Reversal of RNA-dominance by displacement of protein sequestered on triplet repeat RNA (original) (raw)
. Author manuscript; available in PMC: 2014 Jul 24.
Published in final edited form as: Science. 2009 Jul 17;325(5938):336–339. doi: 10.1126/science.1173110
Abstract
Genomic expansions of simple tandem repeats can give rise to toxic RNAs that contain expanded repeats. In myotonic dystrophy the expression of expanded CUG repeats (CUGexp) causes abnormal regulation of alternative splicing and neuromuscular dysfunction. Here we use a transgenic mouse model to show that derangements of myotonic dystrophy are reversed by a morpholino antisense oligonucleotide, CAG25, that binds to CUGexp RNA and blocks its interaction with Muscleblind-like 1 (MBNL1), a CUGexp-binding protein. CAG25 disperses nuclear foci of CUGexp RNA and reduces the overall burden of this toxic RNA. As MBNL1 is released from sequestration, the defect of alternative splicing regulation is corrected, thereby restoring ion channel function. These findings suggest an alternative use of antisense methods, to inhibit deleterious interactions of proteins with pathogenic RNAs.
Myotonic dystrophy type 1 (DM1) is representative of a group of dominantly-inherited disorders in which expression of a toxic RNA leads to neuromuscular degeneration (1-5). A feature common to these pathogenic RNAs is the presence of an expanded repeat. In DM1 the disease-inducing transcript is the DM protein kinase (DMPK) mRNA containing an expanded CUG repeat in its 3′ untranslated region (UTR) (6). These CUG repeats bind to MBNL1, a splicing regulator, with high affinity (7, 8). Because each mutant transcript typically contains thousands of CUG repeats, the capacity for protein binding is large, causing MBNL1 to become sequestered in ribonucleoprotein complexes. These complexes are observed in DM1 cells as foci of CUGexp-MBNL1 aggregates in the nucleus (7, 9-11).
If sequestration of MBNL1 contributes to symptoms of DM1, inhibitors of MBNL1-CUGexp binding may reverse these effects. To test this possibility we used CAG25, an antisense 25-mer morpholino oligonucleotide composed of CAG repeats. Antisense morpholinos do not trigger cleavage of their target RNAs (12), suggesting their use to bind CUGexp RNA and release sequestered proteins, without risk of degrading other transcripts that contain CUG repeats. A potential obstacle for this approach, however, is that CUGexp RNAs form hairpins with high thermal stability (13, 14) and extensive MBNL1 binding (8), which potentially could limit their access to antisense oligonucleotides. In vitro CAG25 was able to invade CUGexp hairpins and form a stable RNA-morpholino heteroduplex (Fig. 1A, Fig. S1, Fig. S2). CAG25 was also able to block the formation of CUGexp-MBNL1 complexes, and disrupt complexes that had already formed (Fig. 1B-D).
Fig. 1.
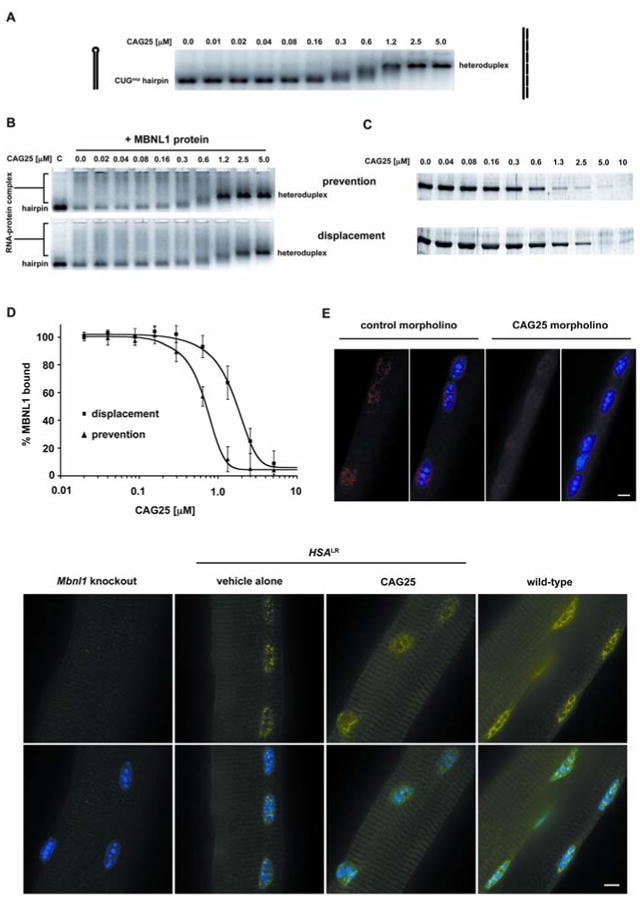
CAG25 inhibits formation of CUGexp-MBNL1 complexes. (A) Gel shift assay demonstrates that addition of CAG25 (indicated concentration) to labeled (CUG)109 (2 nM) results in slower migration of (CUG)109-CAG25 heteroduplex. (B) CAG25 prevents CUGexp-MBNL1 complex formation (upper panel) and displaces MBNL1 protein from pre-formed complex (lower panel). Lane “C” shows migration of labeled (CUG)109 hairpin. Addition of excess MBNL1 produces complexes of variable size that migrate as a broad smear (lane “0.0”). Addition of CAG25 at increasing concentration reconstitutes (CUG)109-CAG25 heteroduplex as the dominant band (27). (C) Microtiter plate/gel assay confirms that CAG25 prevents the formation of (CUG)109-MBNL1 complex (upper panel) and displaces MBNL1 from pre-formed complexes (lower panel). Bands indicate the amount of recombinant MBNL1 protein that remains in ribonucleoprotein complex at the indicated concentration of CAG25 (27). (D) The % MBNL1 bound to CUGexp is expressed as the mean ± SD of protein retained on plate. IC50 for “prevention” is 462 ± 31 nM and for “displacement” is 1032 ± 117 nM. (E) Fluorescence in situ hybridization of single flexor digitorum brevis (FDB) muscle fibers from _HSA_LR mice. Probe (red) binds to _HSA_LR transcripts upstream from CUG repeat, nuclei are blue. CAG25, but not control morpholino, causes dispersal of RNA foci. (F) MBNL1 (immunofluorescence, green) shifts from punctate to diffuse nuclear distribution after injection of FDB with CAG25. Scale bars = 5 μm.
To examine whether CAG25 can influence CUGexp interactions in vivo, we tested its effects in a transgenic mouse model of DM1. _HSA_LR transgenic mice express human skeletal actin transcripts that have (CUG)250 inserted in the 3′ UTR. These mice accumulate CUGexp RNA and MBNL1 protein in nuclear foci in skeletal muscle (1), a process that is thought to depend on CUGexp-MBNL1 interaction (15). We therefore examined the effect of CAG25 on foci in muscle cells. We loaded CAG25 into muscle fibers by intramuscular injection followed by in vivo electroporation. Muscle tissue was examined 1-3 weeks later by fluorescence in situ hybridization, using probes that hybridize to the CUG repeat or to sequences flanking the repeat. Injection of CAG25, but not a control morpholino of unrelated sequence, caused a marked reduction of nuclear foci and a redistribution of MBNL1 protein (Fig. 1E-F, Fig. S3).
To determine whether CAG25 can reverse the biochemical consequences of MBNL1 sequestration, we examined its effects on alternative splicing. _HSA_LR transgenic mice show alternative splicing changes similar to those observed in human DM1 (10). The splicing misregulation is improved when MBNL1 levels are increased (16), aggravated when MBNL1 levels are reduced, and reproduced by ablation of Mbnl1 (17), suggesting that splicing defects in this model are primarily caused by MBNL1 sequestration. For each DM1-affected exon that we examined, the alternative splicing was normalized or nearly corrected at three weeks following injection of CAG25 (Fig. 2). Effects of CAG25 on alternative splicing persisted at fourteen weeks (Fig S4A, B) but not at 8 months following a single injection. By contrast, CAG25 did not correct the misregulated splicing of these same exons in Mbnl1 knockout mice, or alter their splicing patterns in WT mice (Fig S5), indicating that its effects are mediated through CUGexp RNA rather than acting directly on the respective pre-mRNAs. The Capzb and Itgb1 transcripts also show developmentally regulated alternative splicing in skeletal muscle, but they do not depend on MBNL1, they are not misregulated in _HSA_LR mice (10), and CAG25 had no effect on splicing of either transcript.
Fig. 2.
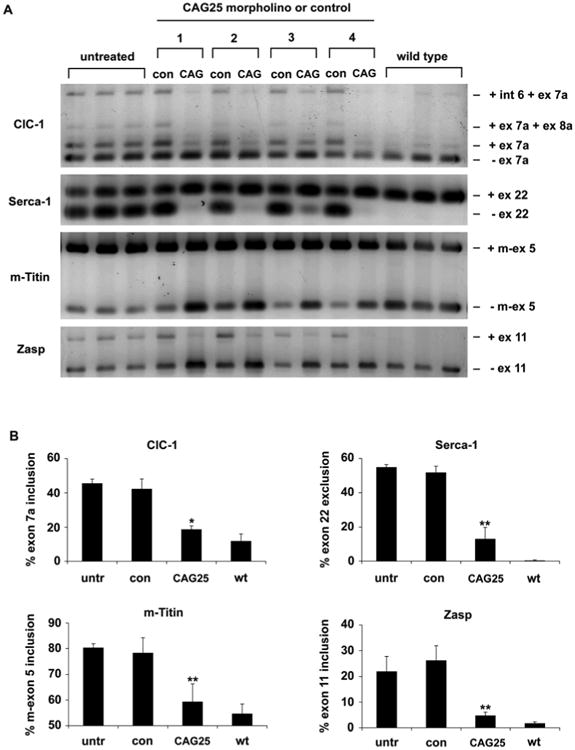
Reversal of misregulated alternative splicing by CAG25. (A) RT-PCR analysis of alternative splicing for chloride channel-1 (ClC-1), Serca-1, m-Titin, and Zasp, 3 weeks after a single injection of CAG25 in tibialis anterior (TA) muscle of _HSA_LR transgenic mice. The contralateral (con) TA was injected with vehicle (saline, mice 1 and 2) or morpholino with inverted sequence (GAC25, mice 3 and 4). Splice products from untreated _HSA_LR transgenic and wild-type TA muscle (n = 3 mice) are shown. Int 6 = intron 6 retention. (B) Quantification of results in A, expressed as the percentage of splice products that include or exclude the indicated exon. *P = 0.003 and ** P < 0.001 for CAG25 vs contralateral (_t_-test). “untr” = untreated _HSA_LR.
To determine whether CAG25 can rescue the physiological deficits of DM1 we examined the expression and function of ClC-1, the muscle-specific chloride channel. Delayed muscle relaxation and repetitive action potentials (myotonia) are cardinal features of DM1, resulting from loss of ClC-1 channels (18, 19). Affected individuals and mouse models show inclusion of an additional exon in the ClC-1 mRNA, resulting in frameshift and loss of channel activity (20). CAG25 corrected the defect of ClC-1 alternative splicing (Fig. 2; Fig. S4A, B) and restored the expression of ClC-1 protein to the surface membrane (Fig. 3A; Fig. S4C). Furthermore, transmembrane chloride ion conductance was normalized (Fig. 3B) and myotonia was markedly reduced (Fig. 3C; Fig. S4D and Fig. S7).
Fig. 3.
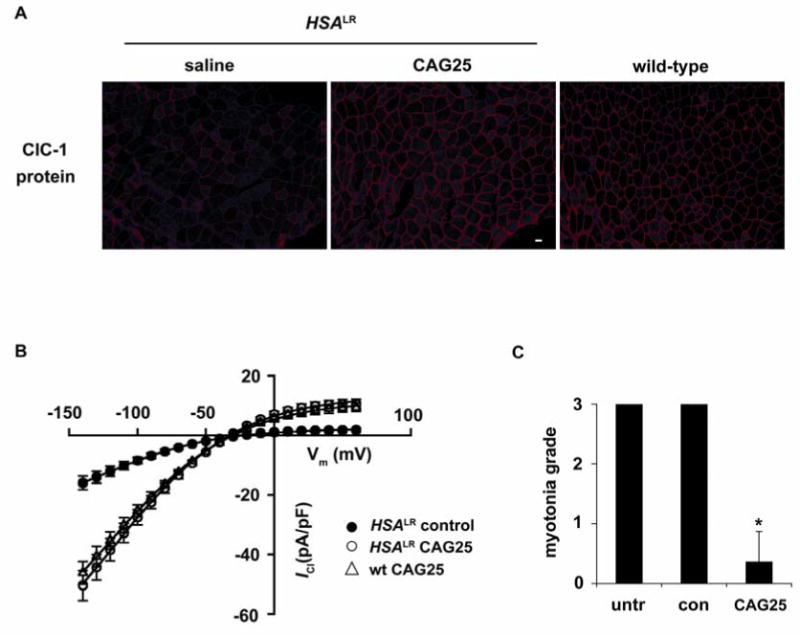
Chloride channel 1 protein expression and function are rescued by CAG25. (A) Immunofluorescence for ClC-1 protein expression in sections from _HSA_LR TA muscle, bar = 20 μm. (B) ClC-1 current density in FDB fibers isolated from 15-day-old _HSA_LR mice injected with CAG25 or control morpholino. Fibers from wild-type mice injected with CAG25 or control morpholino serve as controls (see also Fig. S6). (C) Electromyographic myotonia analysis 3 weeks after injection of CAG25 into TA muscle. The contralateral side was injected with vehicle (saline) alone or control morpholino (27). n = 11 mice examined; *P < 0.0001 for CAG25 vs. saline (_t_-test).
The effects of MBNL1 sequestration on gene expression are not limited to alternative splicing. For example, transcription of Eda2r, Uchl1, and Sarcolipin is highly upregulated in Mbnl1 knockout mice, and similar changes are induced by expression of CUGexp (21). While the mechanism for this effect has not been determined, it was partially reversed by CAG25 (Fig. S6). These data indicate that CAG25 ameliorates both transcriptional and posttranscriptional effects of toxic RNA.
We next determined whether CAG25 can overcome the nuclear retention of CUG-expanded transcripts (22). Despite reducing the overall level of CUGexp RNA in muscle (see below), CAG25 increased the amount of this transcript in the cytoplasm (Fig. 4A and Fig. S8). Moreover, because reduced translation of DMPK mRNA may contribute to cardiac symptoms of DM1 (23), we also tested whether CAG25 could enhance the translation of CUGexp-containing transcripts. We derived transgenic mice that express luciferase mRNA containing (CUG)270 in the 3′ UTR. The luciferase transcript is retained in nuclear foci and basal levels of luciferase activity are accordingly reduced. In vivo bioluminescence imaging showed that CAG25 induced a focal increase of luciferase activity in the injected hindlimb (Fig. 4B), consistent with increased translation of the CUG-expanded mRNA.
Fig. 4.
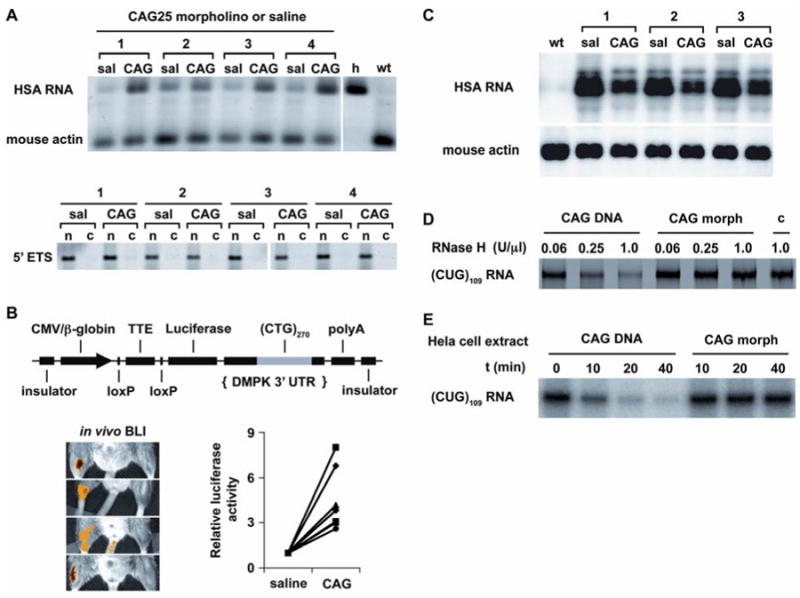
CAG25 increases the cytoplasmic level and translation of CUGexp-containing mRNA, despite reducing the overall level of CUGexp RNA. (A) RT-PCR assay of transgene mRNA in the cytoplasm. Human (transgene) and mouse (endogenous) skeletal actin transcripts were co-amplified by the same primers, species origin was revealed by AluI cleavage (27). “h”, human muscle; “wt”, wild-type mouse. Lower panel shows depletion of nuclear RNA from cytoplasmic fraction (“c”) relative to nuclear pellet (“n”), analyzed by RT-PCR for the 5′ external transcribed spacer (5′ ETS, nuclear-retained) of ribosomal RNA. Numbers refer to different mice treated with CAG25. (B) CAG25 increases luciferase activity in LLC9/Rosa-CreER bitransgenic mice. Upper panel shows LLC9 transgene for conditional expression of CUGexp RNA. Lower panels show in vivo bioluminescence imaging (BLI) of different bitransgenic mice (27). For quantification, luciferase activity (indicated in yellow-orange) in CAG25-injected muscle was normalized to the contralateral side injected with saline or control morpholino (n = 7 mice). (C) Northern blot of total cellular RNA shows decreased _HSA_LR mRNA in muscle injected with CAG25, compared to contralateral muscle injected with vehicle (saline). Mouse actin is loading control. (D) CUGexp-CAG25 heteroduplex is not cleaved by RNase H. (CUG)109 RNA was incubated with CAG25 morpholino or DNA oligonucleotide of identical sequence (CAG25-DNA). The heteroduplex was incubated with the indicated concentration of RNase H, then separated on polyacrylamide gels. Lane “c”, control with (CUG)109 alone. (E) (CUG)109 RNA was incubated with CAG25 morpholino or CAG25-DNA to form heteroduplex as in D, to which HeLa cell extract was added for the indicated time.
CAG25 caused a ∼50% reduction in the overall burden of CUGexp RNA (Fig. 4C). There was no parallel reduction of transgene pre-mRNA (Fig. S9), suggesting that downregulation of this transcript results from accelerated decay. However, previous work has shown that antisense morpholinos do not support RNA cleavage by RNase H (12). Consistent with these observations, CAG25 did not induce cleavage of CUGexp RNA by recombinant RNase H or by HeLa cell extracts in vitro, whereas an equivalent DNA oligonucleotide caused extensive CUGexp degradation (Fig. 4D,E). Moreover, CAG25 did not reduce levels of endogenous CUG-repeat-containing transcripts (Fig. S10), nor did it reduce transgene mRNA in mice that express an equivalent HSA transcript containing a non-expanded CUG repeat (Fig S11). Taken together, these results are consistent with an indirect effect of CAG25 on the accumulation of CUGexp RNA. However, therapeutic effects of CAG25 do not derive solely from downregulation of the repetitive RNA, because the residual levels of CUGexp were not below the threshold for inducing RNA disease (Fig. S12).
DM1 presents a complex phenotype that results from _trans_-dominant effects of mutant RNA on many different transcripts. To intervene at an upstream site and accomplish a general correction of DM1, here we used an antisense methodology to inhibit the protein interactions of expanded RNA repeats. This strategy has several effects that are potentially beneficial, including release of MBNL1 protein from ribonucleoprotein foci, enhanced transport of CUGexp transcripts to the cytoplasm, and reduced burden of CUGexp RNA. The mechanism for the latter effect remains to be determined, but we postulate that it may result from accelerated decay of CUGexp RNA once it has been transported to the cytoplasm. Considering that the extent of MBNL1 sequestration in DM1 is variable in different nuclei from the same individual (10), and that ∼50% of normal MBNL1 levels are sufficient to ensure normal splicing regulation in mice (10, 17), even partial release of sequestered MBNL1 is likely to improve splicing abnormalities in DM1. Notably, the mass of CUGexp RNA in muscle from _HSA_LR mice is 2-to 8-fold higher than in muscle tissue from patients with DM1 (Fig. S13), suggesting that similar therapeutic effects can be achieved in humans, if the antisense can be effectively delivered. Indeed, treatment of DM1 cells in tissue culture with CAG25 led to a reduction of intranuclear RNA foci (Fig. S14). Among several CUGexp-protein interactions that may contribute to DM1 pathogenesis, we have focused on MBNL1 because it has the highest CUGexp-binding affinity (8, 24) and most complete sequestration (11) of any factor so far identified. However, it seems likely that interactions of CUGexp with other RNA binding proteins, such as CUG-binding protein 1 (25), will also be inhibited by this approach, which may favorably impact signaling abnormalities in DM1 (26). Taken together, these data supply proof of concept that agents inhibiting deleterious RNA-protein interactions have therapeutic potential in RNA dominant disorders.
Supplementary Material
Supporting Online Material
Acknowledgments
This work comes from the University of Rochester Wellstone Muscular Dystrophy Cooperative Research Center (U54NS48843) and Center for RNA Biology, with support from the NIH (AR046806, AR/NS48143, NIDCR-T32DE07202 to J. Lueck), the Muscular Dystrophy Association, a postdoctoral fellowship to K.S. from the Foundation for Polish Science, Run America Foundation, and Saunders Family fund. We appreciate help from L. Richardson and S. Leistman. The University of Rochester has applied for a patent based partly on the work described here.
Footnotes
27
Materials and methods are available as supporting material on Science Online.
References
- 1.Mankodi A, et al. Science. 2000;289:1769. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 2.Liquori CL, et al. Science. 2001;293:864. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 3.Jin P, et al. Neuron. 2003;39:739. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 4.Moseley ML, et al. Nat Genet. 2006;38:758. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 5.Li LB, Yu Z, Teng X, Bonini NM. Nature. 2008;453:1107. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brook JD, et al. Cell. 1992;68:799. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 7.Miller JW, et al. Embo J. 2000;19:4439. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan Y, et al. Nucleic Acids Res. 2007;35:5474. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. J Cell Biol. 1995;128:995. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin X, et al. Hum Mol Genet. 2006;15:2087. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 11.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Hum Mol Genet. 2004;13:3079. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 12.Summerton J. Biochim Biophys Acta. 1999;1489:141. doi: 10.1016/s0167-4781(99)00150-5. [DOI] [PubMed] [Google Scholar]
- 13.Napierala M, Krzyzosiak WJ. J Biol Chem. 1997;272:31079. doi: 10.1074/jbc.272.49.31079. [DOI] [PubMed] [Google Scholar]
- 14.Tian B, et al. Rna. 2000;6:79. doi: 10.1017/s1355838200991544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dansithong W, Paul S, Comai L, Reddy S. J Biol Chem. 2005;280:5773. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- 16.Kanadia RN, et al. Proc Natl Acad Sci U S A. 2006;103:11748. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanadia RN, et al. Science. 2003;302:1978. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 18.Mankodi A, et al. Mol Cell. 2002;10:35. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 19.Charlet BN, et al. Mol Cell. 2002;10:45. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 20.Lueck JD, Mankodi A, Swanson MS, Thornton CA, Dirksen RT. J Gen Physiol. 2007;129:79. doi: 10.1085/jgp.200609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osborne RJ, et al. Hum Mol Genet. 2009;18:1471. doi: 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Proc Natl Acad Sci U S A. 1997;94:7388. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berul CI, et al. J Clin Invest. 1999;103:R1. doi: 10.1172/JCI5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warf MB, Berglund JA. RNA. 2007;13:2238. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Timchenko LT, et al. Nucleic Acids Res. 1996;24:4407. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Mol Cell. 2007;28:68. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Online Material