A Novel Nicotinic Acetylcholine Receptor Subtype in Basal Forebrain Cholinergic Neurons with High Sensitivity to Amyloid Peptides (original) (raw)
Abstract
Nicotinic acetylcholine receptors (nAChRs) containing α7 subunits are thought to assemble as homomers. α7-nAChR function has been implicated in learning and memory, and alterations of α7-nAChR have been found in patients with Alzheimer's disease (AD). Here we report findings consistent with a novel, naturally occurring nAChR subtype in rodent, basal forebrain cholinergic neurons. In these cells, α7 subunits are coexpressed, colocalize, and coassemble with β2 subunit(s). Compared with homomeric α7-nAChRs from ventral tegmental area neurons, functional, presumably heteromeric α7β2-nAChRs on cholinergic neurons freshly dissociated from medial septum/diagonal band (MS/DB) exhibit relatively slow kinetics of whole-cell current responses to nicotinic agonists and are more sensitive to the β2 subunit-containing nAChR-selective antagonist, dihydro-β-erythroidine (DHβE). Interestingly, presumed, heteromeric α7β2-nAChRs are highly sensitive to functional inhibition by pathologically relevant concentrations of oligomeric, but not monomeric or fibrillar, forms of amyloid β1–42 (Aβ1–42). Slow whole-cell current kinetics, sensitivity to DHβE, and specific antagonism by oligomeric Aβ1–42 also are characteristics of heteromeric α7β2-nAChRs, but not of homomeric α7-nAChRs, heterologously expressed in Xenopus oocytes. Moreover, choline-induced currents have faster kinetics and less sensitivity to Aβ when elicited from MS/DB neurons derived from nAChR β2 subunit knock-out mice rather than from wild-type mice. The presence of novel, functional, heteromeric α7β2-nAChRs on basal forebrain cholinergic neurons and their high sensitivity to blockade by low concentrations of oligomeric Aβ1–42 suggests possible mechanisms for deficits in cholinergic signaling that could occur early in the etiopathogenesis of AD and might be targeted by disease therapies.
Keywords: nicotinic receptor, basal forebrain, cholinergic neurons, patch clamp, amyloid β, Alzheimer's disease
Introduction
Nicotinic acetylcholine receptors (nAChRs) in mammals exist as a diverse family of channels composed of different, pentameric combinations of subunits derived from at least 16 genes (Lukas et al., 1999; Jensen et al., 2005). Functional nAChRs can be assembled as either heteromers containing α and β subunits or as homomers containing only α subunits (Lukas et al., 1999; Jensen et al., 2005). In the mammalian brain, the most abundant forms of nAChRs are heteromeric α4β2-nAChRs and homomeric α7-nAChRs (Whiting et al., 1987; Flores et al., 1992; Gopalakrishnan et al., 1996; Lindstrom, 1996; Lindstrom et al., 1996). α7-nAChRs appear to play roles in the development, differentiation, and pathophysiology of the nervous system (Z. Liu et al., 2007; Mudo et al., 2007).
nAChRs have been implicated in Alzheimer's disease (AD), in part because significant losses in radioligand binding sites corresponding to nAChRs have been consistently observed at autopsy in a number of neocortical areas and in the hippocampi of patients with AD (Burghaus et al., 2000; Nordberg, 2001). Attenuation of cholinergic signaling is known to impair memory, and nicotine exposure improves cognitive function in AD patients (Levin and Rezvani, 2002). In addition, several studies have suggested that the activation of α7-nAChR function alleviates amyloid-β (Aβ) toxicity. For instance, stimulation of α7-nAChRs inhibits amyloid plaque formation in vitro and in vivo (Geerts, 2005), activates α-secretase cleavage of amyloid precursor protein (APP) (Lahiri et al., 2002), increases acetylcholine (ACh) release and facilitates Aβ internalization (Nagele et al., 2002), inhibits activity of the MAPK/NF-κΒ/c-myc signaling pathway (Q. Liu et al., 2007), and reduces Aβ production and attenuates tau phosphorylation (Sadot et al., 1996). These findings suggest that cholinergic signaling, mediated through α7-nAChRs, not only is involved in cognitive function, but also could protect against a wide variety of insults associated with AD (Sivaprakasam, 2006). Conversely, impairment of α7-nAChR-mediated cholinergic signaling during the early stage(s) of AD might play a pivotal role in AD pathophysiology.
In rat basal forebrain cholinergic neurons, α7 and β2 are the predominant nAChR subunits, and they were found to colocalize (Azam et al., 2003). Thus far, however, there has been no evidence that α7 and β2 subunits coassemble to form functional nAChRs naturally, although functional α7β2-nAChRs have been reported using a heterologous expression system (Khiroug et al., 2002). We asked whether heteromeric α7β2-nAChRs exist in rodent basal forebrain cholinergic neurons and whether such a unique receptor subtype would be sensitive to Aβ. Using patch-clamp electrophysiological, pharmacological, and molecular biological approaches, our findings demonstrate a novel partnership between nAChR α7 and β2 subunits, which likely assemble together to form a unique receptor subtype, and selectively high sensitivity of this novel nAChR subtype to pathologically relevant concentrations of Aβ.
Materials and Methods
All techniques used in this manuscript are standard experimental approaches that are routinely performed in our laboratories, and the details of these techniques are available in our published papers (Wu et al., 2002, 2004a,b).
Acutely dissociated neurons from the CNS and patch-clamp whole-cell current recordings
Neuron dissociation and patch-clamp recordings were performed as described by Wu et al. (2002, 2004b). Briefly, each postnatal 2- to 4-week-old Wistar rat or mouse (wild-type C57BL/6 or nAChR β2 knock-out mice on a C57BL/6 background kindly provided by Dr. Marina Picciotto, Yale University, New Haven, CT) was anesthetized using isoflurane, and the brain was rapidly removed. Several 400 μm coronal slices, which contained the medial septum/diagonal band (MS/DB) or the ventral tegmental area (VTA), were cut using a vibratome (Vibratome 1000 plus; Jed Pella) in cold (2–4°C) artificial CSF (ACSF) and continuously bubbled with carbogen (95% O2–5% CO2). The slices were then incubated in a preincubation chamber (Warner Instruments) and allowed to recover for at least 1 h at room temperature (22 ± 1°C) in oxygenated ACSF. Thereafter, the slices were treated with Pronase (1 mg/6 ml) at 31°C for 30 min and subsequently treated with the same concentration of thermolysin for another 30 min. The MS/DB or VTA region was micropunched out from the slices using a well polished needle. Each punched piece was then dissociated mechanically by using several fire-polished micro-Pasteur pipettes in a 35 mm culture dish filled with well oxygenated, standard external solution [in mm: 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES; pH 7.4 (with Tris-base)]. The separated single cells usually adhered to the bottom of the dish within 30 min. Perforated-patch whole-cell recordings coupled with a U-tube or two-barrel drug application system were used (Wu et al., 2002). Perforated-patch recordings more closely maintain both intracellular divalent cation and cytosolic element composition (Horn and Marty, 1988). In particular, perforated-patch recording was used to maintain the intracellular ATP concentration at a physiological level. To prepare for perforated-patch whole-cell recording, glass microelectrodes (GC-1.5; Narishige) were fashioned on a two-stage vertical pipette puller (P-830; Narishige), and the resistance of the electrode was 3–5 MΩ when filled with the internal solution. A tight seal (>2 GΩ) was formed between the electrode tip and the cell surface, which was followed by a transition from on-cell to whole-cell recording mode due to the partitioning of amphotericin B into the membrane underlying the patch. After whole-cell formation, an access resistance lower than 60 MΩ was acceptable during perforated-patch recordings in current-clamp mode, and an access resistance lower than 30 MΩ was acceptable during voltage-clamp recordings. The series resistance was not compensated in the experiments using dissociated neurons. Under current-clamp configuration, membrane potentials were measured using a patch-clamp amplifier (200B; Axon Instruments). Data were filtered at 2 kHz, acquired at 11 kHz, and digitized on-line (Digidata 1322 series A/D board; Axon Instruments). All experiments were performed at room temperature (22 ± 1°C). The drugs used in the present study were GABA, glutamate, ACh, choline, methyllycaconitine (MLA), dihydro-β-erythroidine (DHβE), muscarine (all purchased from Sigma-Aldrich), RJR-2403 (purchased from Tocris Cookson), and Aβ1–42 and scrambled Aβ1–42 (purchased from rPeptide).
RT-PCR to profile nAChR subunit expression in MS/DB
Riboprobe construction
Templates for in vitro transcription were created using PCR and sense or antisense primers spanning the 5′ SP6 promoter or the 3′ T7 promoter, respectively (α7 subunit: 5′-atttaggtgacactatagaagnggatcatcgtgggcctctcagtg-3′ and 5′-taatacgactcactatagggagagttggcgatgtagcggacctc-3′; β2 subunit: 5′-atttaggtgacactatagaagngtcacggtgttcctgctgctcatct-3′ and 5′-taatacgactcactatagggagatcctccctcacactctggtcatca-3′). Antisense or sense probes were then created by in vitro transcription using SP6 or T7 polymerases, respectively, and by incorporation of biotin-tagged UTP (for β2 subunit probes) or digoxigenin-tagged UTP (for α7 subunit probes; biotin or digoxigenin RNA-labeling mix; Roche Applied Science). The 433 bp or 520 bp products corresponding to mRNA nucleotides 953–1385 for α7 subunits or mRNA nucleotides 1006–1525 for β2 subunits thus produced are highly specific to the individual subunits.
Tissue RT-PCR
RT-PCR assays followed by Southern hybridization with nested oligonucleotides were done as previously described to identify nAChR subunit transcripts and to quantify levels of expression normalized both to housekeeping gene expression and levels of expression in whole brain (Wu et al., 2004), but using primers designed to detect rat nAChR subunits. The Southern hybridization technique coupled with quantitation using electronic isotope counting (Instant Imager, Canaberra Instruments) yielded results equivalent to those obtained using real-time PCR analysis.
Single-cell RT-PCR
Precautions were taken to ensure a ribonuclease-free environment and to avoid PCR product contamination during patch-clamp recording and single-cell collection before execution of RT-PCR. Single-cell RT-PCR was performed using the Superscript III CellDirect RT-PCR system (Invitrogen). Briefly, after whole-cell patch-clamp recording, single-cell content was harvested by suction into the pipette solution (∼3 μl) and immediately transferred to an autoclaved 0.2 ml PCR tube containing 10 μl of cell resuspension buffer and 1 μl of lysis enhancer. Single cells were lysed by heating at 75°C for 10 min. Potential contaminating genomic DNA was removed by DNase I digestion at 25°C for 6 min. After heat-inactivation of DNaseI at 70°C for 6 min in the presence of EDTA, reverse transcription (RT) was performed by adding reaction mix with oligo(dT)20 and random hexamers and SuperScriptIII enzyme mix and then incubating at 25°C for 10 min and 50°C for 50 min. The reaction was terminated by heating the sample to 85°C for 5 min. The PCR primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and nAChR α3, α4, α7, β2, and β4 subunits were designed using the Primer 3 internet server (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) and assuming an annealing temperature of ∼60°C [nearest neighbor]. PCR was performed with 20 μl of hot-start Platinum PCR Supermix (Invitrogen), 3 μl of cDNA template from the RT step, and 1 μl of gene specific primer pairs (5 pmol each) with the following thermocycling parameters: 95°C for 2 min; (95°C for 30 s, 60°C for 30 s, and 72°C for 40 s) × 70 cycles, 72°C for 1 min. PCR products were resolved on 1.5% TBE-agarose gels, and stained gels were used to visualize bands, using digital photography and a gel documentation system to capture images.
Tissue protein extraction, immunoprecipitation, and immunoblotting for confirmation of nAChR α7 and β2 subunit coassembly
Tissues were Dounce homogenized (10 strokes) in ice-cold lysis buffer [1% (v/v) Triton X-100, 150 mm EDTA, 10% (v/v) glycerol, 50 mm Tris-HCl, pH 8.0] containing 1× general protease inhibitor cocktails (Sigma-Aldrich). The lysates were transferred to microcentrifuge tubes and further solubilized for 30 min at 4°C. The detergent extracts (supernatants) were collected by centrifugation at 15,000 × g for 15 min at 4°C, and protein concentration was determined for sample aliquots using bicinchoninic acid (BCA) protein assay reagents (Pierce Chemical). The detergent extracts were then precleared with 50 μl of mixed slurry of protein A-Sepharose and protein G-Sepharose (1:1) (Amersham Biosciences) twice, each for 30 min at 4°C. For each immunoprecipitation, detergent extracts (1 mg) were mixed with 1 μg of rabbit anti-α7 antiserum (H302) or rabbit IgG (as immunological control) (Santa Cruz Biotechnology) and incubated at 4°C overnight with continuous agitation. Protein A-Sepharose and protein G-Sepharose mixtures (50 μl) were added and incubated at 4°C for 1 h. The beads were washed four times with ice-cold lysis buffer containing protease inhibitors. Laemmli sample buffer eluates were resolved by SDS-PAGE. Proteins were transferred onto Hybond ECL nitrocellular membranes (Amersham Biosciences). The membranes were blocked with TBST buffer [20 mm Tris-HCl, pH 7.6, 150 mm NaCl, and 0.1% (v/v) Tween 20] containing 2% (w/v) nonfat dry milk for at least 2 h and incubated with rat monoclonal anti-β2 antibody (mAb270; Santa Cruz) or anti-α7 antiserum (H302), respectively, at 4°C overnight. After three washes in TBST, the membranes were incubated with goat anti-rat or goat anti-rabbit secondary antibodies (1:10,000) (Pierce Chemical) for 1 h and washed. The bound antibodies were detected with SuperSignal chemiluminescent substrate (Pierce Chemical).
Expression of homomeric and heteromeric α7-containing-nAChRs in Xenopus oocytes and two-electrode voltage-clamp recording
cDNAs encoding rat α7 and β2 subunits were amplified by PCR with pfuUltra DNA polymerase and subcloned into an oocyte expression vector, pGEMHE, with T7 orientation and confirmed by automated sequencing. cRNAs were synthesized by standard in vitro transcription with T7 RNA polymerase, confirmed by electrophoresis for their integrity, and quantified based on optical absorbance measurements using an Eppendorf Biophotometer.
Oocyte preparation and cRNA injection
Xenopus laevis (Xenopus I) females were anesthetized using 0.2% MS-222. The ovarian lobes were surgically removed from the frogs and placed in an incubation solution consisting of (in mm): 82.5 NaCl, 2.5 KCl, 1 MgCl2, 1 CaCl2, 1 Na2HPO4, 0.6 theophylline, 2.5 sodium pyruvate, 5 HEPES, 50 mg/ml gentamycin, 50 U/ml penicillin, and 50 μg/ml streptomycin; pH 7.5. The frogs were then allowed to recover from surgery before being returned to the incubation tank. The lobes were cut into small pieces and digested with 0.08 Wunsch U/ml liberase blendzyme 3 (Roche Applied Science) with constant stirring at room temperature for 1.5–2 h. The dispersed oocytes were thoroughly rinsed with incubation solution. Stage VI oocytes were selected and incubated at 16°C before injection. Micropipettes used for injection were pulled from borosilicate glass (Drummond Scientific). cRNAs encoding α7 or β2 at proper dilution were injected into oocytes separately or in different ratios using a Nanoject microinjection system (Drummond Scientific) at a total volume of ∼20–60 nl.
Two-electrode voltage-clamp recording
One to three days after injection, an oocyte was placed in a small-volume chamber and continuously perfused with oocyte Ringer's solution (OR2), consisting of the following (in mm): 92.5 NaCl, 2.5 KCl, 1 CaCl2, 1 MgCl2, and 5 HEPES; pH 7.5. The chamber was grounded through an agarose bridge. The oocytes were voltage clamped at −70 mV to measure ACh (or choline)-induced currents using GeneClamp 500B (Axon Instruments).
Immunocytochemical staining
Dissociated MS/DB neurons were fixed with 4% paraformaldehyde for 5 min, rinsed three times with PBS, and treated with saponin (1 mg/ml) for 5 min as a permeabilizing agent. After rinsing four times with PBS, the neurons were incubated at room temperature in anti-choline acetyltransferase (ChAT) primary antibody (AB305; Chemicon International) diluted 1:400 in HBSS (supplemented with 5% bovine serum albumin as a blocking agent) for 30 min. Following another three rinses with PBS, a secondary antibody (anti-mouse IgG; Sigma-Aldrich) was applied at room temperature for 30 min (diluted 1:100). After rinsing a final three times with PBS, the labeled cells were visualized using a Zeiss fluorescence microscope (Zeiss), and images were processed using Photoshop (Adobe Systems). For double immunolabeling of α7 and β2 subunits of nAChRs on single dissociated MS/DB neurons, we used the following antibodies: a rabbit antibody (AS-5631S, 1:400; R and D) against α7 subunit, a rat antibody against β2 subunit (Ab24698, 1:500; Abcam), Alexa Fluor 594-conjugated anti-rabbit IgG, and Alexa Fluor 488-conjugated anti-rat IgG (1:300; Invitrogen).
Aβ preparation and determination/monitoring of peptide forms
Aβ preparation
Amyloid β peptides (Aβ1–42, scrambled Aβ1–42) were purchased from rPeptide. As previously described (Wu et al., 2004a), some preparations involved reconstitution of Aβ peptides per vendor specifications in distilled water to a concentration of 100 μm, stored at −20°C, and used within 10 d of reconstitution. These thawed peptide stock solutions were used to create working dilutions (1–100 nm) in standard external solution before patch-clamp recording. Working dilutions were used within 4 h before being discarded. Atomic force microscopy (AFM) was used to define and analyze over time the morphology of prepared Aβ1–42. Aliquots of freshly prepared samples of Aβ1–42 diluted in standard external solution were spotted on freshly cleaved mica. After 2 min the mica was washed with 200 μl of deionized water, dried with compressed nitrogen, and completely air-dried under vacuum. Images were acquired in air using a multimode AFM nanoscope IIIA system (Veeco/Digital Instruments) operating in the tapping mode using silicon probes (Olympus).
Protocols to obtain different forms of Aβ1–42
Different conditions were used to specifically prepare monomeric, oligomeric, or fibrillar forms of Aβ1–42.
Monomers.
Aβ1–42 was reconstituted in DMSO to a concentration of 100 μm and stored at −80°C. For each use, an aliquot of stock sample was freshly thawed and diluted into standard extracellular solution as above just before patch recordings and used for no more than 4 h. This protocol yielded a predominant, monomeric form [see supplemental Fig. 1_C_ (left), available at www.jneurosci.org as supplemental material].
Oligomers.
Aβ1–42 reconstituted in distilled water to a concentration of 100 μm and stored at −80°C was used within 7 d of reconstitution. Aliquots diluted in standard extracellular solution and used within 4 h yielded a predominantly oligomeric form [see supplemental Fig. 1_C_ (middle), available at www.jneurosci.org as supplemental material].
Fibrils.
Aliquots of Aβ1–42 stock solution (water dissolved to 100 μm) were thawed and incubated at 37°C for 48 h at low pH (pH = 6.0). Working stocks diluted in standard extracellular solution yielded a predominantly fibrillar form [see supplemental Fig. 1_C_ (right), available at www.jneurosci.org as supplemental material].
Genotyping of the nAChR β2 subunit knock-out mice
Genomic DNA from mice newly born to heterozygotic, nAChR β2 subunit knock-out parents was extracted from mouse tail tips by using the QIAgen DNeasy Blood & Tissue Kit following the manufacture's protocol. PCR amplification of the nAChR β2 subunit or lac-Z (an indicator for the knock-out) were performed using the purified genomic DNA as template and gene specific primer pairs (forward primer: CGG AGC ATT TGA ACT CTG AGC AGT GGG GTC GC; backward primer: CTC GCT GAC ACA AGG GCT GCG GAC; lac-Z forward primer: CAC TAC GTC TGA ACG TCG AAA ACC CG; backward primer: CGG GCA AAT AAT ATC GGT GGC CGT GG) with annealing at 55°C for 1 min and extension at 72°C for 1 min for 30 cycles with GO _Taq_DNA polymerase (Promega). PCR products were resolved on 1% agarose gels and stained for visualization before images were captured using digital photography.
Results
Identification of cholinergic neurons dissociated from basal forebrain
An initial series of experiments identified cholinergic neurons acutely dissociated from rat MS/DB (Fig. 1 A). First, we identified the cholinergic phenotype of acutely dissociated neurons from the MS/DB (Fig. 1 Ba–c) based on published criteria (Henderson et al., 2005; Thinschmidt et al., 2005). In current-clamp mode, MS/DB neurons exhibited spontaneous action potential firing at low frequency (2.3 ± 0.4 Hz, n = 25 from 21 rats). This spontaneous activity was insensitive to the muscarinic acetylcholine receptor agonist, muscarine (1 μm) (Fig. 1 C). Depolarizing pulses induced adaptation of action potential firing (Fig. 1 D), and hyperpolarizing pulses failed to induce “sag”-like membrane potential changes (Fig. 1 E). In some cases, the fluorescent dye lucifer yellow (0.5 mg/ml) was delivered into recorded cells after patch-clamp recordings, and choline acetyltransferase (ChAT) immunocytostaining was used post hoc (Fig. 1 F). The presence of ChAT immunoactivity in recorded, dye-filled neurons confirmed that dissociated MS/DB neurons were cholinergic.
Figure 1.

Identification of cholinergic neurons dissociated from basal forebrain. A, Phase-contrast image of a rat MS/DB brain slice [region confirmed according to the work of Paxinos and Watson (1986)]. MS/DB neurons (phase-contrast images of dissociated neurons; B) exhibited spontaneous action potential firing (C), insensitivity to muscarine (C), and action potential adaptation induced by depolarizing pulses (D) and did not show “sag”-like responses to hyperpolarizing pulses (E), suggesting they were cholinergic. MP, Membrane potential. F, Dissociated neuron (phase contrast, Ph) labeled with lucifer yellow (LY) showed positive ChAT immunostaining following patch-clamp recording.
Naturally occurring nAChRs in rodent forebrain cholinergic neurons
We next tested for the presence of functional nAChRs on MS/DB cholinergic neurons. Under voltage-clamp recording conditions, rapid application of 1 mm ACh induced inward current responses with relatively rapid activation and desensitization kinetics (Fig. 2 A). These ACh-induced responses were mimicked by application of the selective α7-nAChR agonist choline, blocked by the relatively selective α7-nAChR antagonist methyllycaconitine (MLA), and insensitive to the relatively selective α4β2-nAChR agonist RJR-2403 (Fig. 2 A). Thus, the inward current evoked in MS/DB neurons had features similar to receptors containing α7 subunits. In contrast, in acutely dissociated, dopaminergic (DAergic) neurons from the midbrain VTA, ACh-induced currents displayed a mixture of features that could be dissected pharmacologically and with regard to whole-cell current kinetics. Components of responses displaying slow kinetics and sustained, steady-state currents elicited by ACh were mimicked by RJR-2403, suggesting that they were mediated by α4β2-nAChRs, whereas choline only induced transient peak current responses with very fast kinetics that are characteristic of homomeric α7-nAChRs (Fig. 2 B). Interestingly, choline-induced currents in MS/DB cholinergic neurons exhibited relatively slow macroscopic kinetics than observed in VTA DAergic neurons (Fig. 2 C). This impression was confirmed by quantitative analyses, which gave values for current rising time of 72.1 ± 9.1 ms (n = 8) for MS/DB neurons and 29.1 ± 2.9 ms (n = 12) for VTA neurons (p < 0.001) and decay constants (τ, rate of decay from peak to steady-state current) of 28.6 ± 2.8 ms (n = 8) for MS/DB neurons and 10.2 ± 1.5 ms (n = 12) for VTA neurons (p < 0.001). There were no significant differences between either peak current amplitudes or net charge movements for responses elicited by choline in MS/DB or VTA neurons (Fig. 2 D). These results suggested that functional nAChRs naturally expressed on rat MS/DB cholinergic neurons with some features like α7-nAChRs had slower whole-cell current kinetics than found for α7-nAChR-like responses in VTA DAergic neurons.
Figure 2.

Native nAChR-mediated whole-cell current responses. An identified MS/DB cholinergic neuron (no hyperpolarization-induced current, I h) exhibited α7-nAChR-like current responses to 1 mm ACh and 10 mm choline (sensitive to blockade by 1 nm methyllycaconitine; MLA) but not to 0.1 mm RJR-2403, an agonist selective for α4β2-nAChRs (A), whereas an identified VTA DAergic neuron (evident I h) showed both α7-nAChR-like (i.e., choline and MLA-sensitive components) and α4β2-nAChR-like (i.e., RJR-2403-sensitive component) current responses (summed as in the response to ACh) (B). C, Typical traces of 10 mm choline-induced currents in MS/DB and VTA DAergic neurons showing different kinetics for current activation/desensitization with a slower response characteristic of MS/DB neurons. D, Statistical comparisons of kinetics of 10 mm choline-induced currents in MS/DB cholinergic and VTA DAergic neurons. ***p < 0.001.
Subunit partnership for naturally occurring nAChRs in rodent basal forebrain cholinergic neurons
To test the hypothesis that the relatively slow kinetics of α7-nAChR-like responses in MS/DB cholinergic neurons were due to coassembly of α7 with other nAChR subunits, we performed relative quantitative RT-PCR analysis of nAChR subunit expression as messenger RNA in MS/DB compared with whole-brain and VTA tissues. The results demonstrated that nAChR α7 and β2 subunits were among those coexpressed regionally (Fig. 3 A,B). These studies were extended to single-cell RT-PCR analysis of nAChR subunit expression in acutely dissociated neurons from the MS/DB used in patch-clamp recordings (Fig. 3 Ca–c). Quantitative analysis indicated a high frequency of nAChR α7 and β2 subunit coexpression as message in recorded MS/DB neurons (Fig. 3 Cd). Mindful of the current concerns about the specificity of all anti-nAChR subunit antibodies (Moser et al., 2007), we nevertheless showed qualitatively, based on dual-labeling immunofluorescent staining (Fig. 3 D), that α7 and β2 subunits were colocalized in many MS/DB neurons subjected to patch-clamp recording. More direct evidence for coassembly of nAChR α7 and β2 subunit proteins came from coimmunoprecipitation studies using subunit-specific antibodies. Protein extracts from rat MS/DB or VTA tissues (collected from rats aged between 18 and 22 d) were subjected to immunoprecipitation (IP) (Fig. 3 E, left) with a rabbit anti-nAChR α7 subunit antibody (H302) or with rabbit IgG (as an immunological control) followed by immunoblotting (IB) with a rat anti-nAChR β2 subunit monoclonal antibody (mAb270). As indicated, the β2 subunit was readily detected immunologically in anti-α7 immunoprecipitates from MS/DB but not from VTA regions under our experimental conditions (Fig. 3 E, top left, lane 1 vs 2). Reprobing the same blot with the rabbit anti-α7 antibody (H302) verified that similar amounts of α7 subunits were precipitated from both MS/DB and VTA regions (Fig. 3 E, bottom left, lanes 1 and 2). Thus, coprecipitation of nAChR α7 and β2 subunits appeared only in samples from the rat MS/DB but not from the VTA. Collectively, these results suggest that nAChR α7 and β2 subunits are most likely coassembled, perhaps to form a functional nAChR subtype, in rodent basal forebrain cholinergic neurons.
Figure 3.

nAChR α7 and β2 subunits are coexpressed, colocalize, and coassemble in rat forebrain MS/DB neurons. RT-PCR products from whole brain and VTA and MS/DB regions (A) corresponding to the indicated nAChR subunits or to the housekeeping gene GAPDH were resolved on an agarose gel calibrated by the flanking 100 bp ladders (heavy band is 500 bp) and visualized using ethidium staining. Note that the representative gel shown for whole brain did not contain a sample for the nAChR α3 subunit RT-PCR product, which typically is similar in intensity to the sample on the gel for the VTA and MS/DB. B, Quantification of nAChR subunit mRNA levels for RT-PCR amplification followed by Southern hybridization with 32P-labeled, nested oligonucleotides normalized to the GAPDH internal control and to levels of each specific mRNA in whole rat brain (ordinate: ±SEM) for the indicated subunits. C, From 15 MS/DB neurons tested, after patch-clamp recordings (Ca: representative whole-cell current trace), the cell content was harvested and single-cell RT-PCR was performed, and the results show that α7 and β2 were the two major nAChR subunits naturally expressed in MS/DB cholinergic neurons (Cb–Cd). Double immunofluorescence labeling of a MS/DB neuron with anti-α7 and anti-β2 subunit antibodies revealed that α7 and β2 subunit proteins colocalized, and similar results were obtained using 31 neurons from 12 rats (D). Protein extracts from rat MS/DB (lane 1) or rat VTA (lane 2) or from MS/DB from nAChR β2 subunit knock-out (lane 4) or wild-type mice (lane 5) were immunoprecipitated (IP) with a rabbit anti-α7 antibody (Santa Cruz H302; lanes 1, 2, 4, and 5) or rabbit IgG as a control (lane 3). The eluted proteins from the precipitates were analyzed by immunoblotting (IB) with rat monoclonal anti-β2 subunit antibody mAb270 (top) or rabbit anti-α7 antiserum H302 (bottom). The β2 and α7 bands are indicated by arrows (E). All these data suggested that nAChR α7 and β2 nAChR subunits are coassembled in MS/DB neurons.
Pharmacological profiles of functional nAChRs in rat forebrain cholinergic neurons
Pharmacological approaches were used to compare features of functional nAChRs in MS/DB cholinergic or VTA DAergic neurons. The α7-nAChR-selective antagonist, MLA showed similar antagonist potency toward choline-induced currents in either MS/DB (Fig. 4 Aa) or VTA (Fig. 4 Ab) neurons. Analysis of concentration-inhibition curves (Fig. 4 Ac) yielded IC50 values and Hill coefficients of 0.7 nm and 1.1, respectively, for MS/DB neurons (n = 8) and 0.4 nm and 1.2, respectively, for VTA neurons (n = 9, MS/DB vs VTA p > 0.05). However, the β2*-nAChR-selective antagonist, DHβE was ∼500-fold less potent as an inhibitor of choline-induced current in MS/BD neurons (Fig. 4 Ba) than in VTA neurons (Fig. 4 Bb). IC50 values and Hill coefficients for DHβE-induced inhibition were 0.17 μm and 0.9, respectively, for MS/DB neurons (n = 8), and >100 μm and 0.3, respectively, for VTA neurons (n = 7; MS/DB vs VTA, p < 0.001) (Fig. 4 Bc). These results are consistent with the hypothesis that functional α7*-nAChRs on MS/DB cholinergic neurons also contain DHβE-sensitive β2 subunits.
Figure 4.

Antagonist profiles for MS/DB and VTA nAChRs. Concentration-dependent block by MLA (at the indicated concentrations in nm after preexposure for 2 min and continued exposure during agonist application indicated by open bars) of 10 mm choline-induced (applied as indicated by closed bars) whole-cell currents (representative traces shown) in MS/DB (Aa) and VTA (Ab) neurons was not significantly different (p > 0.05, Ac). However, choline-induced currents in MS/DB neurons (Ba) were more sensitive to block by DHβE (at the indicated concentrations in μm after preexposure for 2 min and continued exposure during agonist application indicated by open bars) than in VTA neurons (Bb; concentration–response profile shown in Bc). VH, Holding potential.
Functional nAChRs on rat basal forebrain cholinergic neurons are inhibited by Aβ1–42
Currently, it is not clear why basal forebrain cholinergic neurons are particularly sensitive to degeneration in AD. To test the hypothesis that novel α7β2-nAChRs on MS/DB cholinergic neurons are involved, we determined the effects of Aβ1–42 on these receptors. The experimental protocol involved repeated, acute challenges with 10 mm choline, and control studies in the absence of peptide demonstrated that there was no significant rundown of such responses when spaced at a minimum of 2 min intervals (Fig. 5 Aa). During a continuous exposure to 1 nm Aβ1–42 starting just after an initial choline challenge and continuing for 10 min, responses to choline challenges were progressively inhibited with time, although reversibly so as demonstrated by response recovery after 6 min of peptide washout (Fig. 5 Ab). In contrast, exposure to 1 nm scrambled Aβ1–42 (as a control peptide) had no effect (Fig. 5 Ac). Choline-induced currents in dissociated VTA DAergic neurons were not sensitive to 1 nm Aβ1–42 treatment (Fig. 5 Ad). Quantitative analysis of several replicate experiments (Fig. 5 B) confirmed that Aβ1–42, even at 1 nm concentration, specifically inhibits putative α7β2-nAChR function on MS/DB cholinergic neurons but not function of homomeric α7-nAChRs on VTA DAergic neurons.
Figure 5.

Effects of 1 nm Aβ1–42 on α7β2-nAChRs on MS/DB neurons. Typical whole-cell current traces for responses of MS/DB neurons to 10 mm choline challenge at the indicated times after initial challenge alone show no detectable rundown during repetitive application of agonist (2 s exposure at 2 min intervals; Aa). Choline-induced currents in rat MS/DB neurons were suppressed by 1 nm Aβ1–42 (continuously applied for 10 min, but responses to challenges with choline are shown at the indicated times of Aβ exposure; Ab) but not by 1 nm scrambled Aβ1–42 (as a control; Ac). Choline-induced currents in VTA neurons were not affected by 1 nm Aβ1–42 (Ad). B, Normalized, mean (±SE), peak current responses (ordinate) as a function of time (abscissa, min) during challenges with choline alone (□), in the presence of 1 nm Aβ (▲), or in the presence of control, scrambled Aβ (▼) for the indicated numbers of MS/DB neurons, or during challenges with choline in the presence of 1 nm Aβ for the indicated number of VTA neurons (●) illustrate that only choline-induced currents in rat MS/DB neurons were sensitive to functional inhibition by Aβ. *p < 0.05; **p < 0.01.
Concentration- and form-dependent inhibition by Aβ1–42 of α7β2-nAChR function on basal forebrain cholinergic neurons
Our previous studies indicated that α4β2-nAChRs were more sensitive to Aβ1–42 than homomeric α7-nAChRs (Wu et al., 2004a). Concentration dependence of effects of Aβ1–42 on choline-induced currents in MS/DB neurons was evident, with effects being negligible at 0.1 nm and effects at 1 nm being approximately half of those observed for 10 nm peptide (Fig. 6 A). The magnitude of inhibition apparently had not yet reached maximum after 10 min of peptide exposure. We also asked which form(s) of Aβ1–42 showed the most potent inhibitory effect on choline-induced currents elicited in MS/DB neurons. Using different preparation protocols (see Materials and Methods), we produced Aβ1–42 monomers (peptide dissolved in DMSO), oligomers (peptide dissolved in water), or fibrils (peptides dissolved in water at low pH (pH = 6.0) and incubated at 37°C for 2 d). Peptide forms were defined and monitored using AFM (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). At 1 nm, oligomeric Aβ1–42 exhibited the greatest suppression of choline-induced responses, fibrillar Aβ had weaker inhibitory effect, and monomeric Aβ1–42 failed to suppress choline-induced responses, indicating form-selective, Aβ1–42 inhibition of nAChRs in MS/DB cholinergic neurons. To test whether Aβ1–42 specifically inhibits nAChRs, we also examined the effects of 1 nm Aβ1–42 on GABA- or glutamate-induced currents in rat MS/DB cholinergic neurons, and the results demonstrated that both GABAA receptors and ionotropic glutamate receptors were insensitive to inhibition by 1 nm Aβ1–42 even when peptide effects on ACh-induced current were evident (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Collectively, these results indicate that, under our experimental conditions, pathologically relevant, low nm concentrations of Aβ1–42, especially in an oligomeric form, specifically inhibit function of apparently heteromeric α7β2-nAChRs, but peptides cannot inhibit function of homomeric α7-nAChRs, GABAA, or glutamate receptors on MS/DB cholinergic neurons.
Figure 6.
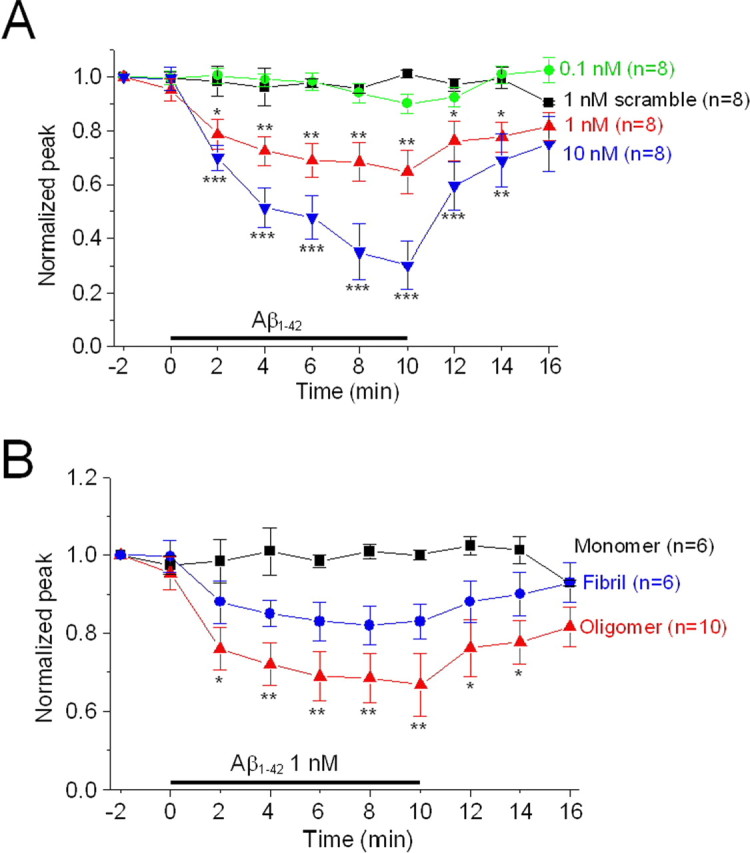
Inhibition of choline-induced currents in dissociated MS/DB neurons by Aβ1–42 was concentration and form dependent. A, Normalized mean (±SE) peak current responses (ordinate) of the indicated numbers of MS/DN neurons as a function of time (abscissa, min) during challenges with choline in the presence of 1 nm scrambled Aβ (■) or in the presence of 0.1 nm (●), 1 nm (▲), or 10 nm (▼) Aβ show concentration dependence of functional block. B, Normalized responses (ordinate) during challenges with choline in the presence of 1 nm monomeric (■), oligomeric (▲), or fibrillar (●) Aβ indicate insensitivity to monomeric Aβ and highest sensitivity to peptide oligomers. *p < 0.05, **p < 0.01, and ***p < 0.001.
Heteromeric α7β2-nAChRs heterologously expressed in Xenopus oocytes display slower current kinetics and high sensitivity to Aβ1–42
To further investigate features of presumed, novel α7β2-nAChRs as naturally expressed in basal forebrain cholinergic neurons, we introduced nAChR α7 subunits alone or in combination with β2 subunits into Xenopus oocytes. Compared with homomeric α7-nAChRs (Fig. 7 Aa), heteromeric α7β2-nAChRs expressed in oocytes injected with rat nAChR α7 and β2 subunit cRNAs at a ratio of 1:1 exhibited smaller peak current responses to choline and slower current decay rates (Fig. 7 Ab). These results are consistent with a previous report indicating that nAChR α7 and β2 subunits can coassemble in the oocyte system to form functional, heteromeric α7β2-nAChRs (Khiroug et al., 2002). As was the case for comparisons between native nAChR responses in rat MS/DB or VTA neurons (Fig. 4), sensitivity to functional blockade by MLA was similar for heterologously expressed α7β2- or α7-nAChR (supplemental Fig. 3_A–C_, available at www.jneurosci.org as supplemental material). Also similar to the case for native nAChR, heterologously expressed α7β2-nAChR were more sensitive to blockade by DHβE than were homomeric α7-nAChR. (Wang et al., 2000)indicates presence of β2 subunits with α7 subunits in rodent MS/DB neurons. We then tested the sensitivity of heterologously expressed α7β2-nAChRs in oocytes to Aβ. As was the case for presumed, native α7β2-nAChRs on MS/DB neurons, heterologously expressed heteromeric α7β2-nAChRs, but not homomeric α7-nAChRs, demonstrated sensitivity to Aβ1–42 (10 nm) and insensitivity to 10 nm scrambled Aβ1–42 (Fig. 7 B). These results obtained using heterologously expressed nAChRs again are consistent with the hypothesis that nAChR α7 and β2 subunits likely coassemble and form a unique α7β2-nAChR that enhances receptor sensitivity to pathologically relevant, low nm concentrations of Aβ1–42.
Figure 7.

Effects of Aβ on heterologously expressed homomeric α7- and heteromeric α7β2-nAChRs in Xenopus oocytes. Choline (10 mm, 2 s exposure at 2 min intervals)-induced whole-cell current responses in oocytes injected with rat α7-nAChR subunit cRNA alone (Aa, black trace) or with α7 and β2 subunit cRNAs at a ratio of 1:1 (Aa, gray trace) show slower decay of elicited currents and a longer decay time constant for heteromeric receptors (Aa, Ab). The calibration bars represent 1 s and 1 μA for the α7-nAChR response (black trace) and 1 s and 100 nA for the α7β2-nAChR response (gray trace), thus also showing that current amplitudes were lower for heteromeric than for homomeric receptors. VH, Holding potential. B, Normalized mean (±SE) peak current responses (ordinate) of the indicated numbers of oocytes heterologously expressing nAChR α7 and β2 subunits (■, ●) or only α7 subunits (▲) as a function of time (abscissa, min) during challenges with choline alone (■) or in the presence of 10 nm Aβ (●, ▲) show sensitivity to functional block by Aβ only for heteromeric receptors. *p < 0.05, **p < 0.01, and ***p < 0.001.
Basal forebrain nAChRs in nAChR β2 subunit-null mice do not show coimmunoprecipitation of nAChR α7 and β2 subunits, exhibit fast whole-cell current kinetics, and show low sensitivity to Aβ1–42
As another test of our hypothesis that basal forebrain cholinergic neurons express novel α7β2-nAChRs, we used wild-type and nAChR β2 subunit knock-out (β2−/−) mice. PCR genotyping was used to identify wild-type or β2−/− mice (Fig. 8 A,B). Using the immunoprecipitation protocol previously described and protein extracts from the MS/DB, nAChR β2 subunits were found to coprecipitate with nAChR α7 subunits only for samples from wild-type but not from β2−/− mice (Fig. 3 E, right). Choline-induced currents in MS/DB cholinergic neurons dissociated from β2−/− mice exhibited higher current amplitude, faster kinetics (Fig. 8 C), and lower sensitivity to DHβE (Fig. 8 Da–c) than responses in cholinergic neurons dissociated from wild-type mice. As expected, 1 nm Aβ1–42 failed to suppress choline-induced currents in MS/DB neurons from β2−/− mice but did suppress choline-induced currents in MS/DB neurons from wild-type mice (Fig. 8 E). These results again strongly support the hypothesis that heteromeric, functional α7β2-nAChRs on basal forebrain MS/DB cholinergic neurons are highly sensitive to a pathologically relevant concentrations of Aβ1–42.
Figure 8.

Kinetics, pharmacology, and Aβ sensitivity of α7-containing-nAChRs in nAChR β2 subunit knock-out mice. Genotype analyses demonstrated that nAChR β2 subunits are not expressed in nAChR β2 knock-out mice (A), whereas Lac-Z (as a marker for the knock-out) was absent in wild-type (WT) mice (B). Kinetic analyses showed that whole-cell current kinetics and amplitudes differed for MS/DB neurons from WT compared with nAChR β2 subunit knock-out homozygote mice (Ca, Cb). VH, Holding potential. Compared with MS/DB neurons from WT mice (Da), choline-induced currents in MS/DB (Db) neurons from β2 knock-outs were insensitive to DHβE but retained sensitivity to MLA (Dc). Aβ1–42 (1 nm) suppressed choline-induced currents in MS/DB neurons from WT (■) but not from β2 knock-out (●) mice (E). “Control” responses (▲) were choline-induced currents in neurons from WT mice without exposure to Aβ1–42. *p < 0.05, **p < 0.01.
Discussion
nAChRs in basal forebrain participate in cholinergic transmission and cognitive processes associated with learning and memory (Levin and Rezvani, 2002; Mansvelder et al., 2006). During the early stages of AD, decreases in nAChR-like radioligand binding sites have been observed (Burghaus et al., 2000; Nordberg, 2001), suggesting that nAChR dysfunction could be involved in AD pathogenesis and cholinergic deficiencies (Nordberg, 2001). Evidence indicates that enhancement of α7-nAChR function protects neurons against Aβ toxicity through any or some combination of a number of different mechanisms, as outlined previously (Sadot et al., 1996; Lahiri et al., 2002; Nagele et al., 2002; Geerts, 2005; Q. Liu et al., 2007). On the other hand, pharmacological interventions or diminished nAChR expression produces learning and memory deficits (Levin and Rezvani, 2002).
The current findings are consistent with the natural expression of a novel, heteromeric, functional α7β2-nAChR subtype on forebrain cholinergic neurons that is particularly sensitive to functional inhibition by a pathologically relevant concentration (1 nm) of Aβ1–42. Some previous studies investigating the acute effects of Aβ1–42 on nAChRs examined receptors on neurons from regions other than the basal forebrain or that were heterologously expressed (Liu et al., 2001; Pettit et al., 2001; Grassi et al., 2003; Wu et al., 2004a; Lamb et al., 2005; Pym et al., 2005) and/or used Aβ peptides at concentrations (between 100 nm and 10 μm) that greatly exceed Aβ concentrations found in AD brain (Kuo et al., 2000; Mehta et al., 2000). Other studies identified α7-nAChR-like, ACh-induced currents in MS/DB cholinergic neurons by using slice-patch recordings (Henderson et al., 2005; Thinschmidt et al., 2005) and characterized functional, non-α7-nAChRs by using acutely dissociated forebrain neurons (Fu and Jhamandas, 2003). Our present study combined whole-cell current recordings from acutely dissociated neurons and investigation of MS/DB cholinergic neuronal nAChRs to identify functional nAChRs that have some features of receptors containing α7 subunits, but we also found high sensitivity of these nAChRs to low concentrations of Aβ1–42.
Heterologous expression of functional α7β2-nAChRs with a unique pharmacological profile also has been reported using the Xenopus oocyte expression system (Khiroug et al., 2002). Our results agree with these previous findings and also indicate that functional α7β2-nAChRs can be heterologously expressed in oocytes. Histological studies have demonstrated coexpression of nAChR α7 and β2 subunits in most forebrain cholinergic neurons (Azam et al., 2003). Our results also are consistent with those observations and show cell-specific, coexpression of nAChR α7 and β2 subunits at both message and protein levels. There are other reports (Yu and Role, 1998; El-Hajj et al., 2007) that nAChR α7 subunits could be coassembled with other subunits to form native, heteromeric, α7*-nAChRs. Our present findings are consistent with those observations.
The notion that the Aβ1–42-sensitive, functional nAChR subtype in MS/DB neurons displaying some features of nAChRs containing α7 subunits, but distinctive from homomeric α7-nAChRs, is composed of α7 and β2 subunits, is supported by the loss of Aβ sensitivity and the conversion of functional nAChR properties to those like homomeric α7-nAChRs in nAChR β2 subunit knock-out animals. It has been reported that there are two isoforms (α7-1 and α7-2) of α7-nAChR transcript in homomeric α7-nAChRs. The α7-2 transcript that contains a novel exon is widely expressed in the brain and showed very slow current kinetics (Saragoza et al., 2003; Severance and Cuevas, 2004; Severance et al., 2004). However, we contend that the heteromeric, presumed α7β2-nAChR described in the present study and expressed in MS/DB neurons is not a homomeric nAChR composed of or containing the α7-2 transcript for three reasons: (1) in β2−/− mice, α7-nAChR-like whole-cell current responses to choline acquire fast kinetic characteristics like those of α7-nAChR responses in VTA neurons, (2) immunoprecipitation–Western blot analyses show coassembly of α7 and β2 subunits from the MS/DB but not from the VTA, nor from the MS/DB of β2−/− mice, and (3) pharmacologically, apparently heteromeric α7β2-nAChRs were sensitive not only to MLA, but also to DHβE.
A recent study suggested that levels of oligomeric forms of Aβ1–42, rather than monomers or Aβ fibrils, most closely correlate with cognitive dysfunction in animal models of AD (Haass and Selkoe, 2007). Our current findings also suggest that Aβ oligomers have the most profound effects on nAChR function, thus extending our earlier studies of Aβ-nAChR interactions (Wu et al., 2004a) and perhaps illuminating why there have been apparent discrepancies in some of the earlier work concerning Aβ-nAChR interactions.
Alzheimer's disease (AD) is a dementing, neurodegenerative disorder characterized by accumulation of amyloid β (Aβ) peptide-containing neuritic plaques, degeneration of basal forebrain cholinergic neurons, and gradually impaired learning and memory (Selkoe, 1999). The extent of learning and memory deficits in AD is proportional to the degree of forebrain cholinergic neuronal degeneration, and the extent of Aβ deposition is used to characterize disease severity (Selkoe, 1999). Processes such as impairment of neurotrophic support and disorders in glucose metabolism have been implicated in cholinergic neuronal loss and AD (Dolezal and Kasparová, 2003). However, clear neurotoxic effects of Aβ across a range of in vivo and in vitro models suggest that Aβ plays potentially causal roles in cholinergic neuronal degeneration and consequent learning and memory deficits (Selkoe, 1999).
Although the “cholinergic hypothesis” of AD etiopathogenesis was established in the 1990s, the exact mechanism(s) by which Aβ accumulation harms cholinergic neurons and results in learning and memory deficits is largely unclear. Seemingly contradictory findings about the effects of Aβ on function of nAChRs (Liu et al., 2001; Pettit et al., 2001; Dineley et al., 2002b; Dougherty et al., 2003; Fu and Jhamandas, 2003; Wu et al., 2004a) have been hard to reconcile, although they may be explained by the use of different experimental preparations and different or non-pathologically relevant concentrations or forms of Aβ. Other studies indicate that α7-nAChR expression is increased in both an AD animal model and in human AD (Dineley et al., 2002a; Counts et al., 2007), suggesting that pathologically relevant concentrations of Aβ1–42 upregulate α7β2-nAChRs, potentially leading to cholinergic neuronal dysfunction due to abnormal accumulation of intracellular Ca2+. Although this all might occur as a consequence of acute suppression of α7β2-nAChR function by Aβ, the present studies did not assess this possibility.
Based on the current findings, we suggest that selective, high-affinity effects of oligomeric Aβ1–42 on basal forebrain, cholinergic neuronal α7β2-nAChRs could acutely contribute to disruption of cholinergic signaling and diminished learning and memory abilities (Yan and Feng, 2004). Moreover, to the extent that basal forebrain cholinergic neuronal health requires activity of α7β2-nAChRs, inhibition of α7β2-nAChR function by oligomeric Aβ1–42 could lead to losses of trophic support for those neurons and/or their targets, and cross-catalyzed spirals of receptor functional loss and neuronal degeneration also could contribute to the progression of AD. Drugs targeting α7β2-nAChRs to protect them against Aβ effects or restoration of α7β2-nAChR function in cholinergic forebrain neurons could be viable tertiary or even primary therapies for AD.
Footnotes
This work was supported by an Arizona Alzheimer's Consortium Pilot grant, the Barrow Neurological Foundation, and the National Institutes of Health (R01 DA015389). We thank Kevin Ellsworth for his assistance in preparing this manuscript and Dr. Marina Picciotto (Yale University) for kindly providing the nAChR β2 knock-out mice.
References
- Azam L, Winzer-Serhan U, Leslie FM. Co-expression of alpha7 and beta2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience. 2003;119:965–977. doi: 10.1016/s0306-4522(03)00220-3. [DOI] [PubMed] [Google Scholar]
- Burghaus L, Schütz U, Krempel U, de Vos RA, Jansen Steur EN, Wevers A, Lindstrom J, Schröder H. Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer patients. Brain Res Mol Brain Res. 2000;76:385–388. doi: 10.1016/s0169-328x(00)00031-0. [DOI] [PubMed] [Google Scholar]
- Counts SE, He B, Che S, Ikonomovic MD, DeKosky ST, Ginsberg SD, Mufson EJ. Alpha7 nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch Neurol. 2007;64:1771–1776. doi: 10.1001/archneur.64.12.1771. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002a;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Bell KA, Bui D, Sweatt JD. beta-Amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem. 2002b;277:25056–25061. doi: 10.1074/jbc.M200066200. [DOI] [PubMed] [Google Scholar]
- Dolezal V, Kasparová J. β-Amyloid and cholinergic neurons. Neurochem Res. 2003;28:499–506. doi: 10.1023/a:1022865121743. [DOI] [PubMed] [Google Scholar]
- Dougherty JJ, Wu J, Nichols RA. β-Amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci. 2003;23:6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hajj RA, McKay SB, McKay DB. Pharmacological and immunological identification of native alpha7 nicotinic receptors: evidence for homomeric and heteromeric alpha7 receptors. Life Sci. 2007;81:1317–1322. doi: 10.1016/j.lfs.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha 4 and beta 2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- Fu W, Jhamandas JH. β-Amyloid peptide activates non-alpha7 nicotinic acetylcholine receptors in rat basal forebrain neurons. J Neurophysiol. 2003;90:3130–3136. doi: 10.1152/jn.00616.2003. [DOI] [PubMed] [Google Scholar]
- Geerts H. Indicators of neuroprotection with galantamine. Brain Res Bull. 2005;64:519–524. doi: 10.1016/j.brainresbull.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan M, Monteggia LM, Anderson DJ, Molinari EJ, Piattoni-Kaplan M, Donnelly-Roberts D, Arneric SP, Sullivan JP. Stable expression, pharmacologic properties and regulation of the human neuronal nicotinic acetylcholine alpha 4 beta 2 receptor. J Pharmacol Exp Ther. 1996;276:289–297. [PubMed] [Google Scholar]
- Grassi F, Palma E, Tonini R, Amici M, Ballivet M, Eusebi F. Amyloid beta(1–42) peptide alters the gating of human and mouse alpha-bungarotoxin-sensitive nicotinic receptors. J Physiol. 2003;547:147–157. doi: 10.1113/jphysiol.2002.035436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Henderson Z, Boros A, Janzso G, Westwood AJ, Monyer H, Halasy K. Somato-dendritic nicotinic receptor responses recorded in vitro from the medial septal diagonal band complex of the rodent. J Physiol. 2005;562:165–182. doi: 10.1113/jphysiol.2004.070300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AA, Frølund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem. 2005;48:4705–4745. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- Khiroug SS, Harkness PC, Lamb PW, Sudweeks SN, Khiroug L, Millar NS, Yakel JL. Rat nicotinic ACh receptor alpha7 and beta2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J Physiol. 2002;540:425–434. doi: 10.1113/jphysiol.2001.013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Kokjohn TA, Watson MD, Woods AS, Cotter RJ, Sue LI, Kalback WM, Emmerling MR, Beach TG, Roher AE. Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. Am J Pathol. 2000;156:797–805. doi: 10.1016/s0002-9440(10)64947-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri DK, Utsuki T, Chen D, Farlow MR, Shoaib M, Ingram DK, Greig NH. Nicotine reduces the secretion of Alzheimer's beta-amyloid precursor protein containing beta-amyloid peptide in the rat without altering synaptic proteins. Ann N Y Acad Sci. 2002;965:364–372. doi: 10.1111/j.1749-6632.2002.tb04178.x. [DOI] [PubMed] [Google Scholar]
- Lamb PW, Melton MA, Yakel JL. Inhibition of neuronal nicotinic acetylcholine receptor channels expressed in Xenopus oocytes by beta-amyloid1–42 peptide. J Mol Neurosci. 2005;27:13–21. doi: 10.1385/JMN:27:1:013. [DOI] [PubMed] [Google Scholar]
- Levin ED, Rezvani AH. Nicotinic treatment for cognitive dysfunction. Curr Drug Targets CNS Neurol Disord. 2002;1:423–431. doi: 10.2174/1568007023339102. [DOI] [PubMed] [Google Scholar]
- Lindstrom J. Neuronal nicotinic acetylcholine receptors. Ion Channels. 1996;4:377–450. doi: 10.1007/978-1-4899-1775-1_10. [DOI] [PubMed] [Google Scholar]
- Lindstrom J, Anand R, Gerzanich V, Peng X, Wang F, Wells G. Structure and function of neuronal nicotinic acetylcholine receptors. Prog Brain Res. 1996;109:125–137. doi: 10.1016/s0079-6123(08)62094-4. [DOI] [PubMed] [Google Scholar]
- Liu Q, Kawai H, Berg DK. beta-Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci U S A. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zhang J, Zhu H, Qin C, Chen Q, Zhao B. Dissecting the signaling pathway of nicotine-mediated neuroprotection in a mouse Alzheimer disease model. FASEB J. 2007;21:61–73. doi: 10.1096/fj.06-5841com. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhang J, Berg DK. Role of endogenous nicotinic signaling in guiding neuronal development. Biochem Pharmacol. 2007;74:1112–1119. doi: 10.1016/j.bcp.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, Le Novère N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- Mansvelder HD, van Aerde KI, Couey JJ, Brussaard AB. Nicotinic modulation of neuronal networks: from receptors to cognition. Psychopharmacology. 2006;184:292–305. doi: 10.1007/s00213-005-0070-z. [DOI] [PubMed] [Google Scholar]
- Mehta PD, Pirttilä T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM. Plasma and cerebrospinal fluid levels of amyloid beta proteins 1–40 and 1–42 in Alzheimer disease. Arch Neurol. 2000;57:100–105. doi: 10.1001/archneur.57.1.100. [DOI] [PubMed] [Google Scholar]
- Moser N, Mechawar N, Jones I, Gochberg-Sarver A, Orr-Urtreger A, Plomann M, Salas R, Molles B, Marubio L, Roth U, Maskos U, Winzer-Serhan U, Bourgeois JP, Le Sourd AM, De Biasi M, Schröder H, Lindstrom J, Maelicke A, Changeux JP, Wevers A. Evaluating the suitability of nicotinic acetylcholine receptor antibodies for standard immunodetection procedures. J Neurochem. 2007;102:479–492. doi: 10.1111/j.1471-4159.2007.04498.x. [DOI] [PubMed] [Google Scholar]
- Mudo G, Belluardo N, Fuxe K. Nicotinic receptor agonists as neuroprotective/neurotrophic drugs. Progress in molecular mechanisms. J Neural Transm. 2007;114:135–147. doi: 10.1007/s00702-006-0561-z. [DOI] [PubMed] [Google Scholar]
- Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1–42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002;110:199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- Nordberg A. Nicotinic receptor abnormalities of Alzheimer's disease: therapeutic implications. Biol Psychiatry. 2001;49:200–210. doi: 10.1016/s0006-3223(00)01125-2. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Orlando, FL: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel JL. β-Amyloid1–42 peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 2001;21(RC120):1–5. doi: 10.1523/JNEUROSCI.21-01-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pym L, Kemp M, Raymond-Delpech V, Buckingham S, Boyd CA, Sattelle D. Subtype-specific actions of beta-amyloid peptides on recombinant human neuronal nicotinic acetylcholine receptors (alpha7, alpha4beta2, alpha3beta4) expressed in Xenopus laevis oocytes. Br J Pharmacol. 2005;146:964–971. doi: 10.1038/sj.bjp.0706403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadot E, Gurwitz D, Barg J, Behar L, Ginzburg I, Fisher A. Activation of m1 muscarinic acetylcholine receptor regulates tau phosphorylation in transfected PC12 cells. J Neurochem. 1996;66:877–880. doi: 10.1046/j.1471-4159.1996.66020877.x. [DOI] [PubMed] [Google Scholar]
- Saragoza PA, Modir JG, Goel N, French KL, Li L, Nowak MW, Stitzel JA. Identification of an alternatively processed nicotinic receptor alpha7 subunit RNA in mouse brain. Brain Res Mol Brain Res. 2003;117:15–26. doi: 10.1016/s0169-328x(03)00261-4. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- Severance EG, Cuevas J. Distribution and synaptic localization of nicotinic acetylcholine receptors containing a novel alpha7 subunit isoform in embryonic rat cortical neurons. Neurosci Lett. 2004;372:104–109. doi: 10.1016/j.neulet.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Severance EG, Zhang H, Cruz Y, Pakhlevaniants S, Hadley SH, Amin J, Wecker L, Reed C, Cuevas J. The alpha7 nicotinic acetylcholine receptor subunit exists in two isoforms that contribute to functional ligand-gated ion channels. Mol Pharmacol. 2004;66:420–429. doi: 10.1124/mol.104.000059. [DOI] [PubMed] [Google Scholar]
- Sivaprakasam K. Towards a unifying hypothesis of Alzheimer's disease: cholinergic system linked to plaques, tangles and neuroinflammation. Curr Med Chem. 2006;13:2179–2188. doi: 10.2174/092986706777935203. [DOI] [PubMed] [Google Scholar]
- Thinschmidt JS, Frazier CJ, King MA, Meyer EM, Papke RL. Medial septal/diagonal band cells express multiple functional nicotinic receptor subtypes that are correlated with firing frequency. Neurosci Lett. 2005;389:163–168. doi: 10.1016/j.neulet.2005.07.038. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Whiting PJ, Schoepfer R, Swanson LW, Simmons DM, Lindstrom JM. Functional acetylcholine receptor in PC12 cells reacts with a monoclonal antibody to brain nicotinic receptors. Nature. 1987;327:515–518. doi: 10.1038/327515a0. [DOI] [PubMed] [Google Scholar]
- Wu J, Chan P, Schroeder KM, Ellsworth K, Partridge LD. 1-Methyl-4-phenylpridinium (MPP+)-induced functional run-down of GABA(A) receptor-mediated currents in acutely dissociated dopaminergic neurons. J Neurochem. 2002;83:87–99. doi: 10.1046/j.1471-4159.2002.01099.x. [DOI] [PubMed] [Google Scholar]
- Wu J, Kuo YP, George AA, Xu L, Hu J, Lukas RJ. beta-Amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J Biol Chem. 2004a;279:37842–37851. doi: 10.1074/jbc.M400335200. [DOI] [PubMed] [Google Scholar]
- Wu J, George AA, Schroeder KM, Xu L, Marxer-Miller S, Lucero L, Lukas RJ. Electrophysiological, pharmacological, and molecular evidence for alpha7-nicotinic acetylcholine receptors in rat midbrain dopamine neurons. J Pharmacol Exp Ther. 2004b;311:80–91. doi: 10.1124/jpet.104.070417. [DOI] [PubMed] [Google Scholar]
- Yan Z, Feng J. Alzheimer's disease: interactions between cholinergic functions and beta-amyloid. Curr Alzheimer Res. 2004;1:241–248. doi: 10.2174/1567205043331992. [DOI] [PubMed] [Google Scholar]
- Yu CR, Role LW. Functional contribution of the alpha5 subunit to neuronal nicotinic channels expressed by chick sympathetic ganglion neurones. J Physiol. 1998;509:667–681. doi: 10.1111/j.1469-7793.1998.667bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]