Monomeric Rhodopsin Is Sufficient for Normal Rhodopsin Kinase (GRK1) Phosphorylation and Arrestin-1 Binding (original) (raw)
Abstract
G-protein-coupled receptor (GPCR) oligomerization has been observed in a wide variety of experimental contexts, but the functional significance of this phenomenon at different stages of the life cycle of class A GPCRs remains to be elucidated. Rhodopsin (Rh), a prototypical class A GPCR of visual transduction, is also capable of forming dimers and higher order oligomers. The recent demonstration that Rh monomer is sufficient to activate its cognate G protein, transducin, prompted us to test whether the same monomeric state is sufficient for rhodopsin phosphorylation and arrestin-1 binding. Here we show that monomeric active rhodopsin is phosphorylated by rhodopsin kinase (GRK1) as efficiently as rhodopsin in the native disc membrane. Monomeric phosphorylated light-activated Rh (P-Rh*) in nanodiscs binds arrestin-1 essentially as well as P-Rh* in native disc membranes. We also measured the affinity of arrestin-1 for P-Rh* in nanodiscs using a fluorescence-based assay and found that arrestin-1 interacts with monomeric P-Rh* with low nanomolar affinity and 1:1 stoichiometry, as previously determined in native disc membranes. Thus, similar to transducin activation, rhodopsin phosphorylation by GRK1 and high affinity arrestin-1 binding only requires a rhodopsin monomer.
Keywords: G Protein-coupled Receptors (GPCR), Phototransduction, Protein Kinases, Protein Phosphorylation, Receptor Desensitization, Rhodopsin, Signal Transduction, Arrestin
Introduction
Visual phototransduction is quenched by a two-step mechanism. First, light-activated rhodopsin (Rh*)3 is phosphorylated multiple times by GRK1. Arrestin-14 binding to active phosphorylated rhodopsin (P-Rh*) blocks further transducin activation (1) by steric exclusion (2). The binding of arrestin-1 to P-Rh* is an important molecular mechanism for signal shut-off (3). However, key details of the requirements for physical interaction of arrestin-1 with rhodopsin remain to be explored. Rhodopsin, which is highly concentrated in photoreceptor membranes, has been observed to form arrays of dimers, thus raising the possibility that the dimer is a functional unit (4). Although evidence of a preferred dimer interface has been reported (5), the functional role of rhodopsin oligomers remains controversial (6, 7). Accumulating evidence with other GPCRs indicates that oligomerization could be widespread (8–10). Several models for rhodopsin dimers (11, 12) and monomers (6, 13) interacting with signaling partners transducin, rhodopsin kinase, and arrestin have been proposed. These models need rigorous experimental testing at different steps of the functional cycle of rhodopsin and other class A GPCRs (reviewed in Refs. 7 and 14).
Modified high density lipoprotein particles (nanodiscs) consist of a phospholipid bilayer stabilized by a membrane scaffold protein (MSP) (15–17). These nanodiscs can be used to selectively isolate monomeric GPCRs imbedded into lipid bilayer. We (18) and others (19, 20) have previously demonstrated that rhodopsin monomers incorporated in nanodiscs are highly functional in signaling to transducin. The same was shown for monomeric rhodopsin purified in detergent (dodecyl maltoside), where rhodopsin and transducin form a 1:1 complex (21). However, depending on experimental conditions, receptor dimers, either in nanodiscs containing two rhodopsin molecules or in detergent micelles, were also shown to be capable of forming a complex with a single G-protein (22, 23). In detergent, the leukotriene receptor BLT1 dimer was reported in a complex with a single G protein, although it remains unclear whether G protein directly interacted with one or both receptors (24); the neurotensin NTS1 receptor dimer activated G protein less efficiently than monomers of the same receptor (25).
Rhodopsin was previously shown to bind to arrestin-1 both in vivo and in isolated rod outer segment membranes, saturating at 1:1 stoichiometry (26). Binding to P-Rh* induced disassembly of all arrestin oligomers, demonstrating that only monomeric arrestin-1 can bind P-Rh* (27). This agrees with the finding that well defined rhodopsin-binding surface (28–30) is shielded by sister subunits in the solution tetramer of arrestin-1 (31). Thus, 1:1 stoichiometry cannot be explained by arrestin-1 dimer binding a P-Rh* dimer. However, all previous experiments used rhodopsin in native disc membranes where it could oligomerize, so the possibility that the physiologically relevant interaction involves two arrestin-1 molecules binding a P-Rh* dimer cannot be excluded. In addition, the ability of GRK1 to phosphorylate monomeric rhodopsin has never been tested. Here we show that GRK1 efficiently phosphorylates monomeric rhodopsin. Moreover, we used two different interaction assays utilizing radiolabeled and purified fluorescently labeled arrestin-1 to show that arrestin-1 binds monomeric P-Rh* with physiologically relevant affinity and stoichiometry.
EXPERIMENTAL PROCEDURES
Materials
Membrane scaffolding protein MSP1E3D1 was expressed and purified as described (32). POPC was obtained from Avanti Polar Lipids (Alabaster, AL), and octyl glucoside was obtained from Anatrace (Maumee, OH). Rod outer segments were isolated from frozen dark-adapted bovine retinas (W. L. Lawson Co., Lincoln, NE), as described (33). Urea-stripped disc membranes containing unphosphorylated rhodopsin or rhodopsin phosphorylated by endogenous GRK1 with >95% purity (supplemental Fig. S1) were prepared as described (33). Bovine arrestin-1 mutants with unique cysteines were expressed in E. coli and purified as described (29, 34).
In Vitro Phosphorylation Assay
Recombinant GRK1 was expressed and purified as described (35). Rhodopsin in native disc membranes or POPC nanodiscs with the indicated fraction of POPS (50 μg/ml), was phosphorylated in 20 mm Tris-HCl, pH 7.4, 2 mm MgCl2, 10 μg/ml BSA, 0.5 mm ATP by purified GRK1 (30 μg/ml) for 10 min at 30 °C. Analytical reactions were performed in 15 μl with [γ-32P]ATP (final specific activity 1,100–1,500 cpm/pmol), stopped by the addition of an equal volume of SDS sample buffer, and resolved on 10% PAGE. Gels were stained with Coomassie Blue, dried, and exposed to x-ray film for 1–3 h. Rhodopsin and GRK1 bands were then excised, and the radioactivity was quantified in a liquid scintillation counter. Preparative reactions were performed in a volume of 200 μl (a-15 μl aliquot was added to [γ-32P]ATP, incubated in parallel, and used to determine phosphorylation stoichiometry). Phosphorhodopsin was regenerated by the addition of 2 μl of 10 mm 11-_cis_-retinal followed by incubation in the dark at room temperature for 60 min. P-Rh with 3.2 ± 0.3 mol of phosphate/mol of receptor was used unless otherwise indicated.
Direct Binding Assay with Radiolabeled Arrestin-1
All arrestins were labeled by incorporation of [3H]leucine and [14C]leucine, as described (36), with the specific activity of the mix 1.5–3 Ci/mmol of leucine, resulting in the specific activity 51–102 Ci/mmol (113–226 dpm/fmol) of bovine arrestin-1, which contains 34 leucines. The translation of every mutant used in this study produced a single labeled protein band with the expected mobility on SDS-PAGE. The relative stability of all mutants used in this study was evaluated as described previously (37) and exceeded 90% of wild type bovine arrestin-1. Translated protein was separated from unincorporated labeled leucine by gel filtration on a 2-ml Sephadex G-75 column. Protein fractions were pooled and used in the binding assay. Radiolabeled arrestin (100 fmol) was incubated in 50 mm Tris-HCl, pH 7.5, 0.5 mm MgCl2, 1.5 mm dithiothreitol, 100 mm potassium acetate, and 7.5 pmol (0.3 μg) of P-Rh* in nanodiscs in a final volume of 50 μl for 5 min at 30 °C under room light. The samples were immediately cooled on ice, and P-Rh*-bound and free arrestin-1 was separated on 2-ml Sephadex G-75 columns equilibrated with 10 mm Tris-HCl, pH 7.5, 100 mm NaCl. Nonspecific binding was determined in the presence of equal amount of empty nanodiscs and subtracted (supplemental Fig. S2). Binding to P-Rh* in native disc membranes was also performed as described (36).
Labeling of MSP
MSP1E3D1 with a cysteine engineered in the N-terminal region (D73C) was expressed in E. coli and purified as described (17, 32). MSP (in 10 mm Tris, pH 7.4, 0.1 m NaCl, 0.01% (w/v) NaN3) was labeled with QXL 610 vinyl sulfone, a dye used as a FRET-based quencher of Texas Red and similar long wavelength-emitting fluorophores, as follows. The MSP preparation (200 μm) was reduced by adding 4 eq of tris(2-carboxyethyl)phosphine (Pierce) from a 10 mm stock in water, pH 7.4. The protein solution was adjusted to pH 7.4, and 2 eq of QXL 610 vinyl sulfone (Anaspec, Fremont, CA) were added from a concentrated stock (15.6 mm) in dimethyl sulfoxide. After 30 min at room temperature, an aliquot was taken, and dye was removed on a desalting cartridge (Hi-Trap, GE Healthcare). The extent of labeling was determined using the QXL 610 absorbance maximum (594 nm, 11,000 m−1 cm−1), MSP extinction at 280 nm (29,900 m−1 cm−1), and a correction factor of 1.45 at 280 nm for the dye. At >95% labeling (∼1 h), DTT was added at a final concentration of 10 mm to quench the labeling reaction, and dye was removed by gel filtration on Sephadex G-25 equilibrated with 10 mm Tris, pH 7.4, 0.1 m NaCl, 0.01% NaN3. The stoichiometry of labeling of the MSP (MSP-Q) was calculated to be 1.08 ± 0.02 dye per MSP.
Arrestin Labeling
Arrestin containing a single cysteine at position 348 was labeled with Texas Red maleimide (Invitrogen) as follows. Arrestin was centrifuged at 116,000 × g for 1 h to remove any aggregates. DTT was removed from the arrestin preparation by repeated dilution and concentration with 10 mm HEPES, pH 7.2, 0.1 m NaCl, 1 mm EDTA. Four equivalents of Texas Red maleimide in dimethyl sulfoxide (40 mm, measured using 112,000 m−1 cm−1 in methanol at 584 nm) were added, and the reaction was allowed to proceed for 1 h at room temperature. Excess dye was removed on a column of Sephadex G-25. The extent of labeling was determined by absorption, using the known extinction coefficient at 593 nm of 82,800 m−1 cm−1 in buffer, a correction factor of 0.18 for the dye at 278 nm in buffer, and arrestin extinction of 28,900 m−1 cm−1 at 278 nm (38).
Nanodisc Self-assembly and Purification
All rhodopsin manipulations were performed under dim red light (Eastman Kodak Co. number 2 or GBX-2 filter). Phosphorylated ROS were solubilized with 150 mm β-octyl glucoside for 5 min on ice and centrifuged for 2 min. Nanodiscs were self-assembled using a molar ratio of 140:1:0.1 phospholipid/MSP/rhodopsin at a phospholipid concentration of 13.5 mm to produce monomeric rhodopsin as described (18). Nanodiscs formed from the MSP-Q behaved normally, having the correct size by size exclusion chromatography, and having the correct stoichiometry of phospholipid to MSP (32). The rhodopsin nanodiscs were separated by size exclusion chromatography using 50 mm Hepes, pH 7.4, 0.1 m NaCl, 0.01% NaN3 as mobile phase (18). When Nanodiscs were self-assembled in the presence of detergent-solubilized rhodopsin using an excess of synthetic phospholipid and membrane scaffold protein, monomers of rhodopsin are produced (18). The absorbance spectrum of a concanavalin A affinity-purified preparation confirmed the presence of one rhodopsin per nanodisc (supplemental Fig. S3). Peak fractions were pooled. Rhodopsin was quantified by change in absorbance at 500 nm (40,600 m−1 cm−1) upon complete bleaching in the presence of hydroxylamine.
Arrestin Binding Measurements by FRET
Binding of arrestin to rhodopsin upon photoactivation was measured using the FRET donor-acceptor pair Texas Red and QXL 610. Binding measurements were made using a Hitachi F-3010 fluorometer equipped with temperature control and stirrer. Photoactivating light was introduced from a 100-watt tungsten lamp equipped with electronic shutter through a 530–590-nm bandpass filter and a fiber optic cable. Texas Red was excited at 600 nm, and emission was measured at 620 nm. Rhodopsin bleaching rates were determined by measuring increase in fluorescence of detergent-solubilized rhodopsin at 330 nm upon release of retinal in the presence of hydroxylamine (39). The bleaching rate due to the 600-nm probe beam was found to be 7 × 10−4/min. The rhodopsin bleaching rate constant by the fiber optic illumination was in the range of 0.05 s−1 and measured before each experiment. Binding of arrestin was measured at 25 °C in a rapidly stirred 5-mm path length quartz cell in a volume of 400 μl. Metarhodopsin was varied by timed exposures to bleaching light. Fluorescence was monitored over time and quantified. A custom-built fluorometer was also used for steady-state and kinetic binding measurements. The instrument utilized a xenon arc lamp as the excitation source and a monochromator for wavelength selection. The stirred sample cuvette was held in a thermostatted holder. A photomultiplier (90° geometry) was used for detection after a CGA-630 long pass filter (Newport). The filter was replaced with a UV bandpass filter (Schott UG2) for calibration of the bleaching rate by the actinic light. Data were digitized and stored using a program written in LabView (National Instruments).
Fluorescence Binding Data Analysis
Data were fit to the following quadratic formula,
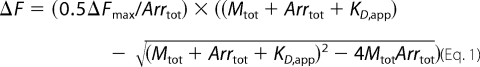
where photoactivated rhodopsin, Mtot, is calculated from the actinic bleaching rate constant, and total arrestin, _Arr_tot, is a known constant. KD and Δ_F_max are fit variables. Bound arrestin is calculated using the equation,

For determination of stoichiometry, data at arrestin ≫ KD were analyzed by plotting 1/(1 − ν) as a function of Rh*/ν/_Arr_tot (40), where ν is the fractional saturation of arrestin, F/Δ_F_max.
RESULTS
Light-dependent Phosphorylation of Monomeric Rhodopsin by Purified GRK1
Rapid and reproducible signal shut-off in vivo requires the phosphorylation of rhodopsin at multiple sites (41). Because the oligomerization state of rhodopsin in native disc membranes remains controversial (4, 6), we compared the efficiency of GRK1 phosphorylation of light-activated rhodopsin from the same preparation in native disc membranes and monomeric Rh* in nanodiscs with varying fractions of negatively charged lipid (Fig. 1). The data show that GRK1, like transducin, does not require negatively charged lipids, phosphorylating monomeric Rh* in nanodiscs containing 0–50% POPS to the same level of ∼3 mol of phosphate/mol, which is required for high affinity arrestin-1 binding in vitro (33) and in vivo (41). If anything, Rh* phosphorylation in native membranes was somewhat less efficient, probably because of rhodopsin crowding in native disc, where it occupies half of the membrane surface, in contrast to rhodopsin monomer in nanodiscs, where each Rh* is fully accessible to GRK1.
FIGURE 1.
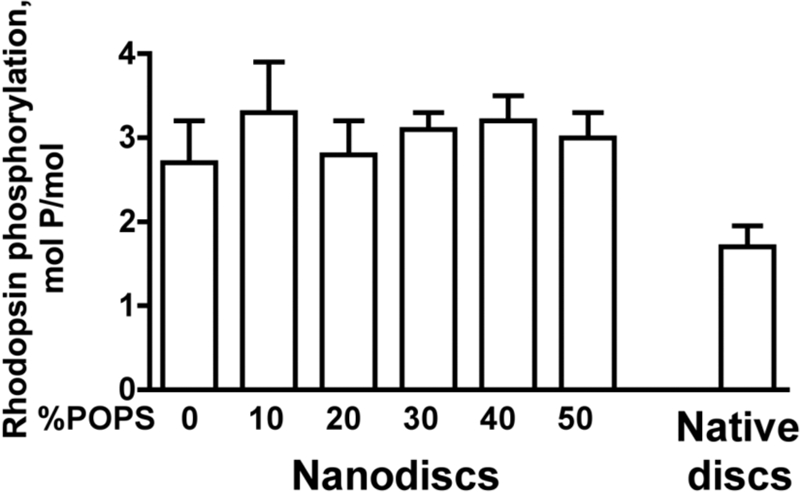
GRK1 efficiently phosphorylates monomeric rhodopsin. Purified rhodopsin from the same batch (50 μg/ml) in original native disc membranes or solubilized and reconstituted into POPC nanodiscs with the indicated fraction of POPS was phosphorylated by purified GRK1 (30 μg/ml) under room light for 10 min at 30 °C. The stoichiometry of phosphorylation was determined, as described under “Experimental Procedures.” Means ± S.D. (error bars) from two experiments performed in duplicate are shown.
Direct Binding Assay with Radiolabeled Arrestin-1
We have previously shown that cell-free translation yields fully functional arrestin-1 that can be radiolabeled by incorporation of [3H]Leu and [14C]Leu (42). Arrestin-1 bound to P-Rh*-containing native disc membranes and free arrestin-1 can be easily separated by gel filtration on Sepharose 2B (43). However, nanodiscs containing a single P-Rh* are much smaller. Thus, to achieve efficient separation of bound and free arrestin-1, we used Sephadex G75 with a much smaller pore size (supplemental Fig. S2). To directly compare arrestin-1 binding to P-Rh* in different preparations, we used the same batch of P-Rh* for direct binding in native disc membranes and nanodiscs. We found that at 0.3 μg of P-Rh* per assay, the binding of arrestin-1 in both preparations was essentially the same (Fig. 2A). Interestingly, when the amount of P-Rh* was reduced, monomeric P-Rh* outperformed native discs (Fig. 2B), with the difference progressively increasing with decreasing P-Rh* concentration. Similar to phosphorylation (Fig. 1), this is probably a kinetic effect because P-Rh* in native discs is crowded, whereas arrestin-1 access to well dispersed monomeric P-Rh* in nanodiscs is unimpeded. At physiological temperature, the active state of rhodopsin, metarhodopsin II, decays to opsin with a half-life of ∼49 s (44). Therefore, arrestin-1 and P-Rh* cannot be incubated long enough for the binding to reach equilibrium (36), so that the resulting binding level reflects the on-rate, which depends on P-Rh* accessibility. Thus, arrestin-1 apparently binds monomeric P-Rh* in nanodiscs at least as efficiently as P-Rh* in native disc membranes.
FIGURE 2.
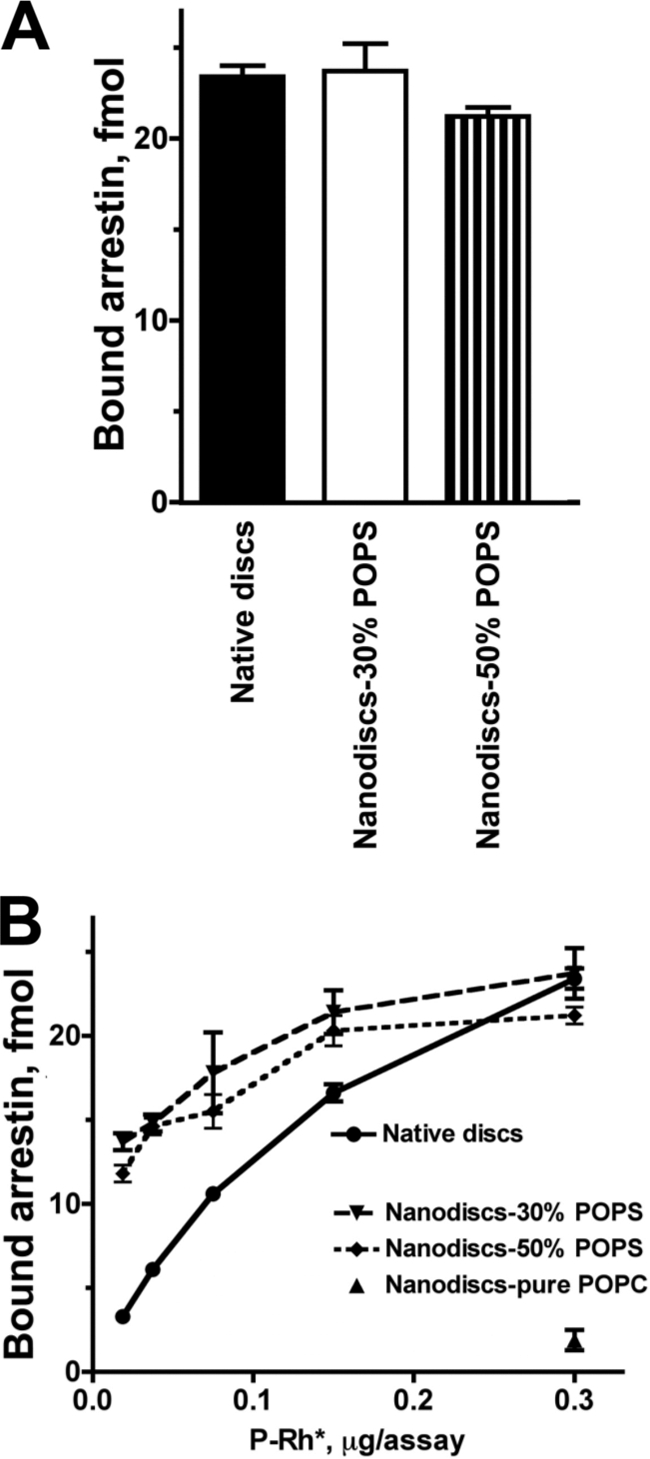
Arrestin-1 binding to monomeric P-Rh*. A, phosphorhodopsin from the same batch (0.3 μg) in native disc membranes or solubilized and reconstituted in POPC nanodiscs with 30 or 50% POPS was incubated with radiolabeled arrestin-1 (100 fmol) in 50 μl at 30 °C. The samples were cooled on ice, and bound and free arrestin-1 was separated, as described under “Experimental Procedures.” B, the same assay was performed with the indicated amounts of P-Rh*. Means ± S.D. from three experiments performed in duplicate are shown.
We also noticed that arrestin-1 binding to P-Rh* in nanodiscs containing pure POPC is much lower than to preparations with negatively charged lipids (Fig. 2B). To determine whether this effect is simply electrostatic or reflects arrestin-1 requirement for specific lipids, we tested the POPS dependence of arrestin-1 binding to monomeric P-Rh* at different ionic strengths (Fig. 3A). The binding reaches 75–100% of maximum at 20–50% POPS, which approximates the content of negatively charged lipids in native discs equivalent to 20–30% POPS. In agreement with our previous findings on arrestin-1 interactions with P-Rh* in native disc membranes (42), salt concentrations greater than 200 mm reduced maximum arrestin-1 binding at 20–50% POPS. Interestingly, increased salt concentration in the assay in the range of 150–450 mm progressively reduced the optimal fraction of negatively charged lipid, with the observed maximum binding gradually shifting from 50 to 20% POPS (Fig. 3B). These data suggest that electrostatic repulsion between arrestin-1, which has multiple positive charges on its rhodopsin-binding surface (reviewed in Ref. 45), and pure POPC membrane is the major contributor to the observed POPS effect on the binding, although we cannot rule out a more specific contribution of negatively charged lipids. To validate our binding data by an independent method and determine precise kinetic parameters of arrestin-1-P-Rh* interaction, we developed a fluorescence-quenching assay with purified arrestin-1 (Fig. 4A).
FIGURE 3.

The dependence of arrestin-1 binding to P-Rh* on negatively charged lipids is reduced by increased ionic strength. A, the binding of radiolabeled arrestin-1 to P-Rh* (0.3 μg) in POPC nanodiscs with the indicated fraction of POPS was performed, as in Fig. 2, at varying total salt concentration (50 mm Tris-HCl, pH 7.4, supplemented with 100, 200, 300, or 400 mm sodium acetate, pH 7.4). Means ± S.D. of two experiments performed in duplicate are shown. B, to facilitate comparison, means from A are plotted as a percentage of maximum binding at each salt concentration (which is reduced by increasing ionic strength).
FIGURE 4.

Fluorescence-based assay of arrestin-1 binding to monomeric P-Rh* in nanodiscs. A, purified recombinant arrestin-1 with unique cysteine in the C-terminal domain (A348C) was covalently labeled with Texas Red maleimide, and the nanodisc MSP is covalently modified with a quenching group. Free arrestin-1 demonstrates bright fluorescence, whereas the signal from arrestin-1 bound to light-activated P-Rh* in nanodisc is quenched by fluorescence energy transfer. B, change of A348C-Texas Red fluorescence upon photoactivation of P-Rh monomer in nanodiscs. Texas Red fluorescence was monitored at 620 nm. Two-second flashes of light were used to photoactivate rhodopsin (asterisks). The addition of hydroxylamine (indicated by the arrow) released bound arrestin-1 and restored fluorescence.
Fluorescence Binding Assays
Recombinant arrestin-1 with a single cysteine in position 348 was produced using a construct in which the three wild-type cysteines were eliminated (29). The cysteine-free recombinant arrestin-1 has been shown to behave identically to wild-type arrestin-1 (29). We chose position A348C (Fig. 4) for labeling because it was previously shown (29) that spin label at A348C does not affect binding affinity. Moreover, the mobility of the nitroxide spin label at A348C upon interaction with P-Rh* does not change, demonstrating that this residue is outside of the arrestin-P-Rh* interface (29). A348C is localized on the C-terminal domain on the solvent-exposed concave face of the β-strand region and therefore also lies near the rhodopsin/nanodisc structure containing the QXL quenching groups on MSP. The calculated stoichiometry of labeling was 1.07 ± 0.02 Texas Red dye per arrestin. The UV fluorescence spectrum of the labeled arrestin exhibited the tyrosine fluorescence characteristic of native non-denatured arrestin (46).
Texas Red was chosen as a long wavelength-absorbing fluorophore to minimize photobleaching of rhodopsin by the probe beam. The QXL 610 absorbance completely overlaps with the maleimide-conjugated arrestin-Texas Red emission, providing a fluorescence-quenching assay for arrestin binding (Fig. 4A). Because two MSPs form the self-assembled nanodisc structure, each nanodisc contains two QXL quenching groups. When a solution of labeled arrestin and nanodiscs containing P-Rh monomer are exposed to short pulses of light, converting it to P-Rh*, the result is a time-dependent decrease in Texas Red fluorescence intensity, as shown in Fig. 4B. Hydroxylamine reacts with the all-_trans_-retinal Schiff base in P-Rh*, releasing retinal oxime and inducing the dissociation of arrestin-1 from opsin (47). The addition of hydroxylamine at the end of the titration resulted in the recovery of fluorescence. No light-dependent fluorescence change was observed with non-phosphorylated rhodopsin, consistent with the requirement of phosphorylation for the high affinity arrestin-1 binding (33, 43, 48, 49).
Affinity of Association and Stoichiometry of the Complex
When arrestin-1 was titrated with photoactivated P-Rh* monomer, binding isotherms with an observed apparent dissociation constant (KD) of 3–4 nm were obtained (Fig. 5A). The second order association rate constant obtained from the kinetic traces (Fig. 5A, inset) after the first light exposure was 1.6 × 106 m−1 s−1. Based on the measured association rate and measured KD of 4 nm, the calculated half-life of the complex is ∼160 s, somewhat longer than the decay time of metarhodopsin II to opsin in intact rods, ∼50 s (45). High arrestin-1 concentration (230 nm), well above the observed KD of 4 nm, was used to determine the stoichiometry of interaction (Fig. 5B). The curve fits a 1:1 complex formed between arrestin-1 and monomeric P-Rh*. Linear transformation of the data (40), shown in the inset, also indicates a 1:1 stoichiometry of interaction. Thus, the binding of monomeric P-Rh* to arrestin-1 demonstrates high affinity and 1:1 stoichiometry characteristic of physiologically relevant interaction of arrestin-1 with P-Rh* in native disc membranes (26, 50, 51).
FIGURE 5.

Affinity and stoichiometry of arrestin-1 interaction with monomeric P-Rh* in nanodiscs. A, arrestin-1 binding isotherms. Measurements were taken at 25 °C with 40 nm total arrestin-1 (A348C mutant labeled with Texas Red as in Fig. 4) and 196 nm rhodopsin monomer in POPC nanodiscs. Calibrated light exposures were used to titrate in photoactivated rhodopsin. Three separate experiments are shown with fitted KD of 2.5 ± 0.3 nm (triangles), 4.2 ± 0.4 nm (open circles), or 4.0 ± 0.2 nm (circles). The inset shows the fluorescence transient from the first light exposure, fit to a second order reaction. B, titration of arrestin with photoactivated rhodopsin. Measurements were made at a total arrestin concentration of 230 nm and P-Rh* monomer nanodiscs at a concentration of 510 nm. Photoactivated P-Rh* was titrated in, using calibrated light exposures. KD from fitting of the curve is 16 nm. The inset shows transformed data with an x intercept of 1.0, representing the number of arrestin binding sites per P-Rh* monomer.
The Role of Negatively Charged Lipids in Arrestin-1 Binding to P-Rh*
We used the same FRET-based assay to identify the binding parameters affected by the negatively charged lipids in nanodiscs (Fig. 6). In native disc membranes (42) and mixed liposomes with physiological distribution of anionic and zwitterionic charged lipids (33), arrestin-1 needs three phosphates per rhodopsin for high affinity binding. Further increase in the level of rhodopsin phosphorylation does not have an appreciable effect on arrestin-1 binding levels (33). To test whether the contribution of additional rhodopsin-attached phosphates can compensate for the deficit of negative charges on lipid headgroups, we used two P-Rh preparations with phosphorylation levels of 2.9 and 4.7 phosphates/rhodopsin. Our analysis revealed that increasing POPS content from 0 to 50% progressively increases the on-rate of arrestin-1 in both cases (Fig. 6B), with a resulting decrease of apparent KD from ∼30 nm in pure POPC to ∼5 nm at 20–50% POPS (Fig. 6A). These data suggest that the primary function of negatively charged lipids in rhodopsin-containing membrane is to accelerate arrestin-1 binding to monomeric P-Rh*. Given that the positively charged surface of arrestin-1 implicated in receptor interaction (28–30, 49, 52, 53) must face the plane of the membrane to allow binding, these results are consistent with the idea that electrostatic repulsion by the cationic choline group at the water-headgroup interface slows down arrestin-1 binding in pure POPC.
FIGURE 6.

The effect of negatively charged lipids on the affinity of arrestin-1 for P-Rh*. A, dissociation constants obtained from the fluorescence binding assay as a function of POPS content of the nanodiscs. Two preparations of rhodopsin with different phosphorylation levels (as determined by [γ-32P]ATP incorporation) were used in the nanodisc assembly. Binding reactions contained 50 nm arrestin and 400 nm rhodopsin. B, pseudo-first order kinetic constants were determined from exponential fit to the binding time course. Reactions contained 25 nm arrestin. 184 nm light-activated rhodopsin in nanodiscs was generated by a 2.5-s light exposure.
Multiple positively charged residues on the receptor-binding arrestin-1 surface were implicated in direct interactions with receptor-attached phosphates and therefore cannot be removed without reducing P-Rh* binding (54–58). However, the removal of Lys14 or Lys15 in the context of the phosphorylation-independent R175E mutant (55, 57), where the polar core (the main phosphate sensor in all arrestins (58–60)) is destabilized by the charge reversal, yields proteins that bind P-Rh* fairly well (56). Therefore, we compared binding dependence on the fraction of POPS for WT arrestin-1, R175E mutant, and combination mutants K14A/R175E and K15A/R175E. We found that “preactivation” by the R175E mutation and elimination of individual exposed positive charges on the receptor-binding surface of arrestin-1 do not dramatically change POPS dependence of its binding to monomeric P-Rh* (supplemental Fig. S4). Arrestin-1 can also be preactivated by the disruption of another intramolecular interaction that anchors the C-tail to the body of the N-domain. This can be achieved by either C-terminal truncation or the detachment of the C-tail by triple alanine substitution of the anchoring hydrophobic residues (37, 42, 43, 53, 56, 61). To test whether structurally distinct phosphorylation-independent arrestin mutants show the same dependence on negatively charged lipids, we compared WT arrestin-1 with two types of polar core mutants, R175E and D296R, in which the same key Arg175–Asp296 salt bridge is destroyed by charge reversals on either end, and two mutants with an intact polar core, in which the C-tail is either deleted or detached (arrestin-1-(1–378) and arrestin-1 (F375A,V376A,F377A) (3A), respectively). Unexpectedly, we found that these two types of mutants have dramatically different phospholipid requirements (Fig. 7A). Both polar core mutations increase arrestin-1 binding in pure POPC from 10% of maximum in WT protein to 20–25%. In contrast, the deletion or detachment of the C-tail increases the binding to ∼60% of maximum and significantly reduces the fraction of POPS required for the maximum binding (Fig. 7B).
FIGURE 7.

Activating mutations differentially change the dependence of arrestin-1 binding to P-Rh* on negatively charged lipids. A, the binding of radiolabeled WT arrestin-1, two polar core mutants (R175E and D296R), and two mutants where the C-tail is either deleted (Tr(1–378)) or detached (arrestin-1 (F375A,V376A,F377A) (3A)) to P-Rh* (0.3 μg) in POPC nanodiscs with the indicated fraction of POPS was performed, as in Fig. 2. Means ± S.D. of two experiments performed in duplicate are shown. B, to facilitate comparison, means are plotted as a percentage of maximum binding of each form of arrestin-1 (which is significantly increased by activating mutations). C, sequence alignment of the C termini of four arrestins with negatively charged residues shown in red. Other highlights are as follows: three bulky hydrophobic residues anchoring the C-tail to the N-domain via β-strand I and α-helix I (olive); arginine that is part of the main phosphate sensor, the polar core (light blue); and the main clathrin-binding site in arrestin-2 (present in all vertebrate non-visual arrestins) (underlined).
In the basal state, the C-tail occupies the cavity on the receptor-binding side of the arrestin N-domain (58–60). Receptor binding displaces the arrestin C-tail (29, 53, 62, 63). The C termini of all arrestin proteins carry multiple negative charges (Fig. 7C) (64), the functional role of which has never been elucidated. Our data suggest that these acidic residues help negatively charged membrane lipids to displace the arrestin C-tail, facilitating receptor binding. In the non-visual arrestin-2, polar core mutation R169E (homologous to R175E in arrestin-1) and the detachment of the C-tail by triple alanine substitution induce similar increases in the C-tail accessibility measured by hydrogen/deuterium exchange (65). Therefore, it appears likely that polar core mutations in arrestin-1 reduce POPS dependence of the binding by indirectly facilitating the release of the C-tail, albeit less efficiently than its direct detachment by the arrestin-1 (F375A,V376A,F377A) mutation.
DISCUSSION
Despite numerous reports that rhodopsin and other class A GPCRs form dimers and higher order oligomers (reviewed in Ref. 11), the biological significance of this phenomenon remains unclear. Because every GPCR has a complex multistep life cycle, its oligomerization state is likely to be quite different at various stages (7, 14). Recent reports from several groups show that monomeric rhodopsin is fully capable of activating its cognate G protein, transducin (18–21), and that forced dimerization of rhodopsin reduces its activity ∼2-fold (18, 20). The next two steps in the physiological sequence of rhodopsin function are its light-dependent phosphorylation by GRK1 followed by arrestin-1 binding to P-Rh*. Therefore, we determined the ability of monomeric rhodopsin to serve as a substrate of GRK1 and to bind arrestin-1.
We found that GRK1 phosphorylates monomeric Rh* in nanodiscs at least as efficiently as Rh* in native disc membranes, ruling out the requirement of rhodopsin dimer for this process. Our results also demonstrate that arrestin-1 binds P-Rh* with the same high affinity in native disc membranes and in monomeric form in nanodiscs. In fact, at lower P-Rh* concentrations, monomeric P-Rh* binds arrestin-1 more efficiently than P-Rh* in native membranes. This is probably due to better accessibility of well dispersed rhodopsin monomer than of overcrowded P-Rh* in the native disc. The binding of fluorescently labeled purified arrestin-1 to P-Rh* monomer demonstrates apparent KD of 4–5 nm. The published apparent KD measured using phosphorylated rhodopsin in native membranes was 20–50 nm (at 4–13.7 °C) (50, 51), which is consistent with the value of 4–5 nm at 25 °C obtained here due to the high activation energy that results in strong temperature dependence (50). Similarly, the association rate constants for arrestin-1 binding to monomeric P-Rh*, which we determined to be ∼106 m−1 s−1 at 25 °C, is consistent with the rate of association measured in native discs (50). A recent study using an extra-metarhodopsin II assay at 10 °C (66) also demonstrated that arrestin-1 binds monomeric P-Rh* in nanodiscs but reported a 2-order of magnitude lower apparent affinity of arrestin-1 for P-Rh* (EC50 ∼1.4 μm). Dr. Hofmann's group previously used an extra-metarhodopsin II assay at low temperatures with native discs and reported a KD of 20 nm (51) or 50 nm (50), consistent with our measurements. Moreover, the group of Dr. Weiss (67) also measured a KD of ∼1 nm for arrestin-1 binding to P-Rh*. Arrestin-1 does not dissociate before rhodopsin decays, eliminating the possibility of rhodopsin reactivation in WT photoreceptors (68). Thus, the half-life of the arrestin-1-P-Rh* complex has to be longer than the half-time of rhodopsin decay. The latter is ∼50 s in vivo (44). Thus, the shortest half-life of the arrestin-1-P-Rh* complex consistent with the biology must be in the range of minutes. Considering that KD = _k_off/_k_on and that _k_on of protein-protein interactions measured here (Fig. 6B) and by others (50, 69) is ∼106 m−1 s−1, KD of ∼3–5 nm yields a half-life of ∼3 min, consistent with the reproducibly observed fidelity of arrestin-1-mediated shut-off of rhodopsin signaling.
The 1:1 stoichiometry of the arrestin-1 complex with monomeric P-Rh* is in agreement with physiologically relevant stoichiometry recently determined in mouse retina and isolated disc membranes containing P-Rh* (26). In that study, the arrestin/rhodopsin expression ratio was varied from 0.4 to 2.7 in genetically modified mice, and the same arrestin/rhodopsin ratios were reproduced with only two purified proteins present in the test tube. In both cases, arrestin-1 saturated rhodopsin at a ∼1:1 molar ratio (26). The well defined receptor-binding surface of arrestin-1 is shielded by sister subunits in the physiological solution tetramer and both possible dimers (31). Indeed, only monomeric arrestin-1 can bind rhodopsin (27). Thus, that study demonstrated that each rhodopsin molecule binds its own arrestin, in contrast to the model where a rhodopsin dimer binds a single arrestin molecule (11). However, in previously reported experiments, in vivo and in vitro, with rhodopsin in native disc membranes, the possibility exists that a rhodopsin dimer participates in arrestin-1 binding. Two models involving dimers could account for the data. First, the P-Rh*/P-Rh* dimer could bind two arrestin-1 molecules rather than one. Second, arrestin binding stoichiometry in native membranes could depend on the fraction of activated rhodopsin, so that at low bleaching levels, a P-Rh/P-Rh* dimer can interact with one arrestin-1 molecule. The data presented here demonstrate that the P-Rh* monomer binds arrestin-1 with nanomolar affinity and 1:1 stoichiometry. Although our results cannot formally exclude the existence of binding modes involving rhodospin dimers, the data presented here rule out the requirement for rhodopsin dimerization for physiologically relevant arrestin-1 binding to P-Rh*.
The importance of negatively charged lipids for arrestin-1 binding was noted previously (47, 66), but the mechanism of their action was not understood. Our analysis of POPS dependence of the binding of a variety of structurally distinct arrestin-1 mutants showed that negative charges on the surface of the membrane assist in pushing the negatively charged arrestin C-tail out of the receptor-binding cavity of the arrestin N-domain, thereby facilitating arrestin-1 association with P-Rh*. This mechanism provides a logical explanation for the abundance of negative charges in the C termini of arrestin proteins from Caenorhabditis elegans to mammals (64). The release of the C-tail is an important part of the receptor binding-induced conformational rearrangement in the arrestin molecule (29, 53). In the case of non-visual arrestin-2 and -3, this event exposes the clathrin and AP2 binding sites in the arrestin C-tail, which serve to recruit the arrestin-receptor complex to the coated pit (reviewed in Ref. 70).
To obtain these results, we have developed a new assay to evaluate GPCR-arrestin interaction, where labeled arrestin binding to monomeric unlabeled receptor is quantified by fluorescence. This assay has proven useful for measuring the affinity, stoichiometry, and kinetics of binding. Our data show that nanodiscs are efficient tools for generating unambiguously monomeric GPCRs for experimental testing of their functional capabilities. This assay system can greatly facilitate detailed exploration of the dependence of binding parameters on the extent of rhodopsin phosphorylation. The same approach can be used for quantitative studies of the interaction of other members of the arrestin family with non-visual GPCRs, other receptor-binding proteins, or any protein-protein interaction where one partner is a membrane protein reconstituted into nanodiscs.
Vertebrate rod photoreceptors demonstrate single photon sensitivity (71). Thus, it makes biological sense that a single light-activated rhodopsin molecule is necessary and sufficient for effective activation of transducin. Rhodopsin signaling in vivo is terminated with sub-second kinetics by rapid phosphorylation followed by high affinity arrestin-1 binding to P-Rh* (41, 72, 73). Arrestin-1 directly competes with transducin for light-activated rhodopsin (1, 2), shutting down the signaling by a simple steric exclusion. Thus, biologically, it also makes sense that the same molecular species, the rhodopsin monomer, is phosphorylated by GRK1 and binds arrestin-1. Interestingly, a recent study showed that in Drosophila, arrestin also binds rhodopsin at 1:1 stoichiometry in vivo (80).
The recent findings that monomeric forms of several other class A GPCRs, such as β2-adrenergic receptor (74, 75) and NTS1 receptor (25), effectively activate their cognate G proteins calls for the exploration of the ability of monomeric receptors to be phosphorylated by their cognate GRKs and interact with non-visual arrestins. Although the crystal structures of three vertebrate GRKs (12, 76–78) and three arrestin subtypes (58–60, 79) show remarkable similarities within each family, additional experiments are necessary to determine whether all GRKs and arrestins interact with monomeric forms of their cognate receptors. As far as rhodopsin is concerned, the monomeric light-activated rhodopsin appears to be the minimal functional unit in G protein-mediated signaling and GRK- and arrestin-mediated desensitization.
Supplementary Material
Supplemental Fig. 1 (.pdf, 864 KB)
Acknowledgments
We thank Dr. Rosalie K. Crouch and the NEI, National Institutes of Health, for 11-cis-retinal and Sharon Fluss for preparing phosphorylated rhodopsin.
*
This work was supported, in whole or in part, by National Institutes of Health Grants EY011500, GM081756, and GM077561 (to V. V. G.), GM033775 (to S. G. S.), and HL071818 (to J. J. T.). This work was also supported by Deutsche Forschungsgemeinschaft Grants Sfb740 and ER 294/1-1 (to O. P. E.).
4
We use systematic names for the arrestin proteins: arrestin-1 (historically called S-antigen, 48-kDa protein, and visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2), and arrestin-4 (cone or X-arrestin).
3
The abbreviations used are:
Rh*
light-activated rhodopsin
Rh
dark (inactive) rhodopsin
P-Rh
phosphorylated Rh
P-Rh*
phosphorylated Rh*
GPCR
G-protein-coupled receptor
MSP
membrane scaffold protein
MSP-Q
QXL 610-labeled MSP1E3D1
POPC
1-palmitoyl-2-oleoyl phosphatidylcholine
POPS
1-palmitoyl-2-oleoyl phosphatidyl-l-serine.
REFERENCES
- 1.Wilden U., Hall S. W., Kühn H. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 1174–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krupnick J. G., Gurevich V. V., Benovic J. L. (1997) J. Biol. Chem. 272, 18125–18131 [DOI] [PubMed] [Google Scholar]
- 3.Xu J., Dodd R. L., Makino C. L., Simon M. I., Baylor D. A., Chen J. (1997) Nature 389, 505–509 [DOI] [PubMed] [Google Scholar]
- 4.Fotiadis D., Liang Y., Filipek S., Saperstein D. A., Engel A., Palczewski K. (2003) Nature 421, 127–128 [DOI] [PubMed] [Google Scholar]
- 5.Kota P., Reeves P. J., Rajbhandary U. L., Khorana H. G. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 3054–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chabre M., le Maire M. (2005) Biochemistry 44, 9395–9403 [DOI] [PubMed] [Google Scholar]
- 7.Gurevich V. V., Gurevich E. V. (2008) Trends Neurosci. 31, 74–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han Y., Moreira I. S., Urizar E., Weinstein H., Javitch J. A. (2009) Nat. Chem. Biol. 5, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pascal G., Milligan G. (2005) Mol. Pharmacol. 68, 905–915 [DOI] [PubMed] [Google Scholar]
- 10.Rivero-Müller A., Chou Y. Y., Ji I., Lajic S., Hanyaloglu A. C., Jonas K., Rahman N., Ji T. H., Huhtaniemi I. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 2319–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fotiadis D., Jastrzebska B., Philippsen A., Müller D. J., Palczewski K., Engel A. (2006) Curr. Opin. Struct. Biol. 16, 252–259 [DOI] [PubMed] [Google Scholar]
- 12.Singh P., Wang B., Maeda T., Palczewski K., Tesmer J. J. (2008) J. Biol. Chem. 283, 14053–14062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurevich V. V., Gurevich E. V. (2004) Trends Pharmacol. Sci. 25, 105–111 [DOI] [PubMed] [Google Scholar]
- 14.Gurevich V. V., Gurevich E. V. (2008) Trends Pharmacol. Sci. 29, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bayburt T. H., Sligar S. G. (2010) FEBS Lett. 584, 1721–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nath A., Atkins W. M., Sligar S. G. (2007) Biochemistry 46, 2059–2069 [DOI] [PubMed] [Google Scholar]
- 17.Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., Sligar S. G. (2009) Methods Enzymol. 464, 211–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayburt T. H., Leitz A. J., Xie G., Oprian D. D., Sligar S. G. (2007) J. Biol. Chem. 282, 14875–14881 [DOI] [PubMed] [Google Scholar]
- 19.Whorton M. R., Jastrzebska B., Park P. S., Fotiadis D., Engel A., Palczewski K., Sunahara R. K. (2008) J. Biol. Chem. 283, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banerjee S., Huber T., Sakmar T. P. (2008) J. Mol. Biol. 377, 1067–1081 [DOI] [PubMed] [Google Scholar]
- 21.Ernst O. P., Gramse V., Kolbe M., Hofmann K. P., Heck M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 10859–10864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jastrzebska B., Golczak M., Fotiadis D., Engel A., Palczewski K. (2009) FASEB J. 23, 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jastrzebska B., Goc A., Golczak M., Palczewski K. (2009) Biochemistry 48, 5159–5170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banères J. L., Parello J. (2003) J. Mol. Biol. 329, 815–829 [DOI] [PubMed] [Google Scholar]
- 25.White J. F., Grodnitzky J., Louis J. M., Trinh L. B., Shiloach J., Gutierrez J., Northup J. K., Grisshammer R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 12199–12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanson S. M., Gurevich E. V., Vishnivetskiy S. A., Ahmed M. R., Song X., Gurevich V. V. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 3125–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanson S. M., Van Eps N., Francis D. J., Altenbach C., Vishnivetskiy S. A., Arshavsky V. Y., Klug C. S., Hubbell W. L., Gurevich V. V. (2007) EMBO J. 26, 1726–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanson S. M., Gurevich V. V. (2006) J. Biol. Chem. 281, 3458–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanson S. M., Francis D. J., Vishnivetskiy S. A., Kolobova E. A., Hubbell W. L., Klug C. S., Gurevich V. V. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 4900–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vishnivetskiy S. A., Hosey M. M., Benovic J. L., Gurevich V. V. (2004) J. Biol. Chem. 279, 1262–1268 [DOI] [PubMed] [Google Scholar]
- 31.Hanson S. M., Dawson E. S., Francis D. J., Van Eps N., Klug C. S., Hubbell W. L., Meiler J., Gurevich V. V. (2008) Structure 16, 924–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denisov I. G., Grinkova Y. V., Lazarides A. A., Sligar S. G. (2004) J. Am. Chem. Soc. 126, 3477–3487 [DOI] [PubMed] [Google Scholar]
- 33.Vishnivetskiy S. A., Raman D., Wei J., Kennedy M. J., Hurley J. B., Gurevich V. V. (2007) J. Biol. Chem. 282, 32075–32083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanson S. M., Francis D. J., Vishnivetskiy S. A., Klug C. S., Gurevich V. V. (2006) J. Biol. Chem. 281, 9765–9772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang C. C., Yoshino-Koh K., Tesmer J. J. (2009) J. Biol. Chem. 284, 17206–17215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gurevich V. V., Benovic J. L. (2000) Methods Enzymol. 315, 422–437 [DOI] [PubMed] [Google Scholar]
- 37.Gurevich V. V. (1998) J. Biol. Chem. 273, 15501–15506 [DOI] [PubMed] [Google Scholar]
- 38.Buczylko J., Palczewski K. (1993) Methods Neurosci. 15, 226–236 [Google Scholar]
- 39.Farrens D. L., Khorana H. G. (1995) J. Biol. Chem. 270, 5073–5076 [DOI] [PubMed] [Google Scholar]
- 40.Lodola A., Spragg S. P., Holbrook J. J. (1978) Biochem. J. 169, 577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mendez A., Burns M. E., Roca A., Lem J., Wu L. W., Simon M. I., Baylor D. A., Chen J. (2000) Neuron 28, 153–164 [DOI] [PubMed] [Google Scholar]
- 42.Gurevich V. V., Benovic J. L. (1993) J. Biol. Chem. 268, 11628–11638 [PubMed] [Google Scholar]
- 43.Gurevich V. V., Benovic J. L. (1992) J. Biol. Chem. 267, 21919–21923 [PubMed] [Google Scholar]
- 44.Shi G., Yau K. W., Chen J., Kefalov V. J. (2007) J. Neurosci. 27, 10084–10093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gurevich V. V., Gurevich E. V. (2006) Pharmacol. Ther. 110, 465–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kotake S., Hey P., Mirmira R. G., Copeland R. A. (1991) Arch. Biochem. Biophys. 285, 126–133 [DOI] [PubMed] [Google Scholar]
- 47.Sommer M. E., Smith W. C., Farrens D. L. (2006) J. Biol. Chem. 281, 9407–9417 [DOI] [PubMed] [Google Scholar]
- 48.Kühn H., Hall S. W., Wilden U. (1984) FEBS Lett. 176, 473–478 [DOI] [PubMed] [Google Scholar]
- 49.Gurevich V. V., Dion S. B., Onorato J. J., Ptasienski J., Kim C. M., Sterne-Marr R., Hosey M. M., Benovic J. L. (1995) J. Biol. Chem. 270, 720–731 [DOI] [PubMed] [Google Scholar]
- 50.Schleicher A., Kühn H., Hofmann K. P. (1989) Biochemistry 28, 1770–1775 [DOI] [PubMed] [Google Scholar]
- 51.Pulvermüller A., Maretzki D., Rudnicka-Nawrot M., Smith W. C., Palczewski K., Hofmann K. P. (1997) Biochemistry 36, 9253–9260 [DOI] [PubMed] [Google Scholar]
- 52.Pulvermüller A., Schroder K., Fischer T., Hofmann K. P. (2000) J. Biol. Chem. 275, 37679–37685 [DOI] [PubMed] [Google Scholar]
- 53.Vishnivetskiy S. A., Francis D., Van Eps N., Kim M., Hanson S. M., Klug C. S., Hubbell W. L., Gurevich V. V. (2010) J. Mol. Biol. 395, 42–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gurevich V. V., Benovic J. L. (1995) J. Biol. Chem. 270, 6010–6016 [DOI] [PubMed] [Google Scholar]
- 55.Gurevich V. V., Benovic J. L. (1997) Mol. Pharmacol. 51, 161–169 [DOI] [PubMed] [Google Scholar]
- 56.Vishnivetskiy S. A., Schubert C., Climaco G. C., Gurevich Y. V., Velez M. G., Gurevich V. V. (2000) J. Biol. Chem. 275, 41049–41057 [DOI] [PubMed] [Google Scholar]
- 57.Vishnivetskiy S. A., Paz C. L., Schubert C., Hirsch J. A., Sigler P. B., Gurevich V. V. (1999) J. Biol. Chem. 274, 11451–11454 [DOI] [PubMed] [Google Scholar]
- 58.Sutton R. B., Vishnivetskiy S. A., Robert J., Hanson S. M., Raman D., Knox B. E., Kono M., Navarro J., Gurevich V. V. (2005) J. Mol. Biol. 354, 1069–1080 [DOI] [PubMed] [Google Scholar]
- 59.Hirsch J. A., Schubert C., Gurevich V. V., Sigler P. B. (1999) Cell 97, 257–269 [DOI] [PubMed] [Google Scholar]
- 60.Han M., Gurevich V. V., Vishnivetskiy S. A., Sigler P. B., Schubert C. (2001) Structure 9, 869–880 [DOI] [PubMed] [Google Scholar]
- 61.Song X., Vishnivetskiy S. A., Gross O. P., Emelianoff K., Mendez A., Chen J., Gurevich E. V., Burns M. E., Gurevich V. V. (2009) Curr. Biol. 19, 700–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palczewski K., Pulvermüller A., Buczyłko J., Hofmann K. P. (1991) J. Biol. Chem. 266, 18649–18654 [PubMed] [Google Scholar]
- 63.Vishnivetskiy S. A., Hirsch J. A., Velez M. G., Gurevich Y. V., Gurevich V. V. (2002) J. Biol. Chem. 277, 43961–43967 [DOI] [PubMed] [Google Scholar]
- 64.Gurevich E. V., Gurevich V. V. (2006) Genome Biol. 7, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carter J. M., Gurevich V. V., Prossnitz E. R., Engen J. R. (2005) J. Mol. Biol. 351, 865–878 [DOI] [PubMed] [Google Scholar]
- 66.Tsukamoto H., Sinha A., DeWitt M., Farrens D. L. (2010) J. Mol. Biol. 399, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang L., Sports C. D., Osawa S., Weiss E. R. (1997) J. Biol. Chem. 272, 14762–14768 [DOI] [PubMed] [Google Scholar]
- 68.Burns M. E., Mendez A., Chen C. K., Almuete A., Quillinan N., Simon M. I., Baylor D. A., Chen J. (2006) J. Neurosci. 26, 1036–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Northrup S. H., Erickson H. P. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 3338–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gurevich V. V., Gurevich E. V. (2003) Structure 11, 1037–1042 [DOI] [PubMed] [Google Scholar]
- 71.Baylor D. A., Lamb T. D., Yau K. W. (1979) J. Physiol. 288, 613–634 [PMC free article] [PubMed] [Google Scholar]
- 72.Krispel C. M., Chen D., Melling N., Chen Y. J., Martemyanov K. A., Quillinan N., Arshavsky V. Y., Wensel T. G., Chen C. K., Burns M. E. (2006) Neuron 51, 409–416 [DOI] [PubMed] [Google Scholar]
- 73.Gross O. P., Burns M. E. (2010) J. Neurosci. 30, 3450–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whorton M. R., Bokoch M. P., Rasmussen S. G., Huang B., Zare R. N., Kobilka B., Sunahara R. K. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yao X. J., Vélez Ruiz G., Whorton M. R., Rasmussen S. G., DeVree B. T., Deupi X., Sunahara R. K., Kobilka B. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 9501–9506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lodowski D. T., Pitcher J. A., Capel W. D., Lefkowitz R. J., Tesmer J. J. (2003) Science 300, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 77.Tesmer V. M., Kawano T., Shankaranarayanan A., Kozasa T., Tesmer J. J. (2005) Science 310, 1686–1690 [DOI] [PubMed] [Google Scholar]
- 78.Lodowski D. T., Tesmer V. M., Benovic J. L., Tesmer J. J. (2006) J. Biol. Chem. 281, 16785–16793 [DOI] [PubMed] [Google Scholar]
- 79.Milano S. K., Pace H. C., Kim Y. M., Brenner C., Benovic J. L. (2002) Biochemistry 41, 3321–3328 [DOI] [PubMed] [Google Scholar]
- 80.Satoh A. K., Xia H., Yan L., Liu C. H., Hardie R. C., Ready D. F. (2010) Neuron 67, 997–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1 (.pdf, 864 KB)