza D, Gillespie HK. Aversive effects of naltrexone in subjects not dependent on opiates. Drug Alcohol Depend. 1981;8:37–41. doi: 10.1016/0376-8716(81)90084-3. [DOI] [PubMed] [Google Scholar]
- 8.Crowley TJ, Wagner JE, Zerbe G, Macdonald M. Naltrexone-induced dysphoria in former opioid addicts. Am J Psychiatry. 1985;142:1081–1084. doi: 10.1176/ajp.142.9.1081. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell JE, Morley JE, Levine AS, Hatsukami D, Gannon M, Pfohl D. High-dose naltrexone therapy and dietary counseling for obesity. Biol Psychiatry. 1987;22:35–42. doi: 10.1016/0006-3223(87)90127-2. [DOI] [PubMed] [Google Scholar]
- 10.Pfohl DN, Allen JI, Atkinson RL, Knopman DS, Malcolm RJ, Mitchell JE, et al. Naltrexone hydrochloride (Trexan): A review of serum transaminase elevations at high dosage. NIDA Res Monogr. 1986;67:66–72. [PubMed] [Google Scholar]
- 11.Lucey MR, Silverman BL, Illeperuma A, O’Brien CP. Hepatic safety of once-monthly injectable extended-release naltrexone administered to actively drinking alcoholics. Alcohol Clin Exp Res. 2008;32:498–504. doi: 10.1111/j.1530-0277.2007.00593.x. [DOI] [PubMed] [Google Scholar]
- 12.Ananthan S, Kezar HS, 3rd, Carter RL, Saini SK, Rice KC, Wells JL, et al. Synthesis, opioid receptor binding, and biological activities of naltrexone-derived pyrido- and pyrimidomorphinans. J Med Chem. 1999;42:3527–3538. doi: 10.1021/jm990039i. [DOI] [PubMed] [Google Scholar]
- 13.Goldstein A, Naidu A. Multiple opioid receptors: Ligand selectivity profiles and binding site signatures. Mol Pharmacol. 1989;36:265–272. [PubMed] [Google Scholar]
- 14.Roberts AJ, Gold LH, Polis I, McDonald JS, Filliol D, Kieffer BL, et al. Increased ethanol self-administration in delta-opioid receptor knockout mice. Alcohol Clin Exp Res. 2001;25:1249–1256. [PubMed] [Google Scholar]
- 15.Ingman K, Salvadori S, Lazarus L, Korpi ER, Honkanen A. Selective delta-opioid receptor antagonist N,N(CH3)2-Dmt-Tic-OH does not reduce ethanol intake in alcohol-preferring AA rats. Addict Biol. 2003;8:173–179. doi: 10.1080/1355621031000117400. [DOI] [PubMed] [Google Scholar]
- 16.Froehlich JC, Badia-Elder NE, Zink RW, McCullough DE, Portoghese PS. Contribution of the opioid system to alcohol aversion and alcohol drinking behavior. J Pharmacol Exp Ther. 1998;287:284–292. [PubMed] [Google Scholar]
- 17.Froehlich JC, Zweifel M, Harts J, Lumeng L, Li TK. Importance of delta opioid receptors in maintaining high alcohol drinking. Psychopharmacology (Berl) 1991;103:467–472. doi: 10.1007/BF02244246. [DOI] [PubMed] [Google Scholar]
- 18.Steensland P, Simms JA, Holgate J, Richards JK, Bartlett SE. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, selectively decreases ethanol consumption and seeking. Proc Natl Acad Sci U S A. 2007;104:12518–12523. doi: 10.1073/pnas.0705368104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simms JA, Steensland P, Medina B, Abernathy K, Chandler LJ, Wise R, Bartlett SE. Intermittent-access to 20% ethanol induces high ethanol consumption in Long-Evans and Wistar rats. Alcohol Clin Exp Res. 2008;32:1–8. doi: 10.1111/j.1530-0277.2008.00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wise RA. Voluntary ethanol intake in rats following exposure to ethanol on various schedules. Psychopharmacologia. 1973;29:203–210. doi: 10.1007/BF00414034. [DOI] [PubMed] [Google Scholar]
- 21.Krishnan-Sarin S, Jing SL, Kurtz DL, Zweifel M, Portoghese PS, Li TK, et al. The delta opioid receptor antagonist naltrindole attenuates both alcohol and saccharin intake in rats selectively bred for alcohol preference. Psychopharmacology (Berl) 1995;120:177–185. doi: 10.1007/BF02246191. [DOI] [PubMed] [Google Scholar]
- 22.Franck J, Lindholm S, Raaschou P. Modulation of volitional ethanol intake in the rat by central delta-opioid receptors. Alcohol Clin Exp Res. 1998;22:1185–1189. [PubMed] [Google Scholar]
- 23.Escher T, Mittleman G. Schedule-induced alcohol drinking: Non-selective effects of acamprosate and naltrexone. Addict Biol. 2006;11:55–63. doi: 10.1111/j.1369-1600.2006.00004.x. [DOI] [PubMed] [Google Scholar]
- 24.Myers RD, Borg S, Mossberg R. Antagonism by naltrexone of voluntary alcohol selection in the chronically drinking macaque monkey. Alcohol. 1986;3:383–388. doi: 10.1016/0741-8329(86)90058-3. [DOI] [PubMed] [Google Scholar]
- 25.Ukai M, Holtzman SG. Effects of intrahypothalamic administration of opioid peptides selective for mu-, kappa, and delta-receptors on different schedules of water intake in the rat. Brain Res. 1988;459:275–281. doi: 10.1016/0006-8993(88)90643-9. [DOI] [PubMed] [Google Scholar]
- 26.Gardell LR, Hubbell CL, Reid LD. Naltrexone persistently reduces rats’ intake of a palatable alcoholic beverage. Alcohol Clin Exp Res. 1996;20:584–588. doi: 10.1111/j.1530-0277.1996.tb01097.x. [DOI] [PubMed] [Google Scholar]
- 27.Asakawa A, Inui A, Momose K, Ueno N, Fujino MA, Kasuga M. Endomorphins have orexigenic and anxiolytic activities in mice. Neuroreport. 1998;9:2265–2267. doi: 10.1097/00001756-199807130-00022. [DOI] [PubMed] [Google Scholar]
- 28.Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- 29.Perrine SA, Hoshaw BA, Unterwald EM. Delta opioid receptor ligands modulate anxiety-like behaviors in the rat. Br J Pharmacol. 2006;147:864–872. doi: 10.1038/sj.bjp.0706686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oguri K, Yamada-Mori I, Shigezane J, Hirano T, Yoshimura H. Potentiation of physical dependence by conjugation at the 6-position of nalorphine. Eur J Pharmacol. 1984;102:229–235. doi: 10.1016/0014-2999(84)90254-1. [DOI] [PubMed] [Google Scholar]
- 31.Berkowitz BA, Spector S, Lee CH. Mechanism of narcotic antagonist and narcotic antagonist analgesic action. In: Ford DH, Clouet DH, editors. Tissues Responses to Addictive Drugs. Jamaica, New York: Spectrum Publications; 1976. pp. 139–153. [Google Scholar]
- 32.Misra AL, Bloch R, Vardy J, Mule SJ, Verebely K. Disposition of (15,16-3H)naltrexone in the central nervous system of the rat. Drug Metab Dispos. 1976;4:276–280. [PubMed] [Google Scholar]
- 33.Wang J, Carnicella S, Phamluong K, Jeanblanc J, Ronesi JA, Chaudhri N, et al. Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: Implications for alcohol drinking behavior. J Neurosci. 2007;27:3593–3602. doi: 10.1523/JNEUROSCI.4749-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Logrip ML, Janak PH, Ron D. Dynorphin is a downstream effector of striatal BDNF regulation of ethanol intake. FASEB J. 2008;22:2393–2404. doi: 10.1096/fj.07-099135. [DOI] [PubMed] [Google Scholar]
- 35.Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, et al. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther. 2000;293:1002–1008. [PubMed] [Google Scholar]
- 36.Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 2001;154:43–49. doi: 10.1007/s002130000622. [DOI] [PubMed] [Google Scholar]
- 37.Becker A, Grecksch G, Kraus J, Loh HH, Schroeder H, Hollt V. Rewarding effects of ethanol and cocaine in mu opioid receptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:296–302. doi: 10.1007/s00210-002-0533-2. [DOI] [PubMed] [Google Scholar]
- 38.Kovacs KM, Szakall I, O’Brien D, Wang R, Vinod KY, Saito M, et al. Decreased oral self-administration of alcohol in kappa-opioid receptor knock-out mice. Alcohol Clin Exp Res. 2005;29:730–738. doi: 10.1097/01.alc.0000164361.62346.d6. [DOI] [PubMed] [Google Scholar]
- 39.Critcher EC, Lin CI, Patel J, Myers RD. Attenuation of alcohol drinking in tetrahydroisoquinoline-treated rats by morphine and naltrexone. Pharmacol Biochem Behav. 1983;18:225–229. doi: 10.1016/0091-3057(83)90367-2. [DOI] [PubMed] [Google Scholar]
- 40.Hyytia P, Kiianmaa K. Suppression of ethanol responding by centrally administered CTOP and naltrindole in AA and Wistar rats. Alcohol Clin Exp Res. 2001;25:25–33. doi: 10.1111/j.1530-0277.2001.tb02123.x. [DOI] [PubMed] [Google Scholar]
- 41.Ciccocioppo R, Martin-Fardon R, Weiss F. Effect of selective blockade of mu(1) or delta opioid receptors on reinstatement of alcohol-seeking behavior by drug-associated stimuli in rats. Neuropsychopharmacology. 2002;27:391–399. doi: 10.1016/S0893-133X(02)00302-0. [DOI] [PubMed] [Google Scholar]
- 42.June HL, McCane SR, Zink RW, Portoghese PS, Li TK, Froehlich JC. The delta 2-opioid receptor antagonist naltriben reduces motivated responding for ethanol. Psychopharmacology (Berl) 1999;147:81–89. doi: 10.1007/s002130051145. [DOI] [PubMed] [Google Scholar]
- 43.Krishnan-Sarin S, Portoghese PS, Li TK, Froehlich JC. The delta 2-opioid receptor antagonist naltriben selectively attenuates alcohol intake in rats bred for alcohol preference. Pharmacol Biochem Behav. 1995;52:153–159. doi: 10.1016/0091-3057(95)00080-g. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell JM, Liang MT, Fields HL. A single injection of the kappa opioid antagonist norbinaltorphimine increases ethanol consumption in rats. Psychopharmacology (Berl) 2005;182:384–392. doi: 10.1007/s00213-005-0067-7. [DOI] [PubMed] [Google Scholar]
- 45.Holter SM, Henniger MS, Lipkowski AW, Spanagel R. Kappa-opioid receptors and relapse-like drinking in long-term ethanol-experienced rats. Psychopharmacology (Berl) 2000;153:93–102. doi: 10.1007/s002130000601. [DOI] [PubMed] [Google Scholar]
- 46.Reid LD, Hunter GA. Morphine and naloxone modulate intake of ethanol. Alcohol. 1984;1:33–37. doi: 10.1016/0741-8329(84)90033-8. [DOI] [PubMed] [Google Scholar]
- 47.Lindholm S, Werme M, Brene S, Franck J. The selective kappa-opioid receptor agonist U50,488H attenuates voluntary ethanol intake in the rat. Behav Brain Res. 2001;120:137–146. doi: 10.1016/s0166-4328(00)00368-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplemental