MED14 Tethers Mediator to the N-Terminal Domain of Peroxisome Proliferator-Activated Receptor γ and Is Required for Full Transcriptional Activity and Adipogenesis (original) (raw)
Abstract
The Mediator subunit MED1/TRAP220/DRIP205/PBP interacts directly with many nuclear receptors and was long thought to be responsible for tethering Mediator to peroxisome proliferator-activated receptor (PPAR)-responsive promoters. However, it was demonstrated recently that PPARγ can recruit Mediator by MED1-independent mechanisms. Here, we show that target gene activation by ectopically expressed PPARγ and PPARα is independent of MED1. Consistent with this finding, recruitment of PPARγ, MED6, MED8, TATA box-binding protein (TBP), and RNA polymerase II (RNAPII) to the enhancer and proximal promoter of the PPARγ target gene Fabp4 is also independent of MED1. Using a small interfering RNA (siRNA)-based approach, we identify MED14 as a novel critical Mediator component for PPARγ-dependent transactivation, and we demonstrate that MED14 interacts directly with the N terminus of PPARγ in a ligand-independent manner. Interestingly, MED14 knockdown does not affect the recruitment of PPARγ, MED6, and MED8 to the Fabp4 enhancer but does reduce their occupancy of the Fabp4 proximal promoter. In agreement with the necessity of MED14 for PPARγ transcriptional activity, we show that knockdown of MED14 impairs adipogenesis of 3T3-L1 cells. Thus, MED14 constitutes a novel anchoring point between Mediator and the N-terminal domain of PPARγ that is necessary for functional PPARγ-mediated recruitment of Mediator and transactivation of PPARγ subtype-specific target genes.
The peroxisome proliferator-activated receptors (PPARs) belong to the subfamily of nuclear receptors that heterodimerize with retinoid X receptors (RXRs). To date, three different PPAR subtypes, PPARα, PPARδ, and PPARγ, have been identified. PPARγ is essential for triglyceride storage in adipose tissue and is a dominant regulator of adipogenesis (50). In addition, PPARγ regulates the expression of genes involved in differentiation, cellular signaling, and fatty acid handling in cells like macrophages (5, 55), vascular smooth muscle cells (17, 38), and osteoclasts (61). PPARα is responsible for the induction of genes involved in lipid catabolism and ketogenesis in the liver in response to fasting (26, 30) and is a general inducer of fatty acid oxidation in cells like brown adipocytes (2) and cardiomyocytes (1). PPARδ has many of the same target gene specificities as PPARα and is particularly important for the activation of fatty acid oxidation in muscle (63).
The PPAR-RXR heterodimers bind specifically to PPAR response elements (PPREs), which are direct repeats (DRs) of 5′-AGGTCA separated by one or, in a few cases, two nucleotide(s) (10, 25, 43). In the heterodimer, PPAR occupies the 5′ repeat, whereas RXR occupies the 3′ repeat (21). The activation of transcription by the PPARs relies on two activation domains, i.e., activation function one (AF-1) located in the N terminus and activation function two (AF-2) located in the C-terminal ligand binding domain (LBD). The activity of AF-2 is regulated by the binding of ligands, such as fatty acids, fatty acid derivatives, and a number of synthetic agonists and antagonists (64). The binding of agonists leads to a conformational change of the C-terminal domain, the AF-2 helix in particular, that favors interactions with a large number of transcriptional coactivators (41). The N-terminal AF-1 displays ligand-independent transactivation, and a few coactivators have been shown to interact directly with this domain (4, 13, 58). We have recently shown that the N terminus of PPARγ is specifically involved in the activation of a subset of PPARγ target genes that are implicated in fatty acid accumulation (3), emphasizing that the N-terminal domain contributes not only to overall receptor activity but also to receptor specificity.
Mediator is an evolutionarily conserved complex that serves as a regulatory link, relying primarily on protein-protein interactions, between DNA-bound transcription factors and the basal transcription machinery (37). The Mediator complex is essential for the regulation of several transcription factors that include members of the nuclear receptor family, SREBP, Sp1, p53, and NF-κB and is recruited to promoters in a gene- and transcription factor-specific manner by mechanisms dependent on different Mediator subunits (31). The Mediator subunit MED1/TRAP220/PBP/DRIP205 interacts through LXXLL motifs in a ligand-dependent manner with a number of nuclear receptors, such as estrogen receptor (ER) (70), thyroid hormone receptor (TR) (69), vitamin D receptor (VDR) (47), RXR (68), and PPARα and -γ (11, 57, 68, 71). MED1 is also necessary for the activity of other transcription factors, such as the GATA family and C/EBPβ (32, 51), but is not required for transcriptional activation by VP16 and p53 (36). Knockout (KO) of MED1 is embryonic lethal, and primary mouse embryonic fibroblasts (MEFs) from MED1 KO mice have impaired cell growth and cell cycle progression. MED1 depletion abrogates TR and ER transactivation (36, 70), and it has been reported that the liver-specific KO of MED1 impairs the ligand-dependent activation of PPARα target genes (24). Furthermore, MEFs derived from MED1 KO embryos show reduced PPARγ-mediated adipogenesis (12). Interestingly, however, it was recently shown that PPARγ can induce adipogenesis independently of the reported LXXLL motifs of MED1 (11), suggesting that the Mediator complex can be recruited to PPARγ target promoters independently of MED1. Whether this occurs through direct interaction of PPARγ with other subunits of the Mediator complex or through interaction with other transcription factors occupying PPARγ-responsive promoters remains to be shown.
Several potential mechanisms for MED1-independent Mediator recruitment to nuclear receptors have been suggested. Retinoic acid receptor (RAR) interacts with MED25/ARC92, which interacts with MED1 and, potentially, the rest of the Mediator complex (28). Cell cycle and apoptosis regulator 1 (CCAR1) interacts with the coiled-coil coactivator (CoCoA), which binds the p160 family of coactivators anchored to nuclear receptor LBDs through LXXLL motifs, and CCAR1 depletion impairs Mediator recruitment to glucocorticoid receptor (GR)- and ER-responsive promoters (27). MED14 interacts with the N terminus of GR (20), and depletion of MED14 results in reduced activation of some but not all promoters by GR (7). Interestingly, some MED14-dependent GR target promoters can be activated independently of MED1, whereas MED1-dependent promoters are activated independently of MED14 (7). These findings indicate that different promoters may have different critical requirements for Mediator components, a notion which is supported by the results of genetic studies in yeast (59).
Here, we use a previously described adenovirus (Ad)-based PPAR expression system (42) to acutely express PPARγ in cells depleted of different Mediator subunits. We show that depletion of MED1 affects neither PPARγ-dependent activation of target genes nor PPARγ-dependent recruitment of Mediator to target promoters. Using a small interfering RNA (siRNA)-based screen, we identify MED14 as a critical component for PPARγ-dependent transactivation and Mediator recruitment. We show that MED14 interacts with the N-terminal domain of PPARγ in a ligand-independent manner both in vitro and in cell culture. Interestingly, knockdown of MED14 leads to reduced PPARγ-mediated activation of a subset of target genes involved in fatty acid storage. The results of chromatin immunoprecipitation (ChIP) assays demonstrate that recruitment of PPARγ, MED6, MED8, and RNA polymerase II (RNAPII) to the Fabp4 promoter is impaired as a result of reduced MED14 levels. In keeping with the importance of MED14 for PPARγ activity, we show that MED14 knockdown leads to impaired adipogenesis of 3T3-L1 cells.
MATERIALS AND METHODS
Cell culture, adipocyte differentiation, and retrovirus and adenovirus transduction.
Phoenix cells, 293T cells, and mouse embryonic fibroblasts (MEFs) derived from wild-type (WT) and MED1 KO embryos were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco). Mouse AML-12 hepatocytes (ATCC CRL-2254) were cultured in Ham's nutrient mixture F-12-DMEM (1:1) with 2.5 mM l-glutamine, 1.2 g/liter sodium bicarbonate, 15 mM HEPES, and 0.5 mM sodium pyruvate (Gibco) supplemented with 10% FBS (Gibco) (5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml selenium [Sigma-Aldrich]) and 0.1 μM dexamethasone (Sigma-Aldrich). Retrovirus expressing the coxsackie adenovirus receptor (CARΔ1) were generated using Phoenix cells, and retroviral transduction of WT and KO MEFs was performed as described previously (42). Adenoviral transduction of AML-12 cells and MEFs by adenovirus expressing hemagglutinin (HA)-tagged PPARγ2 (AdHA-PPARγ2) and AdHA-PPARγ-CDE (expressing the HA-tagged PPARγ C, D, and E domains) was performed as previously described (3, 42). In short, 80- to 90%-confluent cells were incubated with adenovirus for 2 h, after which the medium was changed for medium containing PPARγ ligand (1 μM rosiglitazone). Cells were harvested for protein, mRNA, and ChIP analyses after another 6 h and for fatty acid oxidation and mRNA analyses after 22 h. 3T3-L1 cells were cultured and differentiated into adipocytes as previously described (19).
Cloning of shRNA, lentivirus production, and transduction.
Oligonucleotides coding for short hairpin RNA (shRNA) against MED1 and MED14 were designed with HpaI and XhoI overhangs using an algorithm described by Reynolds et al. (49). The sequences of the shRNA oligonucleotides were as follows: MED1#1 sense, 5′-TGCAGAAGGCTCTCAAAGTATTCAAGAGATACTTTGAGAGCCTTCTGCTTTTTTC; shRNA MED1#3 sense, 5′-TGGACTTCAGTATTATATCATTCAAGAGATGATATAATACTGAAGTCCTTTTTTC; shRNA LacZ sense, 5′-TGAAGGCCAGACGCGAATTATTCAAGAGATAATTCGCGTCTGGCCTTCTTTTTTC; shRNA MED14#1 sense, 5′-TGCAATTCGCTTATTAAAGATTCAAGAGATCTTTAATAAGCGAATTGCTTTT TTC; and shRNA MED14#2 sense, 5′-TGACCCTAGTTCTCCATATATTCAAGAGATATATGGAGAACTAGGGTCTTTTTTC.
The oligonucleotides were annealed and cloned into HpaI-XhoI-digested pSicoR-GFP or pSico-puro (60). Lentivirus was produced using a 3rd-generation packaging system that was previously described (9). In short, 80- to 90%-confluent 293T cells were cotransfected with pSicoR-GFP and each of the lentiviral packaging vectors pMDLg/pRRE, pRSV-Rev, and pMD2.G using polyethylenimine. Medium containing lentivirus was harvested after 48 h, centrifuged to remove cellular debris, and passed through a 0.45 uM polyvinylidene difluoride (PVDF) filter. AML-12 cells or 3T3-L1 cells at 50% confluence were transduced with a 1:1 dilution of lentivirus supernatant and fresh growth medium in the presence of 6 μg/ml Polybrene (Sigma-Aldrich). The medium was replaced with normal growth medium the day after transduction. The lentiviral transduction efficiency was estimated by the level of green fluorescent protein (GFP) expression using microscopy.
RNA purification and quantitative RT-PCR.
RNA was purified using Trizol (Invitrogen) according to the manufacturer's protocol. Purified RNA was subjected to DNase I (Invitrogen) treatment, and cDNA was synthesized using random deoxynucleic acid hexamers and reverse transcriptase (First-Strand kit; Invitrogen) as previously described (42). cDNA was quantified by real-time reverse transcription (RT)-PCR (MX-3000; Stratagene) using SYBR green master mix and primers recognizing the following transcripts: perilipin (Plin), cell death-inducing DFFA-like effector c (Cidec), adipose differentiation-related protein (Adrp), aquaporin 7 (Aqp7), lipoprotein lipase (Lpl), enoyl coenzyme A, hydratase/3-hydroxyacyl coenzyme A dehydrogenase (Ehhadh), pyruvate dehydrogenase kinase, isozyme 4 (Pdk4), fatty acid binding protein 4 (Fabp4), Cd36, carnitine palmitoyltransferase 2 (Cpt2), long-chain acyl coenzyme A dehydrogenase (Lcad), acyl coenzyme A oxidase 1 (Acox1), glucocorticoid-induced leucine zipper (Gilz), CCAAT/enhancer binding protein beta (C/EBPβ), CCAAT/enhancer binding protein delta (_C/EBP_δ), peroxisome proliferator-activated receptor gamma (_PPAR_γ), and general transcription factor IIB (Gtf2b) (3, 42). Experiments were performed in triplicates or duplicates as indicated in the figure legends.
Immunoblotting.
Adenovirally transduced cells were harvested in a hypotonic sodium dodecyl sulfate (SDS) sample buffer as described previously (42) and separated by SDS-polyacrylamide gel electrophoresis (PAGE). Proteins were blotted onto PVDF membranes (Millipore Corp.) and probed with specific antibodies. The following primary antibodies were used: anti-Flag (M2; Sigma-Aldrich), anti-PPAR (sc-7273; Santa Cruz), anti-TFIIB (sc-225; Santa Cruz), anti-TRAP220 (sc-8998; Santa Cruz), anti-MED14 (36), and anti-HA (42). The secondary antibodies used were horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (P0447; Dako) and swine anti-rabbit immunoglobulin G (P0339; Dako). Enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech) was used for detection.
Cloning of Fabp4 promoter constructs and transient transfections.
The pGL3basic-Fabp4(−7900/+1) vector has been described previously (58), and the truncated Fabp4 upstream promoter construct, pGL3basic-Fabp4(−4500/+1), was generated by digesting pGL3basic-Fabp4(−7900/+1) with BglII and cloning the BglII-BglII 4.5-kb upstream promoter sequence into pGL3basic. A point mutation of the proximal DR-1 element was introduced into the Fabp4 promoter by PCR-mediated overlap extension as described by Heckman and Pease (18), creating a unique ApaI site. The PCR product containing the mutated sequence was digested with PflMI and NarI and cloned into pGL3basic. Mutation was confirmed by sequencing. For Fabp4 promoter analysis, NIH 3T3 cells were transfected using MetafectenePro (Biontex) according to the manufacturer's protocol with the above-mentioned pGL3-based luciferase reporter constructs together with pShuttle-CMV-PPARγ2 (42), pShuttle-CMV-RXRα (48), and simian virus 40 (SV40) β-galactosidase (Promega) in the presence or absence of 1 μM rosiglitazone and 200 nM LG100268. The luciferase and β-galactosidase activities were measured at 24 h posttransfection. For MED14 coactivation analysis, MEFs were transfected with pM-PPARγ2-AB (expressing the PPARγ2 A/B domain fused to the Gal4 DNA binding domain), p4xUAS-Luc, pcDNA3-MED14 (15), and SV40 β-galactosidase. The luciferase and β-galactosidase activities were measured 24 h posttransfection.
In vitro translation and GST pulldown.
MED1 and MED14 were translated in vitro from pSG5-MED1 (71) and pcDNA3-MED14 in the presence of [35S]methionine using a TnT coupled transcription/translation system (Promega). The various domains of mouse PPARγ2 were PCR cloned into pGEX (GE Healthcare), and the glutathione _S_-transferase (GST) fusion proteins were expressed in Escherichia coli, coupled to glutathione Sepharose 4B (GE Healthcare) according to the manufacturer's protocol, washed in phosphate-buffered saline (PBS), and stored in buffer at 4°C. The amounts of coupled GST fusion proteins were evaluated by SDS-PAGE, and equal amounts were incubated with _in vitro_-translated MED1 or MED14 in NET-N buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 10 mM EDTA, 0.5% NP-40, 10 mM dithioerythritol [DTE], 1% milk, and protease inhibitors) in the absence or presence of 1 μM rosiglitazone for 2 h at 4°C. Bound MED14 and MED1 were washed three times in NET-N buffer without milk, followed by incubation in SDS-PAGE sample buffer for 5 min at 95°C. Proteins were separated by SDS-PAGE, and enriched MED1 and MED14 were evaluated using a Typhoon TRIO scanner (Amersham Biosciences).
Coimmunoprecipitation.
293T cells were transfected with pShuttle-CMV-PPARγ2 (42) or pShuttle-CMV-PPARγ-CDE (3) together with pcDNA3-Flag-MED14 (46) using polyethylenimine. Cells were lysed in IP buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 0.1% sodium deoxycholate, and protease inhibitors) followed by 4 h of incubation with anti-Flag agarose (Sigma-Aldrich) at 4°C. Immunoprecipitate was washed three times in IP buffer and two times in Tris-EDTA buffer and eluted with Flag peptide (Sigma-Aldrich). Eluates were incubated in SDS-PAGE sample buffer for 5 min at 95°C, and proteins were separated by SDS-PAGE. For immunoprecipitation of endogenous MED14 in 3T3-L1 adipocytes, cells were washed in buffer A (20 mM Tris-HCl, pH 7.5, 20 mM NaCl, 0.2 mM EDTA) and subsequently lysed in buffer A with 0.04% NP-40 for 10 min to produce intact nuclei. NP-40 was removed by washing in buffer A, after which the proteins from the nuclei were extracted using a hypertonic buffer (20 mM Tris-HCl, pH 7.5, 25% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, and 400 mM NaCl) for 30 min at 4°C. Nuclear extracts were diluted in 1.5 volumes of buffer B (20 mM Tris-HCl, pH 7.5, 0.2 mM EDTA, and 1.67% NP-40). Nuclear debris was removed by centrifugation, and nuclear extracts were incubated with antibody against PPARγ (sc-7196; Santa Cruz) overnight. Immunocomplexes were recovered by using protein A beads (GE Healthcare) and washed five times in IP buffer (20 mM Tris-HCl, pH 7.5, 10% glycerol, 0.2 mM EDTA, 150 mM NaCl, and 1% NP-40). Washed immunocomplexes were incubated in SDS-PAGE sample buffer for 5 min at 95°C, and proteins were separated by SDS-PAGE.
ChIP.
Chromatin immunoprecipitation (ChIP) was performed as previously described (42) with minor modifications. In short, cells were cross-linked with 1% formaldehyde for 10 min. Cross-linking was stopped by the addition of glycine to a final concentration of 0.125 M. Cross-linked cells were removed from the plate, washed twice in cold PBS, and resuspended in lysis buffer (1% Triton X-100, 0.1% SDS, 150 mM NaCl, 2 mM EDTA, 20 mM HEPES, pH 7.6, and protease inhibitors). Lysed cells were sonicated by using a Bioruptor (Diagenode) according to the manufacturer's protocol, and chromatin was immunoprecipitated with antibodies against HA (42), TATA box-binding protein (TBP) (42), RNAPII (Covance), MED6, MED8 (sc-103619; Santa Cruz), or TRAP220 (sc-5334; Santa Cruz) overnight at 4°C in the presence of protein A or G beads (GE Healthcare) at 4°C. Beads were washed twice with IP wash buffer 1 (1% Triton X-100, 0.1% SDS, 150 mM NaCl, 2 mM EDTA, 20 mM HEPES, pH 7.6), once with IP wash buffer 2 (1% Triton X-100, 0.1% SDS, 500 mM NaCl, 2 mM EDTA, 20 mM HEPES, pH 7.6), once with IP wash buffer 3 (0.25 M LiCl, 1% NP-40, 1% deoxycholic acid, 1 mM EDTA, 20 mM HEPES, pH 7.6), and finally, twice with HEPES-EDTA buffer, all at 4°C. DNA-protein complexes were eluted with 1% SDS and 0.1 M NaHCO3 and de-cross-linked by adding 0.2 M NaCl and incubating overnight at 65°C. DNA was purified by phenol-chloroform extraction, precipitated in ethanol with sodium acetate, and dissolved in water. DNA enrichment was quantified by real-time PCR (MX-3000; Stratagene) using SYBR green Master Mix (Sigma-Aldrich) and the following primers spanning the Fabp4 PPREs at −5500 (42): Fabp4 −2500 (5′-GGATGGCCTTGGACTCACTC and 5′-AGAAACACCACAGGAGGCTGA), Fabp4 −200 (5′-CATTGCCAGGGAGAACCAA and 5′-TCCTTCATGACCAGACCCTGT), Fabp4 +500 (5′-CAGGTGAACCCGCAAGAAAG and 5′-GCTTGGCAAAGAAGGCCAC), and Fabp4 +3500 (5′-GTGCAGAAGTGGGATGGAAAG and 5′-TGCAGCGTAACTCACCACCA). Numbers indicate positions in bp relative to the transcriptional start site (TSS).
siRNA transfection.
MEFs and 3T3-L1 preadipocytes were transfected with 20 to 50 nM siRNA against various Mediator subunits using RNAiMAX (Invitrogen) as previously described (3). In short, cells were plated at a density of 30 to 50% and subsequently transfected with siRNA in Opti-MEM (Gibco) without antibiotics. The medium was replaced with normal growth medium after 6 h. At 24 h posttransfection, cells were transduced with HA-PPARγ2 adenovirus or differentiated into adipocytes. siRNA was ordered through the predesigned Mission siRNA library (Sigma-Aldrich). Sequences are available upon request.
RESULTS
PPARγ activates target genes independently of MED1.
MED1 is known to mediate strong ligand-dependent interactions between the Mediator complex and many nuclear receptors, including members of the PPAR family. In keeping with this, it was demonstrated that PPARγ-mediated adipogenesis depends on MED1 (12). However, it was shown recently that PPARγ-induced adipogenesis in MEFs is dependent exclusively on a conserved MED1 N-terminal domain that lacks the LXXLL motifs and does not interact with PPARγ (11). These results suggest that the Mediator complex can be recruited to PPARγ or PPARγ-activated promoters by MED1-independent mechanisms but that the N-terminal part of MED1 plays a key role in PPARγ-induced adipogenesis.
We have previously described an adenoviral PPAR expression system to investigate mechanisms involved in acute target gene activation by the PPAR subtypes (42). Using this system, we investigated the requirement of MED1 for acute target gene activation by PPARγ. MEFs derived from WT and MED1 KO embryos (23) were initially transduced with the coxsackie adenovirus receptor (CAR) to ensure efficient adenoviral uptake (results not shown). PPARγ was acutely overexpressed to similar levels in WT and MED1 KO MEFs by transduction with recombinant adenovirus expressing HA-tagged PPARγ2 (Fig. 1A). The transcription of well-documented PPARγ target genes, such as Fabp4, Cidec, Aqp7, and Cd36, was potently induced upon forced PPARγ expression in MEFs derived from wild-type mice (Fig. 1B), in agreement with the results in our previous report (42). Notably, forced expression of PPARγ2 in MEFs derived from MED1 KO mice resulted in similar induction of PPARγ target genes, thereby demonstrating that PPARγ has the ability to activate target genes independently of MED1 (Fig. 1B; see also Figure 1 posted at www.sdu.dk/susannemandrup/mcb2010). The level to which some target genes were induced differed between the two MEF cell lines (e.g., Aq7 and Cidec were induced to a higher level in the absence of MED1) both with and without agonist (Fig. 1B) and in time course experiments (see Figure 1 at the URL listed above). This is most likely due to indirect effects as a result of the MED1 knockdown or the fact that two independently derived cell lines were used.
FIG. 1.
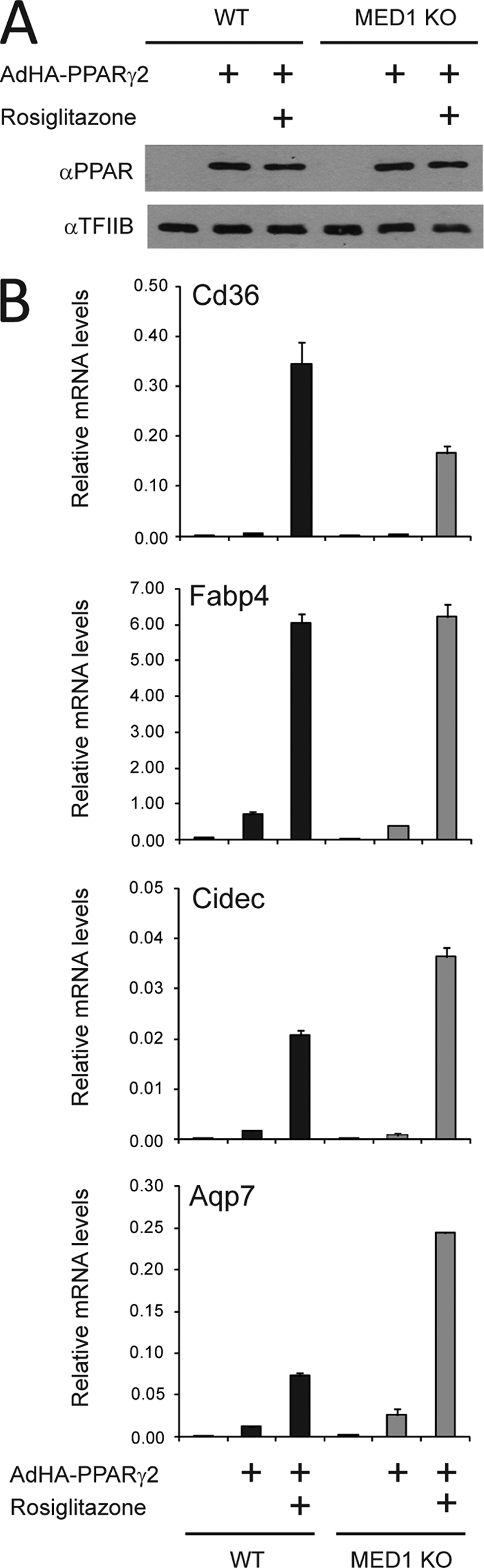
PPARγ activates target genes independently of MED1. WT and MED1 KO MEFs were transduced with AdHA-PPARγ2 in the presence or absence of 1 μM rosiglitazone. Whole-cell lysates and RNA were prepared 8 h after transduction. (A) Proteins from whole-cell extracts were separated by SDS-PAGE and analyzed by immunoblotting with antibodies against PPAR and TFIIB. α, anti. (B) RNA was quantified by real-time PCR with primers against Gtf2b, Fabp4, Cd36, Cidec, or Aqp7. Threshold cycle (CT) values were normalized to CT values from Gtf2b and visualized as relative mRNA levels. Error bars indicate the ranges of the results of experiments performed in duplicate. Results are representative of a minimum of three independent experiments.
Interestingly, similar experiments carried out with PPARα showed that MED1 was also dispensable for acute activation of target genes (see Figure 2A and B posted at www.sdu.dk/susannemandrup/mcb2010) and for the induction of fatty acid β-oxidation (see Figure 2B at the URL listed above) in response to forced PPARα expression. In order to investigate whether the MED1-independent activation of target genes by PPARγ and PPARα was specific for fibroblasts, we knocked down MED1 in AML-12 hepatocytes using lentivirus-delivered shRNA (see Figure 3A at the URL listed above). Subsequent transduction with adenovirus expressing PPARγ2 or PPARα (see Figure 3B at the URL listed above) showed that PPAR-mediated transactivation in hepatocytes is also independent of MED1 (see Figure 3C and D at the URL listed above). Thus, acute activation of transcription by PPARγ and PPARα is mediated through mechanisms that are independent of the presence of MED1.
FIG. 2.

PPARγ occupies the proximal promoter of the Fabp4 gene indirectly. (A) Representation of the Fabp4 gene loci with the relative position of the reported PPREs. Primer pairs for ChIP analysis are indicated below the diagram. Numbers refer to positions relative to TSS. (B) ChIP-PCR confirmation of PPARγ occupancy of the proximal promoter of the Fabp4 gene. ChIP was performed during adipogenesis of 3T3-L1 adipocytes. PPAR occupancy of the Fabp4 enhancer and proximal promoter was investigated by real-time PCR with the indicated primers. (C) NIH 3T3 cells were transfected with the indicated luciferase (Luc) reporter constructs [pGL3basic-Fabp4(−4500/+1), pGL3basic-Fabp4(−7900/+1)DR1mut, pGL3basic-Fabp4(−7900/+1), and pGL3basic] together with pShuttle-CMV-PPARγ2, pShuttle-CMV-RXRα, and SV40 β-galactosidase in the presence or absence of 1 μM rosiglitazone (Rosi) and 200 nM LG100268. Luciferase and β-galactosidase activities were measured 24 h posttransfection, and luciferase activity was plotted as relative luciferase activity normalized to β-galactosidase activity. Data are represented as means ± standard errors of three independent experiments performed in triplicate. DMSO, dimethyl sulfoxide.
FIG. 3.

PPARγ-induced recruitment of Mediator at the Fabp4 promoter and enhancer is independent of MED1. WT and MED1 KO MEFs were transduced with adenovirus without an insert (empty) or AdHA-PPARγ2 in the presence of 1 μM rosiglitazone (Rosi). Whole-cell extracts and chromatin were prepared 8 h after transduction. (A) Proteins from whole-cell extracts were separated by SDS-PAGE and analyzed by immunoblotting with antibodies against PPAR and TFIIB. α, anti. (B) Representation of the Fabp4 gene loci and of the relative positions of the primer pairs for ChIP-PCR analysis are indicated below the diagram. (C and D) ChIP was performed using antibodies against the HA tag, TBP, RNAPII, MED1, MED6, and MED8. Enriched DNA was quantified using real-time PCR with the primer pairs (shown in panel B) indicated and plotted as the amount of DNA recovered relative to the amount of quantified DNA from input chromatin. Error bars indicate the ranges of the results of experiments performed in duplicates. Results are representative of a minimum of two independent experiments.
PPARγ recruits the Mediator complex to promoters independently of MED1.
To investigate Mediator assembly on PPARγ-responsive promoters in the absence of MED1, we chose to focus on the well-described PPARγ-responsive gene Fabp4/A-FABP/aP2. Activation of the Fabp4 gene by PPARγ is mediated through direct PPARγ interactions with two PPREs located approximately 5,500 bp upstream of the transcriptional start site (TSS) (14, 54). We have shown previously that acute activation of Fabp4 by PPARγ is associated with PPARγ and MED1 recruitment to the Fabp4 enhancer PPREs within 1 h after forced PPARγ protein expression in fibroblasts (42). Interestingly, in a global PPARγ-RXR genome interaction profile, we have shown that PPARγ and RXR occupy not only the enhancer PPRE but also the proximal promoter in differentiated adipocytes (43) (see Figure 4 posted at www.sdu.dk/susannemandrup/mcb2010). We applied chromatin immunoprecipitation (ChIP) against PPARγ, quantified enriched DNA by quantitative PCR using primers across the Fabp4 promoter (Fig. 2A), and confirmed that PPARγ specifically interacts with the enhancer and proximal promoter during 3T3-L1 adipocyte differentiation (Fig. 2B). The proximal promoter contains a putative DR-1, suggesting a direct PPARγ/RXR interaction with the proximal promoter. To investigate whether this DR-1 is a functional PPRE, we constructed a luciferase reporter controlled by a Fabp4 5′ upstream region that includes both the proximal promoter DR-1 and the well-established enhancer PPREs that lie 5.5 kb upstream of the TSS. As reported previously (58), PPARγ and RXRα activated this reporter in NIH 3T3 cells in a ligand-dependent manner (Fig. 2C). Deletion of the enhancer PPREs completely abolished ligand-dependent activation of the promoter, whereas mutation of DR-1 in the proximal promoter had no effect (Fig. 2C). These results indicate that the DR-1 is not a functional PPRE and that the weak binding of PPARγ to the proximal promoter is probably due to indirect PPARγ binding. Such indirect binding of PPARγ to the proximal promoter is likely to be a consequence of juxtaposition of the enhancer with the promoter.
FIG. 4.
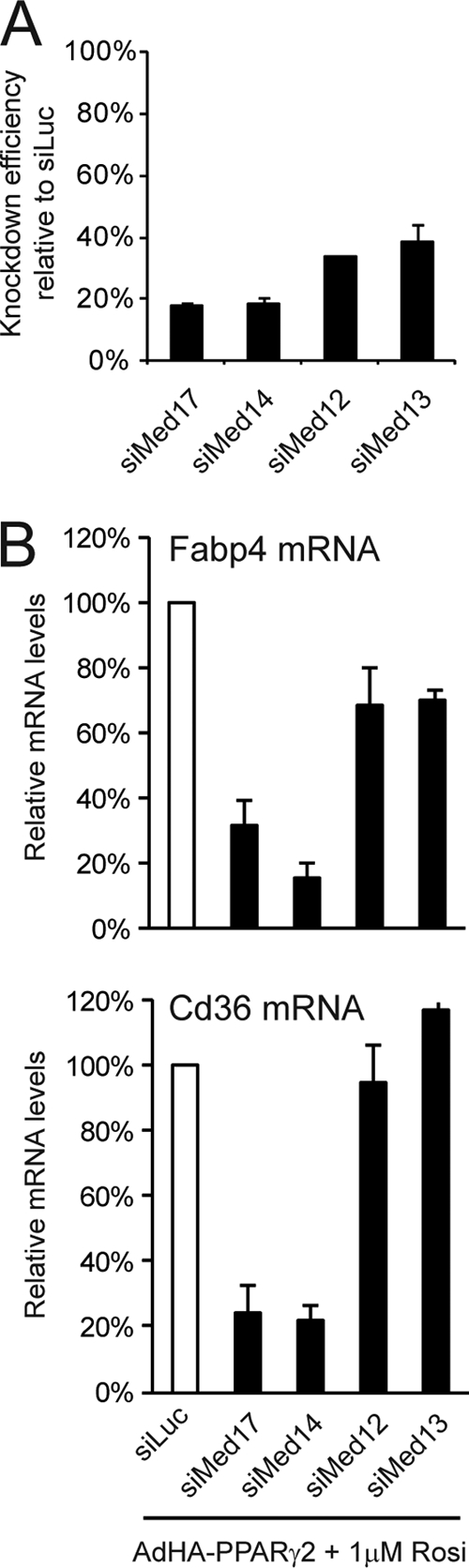
Knockdown of specific Mediator subunits reduces the transcriptional activity of PPARγ. WT MEFs were transfected with siRNA (20 nM) against luciferase (Luc), MED17, MED14, MED12, or MED13 for 24 h, followed by transduction with AdHA-PPARγ2 in the presence of 1 μM rosiglitazone (Rosi). (A) Knockdown efficiency of the different siRNAs against Mediator subunits relative to results for cells transfected with siRNA against luciferase. RNA expression levels were quantified by real-time PCR, normalized to the level of Gtf2b, and visualized as the percentage of the expression level in cells transfected with siRNA against luciferase. (B) RNA levels of the PPARγ target genes Cd36 and Fabp4 relative to that of Gtf2b were determined by real-time PCR and visualized as the percentage of the expression level in cells transfected with siRNA against luciferase. Error bars indicate the ranges of the results of experiments performed in duplicates. Results are representative of a minimum of three independent experiments.
In keeping with the observations for 3T3-L1 adipocytes, acute expression of PPARγ2 in WT MEFs resulted in PPARγ occupancy of the enhancer PPREs, as well as the proximal promoter (Fig. 3C). MED6 and MED8, which are part of the Mediator head module, and MED1 showed similar patterns of occupancy on the Fabp4 locus (Fig. 3C). In addition, we observed that TATA box-binding protein (TBP) and RNA polymerase II (RNAPII) occupied not only the proximal promoter but also the enhancer in response to PPARγ expression, thereby supporting the model that PPARγ induces juxtaposition of the −5,500-bp-upstream enhancer with the proximal promoter of Fabp4 (Fig. 3C). Acute expression of PPARγ to a similar level in MEFs depleted of MED1 (Fig. 3A) resulted in the recruitment of PPARγ, TBP, RNAPII, MED6, and MED8 to both enhancer and proximal promoter regions at levels similar to those observed in the WT MEFs (Fig. 3D). In keeping with the absence of MED1, we observed no recruitment of MED1 to the Fabp4 promoter or enhancer. In summary, these results show that MED1 is dispensable for PPARγ-induced Mediator assembly on the Fabp4 enhancer and proximal promoter and further indicate that enhancer-promoter interactions can occur in the absence of MED1.
Knockdown of MED14 interferes with PPARγ-mediated transactivation.
Like PPARγ, GR has been shown to interact directly with MED1 in a manner dependent on the two LXXLL motifs, and the activity of GR is only slightly reduced when the LXXLL motifs are mutated (6). In addition, only a subset of GR target genes requires the presence of MED1 in order to be activated by dexamethasone (6, 7), clearly demonstrating that GR is able to activate target genes independently of MED1. Interestingly, the N-terminal domain of GR has been shown to interact with the Mediator subunit MED14 (20), and a reduced level of MED14 has been demonstrated to obstruct the activation of MED1-independent target genes by GR (6, 7). We speculated that MED14 might also be involved in MED1-independent activation of target genes by PPARγ, and in order to investigate this, we knocked down MED14 using siRNA prior to forced PPARγ expression. Knockdown of the essential Mediator subunit MED17, which has been shown to be necessary for Mediator function in yeast (34), served as a positive control for the necessity of Mediator. Knockdowns of MED12 and MED13 served as putative negative controls, as these subunits have been found to act primarily as repressive subunits of the Mediator complex (52). Following validation of the knockdown at the mRNA level (Fig. 4A), we analyzed the ability of PPARγ to activate Fabp4 and Cd36. In keeping with the importance of MED17 in the Mediator complex, we observed that the PPARγ-mediated activation of Cd36 and Fabp4 was reduced to 30% following MED17 knockdown, whereas knockdowns of MED12 or MED13 only modestly reduced the ability of PPARγ to activate Fabp4 and had no effect on PPARγ activation of Cd36. Interestingly, knockdown of MED14 resulted in a reduction of PPARγ-mediated expression of Fabp4 and Cd36 to 20% of the induced level in control cells. These results indicate that the Mediator complex per se is required for PPARγ-mediated transcriptional activation of endogenous target genes and that the presence of MED14 is necessary for the full transcriptional activity of PPARγ.
The N-terminal domain of PPARγ interacts with MED14.
To investigate whether MED14 and PPARγ interact directly, we performed a GST pulldown with full-length PPARγ2 and _in vitro_-translated MED14. As shown in Fig. 5B, PPARγ interacts with MED14 in a ligand-independent manner, in contrast to the ligand-dependent interaction between MED1 and PPARγ. Interestingly, and in keeping with the ligand independency, the interaction between PPARγ and MED14 relies on the N-terminal A/B domain of PPARγ, whereas the interaction with MED1 depends entirely on the LBD (Fig. 5C). To confirm the interaction between MED14 and PPARγ and the importance of the A/B domain in a cellular context, we performed coimmunoprecipitation experiments in 293T cells after ectopically expressing PPARγ2 or PPARγ-CDE in combination with Flag-tagged MED14. The results showed that MED14 and PPARγ interact in cells and that this interaction is strictly dependent on the PPARγ A/B domain (Fig. 5D). Importantly, MED14 can also be immunoprecipitated with a PPARγ antibody in 3T3-L1 adipocytes, demonstrating that endogenous PPARγ and MED14 interact in living cells (Fig. 5E). In keeping with this, MED14 was able to coactivate a Gal4-PPARγ A/B domain fusion construct in transient transfections (Fig. 5F). Thus, these results show that MED14 interacts directly with the PPARγ2 A/B domain and present MED14 as a potential candidate for anchoring the Mediator complex to PPARγ-responsive promoters in a MED1-independent manner.
FIG. 5.

MED14 interacts with the N-terminal domain of PPARγ. (A) PPARγ consists of four functional domains. The N-terminal A/B domain (amino acids [aa] 1 to 138) contains the ligand-independent transactivation function, the C (aa 138 to 203) and D (aa 203 to 279) domains are involved in DNA binding, and the E domain (aa 279 to 505) constitutes the ligand binding domain and has ligand-dependent transactivity. (B) MED14 interacts with GST-tagged full-length PPARγ2 in a ligand-independent manner. _In vitro_-translated and 35S-labeled MED14 and MED1 were incubated with immobilized GST-tagged PPARγ2 in the presence or absence of 1 μM rosiglitazone (Rosi). Bound proteins were separated by SDS-PAGE. (C) MED14 specifically interacts with the N-terminal domain of PPARγ2 _in vitro. In vitro_-translated and 35S-labeled MED14 and MED1 were incubated with immobilized GST-tagged PPARγ domains (represented in panel A) in the presence of 1 μM rosiglitazone. Bound proteins were separated by SDS-PAGE. Similar amounts of the GST-tagged PPARγ domains were verified by Coomassie staining. (D) MED14 interaction with PPARγ in cells is dependent on the N-terminal domain of PPARγ. Flag-tagged MED14, PPARγ2, or N-terminally truncated PPARγ was transfected into 239T cells, and cellular extracts were immunoprecipitated with antibody against the Flag epitope. Immunoprecipitated proteins were separated by SDS-PAGE and detected by immunoblotting with antibodies against Flag and PPAR. Amounts of 5% of the cellular extracts from transfected cells were immunoblotted with PPAR antibody to verify similar levels of expression of full-length and truncated PPARγ. WB, Western blot. (E) Endogenous PPARγ interacts with endogenous MED14 in 3T3-L1 adipocytes. Nuclear extracts from 3T3-L1 day 6 adipocytes were used for immunoprecipitation with antibody against HA or PPARγ. Immunoprecipitates were separated by SDS-PAGE (right) together with 10% input nuclear extract (left) and immunoblotted with antibodies against MED14 or PPARγ. α, anti. (F) MED14 coactivates the N-terminal transactivation domain of PPARγ. MEFs were transfected with a Gal4-responsive luciferase reporter (UAS-Luc) and a plasmid encoding the N-terminal A/B domain of PPARγ2 fused to the Gal4 DNA binding domain in the presence or absence of an expression plasmid encoding MED14. SV40 β-galactosidase was used for normalization. Error bars represent standard deviations of the results of experiments performed in triplicates. All results are representative of a minimum of two independent experiments.
MED14 is necessary for full PPARγ transcriptional activity of a subset of endogenous target genes.
MED14 is necessary only for the activation of a subset of GR target genes in response to dexamethasone (7). In order to investigate whether some genes are activated by PPARγ independently of MED14, we analyzed the expression level of a range of PPARγ target genes following MED14 knockdown and forced PPARγ2 expression in the presence and absence of ligand. Knockdown of MED14 using siRNA followed by equal PPARγ2 overexpression (Fig. 6A) resulted in significantly reduced PPARγ-mediated activation of genes involved in lipid storage, including Fabp4, Cd36, and Cidec, both in the absence and in the presence of agonist (Fig. 6B). In contrast, knockdown of Med14 resulted in only a modest reduction of PPARγ-mediated induction of target genes, such as Lpl, Acox1, and Cpt-2 (Fig. 6B). Interestingly, we have recently shown that the N-terminal transactivation function of PPARγ is specifically required for the full activation of PPARγ-selective genes involved in lipid storage, such as Fabp4, Cd36, Plin, Lpl, and Cidec (3). Our results here show that these genes require MED14 for PPARγ transactivation. In contrast, PPARγ activation of genes such as Acox1, Pex11a, Cpt-2, and Irs2 did not require the N-terminal domain (3) and is much less sensitive to MED14 knockdown. Notably, PPARγ-mediated activation of Lpl, which is PPARγ selective and dependent on the PPARγ N terminus (3), does not require the presence of Med14, indicating that on some PPARγ-selective target promoters, MED14 is not important for transactivation.
FIG. 6.

Knockdown of MED14 compromises the ability of both full-length and A/B domain-truncated PPARγ to activate a subset of target genes. WT MEFs were transfected with siRNA (50 nM) against luciferase (Luc) or MED14 and, 24 h thereafter, transduced with AdHA-PPARγ2 (expressing full-length PPARγ2 [amino acids 1 to 505]) or AdHA-PPARγ-CDE (expressing truncated PPARγ [amino acids 138 to 505]), respectively, in the absence or presence of 1 μM rosiglitazone (Rosi). (A and C) Eight hours after adenoviral transduction, whole-cell extracts were prepared and analyzed by immunoblotting with antibodies against PPAR and TFIIB. α, anti. (B and D) RNA was prepared 8 h after transduction, and RNA levels of Gtf2b, Fabp4, Cidec, Cd36, Lpl, Acox1, and Cpt2 were determined by real-time PCR, normalized to the levels of Gtf2b, and visualized as relative mRNA levels. Error bars represent standard deviations of results of experiments performed in triplicates. Results are representative of a minimum of three independent experiments.
In order to investigate whether the function of MED14 in PPARγ transactivation is dependent on the interaction with the PPARγ A/B domain, we expressed full-length PPARγ and N-terminally truncated PPARγ in MEFs transfected with siRNA against luciferase or MED14 (Fig. 6C). Interestingly, we observed that knockdown of MED14 reduces the transcriptional activity of the truncated PPARγ, as well as that of full-length PPARγ (Fig. 6D). These data indicate that MED14 is not only involved as a PPARγ-tethering component of Mediator but is also important for Mediator activity per se on PPARγ-responsive genes.
MED14 is required for Mediator recruitment to a PPARγ-responsive promoter.
To evaluate the effect of reduced MED14 levels on the assembly of Mediator on the Fabp4 enhancer and promoter, MEFs were transfected with siRNA against MED14 and then transduced with adenovirus expressing HA-PPARγ2 for 8 h to obtain equal levels of PPARγ expression (Fig. 7A). Subsequent ChIP analyses showed that knockdown of MED14 did not affect PPARγ recruitment to the enhancer PPRE; however, occupancy of the proximal promoter was significantly impaired, suggesting that MED14 is required for indirect association of PPARγ with the proximal promoter (Fig. 7B). Furthermore, reduced levels of MED14 resulted in depletion of MED1 on the enhancer and proximal promoter, thus indicating a functional interaction between these two subunits (Fig. 7B). This is supported by the results of a yeast Mediator protein interaction study, which demonstrated that MED14 interacts with MED1 and MED10 (16). Interestingly, reduced levels of MED14 did not change the occupancy of MED6 and MED8 on the enhancer PPRE (Fig. 7B), suggesting that the Mediator head module is recruited to the enhancer in the absence of MED1 and MED14. In contrast, MED6 and MED8 occupancy of the proximal promoter was reduced upon MED14 knockdown (Fig. 7B), indicating that Mediator recruitment to the TSS is dependent on MED14 interaction with PPARγ. In addition, a reduced level of MED14 resulted in depletion of RNAPII at the enhancer and proximal promoter (Fig. 7B). Thus, these results indicate that the presence of MED14 in the Mediator complex supports PPARγ-induced Mediator assembly at the enhancer and is required for juxtaposition of the enhancer and promoter and RNAPII recruitment (Fig. 7C). The observed PPARγ-induced recruitment of MED6 and MED8 to the enhancer in the absence of MED14 indicates that parts of the Mediator complex can be recruited to PPARγ by redundant mechanisms but that productive Mediator assembly is dependent on MED14.
FIG. 7.

MED14 is necessary for Mediator recruitment to the proximal promoter of Fabp4. WT MEFs were transfected with siRNA (50 nM) against luciferase or MED14 and, 24 h thereafter, transduced with AdHA-PPARγ2 in the presence of 1 μM rosiglitazone. Whole-cell extracts and chromatin were prepared 8 h after adenoviral transduction. (A) Proteins from whole-cell extracts were separated by SDS-PAGE and analyzed by immunoblotting with antibodies against PPAR and TFIIB. α, anti. (B) ChIP-PCR was used to determine the relative levels of occupancy of PPARγ, MED1, RNAPII, MED6, and MED8 in the Fabp4 locus. Relative levels of occupancy, i.e., levels recovered with ChIP-PCR from cells overexpressing PPARγ2 relative to levels recovered from untransduced cells, are indicated. The figure shows the mean results of two independent experiments, with the ranges indicated by error bars. (C) In cells with normal expression of MED14, Mediator is recruited to an enhancer occupied by PPARγ:RXR through direct interaction between MED14 and the N-terminal A/B domain of PPARγ and between MED1 and the ligand binding domain (LBD) of PPARγ. This facilitates juxtaposition of the enhancer with the proximal promoter and, subsequently, recruitment of RNAPII (Pol II) to the promoter. In the absence of MED14, residual Mediator can still be recruited to the enhancer; however, functional interaction with the proximal promoter and recruitment of RNAPII is compromised.
3T3-L1 adipogenesis requires MED14.
PPARγ is a key regulator of adipogenesis (50), and given that MED14 is necessary for the maximum activation of genes involved in lipid storage by PPARγ, we predicted that MED14 would also be involved in adipogenesis. We therefore constructed two lentiviral shRNA vectors to target MED14 in 3T3-L1 preadipocytes. Lentiviral delivery was visualized by GFP expression from the shRNA expression cassette (60), and the results showed that the majority of cells had been transduced with virus (Fig. 8A). The knockdown of MED14 expression at the RNA level was confirmed for the MED14#1 shRNA construct, whereas the MED14#2 construct did not result in any significant knockdown (Fig. 8B). Six days after transduction, cells were induced to differentiate, and the ability to undergo adipogenesis was assessed by the determination of lipid accumulation (Fig. 8C) and of the mRNA levels of adipocyte-specific genes (Fig. 8D). Lentiviral expression of LacZ or MED14#2 shRNA did not affect adipocyte differentiation, whereas knockdown of MED14 by MED14#1 shRNA significantly impaired adipocyte differentiation. The importance of MED14 for 3T3-L1 adipogenesis was further demonstrated by showing that siRNA-mediated knockdown of MED14 resulted in impairment of adipogenesis compared to that in cells expressing siRNA against luciferase (Fig. 9A; also see Figure 5 posted at www.sdu.dk/susannemandrup/mcb2010). For comparison, we knocked down MED12 and MED13, which are both dispensable for PPARγ transactivation in MEFs (Fig. 4B). Interestingly, whereas siRNA-mediated knockdown of MED13 did not interfere with normal differentiation, knockdown of MED12 abolished adipogenesis (see Fig. 5 at the URL listed above). Thus, MED12 is likely to be necessary for the activity of other transcription factors involved in adipogenesis. Collectively, these data clearly demonstrate that MED14 is required for efficient adipose conversion of 3T3-L1 cells. This observation is in keeping with the importance of MED14 for the PPARγ-dependent activation of target genes; however, it is evident that MED14 may also be important for the function of several other adipogenic transcription factors.
FIG. 8.

MED14 is essential for adipogenesis of 3T3-L1 cells. Proliferating 3T3-L1 cells were transduced with lentivirus expressing shRNAs targeting either LacZ or MED14. Cells were allowed to proliferate for 4 days and were then cultured to confluence and induced to differentiate. (A) Confluent undifferentiated cells were analyzed by epifluorescence microscopy to detect GFP expression. (B) RNA levels of MED14 in undifferentiated cells were determined by real-time PCR and normalized to that of Gtf2b. Error bars indicate the ranges of the results of experiments performed in duplicates. (C) Cells were stained with oil red O to visualize the accumulation of lipid droplets at days 0 and 6 of differentiation. (D) RNA levels of Gtf2b, Fabp4, and _Ppar_γ were determined by real-time PCR at day 0 and day 6 of differentiation. Levels were normalized to levels of Gtf2b and visualized as relative mRNA levels. Error bars indicate the ranges of the results of experiments performed in duplicates. Results are representative of a minimum of two independent experiments.
FIG. 9.

Preconfluent 3T3-L1 cells were transfected with siRNA against luciferase or MED14. Transfected cells were grown to confluence and subsequently differentiated into adipocytes by treatment with a cocktail of dexamethasone, methyl-isobutyl xanthine, and insulin (DMI). (A) Lipid accumulation was evaluated by oil red O staining at day 6 of differentiation. (B and C) RNA was purified 0, 6, 12, 24, and 48 h after addition of the adipogenic cocktail, and RNA expression levels of Gtf2b and Med14 (B) or C/EBPβ, _C/EBP_δ, Gilz, _PPAR_γ, Fabp4, and Cidec (C) were determined by real-time PCR. Levels were normalized to levels of Gtf2b and visualized as relative mRNA levels. Error bars indicate the ranges of the results of experiments performed in duplicates. Results are representative of two independent experiments.
The induction of adipogenesis in 3T3-L1 cells is initiated by a hormonal cocktail consisting of insulin, the AMP-elevating drug methyl isobutylxanthine, and the synthetic glucocorticoid dexamethasone (35). As MED14 has been previously shown to be necessary for GR activity, we investigated the effect of MED14 knockdown on the early transcriptional events of 3T3-L1 adipogenesis. Dexamethasone treatment of 3T3-L1 cells has previously been shown to acutely activate the expression of C/ebpβ and _C/ebp_δ (44, 65), both of which are central transcription factors driving early events of 3T3-L1 adipogenesis (40). Moreover, dexamethasone has also been shown to acutely activate the GR target gene Gilz in 3T3-L1 cells (44). Using siRNA-mediated knockdown, we demonstrated that the induction of both C/ebpβ and Gilz during the first 12 h following the addition of dexamethasone is reduced by the knockdown of MED14 (Fig. 9B and C). In contrast, there is little effect on the rather modest induction of C/_ebp_δ during that time. These results suggest that the activity of GR on some target genes is reduced by the knockdown of MED14. In keeping with the importance of MED14 for PPARγ transactivation, the induction of the highly PPARγ-dependent target genes Fabp4 and Cidec at early stages of adipocyte differentiation was severely compromised by the MED14 knockdown. Interestingly, we also showed that knockdown of MED14 led to a significant reduction of _Ppar_γ gene expression itself as early as 6 h following induction by the hormonal cocktail. Transcription of the PPARγ gene is known to be activated by members of the C/EBP family (8, 66) and, probably, by PPARγ itself (43) but not by dexamethasone activation of GR alone (44). Therefore, it is possible that the expression of PPARγ at very early stages of adipocyte differentiation is dependent on MED14 for coactivation of other transcription factors, such as C/EBPβ, that act early in differentiation to activate the PPARγ gene, whereas MED14 coactivation of PPARγ itself may play a role in the activation of the PPARγ gene from day 1 and onwards.
DISCUSSION
Recent reports have accumulated evidence for Mediator recruitment to nuclear receptor-responsive promoters through Mediator subunits other than MED1 (7, 11, 27, 28). In this report, we demonstrate that MED1 is dispensable for acute PPARγ activation of target genes in MEFs and hepatoma cells. Similarly, MED1 is dispensable for the PPARα-mediated activation of target genes in both cell types. In keeping with this, we demonstrated by ChIP analysis that PPARγ, Mediator subunits MED6 and MED8, TBP, and RNAPII are recruited to the Fabp4 enhancer PPRE and the proximal promoter in a MED1-independent manner. To investigate which Mediator subunits may be involved in the PPAR-dependent recruitment of Mediator to target genes, we applied an RNAi approach. These analyses revealed that MED14 is necessary for full acute activation of the majority of PPARγ subtype-specific target genes involved in fatty acid accumulation. Using in vitro and in vivo interaction studies, we demonstrate that MED14 interacts directly with the N-terminal ligand-independent activation domain of PPARγ. In keeping with the importance of MED14 for PPARγ transcriptional activity, we show that 3T3-L1 adipogenesis depends on MED14 and that MED14 is necessary for PPARγ-induced Mediator assembly on the proximal promoter of Fabp4 and for the recruitment of RNAPII.
Recruitment of Mediator to PPAR target promoters in the absence of MED1.
Since the cloning of MED1 as a member of the Mediator complex and as a ligand-dependent direct interaction partner of nuclear receptors through two LXXLL motifs, MED1 has been suggested to anchor the Mediator complex to promoters activated by nuclear receptors. Physical interaction studies, transcription assays, and in vitro transcription systems have shown that PPARγ interacts directly with MED1 through mechanisms that depend on these two LXXLL motifs and that MED1 coactivation depends on this interaction (11, 12, 57, 71). However, this interaction seems to be dispensable for PPARγ activation of endogenous genes in cultured cells, as MED1 lacking LXXLL motifs is able to support PPARγ-induced adipogenesis (11). In keeping with this, we show that PPARγ is able to recruit Mediator to the endogenous target promoter of the Fabp4 gene and to acutely activate target genes in the absence of MED1. Using ChIP to assess factor occupancy of the Fabp4 gene locus, we show that direct PPARγ recruitment to enhancer PPREs most likely promotes juxtaposition of the enhancer with the proximal promoter and that this process is independent of MED1. This MED1 independency contrasts with what has been reported for the TR, where the Mediator complex, and MED1 in particular, have been shown to be essential for the juxtaposition of regulatory elements in the Crabp1 promoter in P19 cells in response to thyroid hormone treatment (45).
Identification of MED14 as a Mediator subunit important for PPARγ activity.
Depletion of MED1 is known to leave the Mediator complex relatively intact (36), and we therefore considered it likely that other subunits in the residual Mediator complex were responsible for Mediator recruitment to PPAR-responsive promoters through direct interaction between Mediator subunits and PPAR, RXR, and/or other transcription factors that co-occupy promoters together with PPAR. We demonstrate here that knockdown of MED14 results in reduced activation of a subset of genes by PPARγ. Similarly, MED14 has been shown to be required for GR-mediated transactivation of a subset of target genes (7), indicating that MED14 is involved in gene-selective activation of target genes by nuclear receptors. Interestingly, we show that MED14 interacts directly with the N-terminal domain of PPARγ2 both in vitro and in cells and that MED14 is able to coactivate the N-terminal PPARγ ligand-independent transactivation domain. These results indicate that MED14 is not only necessary for PPARγ function but also interacts directly with PPARγ to recruit Mediator to PPARγ-responsive promoters. Ge et al. have previously observed that Mediator association with PPARγ in GST pulldown assays is lost when MED1 is absent (11), indicating that the strong in vitro Mediator interaction is MED1 dependent. Here we show that PPARγ is able to interact with MED14 in a GST pulldown assay, although it is evident that the in vitro interaction of MED14 with PPARγ is weaker than the interaction of MED1 with PPARγ (Fig. 4A and B). Thus, strong in vitro interactions between Mediator and PPARγ do not necessarily reflect functionality on endogenous promoters. The possibility that the in vivo interaction between PPARγ and MED14 reflects a relatively weak interaction that is supported by additional protein-protein interactions cannot be excluded. However, it is tempting to speculate that the in vivo strength of the PPARγ-MED14 interaction is underestimated by in vitro assays, because the A/B domain is relatively unstructured and, therefore, may not adopt the proper confirmation for interaction with MED14.
We previously showed that the N terminus of PPARγ is necessary for full transcriptional activation of a subset of PPARγ target genes involved in lipid storage (3). We further showed that the A/B domain is required for CBP and p300 recruitment to PPARγ target sites associated with these genes. Here, we show that MED14 is necessary for transactivation of the Fabp4, Cidec, and Cd36 genes, all of which are dependent on the N-terminal A/B domain of PPARγ for full transactivation. These results suggest that compromised ability to interact with MED14 may contribute to the reduced transcriptional activity of N-terminally truncated PPARγ. Notably, however, knockdown of MED14 also compromises the transcriptional activity of a version of PPARγ with the A/B domain deleted, indicating that MED14, independent of its ability to interact with PPARγ, is important for Mediator activity per se on PPARγ-responsive genes. Since the interaction of MED14 with PPARγ is dependent on the A/B domain, this suggests that another Mediator subunit(s) tethers Mediator to PPARγ in an A/B domain-independent manner. These results are in keeping with our previous results showing that Mediator (as evaluated by MED1) recruitment to PPARγ target sites is not grossly affected by deletion of the A/B domain (3).
Mediator assembly on a PPARγ-responsive promoter.
The Mediator complex is recruited to promoters by transcription factors through different subunits located primarily in the Mediator tail module (37). Our interaction studies suggest that the complex is recruited to PPARγ-responsive promoters through MED14, which is located in the tail module. We show that reduced levels of MED14 result in impaired recruitment of PPARγ, MED6, and MED8 to the proximal promoter of Fabp4, and we demonstrate that MED1 and RNAPII are recruited neither to the enhancer nor to the proximal promoter when MED14 is knocked down. Interestingly, knockdown of MED14 did not change MED6 or MED8 recruitment to the PPARγ-occupied enhancer, indicating that residual Mediator is recruited to the enhancer in a PPARγ-dependent manner but, given the loss of transcriptional activity, that this partial Mediator recruitment is insufficient for cross talk between the enhancer and the proximal promoter. This result indicates that at least part of the Mediator complex is assembled on the enhancer by mechanisms that do not depend on MED1 or MED14 and, further, suggests that PPARγ and/or other transcription factors occupying the Fabp4 enhancer may facilitate the recruitment of a nonfunctional Mediator complex.
The unproductive Mediator recruitment induced by direct binding of PPARγ to the Fabp4 enhancer indicates that PPARγ or other transcription factors recruit part of the Mediator complex independently of MED14. It has been shown in adipocytes that the majority of PPARγ binding sites, including the Fabp4 enhancer, are co-occupied by members of the C/EBP family (29, 43). Interestingly, it has been shown that C/EBPβ interacts directly with MED23 (39), which is part of a Mediator subcomplex consisting of MED23, MED24, and MED16 (22), and that the transcriptional activity of PPARγ also relies on MED24 (11). These results suggest that the MED23, MED24, and MED16 submodule of Mediator may be important for PPARγ activity through cooperative transcriptional activation with members of the C/EBP family. In keeping with this, knockdown of MED16, MED23, or MED24 also results in reduced transcriptional activity of PPARγ (L. Grøntved and S. Mandrup, unpublished data). Moreover, the yeast orthologs of MED16/Sin4, MED14/Rgr1, and MED15 comprise a subcomplex of the yeast Mediator complex (33), suggesting a structural and functional link between the mammalian MED16-MED23-MED24 submodule and MED14.
Alternatively, the Mediator complex may be recruited through transcriptional coregulators that are not part of the conventional purified Mediator complex but that function as intermediate subunits forming a bridge between Mediator and PPARγ. PRIC285, which was cloned as a direct interaction partner with PPARα, is found in a complex with Mediator components like MED1, MED24, MED23, and MED12 (53). Another example is CCAR1, which interacts directly with CoCoA, an interaction partner of the p160 coactivators. CCAR1 has been demonstrated to interact with MED1 and is required for Mediator recruitment to ER- and GR-responsive promoters (27). Thus, there exist numerous possible mechanisms for Mediator recruitment to promoters, indicating that Mediator recruitment is not restricted to a few protein-protein interactions between a transcription factor and Mediator.
Requirement for Mediator subunits during adipogenesis.
The finding that MED1 is required for adipogenesis in vitro (11, 12), despite the fact that ligand activation of PPARγ (11) and acute transactivation by ectopically expressed PPARγ (this study) are independent of MED1, indicates that MED1 is involved in transactivation by other adipogenic transcription factors than PPARγ. A potential candidate could be GR, since GR-mediated transactivation of at least some target genes has been shown to be sensitive to MED1 knockdown (6, 7). However, the fact that the LXXLL motifs are not required for adipogenesis indicates that adipogenic transcription factors other than nuclear receptors require MED1. One potential candidate is C/EBPβ (12, 32).
Here, we demonstrate that MED14 is required for adipogenesis in vitro, which is in keeping with our finding that knockdown of MED14 compromises PPARγ transactivation. In addition, our data indicate that inhibition of GR transactivation may also contribute to inhibition of adipogenesis, possibly along with other adipogenic transcription factors, such as SREBP-1c, the transactivation of which has also been shown to be MED14 dependent (56). It is likely that many other Mediator subunits are necessary for the adipogenic process. In this regard, we show here that MED12 is required for adipogenesis, whereas others have shown recently that MED23 is required for the early transcriptional events during adipogenesis (62) and, probably, also for optimal PPARγ transcriptional activity (11). In Caenorhabditis elegans, MED15 has been shown to be required for intestinal fat storage (67).
In conclusion, in this report, we have identified MED14 as a novel PPARγ-interacting protein that interacts directly and specifically with the N-terminal A/B domain of PPARγ. Furthermore, we show that MED14 is involved in tethering functional Mediator to PPARγ at PPARγ-selective lipogenic genes and is required for activation of these genes. In keeping with this, we show that MED14 is necessary for Mediator assembly on the Fabp4 proximal promoter and that MED14 is obligate for 3T3-L1 adipogenesis.
Acknowledgments
We thank P. Sauerberg (Novo Nordisk A/S) for the rosiglitazone/BRL49653 ligand and J. K. Reddy, C. M. Horvath, and R. M. Evans for plasmids.
This study was supported by grants from the Danish Natural Science Research Council, grants to the EU FP6 STREP project X-TRA-NET, and National Institutes of Health grant DK071900 (to R.G.R.). M.S.M. was supported by the Novo Scholarship program.
Footnotes
▿
Published ahead of print on 1 March 2010.
REFERENCES
- 1.Aoyama, T., J. M. Peters, N. Iritani, T. Nakajima, K. Furihata, T. Hashimoto, and F. J. Gonzalez. 1998. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem. 273**:**5678-5684. [DOI] [PubMed] [Google Scholar]
- 2.Barbera, M. J., A. Schluter, N. Pedraza, R. Iglesias, F. Villarroya, and M. Giralt. 2001. Peroxisome proliferator-activated receptor alpha activates transcription of the brown fat uncoupling protein-1 gene: a link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. J. Biol. Chem. 276**:**1486-1493. [DOI] [PubMed] [Google Scholar]
- 3.Bugge, A., L. Grontved, M. M. Aagaard, R. Borup, and S. Mandrup. 2009. The PPAR{gamma}2 A/B-domain plays a gene specific role in transactivation and co-factor recruitment. Mol. Endocrinol. 23**:**794-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castillo, G., R. P. Brun, J. K. Rosenfield, S. Hauser, C. W. Park, A. E. Troy, M. E. Wright, and B. M. Spiegelman. 1999. An adipogenic cofactor bound by the differentiation domain of PPARgamma. EMBO J. 18**:**3676-3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chawla, A., W. A. Boisvert, C. H. Lee, B. A. Laffitte, Y. Barak, S. B. Joseph, D. Liao, L. Nagy, P. A. Edwards, L. K. Curtiss, R. M. Evans, and P. Tontonoz. 2001. A PPAR[gamma]-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 7**:**161-171. [DOI] [PubMed] [Google Scholar]
- 6.Chen, W., and R. G. Roeder. 2007. The Mediator subunit MED1/TRAP220 is required for optimal glucocorticoid receptor-mediated transcription activation. Nucleic Acids Res. 35**:**6161-6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, W., I. Rogatsky, and M. J. Garabedian. 2006. MED14 and MED1 differentially regulate target-specific gene activation by the glucocorticoid receptor. Mol. Endocrinol. 20**:**560-572. [DOI] [PubMed] [Google Scholar]
- 8.Clarke, S. L., C. E. Robinson, and J. M. Gimble. 1997. CAAT/enhancer binding proteins directly modulate transcription from the peroxisome proliferator-activated receptor gamma 2 promoter. Biochem. Biophys. Res. Commun. 240**:**99-103. [DOI] [PubMed] [Google Scholar]
- 9.Dull, T., R. Zufferey, M. Kelly, R. J. Mandel, M. Nguyen, D. Trono, and L. Naldini. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72**:**8463-8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fontaine, C., G. Dubois, Y. Duguay, T. Helledie, N. Vu-Dac, P. Gervois, F. Soncin, S. Mandrup, J. C. Fruchart, J. Fruchart-Najib, and B. Staels. 2003. The orphan nuclear receptor Rev-Erbalpha is a peroxisome proliferator-activated receptor (PPAR) gamma target gene and promotes PPARgamma-induced adipocyte differentiation. J. Biol. Chem. 278**:**37672-37680. [DOI] [PubMed] [Google Scholar]
- 11.Ge, K., Y. W. Cho, H. Guo, T. B. Hong, M. Guermah, M. Ito, H. Yu, M. Kalkum, and R. G. Roeder. 2008. Alternative mechanisms by which mediator subunit MED1/TRAP220 regulates peroxisome proliferator-activated receptor gamma-stimulated adipogenesis and target gene expression. Mol. Cell Biol. 28**:**1081-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ge, K., M. Guermah, C. X. Yuan, M. Ito, A. E. Wallberg, B. M. Spiegelman, and R. G. Roeder. 2002. Transcription coactivator TRAP220 is required for PPAR gamma 2-stimulated adipogenesis. Nature 417**:**563-567. [DOI] [PubMed] [Google Scholar]
- 13.Gelman, L., G. Zhou, L. Fajas, E. Raspe, J. C. Fruchart, and J. Auwerx. 1999. p300 interacts with the N- and C-terminal part of PPARgamma 2 in a ligand-independent and -dependent manner, respectively. J. Biol. Chem. 274**:**7681-7688. [DOI] [PubMed] [Google Scholar]
- 14.Graves, R. A., P. Tontonoz, K. A. Platt, S. R. Ross, and B. M. Spiegelman. 1992. Identification of a fat cell enhancer: analysis of requirements for adipose tissue-specific gene expression. J. Cell. Biochem. 49**:**219-224. [DOI] [PubMed] [Google Scholar]
- 15.Gu, W., S. Malik, M. Ito, C. X. Yuan, J. D. Fondell, X. Zhang, E. Martinez, J. Qin, and R. G. Roeder. 1999. A novel human SRB/MED-containing cofactor complex, SMCC, involved in transcription regulation. Mol. Cell 3**:**97-108. [DOI] [PubMed] [Google Scholar]
- 16.Guglielmi, B., N. L. van Berkum, B. Klapholz, T. Bijma, M. Boube, C. Boschiero, H. M. Bourbon, F. C. Holstege, and M. Werner. 2004. A high resolution protein interaction map of the yeast Mediator complex. Nucleic Acids Res. 32**:**5379-5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halabi, C. M., A. M. Beyer, W. J. de Lange, H. L. Keen, G. L. Baumbach, F. M. Faraci, and C. D. Sigmund. 2008. Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab. 7**:**215-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heckman, K. L., and L. R. Pease. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2**:**924-932. [DOI] [PubMed] [Google Scholar]
- 19.Helledie, T., L. Grontved, S. S. Jensen, P. Kiilerich, L. Rietveld, T. Albrektsen, M. S. Boysen, J. Nohr, L. K. Larsen, J. Fleckner, H. G. Stunnenberg, K. Kristiansen, and S. Mandrup. 2002. The gene encoding the Acyl-CoA-binding protein is activated by peroxisome proliferator-activated receptor gamma through an intronic response element functionally conserved between humans and rodents. J. Biol. Chem. 277**:**26821-26830. [DOI] [PubMed] [Google Scholar]
- 20.Hittelman, A. B., D. Burakov, J. A. Iniguez-Lluhi, L. P. Freedman, and M. J. Garabedian. 1999. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J. 18**:**5380-5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.IJpenberg, A., E. Jeannin, W. Wahli, and B. Desvergne. 1997. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J. Biol. Chem. 272**:**20108-20117. [DOI] [PubMed] [Google Scholar]
- 22.Ito, M., H. J. Okano, R. B. Darnell, and R. G. Roeder. 2002. The TRAP100 component of the TRAP/Mediator complex is essential in broad transcriptional events and development. EMBO J. 21**:**3464-3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito, M., C. X. Yuan, H. J. Okano, R. B. Darnell, and R. G. Roeder. 2000. Involvement of the TRAP220 component of the TRAP/SMCC coactivator complex in embryonic development and thyroid hormone action. Mol. Cell 5**:**683-693. [DOI] [PubMed] [Google Scholar]
- 24.Jia, Y., C. Qi, P. Kashireddi, S. Surapureddi, Y. J. Zhu, M. S. Rao, R. D. Le, P. Chambon, F. J. Gonzalez, and J. K. Reddy. 2004. Transcription coactivator PBP, the peroxisome proliferator-activated receptor (PPAR)-binding protein, is required for PPARalpha-regulated gene expression in liver. J. Biol. Chem. 279**:**24427-24434. [DOI] [PubMed] [Google Scholar]
- 25.Juge-Aubry, C., A. Pernin, T. Favez, A. G. Burger, W. Wahli, C. A. Meier, and B. Desvergne. 1997. DNA binding properties of peroxisome proliferator-activated receptor subtypes on various natural peroxisome proliferator response elements. Importance of the 5′-flanking region. J. Biol. Chem. 272**:**25252-25259. [DOI] [PubMed] [Google Scholar]
- 26.Kersten, S., J. Seydoux, J. M. Peters, F. J. Gonzalez, B. Desvergne, and W. Wahli. 1999. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 103**:**1489-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim, J. H., C. K. Yang, K. Heo, R. G. Roeder, W. An, and M. R. Stallcup. 2008. CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol. Cell 31**:**510-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, H. K., U. H. Park, E. J. Kim, and S. J. Um. 2007. MED25 is distinct from TRAP220/MED1 in cooperating with CBP for retinoid receptor activation. EMBO J. 26**:**3545-3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lefterova, M. I., Y. Zhang, D. J. Steger, M. Schupp, J. Schug, A. Cristancho, D. Feng, D. Zhuo, C. J. Stoeckert, Jr., X. S. Liu, and M. A. Lazar. 2008. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 22**:**2941-2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leone, T. C., C. J. Weinheimer, and D. P. Kelly. 1999. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. U. S. A. 96**:**7473-7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis, B. A., and D. Reinberg. 2003. The mediator coactivator complex: functional and physical roles in transcriptional regulation. J. Cell Sci. 116**:**3667-3675. [DOI] [PubMed] [Google Scholar]
- 32.Li, H., P. Gade, S. C. Nallar, A. Raha, S. K. Roy, S. Karra, J. K. Reddy, S. P. Reddy, and D. V. Kalvakolanu. 2008. The Med1 subunit of transcriptional mediator plays a central role in regulating CCAAT/Enhancer-binding protein-{beta}-driven transcription in response to interferon-{gamma}. J. Biol. Chem. 283**:**13077-13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, Y., S. Bjorklund, Y. W. Jiang, Y. J. Kim, W. S. Lane, D. J. Stillman, and R. D. Kornberg. 1995. Yeast global transcriptional regulators Sin4 and Rgr1 are components of mediator complex/RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. U. S. A. 92**:**10864-10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linder, T., X. Zhu, V. Baraznenok, and C. M. Gustafsson. 2006. The classical srb4-138 mutant allele causes dissociation of yeast Mediator. Biochem. Biophys. Res. Commun. 349**:**948-953. [DOI] [PubMed] [Google Scholar]
- 35.MacDougald, O. A., and S. Mandrup. 2002. Adipogenesis: forces that tip the scales. Trends Endocrinol. Metab. 13**:**5-11. [DOI] [PubMed] [Google Scholar]
- 36.Malik, S., M. Guermah, C. X. Yuan, W. Wu, S. Yamamura, and R. G. Roeder. 2004. Structural and functional organization of TRAP220, the TRAP/Mediator subunit that is targeted by nuclear receptors. Mol. Cell. Biol. 24**:**8244-8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malik, S., and R. G. Roeder. 2005. Dynamic regulation of pol II transcription by the mammalian Mediator complex. Trends Biochem. Sci. 30**:**256-263. [DOI] [PubMed] [Google Scholar]
- 38.Meredith, D., M. Panchatcharam, S. Miriyala, Y. S. Tsai, A. J. Morris, N. Maeda, G. A. Stouffer, and S. S. Smyth. 2009. Dominant-negative loss of PPARgamma function enhances smooth muscle cell proliferation, migration, and vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 29**:**465-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mo, X., E. Kowenz-Leutz, H. Xu, and A. Leutz. 2004. Ras induces mediator complex exchange on C/EBP beta. Mol. Cell 13**:**241-250. [DOI] [PubMed] [Google Scholar]
- 40.Morrison, R. F., and S. R. Farmer. 1999. Insights into the transcriptional control of adipocyte differentiation. J. Cell. Biochem. 33(Suppl. 32)**:**59-67. [DOI] [PubMed] [Google Scholar]
- 41.Nagy, L., and J. W. Schwabe. 2004. Mechanism of the nuclear receptor molecular switch. Trends Biochem. Sci. 29**:**317-324. [DOI] [PubMed] [Google Scholar]
- 42.Nielsen, R., L. Grontved, H. G. Stunnenberg, and S. Mandrup. 2006. Peroxisome proliferator-activated receptor subtype- and cell-type-specific activation of genomic target genes upon adenoviral transgene delivery. Mol. Cell Biol. 26**:**5698-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nielsen, R., T. A. Pedersen, D. Hagenbeek, P. Moulos, R. Siersbaek, E. Megens, S. Denissov, M. Borgesen, K. J. Francoijs, S. Mandrup, and H. G. Stunnenberg. 2008. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 22**:**2953-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pantoja, C., J. T. Huff, and K. R. Yamamoto. 2008. Glucocorticoid signaling defines a novel commitment state during adipogenesis in vitro. Mol. Biol. Cell 19**:**4032-4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park, S. W., G. Li, Y. P. Lin, M. J. Barrero, K. Ge, R. G. Roeder, and L. N. Wei. 2005. Thyroid hormone-induced juxtaposition of regulatory elements/factors and chromatin remodeling of Crabp1 dependent on MED1/TRAP220. Mol. Cell 19**:**643-653. [DOI] [PubMed] [Google Scholar]
- 46.Rachez, C., B. D. Lemon, Z. Suldan, V. Bromleigh, M. Gamble, A. M. NAAR, H. Erdjument-Bromage, P. Tempst, and L. P. Freedman. 1999. Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature 398**:**824-828. [DOI] [PubMed] [Google Scholar]
- 47.Rachez, C., Z. Suldan, J. Ward, C. P. B. Chang, D. Burakov, H. Erdjument-Bromage, P. Tempst, and L. P. Freedman. 1998. A novel protein complex that interacts with the vitamin D3 receptor in a ligand-dependent manner and enhances VDR transactivation in a cell-free system. Genes Dev. 12**:**1787-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ravnskjaer, K., M. Boergesen, B. Rubi, J. K. Larsen, T. Nielsen, J. Fridriksson, P. Maechler, and S. Mandrup. 2005. Peroxisome proliferator-activated receptor alpha (PPARalpha) potentiates, whereas PPARgamma attenuates, glucose-stimulated insulin secretion in pancreatic beta-cells. Endocrinology 146**:**3266-3276. [DOI] [PubMed] [Google Scholar]
- 49.Reynolds, A., D. Leake, Q. Boese, S. Scaringe, W. S. Marshall, and A. Khvorova. 2004. Rational siRNA design for RNA interference. Nat. Biotech. 22**:**326-330. [DOI] [PubMed] [Google Scholar]
- 50.Rosen, E. D., P. Sarraf, A. E. Troy, G. Bradwin, K. Moore, D. S. Milstone, B. M. Spiegelman, and R. M. Mortensen. 1999. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 4**:**611-617. [DOI] [PubMed] [Google Scholar]
- 51.Stumpf, M., C. Waskow, M. Krotschel, D. van Essen, P. Rodriguez, X. Zhang, B. Guyot, R. G. Roeder, and T. Borggrefe. 2006. The mediator complex functions as a coactivator for GATA-1 in erythropoiesis via subunit Med1/TRAP220. Proc. Natl. Acad. Sci. U. S. A. 103**:**18504-18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun, X., Y. Zhang, H. Cho, P. Rickert, E. Lees, W. Lane, and D. Reinberg. 1998. NAT, a human complex containing Srb polypeptides that functions as a negative regulator of activated transcription. Mol. Cell 2**:**213-222. [DOI] [PubMed] [Google Scholar]
- 53.Surapureddi, S., S. Yu, H. Bu, T. Hashimoto, A. V. Yeldandi, P. Kashireddy, M. Cherkaoui-Malki, C. Qi, Y. J. Zhu, M. S. Rao, and J. K. Reddy. 2002. Identification of a transcriptionally active peroxisome proliferator-activated receptor alpha-interacting cofactor complex in rat liver and characterization of PRIC285 as a coactivator. Proc. Natl. Acad. Sci. U. S. A. 99**:**11836-11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tontonoz, P., E. Hu, R. A. Graves, A. I. Budavari, and B. M. Spiegelman. 1994. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 8**:**1224-1234. [DOI] [PubMed] [Google Scholar]
- 55.Tontonoz, P., L. Nagy, J. G. A. Alvarez, V. A. Thomazy, and R. M. Evans. 1998. PPAR[gamma] promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93**:**241-252. [DOI] [PubMed] [Google Scholar]
- 56.Toth, J. I., S. Datta, J. N. Athanikar, L. P. Freedman, and T. F. Osborne. 2004. Selective coactivator interactions in gene activation by SREBP-1a and -1c. Mol. Cell. Biol. 24**:**8288-8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Treuter, E., L. Johansson, J. S. Thomsen, A. Warnmark, J. Leers, M. Pelto-Huikko, M. Sjoberg, A. P. Wright, G. Spyrou, and J. A. Gustafsson. 1999. Competition between thyroid hormone receptor-associated protein (TRAP) 220 and transcriptional intermediary factor (TIF) 2 for binding to nuclear receptors. Implications for the recruitment of TRAP and p160 coactivator complexes. J. Biol. Chem. 274**:**6667-6677. [DOI] [PubMed] [Google Scholar]
- 58.van Beekum, N. L., A. B. Brenkman, L. Grontved, N. Hamers, N. J. van den Broek, R. Berger, S. Mandrup, and E. Kalkhoven. 2008. The adipogenic acetyltransferase Tip60 targets activation function 1 of peroxisome proliferator-activated receptor {gamma}. Endocrinology 149**:**1840-1849. [DOI] [PubMed] [Google Scholar]
- 59.van de Peppel, J., N. Kettelarij, H. van Bakel, T. T. Kockelkorn, D. van Leenen, and F. C. Holstege. 2005. Mediator expression profiling epistasis reveals a signal transduction pathway with antagonistic submodules and highly specific downstream targets. Mol. Cell 19**:**511-522. [DOI] [PubMed] [Google Scholar]
- 60.Ventura, A., A. Meissner, C. P. Dillon, M. McManus, P. A. Sharp, L. Van Parijs, R. Jaenisch, and T. Jacks. 2004. Cre-lox-regulated conditional RNA interference from transgenes. Proc. Natl. Acad. Sci.U. S. A. 101**:**10380-10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wan, Y., L. W. Chong, and R. M. Evans. 2007. PPAR-gamma regulates osteoclastogenesis in mice. Nat. Med. 13**:**1496-1503. [DOI] [PubMed] [Google Scholar]
- 62.Wang, W., L. Huang, Y. Huang, J. W. Yin, A. J. Berk, J. M. Friedman, and G. Wang. 2009. Mediator MED23 links insulin signaling to the adipogenesis transcription cascade. Dev. Cell 16**:**764-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang, Y. X., C. H. Lee, S. Tiep, R. T. Yu, J. Ham, H. Kang, and R. M. Evans. 2003. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 113**:**159-170. [DOI] [PubMed] [Google Scholar]
- 64.Willson, T. M., P. J. Brown, D. D. Sternbach, and B. R. Henke. 2000. The PPARs: from orphan receptors to drug discovery. J. Med. Chem. 43**:**527-550. [DOI] [PubMed] [Google Scholar]
- 65.Wu, Z., N. L. Bucher, and S. R. Farmer. 1996. Induction of peroxisome proliferator-activated receptor gamma during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPbeta, C/EBPdelta, and glucocorticoids. Mol. Cell. Biol. 16**:**4128-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu, Z., Y. Xie, N. L. Bucher, and S. R. Farmer. 1995. Conditional ectopic expression of C/EBP beta in NIH-3T3 cells induces PPAR gamma and stimulates adipogenesis. Genes Dev. 9**:**2350-2363. [DOI] [PubMed] [Google Scholar]
- 67.Yang, F., B. W. Vought, J. S. Satterlee, A. K. Walker, Z. Y. Jim Sun, J. L. Watts, R. DeBeaumont, R. M. Saito, S. G. Hyberts, S. Yang, C. Macol, L. Iyer, R. Tjian, S. van den Heuvel, A. C. Hart, G. Wagner, and A. M. Naar. 2006. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 442**:**700-704. [DOI] [PubMed] [Google Scholar]
- 68.Yang, W., C. Rachez, and L. P. Freedman. 2000. Discrete roles for peroxisome proliferator-activated receptor gamma and retinoid x receptor in recruiting nuclear receptor coactivators. Mol. Cell. Biol. 20**:**8008-8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan, C. X., M. Ito, J. D. Fondell, Z. Y. Fu, and R. G. Roeder. 1998. The TRAP220 component of a thyroid hormone receptor-associated protein (TRAP) coactivator complex interacts directly with nuclear receptors in a ligand-dependent fashion. Proc. Natl. Acad. Sci. U. S. A. 95**:**7939-7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang, X., A. Krutchinsky, A. Fukuda, W. Chen, S. Yamamura, B. T. Chait, and R. G. Roeder. 2005. MED1/TRAP220 exists predominantly in a TRAP/Mediator subpopulation enriched in RNA polymerase II and is required for ER-mediated transcription. Mol. Cell 19**:**89-100. [DOI] [PubMed] [Google Scholar]
- 71.Zhu, Y., C. Qi, S. Jain, M. S. Rao, and J. K. Reddy. 1997. Isolation and characterization of PBP, a protein that interacts with peroxisome proliferator-activated receptor. J. Biol. Chem. 272**:**25500-25506. [DOI] [PubMed] [Google Scholar]