Decreased Pulmonary Inflammation Following Ethanol and Burn Injury in Mice Deficient in TLR4 but not TLR2 Signaling (original) (raw)
. Author manuscript; available in PMC: 2011 Oct 1.
Abstract
Background
Clinical and laboratory evidence suggests that alcohol consumption prior to burn injury leads to dysregulated immune function and subsequent higher rates of morbidity and mortality. Our laboratory previously observed higher levels of pro-inflammatory cytokines and leukocyte infiltration in the lungs of mice following ethanol and burn injury. To understand the mechanism of the increased inflammatory response, we looked at different signaling initiators of inflammation including toll-like receptors 2 and 4 (TLR2 and 4) pathways.
Methods
Wild-type, TLR2, and TLR4 knockout mice were treated with vehicle or a single binge dose of ethanol (1.11 g/kg) and subsequently given a sham or burn injury. Twenty-four hours postinjury, systemic and pulmonary levels of pro-inflammatory cytokines were quantified, and differences in neutrophil infiltration were determined by histological examination.
Results
Higher numbers of neutrophils were observed in the lungs of wild-type mice following the combined insult of ethanol and burn injury relative to either injury alone. This increase in leukocyte accumulation was absent in the TLR4 knockout mice. Circulating levels of IL-6 and tumor necrosis factor-α were also elevated in wild-type mice but not in TLR4 knockout mice. Consistent with these findings, pulmonary levels of KC and IL-6 were increased in wild-type mice following burn and ethanol compared to burn injury alone as well as to their TLR4 knockout counterparts. In contrast, TLR2 knockout mice displayed similar levels, to wild-type mice, of neutrophil infiltration as well as IL-6 and KC in the lung.
Conclusions
These data suggest that TLR4 signaling is a crucial contributory component in the exuberant inflammation after ethanol and burn injury. However, TLR2 does not appear to play a vital role in the aberrant pulmonary inflammation.
Keywords: Lungs, Toll-Like Receptor, Inflammation, Ethanol, Burn
Clinical and animal model studies demonstrate that burn injury results in an enhanced systemic inflammatory response leading to multiple organ dysfunction syndrome and multiple organ failure (MOF) (Marshall, 2000; Moore and Moore, 1995). This dysregulated immune response in humans and mice can be characterized by higher levels of interleukin-6 (IL-6), interleukin-1_β_ (IL-1_β_), tumor necrosis factor-α (TNF-α), prostaglandin-E2 (PGE2), and lower delayed-type hypersensitivity response and lymphocyte proliferation (Drost et al., 1993; Kowal-Vern et al., 1997; Zhou et al., 1992). The suppression of the immune response observed after burn is exaggerated with the addition of alcohol in patients (Howland and Hingson, 1987; Kowal-Vern et al., 1994; McGill et al., 1995) and also in animal models (Colantoni et al., 2000; Faunce et al., 1997, 1998; Fontanilla et al., 2000; Messingham et al., 2000). Currently, the major postinjury complications result from pulmonary failure (Solomkin, 1990). This phenomenon is likely a result of bacteria and endotoxin leaking from the gut to the lungs (Magnotti and Deitch, 2005), the increased risk of contact with pathogens from both the circulation and the airway, and the delicate architecture of the lung itself. In support of this hypothesis, the combined insult of ethanol and burn injury was shown to augment intestinal permeability and bacterial translocation (Choudhry et al., 2002; Kavanaugh et al., 2005).
In animal models, increased numbers of neutrophils were observed in the lungs of both mice and rats following burn injury (Mester et al., 1994; Stengle et al., 1996) and also in animals receiving ethanol prior to burn (Bird et al., 2010; Li et al., 2007; Patel et al., 1999). Previous studies in this laboratory demonstrated increased pulmonary inflammation after ethanol and burn injury consisting of higher levels of neutrophil accumulation, edema, and chemokine production in the lungs (Bird et al., 2010; Patel et al., 1999), as well as in a model with acute ethanol, burn injury, and infection with Pseudomonas aeruginosa (Murdoch et al., 2008).
Toll-like receptors (TLRs) recognize specific molecular patterns from both invading pathogens and host tissue proteins that are released at times of injury. Previous studies using other models of lung injury and/or infection demonstrated decreased inflammatory responses in mice lacking certain TLRs and the adaptor molecules used in their signaling pathways. Mice genetically deficient in TLR2 and TLR4 showed a decreased inflammatory response in the lung after bleomycin-induced injury compared to wild-type mice (Jiang et al., 2006). After burn injury, it was observed that stimulation of total splenocytes or purified splenic macrophages with TLR2 and TLR4 ligands, peptidoglycan, and lipopolysaccharide (LPS), respectively, resulted in enhanced production of pro-inflammatory cytokines (Cairns et al., 2008; Paterson et al., 2003). Administration of LPS to mice after burn resulted in increased pulmonary neutrophil accumulation, cytokine production, and mortality (Murphy et al., 2005). Microvascular leakage after burn injury was also attenuated in TLR4 knockout mice relative to their wild-type counterparts (Breslin et al., 2008).
In addition to TLR4, TLR2 was shown to play a role in pulmonary inflammation. It was recently shown that blocking TLR2 signaling, using both TLR2 knockout mice and a neutralizing antibody against the receptor, decreased pulmonary inflammation and fibrosis compared to wild-type mice after intratracheal instillation of bleomycin (Yang et al., 2009) as well as in a model of pneumocystis pneumonia (Wang et al., 2008). Using a mouse model of blunt chest trauma injury, Hoth and colleagues demonstrated decreased neutrophil infiltrate and pro-inflammatory cytokine production in the lungs of TLR2 knockout mice (Hoth et al., 2007). Additionally, biologically relevant doses of alcohol (25 to 50 mM) were shown to up-regulate TLR2 in human airway epithelial cells in vitro, which in turn lead to significantly higher production of the neutrophil chemoattractant, IL-8 (Bailey et al., 2009). Therefore, it appears that TLR2 may also be important in the distal organ inflammation after injury and/or infection.
The studies described later examine the role of TLR signaling in the aberrant pulmonary inflammation observed in mice treated with ethanol and burn injury. We observed increased neutrophil infiltration as well as increased production of neutrophil chemoattractant, KC, in the lungs of C57BL/6 (wild-type) mice at 24 hours after ethanol and burn injury compared to sham and burn-injured mice. Elevated pulmonary levels of pro-inflammatory mediators, IL-6 and KC, were also seen at this time point. Mice genetically deficient in TLR4 were protected against the excessive pulmonary inflammation seen in mice given ethanol and burn injury relative to either insult alone. In contrast, mice deficient in TLR2 had similar levels of leukocytes and chemokine production in their lungs as their wild-type counterparts after the combined insult. These data suggest a role for TLR4 signaling and not TLR2 in the inflammatory response in the lung after ethanol and burn injury.
MATERIALS AND METHODS
Mice
Male Wild-type (C57BL/6), TLR4 KO (B6.B10ScN-Tlr4lps-del/JthJ), and TLR2 KO (B6.129-Tlr2tm1Kir/J) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were housed in sterile microisolator cages under specific pathogen-free conditions in the Loyola University Medical Center Comparative Medicine facility. All experiments were conducted in accordance with the Institutional Animal Care and Use Committee.
Murine Model of Ethanol and Burn Injury
A murine model of a single (acute) ethanol exposure and burn injury was employed as described previously (Faunce et al., 1997). Briefly, mice were given a single dose of 150 _μ_l of 20%(v/v) ethanol solution (1.11 g/kg) given intraperitoneally that resulted in a blood ethanol level of 150 mg/dl at 30 minutes. The mice were then anesthetized, their dorsum shaved, and placed in a plastic template exposing 15% of the total body surface area (Spector et al., 1965) and subjected to a scald injury in a 92 to 95°C water bath or a sham injury in room-temperature water. The scald injury resulted in an insensate, full-thickness burn injury of approximately 15% total body surface area (Faunce et al., 1999). To model clinical conditions, the mice were then resuscitated with 1.0 ml saline and allowed to recover on warming pads.
Histopathologic Examination of the Lungs
At 24-hour postinjury, mice were euthanized and the lungs were harvested. The upper right lobe was inflated with 10% formalin and fixed overnight as described previously (Bird et al., 2010; Patel et al., 1999). The lung was then embedded in paraffin, sectioned at 5 _μ_m, and stained with hematoxylin and eosin (H & E). The sections were then analyzed microscopically in a blinded fashion for pathologic changes, and the numbers of neutrophils in 10 high-power fields were counted as a marker of inflammation (Bird et al., 2010; Patel et al., 1999).
Cytokine Analysis of Lung Homogenates
In the same animals, the middle right lung lobe was snap-frozen in liquid nitrogen. The tissues were then homogenized in 1 ml of BioPlex cell lysis buffer according to manufacturer’s instructions (BioRad, Hercules, CA). The homogenates were filtered and analyzed for cytokine production using BioRad multiplex assay or ELISA. The results were normalized to total protein present in the homogenate using the BioRad protein assay (BioRad).
Analysis of Myeloperoxidase (MPO) Content in the Lungs
To confirm the neutrophil counts, MPO activity was determined in the lungs of wild-type mice as described previously (Li et al., 2007). Briefly, an additional lobe of the lung was homogenized in phosphate buffer containing 0.5% hexadecyl-trimethylammonium. The samples were then sonicated and the supernatants cleared by centrifugation. MPO content was then determined following incubation with _o_-dianisidine hydrochloride and hydrogen peroxide. MPO content in the samples was determined based on optical density readings from the MPO standard (Sigma Aldrich, St Louis, MO), which was run in parallel. Data are listed asMPO activity per mg of tissue.
Immunofluorescent Staining of Neutrophils in the Lung
The lower left lobe was inflated with 25% optimal cutting temperature (OCT) freezing medium in phosphate-buffered saline, then embedded in OCT, and frozen for immunofluorescent staining of cells as described previously (Bird et al., 2010; Nomellini et al., 2008). The lung was then sectioned (5 um) and stained using rat anti-Gr1 (Invitrogen, Carlsbad, CA) followed by goat anti-rat IgA conjugated to Alexa Fluor 488 (Invitrogen) to detect Gr-1-positive neutrophils. The sections were also stained with biotinylated anti-MOMA-2 antibody (BMA Biomedicals, Augst, Switzerland), a pan-macrophage marker, and detected with streptavidin conjugated to Alexa Fluor 555 (Invitrogen).
Detection of Circulating Cytokine Levels
Blood was collected from cardiac puncture, and serum was obtained by centrifugation after clotting. Cytokine production was then determined using an Invitrogen multiplex bead array according to manufacturer’s instructions (Invitrogen).
Determination of Circulating Leukocytes
Approximately 100 _μ_l of whole blood was collected in an EDTA-treated microcuvette (Sarstedt, Nümbrecht Germany). Total and differential counts were then obtained using a Hemavet 950 machine (Drew Scientific Inc., Oxford, CT) according to manufacturer’s instructions. Data are shown as total cell counts in thousands of cells per microliter.
Statistical Analysis
Statistical comparisons were made between wild-type and knockout animals in the sham vehicle, sham ethanol, burn vehicle, and burn ethanol treatment groups, resulting in 8 total groups analyzed. One-way analysis of variance was used to determine differences between treatment responses, and Tukey’s post-hoc test once significance was achieved (p < 0.05).
RESULTS
TLR4 Is Involved in Systemic Inflammation After Ethanol and Burn Injury
Clinical studies have correlated increased levels of IL-6 in the blood with poor outcomes in patients after injury (Biffl et al., 1996). Consistent with this study and our previous data, we observed relatively low or nondetectable levels of IL-6 in the serum of sham animals; however, there was an 8-fold increase in IL-6 present in the serum of wild-type mice (p < 0.05) after ethanol and burn injury compared to mice receiving burn injury alone (Fig. 1_A_). In contrast, the elevated IL-6 was significantly diminished in mice deficient in TLR4 after burn alone and the combined injury compared to their wild-type counterparts (p < 0.05). As shown in Fig. 1_B_, TNF-α was also elevated in the blood at 24 hours after ethanol and burn injury in wild-type mice compared to animals receiving sham injury or burn injury alone (p < 0.01). This increase in TNF-α was again blunted in the TLR4 KO mice (p < 0.01). These data suggest that TLR4 is important for the systemic inflammatory process after ethanol and burn injury.
Fig. 1.
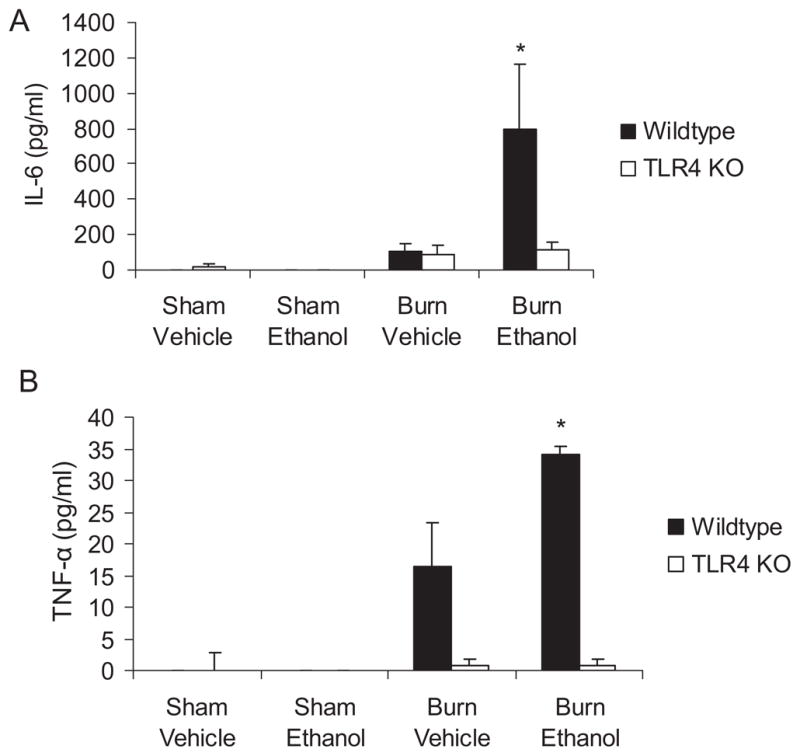
Decreased levels of circulating pro-inflammatory cytokines in TLR4 knockout mice. Levels of tumor necrosis factor-α and IL-6 in serum were quantified by Invitrogen multiplex assay. *p < 0.05 compared to all other groups using 1-way ANOVA. N = 3 per group. Representative data from 1 of 2 studies performed are shown as mean concentration ± SEM.
Increased Pulmonary Inflammation After Ethanol and Burn Injury
To determine whether TLR signaling is responsible for increased pulmonary inflammation after ethanol and burn injury, formalin-fixed lung sections were stained with H & E and examined by light microscopy. Consistent with our previous studies (Patel et al., 1999), the lungs of animals exposed to ethanol and burn injury displayed increased leukocyte infiltration and thickened alveolar walls (Fig. 2_D_) compared to their sham counterparts and animals receiving burn injury alone (Fig. 2_A_ and 2_B_). We next examined whether the pulmonary inflammation observed after ethanol and burn injury was ameliorated in mice genetically deficient in TLR activation, namely TLR2 and TLR4 knockout mice. There was substantially less inflammation observed in the lungs of TLR4 KO mice after the combined insult (Fig. 2_H_) compared to wild-type mice. In contrast, TLR2 knockout mice did have thickened alveolar walls and leukocyte infiltration similar to wild-type mice after ethanol and burn injury (Fig. 2_L_), suggesting that TLR2 is not responsible for the aberrant pulmonary inflammation after ethanol and burn injury.
Fig. 2.

Decreased pulmonary inflammation following ethanol and burn injury in TLR4 KO mice. Lung sections from WT, TLR4 KO, and TLR2 KO mice were examined for degree of inflammation 24 hours after ethanol and burn injury. Representative micrographs of hematoxylin and eosin (H & E) lung sections are shown. All images are at 200× magnification.
Decreased Leukocyte Infiltration in the Lungs of TLR4 KO Mice
Upon closer examination of the H & E sections under higher power (400×), it was determined that the majority of the infiltrating leukocytes after ethanol and burn injury in wild-type mice were neutrophils based on nuclear morphology. The neutrophils were observed to be located in capillaries or the interstitium of the lung, but not in the alveolar spaces (Fig. 3_A_). Similar to what was observed under low power, lungs from TLR4 KO mice had less cellular infiltrate observed compared to that of wild-type mice (Fig. 3_B_) when observed under high magnification (400×), while TLR2 KO lungs resembled wild-type lungs after ethanol and burn (Fig. 3_C_). These neutrophils were then counted in 10 high-power fields of the H & E sections. Consistent with the observed pathological changes observed in the lungs of wild-type mice after burn injury alone and after ethanol and burn injury, there were significantly higher numbers of neutrophils (59 ± 4 neutrophils in 10 high-power fields) present in the lungs of these animals compared to those in sham animals (2 ± 1 neutrophils in 10 high-power fields) (Table 1). Additionally, lungs from burn ethanol mice contained almost 4-fold more neutrophils than lungs from mice given burn alone. The increased neutrophils present in the lungs of wild-type mice were confirmed using both immunohistochemistry (IHC) and detection of MPO activity (Fig. 4). The number of neutrophils present in the H & E sections of lung from wild-type mice (Fig. 4_A_) correlated strongly with the amount of Gr-1-positive neutrophils in IHC-stained sections (Fig. 4_A_) as well as MPO activity (Fig. 4_C_).
Fig. 3.

Increased leukocyte infiltration in the lungs following ethanol and burn injury of WT and TLR2 KO mice. Enlarged representative images of lungs WT, TLR4 KO, and TLR2 KO mice 24 hours after exposure to ethanol and burn injury (400×). Neutrophils are indicated by arrows.
Table 1.
Neutrophils in the Lungs After Ethanol and Burn Injurya
| Sham vehicle | Sham ethanol | Burn vehicle | Burn ethanol | |
|---|---|---|---|---|
| Study #1 | ||||
| Wild-type | 2 ± 1 | 5 ± 3 | 15 ± 3 | 59 ± 4* |
| TLR4 KO | 10 ± 4 | 3 ± 0 | 11 ± 8 | 27 ± 4** |
| Study #2 | ||||
| Wild-type | 5 ± 2 | 4 ± 2 | 28 ± 6*** | 57 ± 12**** |
| TLR2 KO | 7 ± 5 | 2 ± 1 | 29 ± 7*** | 41 ± 6**** |
Fig. 4.

Increased Gr-1-positive cells and myeloperoxidase (MPO) activity after ethanol and burn injury in the lungs of wild-type mice. (A) Lung sections from wild-type mice were stained by flurochrome-conjugated antibodies against Gr-1 (green) to detect the presence of neutrophils. Representative micrographs from all 4 treatment groups are shown. All images are at 200× magnification. (B) Representative lung sections from wild-type (WT), TLR4 KO, and TLR2 KO mice treated with ethanol and burn injury. Green staining indicates neutrophils. (C) MPO activity was measured in lung homogenates from wild-type animals. The data are represented as MPO activity per mg of protein. N = 7 to 12 per group. *p < 0.05 compared to sham animals, #p < 0.05 compared to burn vehicle animals.
Consistent with the histological observations detailed previously, neutrophil counts (Table 1) were decreased in TLR4 knockout mice compared to their wild-type counterparts after the combined injury (p < 0.01) but not after burn injury alone. This decrease in neutrophil counts was consistent with decreased numbers of Gr-1-positive neutrophils in IHC sections (Fig. 4_B_). Similar to wild-type mice, TLR2 knockout mice also had higher numbers of neutrophils present in the lung interstitium after ethanol and burn compared to sham animals (Table 1), which was consistent with the histopathology and IHC described earlier. These results suggest that TLR4 signaling may play a dominant role in the increased pulmonary inflammation after ethanol and burn injury.
Interestingly, the decreased numbers of neutrophils observed in the lungs of TLR4 KO mice were not simply attributable to an overall drop in circulating leukocytes. We obtained total and differential blood cell counts at 24 hours after injury. Numbers of total white blood cells and neutrophils were increased after both burn alone and the combined injury in wild-type mice. This elevation in circulating neutrophils was also observed in both TLR2 KO (data not shown) and TLR4 KO mice after ethanol and burn (Fig. 5).
Fig. 5.

No differences in circulating white blood cells in TLR4 knockout mice after ethanol and burn injury. Total and differential cell counts in whole blood were obtained using a Hemavet 750 machine. Mean values are listed as thousands of cells per microliter of blood ± SEM. *p < 0.05 compared to all sham animals, #p < 0.05 compared to sham ethanol wild-type only by 1-way ANOVA. N = 3 to 6 per group.
Decreased Pulmonary Pro-Inflammatory Mediators in TLR4 KO Mice
To determine whether the significant reduction in neutrophil accumulation observed in the TLR4 KO mice could be attributed to decreased pulmonary content of neutrophil chemokines, we quantitated the levels of KC, a murine neutrophil chemoattractant, in frozen lung homogenates by multiplex bead assay (BioRad). We observed almost 4-fold higher levels of KC in the lungs of wild-type mice given ethanol and burn injury compared to animals given burn injury alone (p < 0.001) (Fig. 6_A_), which is consistent with the increased numbers of neutrophils in the lungs of these animals. In contrast to these observations, KC levels were lower (p < 0.01) in TLR4 KO animals after ethanol and burn injury. Similarly, tissue levels of IL-6 were also (5-fold, p < 0.01) higher in the wild-type mice compared to their knockout counterparts (Fig. 6_B_) after the combined injury, further suggesting an important role for TLR4 in the aberrant inflammation after ethanol and burn.
Fig. 6.

Decreased levels of pro-inflammatory cytokines and chemokines in TLR4 knockout mice. Levels of KC and IL-6 in lung homogenates were quantified by multiplex bead array. Cytokine concentrations were normalized to total protein in the sample and shown as mean concentration ± SEM. *p < 0.05 compared to all groups using 1-way ANOVA. N = 3 to 6 per group.
Elevated levels of IL-6 were observed in TLR2 KO mice after burn injury alone (55.7 ± 22.6 pg/mg protein) and the combined insult (35.6 ± 7.3 pg/mg protein) compared to sham animals (7.7 ± 2.11 pg/mg protein) (p < 0.05); however, there were no significant differences in the amount of IL-6 in the lungs of knockout animals compared to those in wild-type mice in any of the treatment groups including after burn injury (Fig. 7_A_). Similarly, levels of pulmonary KC were significantly increased in mice given burn or burn plus ethanol (241.3 ± 61.3 and 283.7 ± 60.5 pg/mg protein, respectively) over sham animals (12.7 ± 3.4 pg/mg protein) (Fig. 7_B_). However, there were no differences between the levels of KC observed in the lungs of TLR2 KO mice and those seen in wild-type animals.
Fig. 7.

No difference in pulmonary inflammation between wild-type and TLR2 knockout mice after ethanol and burn injury. Levels of KC (A) and IL-6 (B) were quantified in lung homogenates by multiplex bead array. Cytokine concentrations were normalized to total protein in the sample and shown as mean concentration ± SEM. *p < 0.05 compared to sham groups by 1-way ANOVA comparing all 8 groups. N = 4 to 10 per group.
DISCUSSION
Prior to recent advances in antibiotics and standard care practices, infection in the wound bed was the major cause of morbidity and mortality in patients after burn injury (Tompkins and Burke, 1986; Tompkins et al., 1988). Currently, the major postinjury complications result from sepsis and MOF (Solomkin, 1990), with lungs being one of the first organs to fail. In humans, chronic ethanol consumption was linked with increased risk of acute respiratory distress syndrome (Moss et al., 1996). While the exact mechanism by which ethanol increases the pulmonary dysfunction after ethanol and burn injury has yet to be determined, several possibilities include leakage of bacteria and endotoxin from the gut to the lungs, the increased risk of contact with pathogens from both the circulation and the airway, and the delicate architecture of the lung itself. Burn and other injuries were shown to cause intestinal bacterial translocation resulting in bacteria and pro-inflammatory mediators (i.e., cytokines and endotoxin) being carried from the gut to distal organs, primarily the lung (Magnotti and Deitch, 2005). Animal models have shown that these pro-inflammatory factors released from the gut after burn lead to neutrophil activation, endothelial cell activation and damage, and acute lung injury (Magnotti and Deitch, 2005). These increases in transport of pro-inflammatory factors and bacteria from the gut may then signal through TLRs leading to the observed lung inflammation. Increased translocation of bacteria from the intestine to the mesenteric lymph node has been observed after ethanol and burn injury (Choudhry et al., 2002). This elevation in bacterial translocation may be a result of the increased systemic inflammatory response after the combined injury leads to damage of the intestinal barrier allowing for the gut leakiness. This hypothesis is supported by the fact that pro-inflammatory cytokines, such as TNF-α, were shown to cause tight junction disruption (Clayburgh et al., 2005). As TLR4 KO mice had decreased levels of pro-inflammatory cytokines both in the serum (Fig. 1) and in intestine (data not shown) as well as decreased levels of bacteria present in the mesenteric lymph nodes (data not shown), one may hypothesize that blocking of TLR4 signaling may lead to decreased levels of bacteria leaking into the lymphatics and subsequent pulmonary inflammation.
Our laboratory previously demonstrated that ethanol prior to burn injury results in increased pulmonary edema and neutrophil accumulation beginning as early as 2 hours after injury and lasts beyond 48 hours (Patel et al., 1999). Histochemical analysis of H & E-stained lung sections at 24-hour postinjury showed increased leukocyte migration into the lungs of mice given ethanol and burn injury compared to mice given burn alone. Neutrophil counts revealed significantly higher numbers of infiltrating cells at 2-, 8-, 12-, and 24-hour postinjury in the lungs of ethanol-treated mice relative to shams. Levels of neutrophil chemoattractant factor, MIP-2, were also found to be elevated in the lung at those same time points. However, levels of KC were not observed to be different in burn ethanol mice. More recently, we have shown increased KC in the lung of ethanol- and burn-treated mice at 24-hour postinjury, which correlated with increased neutrophil accumulation (Bird et al., 2010). Interestingly, pulmonary edema (as measured by lung wet weight) was higher in mice given burn and ethanol, suggesting a possible increase in vascular leakiness (Patel et al., 1999). Using a rat model, Li et al. also showed increased levels of neutrophil infiltration, neutrophil chemoattractants, and pulmonary edema after the combined injury compared to ethanol or burn alone (Li et al., 2007). In this study, we observed a similar pattern of increased neutrophil infiltration (Table 1, Fig. 4_A_ and 4_C_), as well as significantly higher levels of KC at 24-hour postinjury in the wild-type mice exposed to ethanol and burn injury (Fig. 6_A_). We also demonstrated decreased systemic inflammation in the TLR4 KO mice after ethanol and burn injury as evidenced by lower levels of circulating IL-6 and TNF-α (Fig. 1).
The exact mechanism for the aberrant pulmonary inflammation after ethanol and burn injury is still poorly understood. Many studies of lung injury have shown that TLR4 is responsible for the increased inflammation (Breslin et al., 2008; Frink et al., 2007; Hoth et al., 2009; Imai et al., 2008; Lv et al., 2009; Murphy et al., 2005; Paterson et al., 2003; Shimamoto et al., 2006; Smith et al., 2008). Burn injury appears to “prime” innate immune cells to produce augmented levels of pro-inflammatory cytokines after stimulation with LPS (Paterson et al., 2003). Furthermore, LPS was shown to lead to increased mortality, neutrophil sequestration, and increased pro-inflammatory cytokine production in burn animals (Murphy et al., 2005). Increased expression of TLR2 and TLR4 was observed on splenic macrophages after burn along with spontaneous production of pro-inflammatory cytokines (Cairns et al., 2008). TLR2 and TLR4 can also recognize endogenous host proteins, such as hyaluronan which is present in a variety of tissues including skin and has been linked to inflammation after injury in the lung (reviewed in (Jiang et al., 2006)). Thus, it is possible that endogenous proteins such as hyaluronan, surfactant protein A, fibrinogen, and fibronectin that are normally sequestered from immune cells are released after injury resulting in enhanced TLR signaling.
After binding its ligand, TLR4 signals through 2 different cascades: one using MyD88 and the other using another adaptor protein, TIR-domain-containing adapter-inducing IFN-β (TRIF). Both pathways result in the translocation of NF-_κ_B to the nucleus and the increased production of pro-inflammatory cytokines, including IL-6 and KC. Increased levels of KC will translate into increased neutrophil migration to the lungs. While IL-6 has not been linked to KC production, it was shown to play a role in pulmonary neutrophil accumulation during pneumonia in a STAT1- and STAT3- dependent pathway (Jones et al., 2006). In our model, it is still unclear which pathway is more important. We also examined the role of TRIF in lung inflammation after ethanol and burn injury, and mice genetically deficient in TRIF (C57BL/6J-Ticam1< Lps2>/J) do display lower numbers of neutrophils in the lung and decreased levels of KC and IL-6 (data not shown). However, these mice are not complete knockouts but instead have a distal frameshift mutation that renders them more resistant to LPS (Hoebe et al., 2003). With that caveat, even though our results were not always consistent, in 3 of 5 experiments, we did see the lung neutrophil accumulation and cytokine production decreased by 50% in the TRIF-defective mice compared to wild-types after ethanol and burn injury (data not shown). Still, these decreases are not as great as observed in the TLR4 knockout mice, suggesting that the MyD88-dependent pathway may be more important for the excessive pulmonary inflammation observed in our model.
Other TLRs, such as TLR2, were shown to play a role in pulmonary inflammation. It was recently shown that bleomycin, a well-characterized inducer of lung inflammation and fibrosis, is also a ligand for TLR2 (Razonable et al., 2006; Yang et al., 2009). By blocking TLR2 signaling either through the use of knockout mice or antibody, Yang and colleagues showed decreased pulmonary inflammation and fibrosis compared to wild-type mice after intratracheal administration of bleomycin (Yang et al., 2009). In addition to burn, other injuries, such as blunt chest trauma, result in pulmonary complications. As mentioned previously, decreased neutrophil infiltrate and pro-inflammatory cytokine production was observed in the lungs of TLR2 knockout mice compared to that of wild-type mice following injury (Hoth et al., 2007). In models of infection, TLR2-deficient mice infected with Pneumocystis pneumonii displayed decreased inflammatory responses in the lung with decreased macrophage infiltrate and TNF-α production; however, these mice were unable to control the infection, suggesting that some level of TLR2 signaling is important (Wang et al., 2008). Additionally, acute doses of alcohol were shown to up-regulate TLR2 in human airway epithelial cells in vitro, which in turn produced significantly higher levels of IL-8 (Bailey et al., 2009). Therefore, based on the literature it appears TLR2 may also be important in distal organ inflammation after injury. However, this seems to be dependent on the type of injury as our data suggest that TLR2 is not involved in the aberrant pulmonary inflammation after ethanol and burn injury or even burn injury alone because TLR2 KO mice had similar levels of neutrophils in the lung interstitium as well as similar levels of IL-6 and KC in the lung at 24-hour postinjury. Whether this is simply attributed to some redundancy in the knockout animals remains to be determined. It may be possible that the bacterial products and/or host proteins from the burn site are specific for TLR4, and this is an avenue of research that is ongoing in our laboratory.
Interestingly, ethanol by itself was demonstrated to decrease the ability of splenic macrophages to respond to TLR2 and TLR4 ligands (Goral and Kovacs, 2005), as well as inhibit TLR4 signaling through the disruption of receptor clustering in lipid rafts on the surface of cells, in particular macrophages resulting in diminished pro-inflammatory cytokine production (Dolganiuc et al., 2006; Szabo et al., 2007). However, previous studies showed increased TNF-α in the presence of dual stimulation with TLR2 and TLR4 ligands after acute ethanol by monocytes (Oak et al., 2006). Our data suggest that the combined insult of acute ethanol and burn injury augments TLR4-induced cytokine production. It may be possible that, when combined with an injury, the effects of ethanol are no longer inhibitory and instead ethanol works synergistically with the injury to result in higher levels of inflammation compared to burn injury alone.
The actual mechanism by which ethanol and burn injury increases TLR4 signaling is still under investigation. Whether the combined insult causes perturbations in the signaling molecules themselves, or whether there is simply an overabundance of endotoxin or endogenous ligand present in the lung remains to be determined. While endotoxin levels are likely to play a role, we cannot ignore that both ethanol and burn injury were shown to affect activation states of proteins involved in TLR4 signaling. For instance, the increased TNF-α in the presence of TLR2 and TLR4 stimulation after acute ethanol by monocytes was mediated through increased IRAK-1 activation and JNK phosphorylation (Oak et al., 2006). While there were similar levels of MAPK protein in macrophages from burn animals, increased phosphorylated p38 after LPS stimulation was observed (Maung et al. 2005). Therefore, it would seem likely that the combined insult does cause changes in the signaling cascade of TLR4 resulting in enhanced levels of pro-inflammatory mediators and the subsequent leukocyte infiltration.
In summary, these data suggest that the aberrant pulmonary inflammation observed after the combined insult of acute ethanol and burn injury may be the result of TLR4 signaling in the lung. While the exact ligand responsible for induction of this signaling is unknown, we believe that signaling through TLR4 may then cause increased production of pro-inflammatory cytokines and chemokines resulting in elevated levels of neutrophils migrating into the lung capillaries and interstitium.
Acknowledgments
The authors thank Bartlomiej Posnik for technical assistance, Marykay Olson for help with histology, and Dr. Henry Brown for thoughtful discussions about lung pathology. This work was supported by R01AA012034 (EJK), T32AA013527 (EJK), F32AA018068 (MDB), an Illinois Excellence in Academic Medicine Grant, The Margaret A. Baima Endowment Fund for Alcohol Research, and the Dr. Ralph and Marian C. Falk Medical Research Trust.
References
- Bailey KL, Wyatt TA, Romberger DJ, Sisson JH. Alcohol functionally upregulates Toll-like receptor 2 in airway epithelial cells. Alcohol Clin Exp Res. 2009;33:499–504. doi: 10.1111/j.1530-0277.2008.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffl WL, Moore EE, Moore FA, Peterson VM. Interleukin-6 in the injured patient. Marker of injury or mediator of inflammation? Ann Surg. 1996;224:647–664. doi: 10.1097/00000658-199611000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird MD, Morgan MO, Ramirez L, Yong S, Kovacs EJ. Decreased pulmonary inflammation following ethanol and burn injury in ICAM-1 knockout mice. J Burn Care Res. 2010 doi: 10.1097/BCR.0b013e3181e4c58c. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslin JW, Wu MH, Guo M, Reynoso R, Yuan SY. Toll-like receptor 4 contributes to microvascular inflammation and barrier dysfunction in thermal injury. Shock. 2008;29:349–355. doi: 10.1097/shk.0b013e3181454975. [DOI] [PubMed] [Google Scholar]
- Cairns BA, Barnes CM, Mlot S, Meyer AA, Maile R. Toll-like receptor 2 and 4 ligation results in complex altered cytokine profiles early and late after burn injury. J Trauma. 2008;64:1069–1077. doi: 10.1097/TA.0b013e318166b7d9. discussion 1077–1078. [DOI] [PubMed] [Google Scholar]
- Choudhry MA, Fazal N, Goto M, Gamelli RL, Sayeed MM. Gutassociated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am J Physiol Gastrointest Liver Physiol. 2002;282:G937–G947. doi: 10.1152/ajpgi.00235.2001. [DOI] [PubMed] [Google Scholar]
- Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantoni A, Duffner LA, De Maria N, Fontanilla CV, Messingham KA, Van Thiel DH, Kovacs EJ. Dose-dependent effect of ethanol on hepatic oxidative stress and interleukin-6 production after burn injury in the mouse. Alcohol Clin Exp Res. 2000;24:1443–1448. [PubMed] [Google Scholar]
- Dolganiuc A, Bakis G, Kodys K, Mandrekar P, Szabo G. Acute ethanol treatment modulates Toll-like receptor-4 association with lipid rafts. Alcohol Clin Exp Res. 2006;30:76–85. doi: 10.1111/j.1530-0277.2006.00003.x. [DOI] [PubMed] [Google Scholar]
- Drost AC, Larsen B, Aulick LH. The effects of thermal injury on serum interleukin 1 activity in rats. Lymphokine Cytokine Res. 1993;12:181–185. [PubMed] [Google Scholar]
- Faunce DE, Gregory MS, Kovacs EJ. Effects of acute ethanol exposure on cellular immune responses in a murine model of thermal injury. J Leukoc Biol. 1997;62:733–740. doi: 10.1002/jlb.62.6.733. [DOI] [PubMed] [Google Scholar]
- Faunce DE, Gregory MS, Kovacs EJ. Glucocorticoids protect against suppression of T cell responses in a murine model of acute ethanol exposure and thermal injury by regulating IL-6. J Leukoc Biol. 1998;64:724– 732. doi: 10.1002/jlb.64.6.724. [DOI] [PubMed] [Google Scholar]
- Faunce DE, Llanas JN, Patel PJ, Gregory MS, Duffner LA, Kovacs EJ. Neutrophil chemokine production in the skin following scald injury. Burns. 1999;25:403–410. doi: 10.1016/s0305-4179(99)00014-5. [DOI] [PubMed] [Google Scholar]
- Fontanilla CV, Faunce DE, Gregory MS, Messingham KA, Durbin EA, Duffner LA, Kovacs EJ. Anti-interleukin-6 antibody treatment restores cell-mediated immune function in mice with acute ethanol exposure before burn trauma. Alcohol Clin Exp Res. 2000;24:1392–1399. [PubMed] [Google Scholar]
- Frink M, Hsieh YC, Thobe BM, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. TLR4 regulates Kupffer cell chemokine production, systemic inflammation and lung neutrophil infiltration following traumahemorrhage. Mol Immunol. 2007;44:2625–2630. doi: 10.1016/j.molimm.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Goral J, Kovacs EJ. In vivo ethanol exposure down-regulates TLR2-, TLR4-, and TLR9-mediated macrophage inflammatory response by limiting p38 and ERK1/2 activation. J Immunol. 2005;174:456–463. doi: 10.4049/jimmunol.174.1.456. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- Hoth JJ, Hudson WP, Brownlee NA, Yoza BK, Hiltbold EM, Meredith JW, McCall CE. Toll-like receptor 2 participates in the response to lung injury in a murine model of pulmonary contusion. Shock. 2007;28:447–452. doi: 10.1097/shk.0b013e318048801a. [DOI] [PubMed] [Google Scholar]
- Hoth JJ, Wells JD, Brownlee NA, Hiltbold EM, Meredith JW, McCall CE, Yoza BK. Toll-like receptor 4-dependent responses to lung injury in a murine model of pulmonary contusion. Shock. 2009;31:376–381. doi: 10.1097/SHK.0b013e3181862279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland J, Hingson R. Alcohol as a risk factor for injuries or death due to fires and burns: review of the literature. Public Health Rep. 1987;102:475–483. [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Liang J, Li Y, Noble PW. The role of Toll-like receptors in non-infectious lung injury. Cell Res. 2006;16:693–701. doi: 10.1038/sj.cr.7310085. [DOI] [PubMed] [Google Scholar]
- Jones MR, Quinton LJ, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. Roles of interleukin-6 in activation of STAT proteins and recruitment of neutrophils during Escherichia coli pneumonia. J Infect Dis. 2006;193:360–369. doi: 10.1086/499312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh MJ, Clark C, Goto M, Kovacs EJ, Gamelli RL, Sayeed MM, Choudhry MA. Effect of acute alcohol ingestion prior to burn injury on intestinal bacterial growth and barrier function. Burns. 2005;31:290–296. doi: 10.1016/j.burns.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Kowal-Vern A, Walenga JM, Hoppensteadt D, Sharp-Pucci M, Gamelli RL. Interleukin-2 and interleukin-6 in relation to burn wound size in the acute phase of thermal injury. J Am Coll Surg. 1994;178:357–362. [PubMed] [Google Scholar]
- Kowal-Vern A, Walenga JM, Sharp-Pucci M, Hoppensteadt D, Gamelli RL. Postburn edema and related changes in interleukin-2, leukocytes, platelet activation, endothelin-1, and C1 esterase inhibitor. J Burn Care Rehabil. 1997;18:99–103. doi: 10.1097/00004630-199703000-00002. [DOI] [PubMed] [Google Scholar]
- Li X, Kovacs EJ, Schwacha MG, Chaudry IH, Choudhry MA. Acute alcohol intoxication increases interleukin-18-mediated neutrophil infiltration and lung inflammation following burn injury in rats. Am J Physiol Lung CellMol Physiol. 2007;292:L1193–L1201. doi: 10.1152/ajplung.00408.2006. [DOI] [PubMed] [Google Scholar]
- Lv T, Shen X, Shi Y, Song Y. TLR4 is essential in acute lung injury induced by unresuscitated hemorrhagic shock. J Trauma. 2009;66:124–131. doi: 10.1097/TA.0b013e318181e555. [DOI] [PubMed] [Google Scholar]
- Magnotti LJ, Deitch EA. Burns, bacterial translocation, gut barrier function, and failure. J Burn Care Rehabil. 2005;26:383–391. doi: 10.1097/01.bcr.0000176878.79267.e8. [DOI] [PubMed] [Google Scholar]
- Marshall JC. SIRS and MODS: what is their relevance to the science and practice of intensive care? Shock. 2000;14:586–589. [PubMed] [Google Scholar]
- Maung AA, Fujimi S, Miller ML, Mac Conmara MP, Mannick JA, Lederer JA. Enhanced TLR4 reactivity following injury is mediated by increased p38 activation. J Leukoc Biol. 2005;78:565–573. doi: 10.1189/jlb.1204698. [DOI] [PubMed] [Google Scholar]
- McGill V, Kowal-Vern A, Fisher SG, Kahn S, Gamelli RL. The impact of substance use on mortality and morbidity from thermal injury. J Trauma. 1995;38:931–934. doi: 10.1097/00005373-199506000-00019. [DOI] [PubMed] [Google Scholar]
- Messingham KA, Fontanilla CV, Colantoni A, Duffner LA, Kovacs EJ. Cellular immunity after ethanol exposure and burn injury: dose and time dependence. Alcohol. 2000;22:35–44. doi: 10.1016/s0741-8329(00)00100-2. [DOI] [PubMed] [Google Scholar]
- Mester M, Carter EA, Tompkins RG, Gelfand JA, Dinarello CA, Burke JF, Clark BD. Thermal injury induces very early production of interleukin-1 alpha in the rat by mechanisms other than endotoxemia. Surgery. 1994;115:588–596. [PubMed] [Google Scholar]
- Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am. 1995;75:257–277. doi: 10.1016/s0039-6109(16)46587-4. [DOI] [PubMed] [Google Scholar]
- Moss M, Bucher B, Moore FA, Moore EE, Parsons PE. The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA. 1996;275:50–54. [PubMed] [Google Scholar]
- Murdoch EL, Brown HG, Gamelli RL, Kovacs EJ. Effects of ethanol on pulmonary inflammation in postburn intratracheal infection. J Burn Care Res. 2008;29:323–330. doi: 10.1097/BCR.0b013e3181667599. [DOI] [PubMed] [Google Scholar]
- Murphy TJ, Paterson HM, Kriynovich S, Zang Y, Kurt-Jones EA, Mannick JA, Lederer JA. Linking the “two-hit” response following injury to enhanced TLR4 reactivity. J Leukoc Biol. 2005;77:16–23. doi: 10.1189/jlb.0704382. [DOI] [PubMed] [Google Scholar]
- Nomellini V, Faunce DE, Gomez CR, Kovacs EJ. An age-associated increase in pulmonary inflammation after burn injury is abrogated by CXCR2 inhibition. J Leukoc Biol. 2008;83:1493–1501. doi: 10.1189/jlb.1007672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oak S, Mandrekar P, Catalano D, Kodys K, Szabo G. TLR2- and TLR4-mediated signals determine attenuation or augmentation of inflammation by acute alcohol in monocytes. J Immunol. 2006;176:7628–7635. doi: 10.4049/jimmunol.176.12.7628. [DOI] [PubMed] [Google Scholar]
- Patel PJ, Faunce DE, Gregory MS, Duffner LA, Kovacs EJ. Elevation in pulmonary neutrophils and prolonged production of pulmonary macrophage inflammatory protein-2 after burn injury with prior alcohol exposure. Am J Respir CellMol Biol. 1999;20:1229–1237. doi: 10.1165/ajrcmb.20.6.3491. [DOI] [PubMed] [Google Scholar]
- Paterson HM, Murphy TJ, Purcell EJ, Shelley O, Kriynovich SJ, Lien E, Mannick JA, Lederer JA. Injury primes the innate immune system for enhanced Toll-like receptor reactivity. J Immunol. 2003;171:1473–1483. doi: 10.4049/jimmunol.171.3.1473. [DOI] [PubMed] [Google Scholar]
- Razonable RR, Henault M, Paya CV. Stimulation of toll-like receptor 2 with bleomycin results in cellular activation and secretion of pro-inflammatory cytokines and chemokines. Toxicol Appl Pharmacol. 2006;210:181–189. doi: 10.1016/j.taap.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Shimamoto A, Pohlman TH, Shomura S, Tarukawa T, Takao M, Shimpo H. Toll-like receptor 4 mediates lung ischemia-reperfusion injury. Ann Thorac Surg. 2006;82:2017–2023. doi: 10.1016/j.athoracsur.2006.06.079. [DOI] [PubMed] [Google Scholar]
- Smith LS, Kajikawa O, Elson G, Wick M, Mongovin S, Kosco-Vilbois M, Martin TR, Frevert CW. Effect of Toll-like receptor 4 blockade on pulmonary inflammation caused by mechanical ventilation and bacterial endotoxin. Exp Lung Res. 2008;34:225–243. doi: 10.1080/01902140802022492. [DOI] [PubMed] [Google Scholar]
- Solomkin JS. Neutrophil disorders in burn injury: complement, cytokines, and organ injury. J Trauma. 1990;30:S80–S85. doi: 10.1097/00005373-199012001-00019. [DOI] [PubMed] [Google Scholar]
- Spector WG, Walters MN, Willoughby DA. Venular and capillary permeability in thermal injury. J Pathol Bacteriol. 1965;90:635–640. doi: 10.1002/path.1700900233. [DOI] [PubMed] [Google Scholar]
- Stengle J, Meyers R, Pyle J, Dries DJ. Neutrophil recruitment after remote scald injury. J Burn Care Rehabil. 1996;17:14–18. doi: 10.1097/00004630-199601000-00006. [DOI] [PubMed] [Google Scholar]
- Szabo G, Dolganiuc A, Dai Q, Pruett SB. TLR4, ethanol, and lipid rafts: a new mechanism of ethanol action with implications for other receptor- mediated effects. J Immunol. 2007;178:1243–1249. doi: 10.4049/jimmunol.178.3.1243. [DOI] [PubMed] [Google Scholar]
- Tompkins RG, Burke JF. Burn therapy 1985: acute management. Intensive CareMed. 1986;12:289–295. doi: 10.1007/BF00261738. [DOI] [PubMed] [Google Scholar]
- Tompkins RG, Remensnyder JP, Burke JF, Tompkins DM, Hilton JF, Schoenfeld DA, Behringer GE, Bondoc CC, Briggs SE, Quinby WC., Jr Significant reductions in mortality for children with burn injuries through the use of prompt eschar excision. Ann Surg. 1988;208:577–585. doi: 10.1097/00000658-198811000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SH, Zhang C, Lasbury ME, Liao CP, Durant PJ, Tschang D, Lee CH. Decreased inflammatory response in Toll-like receptor 2 knockout mice is associated with exacerbated Pneumocystis pneumonia. Microbes Infect. 2008;10:334–341. doi: 10.1016/j.micinf.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HZ, Cui B, Liu HZ, Chen ZR, Yan HM, Hua F, Hu ZW. Targeting TLR2 attenuates pulmonary inflammation and fibrosis by reversion of suppressive immune microenvironment. J Immunol. 2009;182:692–702. doi: 10.4049/jimmunol.182.1.692. [DOI] [PubMed] [Google Scholar]
- Zhou DH, Munster AM, Winchurch RA. Inhibitory effects of interleukin 6 on immunity. Possible implications in burn patients. Arch Surg. 1992;127:65–68. doi: 10.1001/archsurg.1992.01420010079011. discussion 68–69. [DOI] [PubMed] [Google Scholar]