TGF-β-induced IRAK-M Expression in Tumor Associated Macrophages Regulates Lung Tumor Growth (original) (raw)
. Author manuscript; available in PMC: 2011 Nov 26.
Published in final edited form as: Oncogene. 2011 Jan 31;30(21):2475–2484. doi: 10.1038/onc.2010.619
Abstract
Tumor associated macrophages (TAMs) constitute a major component of the immune cell infiltrate observed in the tumor microenvironment (TME). Factors present in the TME including TGF-β, allow tumors to circumvent host mediated immune responses to promote tumor progression. However, the molecular mechanism(s) involved are not clear. Toll-like receptors (TLRs) are important mediators of innate immune responses by immune cells, whose activation triggers the production of molecules required for anti-tumoral responses. Interleukin receptor associated kinase (IRAK)-M is an inactive serine/threonine kinase, predominantly expressed in macrophages and is a potent negative regulator of TLR signaling. Here we show that TAMs express significantly higher levels of IRAK-M compared to peritoneal macrophages (PEMs) in a syngeneic mouse model of lung cancer. Subcutaneous implantation of LLC cells in IRAK-M−/− mice resulted in a five-fold reduction in tumor growth, as compared to tumors in wild type animals. Furthermore, compared to WT TAMs, TAMs isolated from IRAK-M−/− mice displayed features of a classically activated (M1) rather than alternatively activated (M2) phenotype, as manifest by greater expression of IL-12, IFN-γ, and iNOS. Human lung cancer cells induced IRAK-M expression in human PBMCs when co-cultured together. Tumor cell-induced expression of IRAK-M was dependent on the activation of TGF-β pathway. Similarly, treatment of human PBMCs or mouse macrophage cell line, RAW 264.4, with TGF-β, induced IRAK-M expression. Interestingly, IRAK-M gene expression in 439 human lung adenocarcinoma tumors correlated with poor survival in patients with lung cancer. Together, our data demonstrates that TGF-β-dependent induction of IRAK-M expression is an important, clinically relevant mechanism by which tumors may circumvent anti-tumor responses of macrophages.
Keywords: IRAK-M, TGF-β, lung cancer, tumor-associated macrophages, toll-like receptors
INTRODUCTION
The phenomenon of tolerance has evolved to allow the immune system to distinguish between self from non-self, largely to prevent pathologic reactivity against self antigens (Pardoll, 2003). However, tumors cells can activate tolerance signaling pathways that are critical for normal homeostasis of the immune system, resulting in immune tolerance to cancer cells. Dynamic interactions between cancer cells and tumor associated immune cells initiate and maintain tumor immune tolerance, which eventually predominates and mitigates effective host immune response (Elgert et al., 1998; Pardoll, 2003). Among immune cells, macrophages constitute one of the major components of immune cell infiltrate observed in the tumor microenvironment (TME) of many types of malignancies, including lung cancer (Bingle et al., 2002). Although macrophages can mediate tumor cytotoxicity and present tumor associated antigens to stimulate anti-tumor lymphocyte responses, cancer cells routinely circumvent these macrophage-mediated immune responses. Tumor-associated macrophages (TAMs) are a distinct macrophage phenotype mimicking the phenotype of alternatively activated or M2 macrophages (Mantovani et al., 2002). These M2 cells have poor antigen presenting capability, produce factors that suppress T-cell proliferation and activity, and are generally better adapted to scavenging debris, promoting angiogenesis and remodeling wounded tissues.
Accumulating evidence demonstrates that cancer cells not only overcome anti-tumor activities of macrophages, but also actively redirect TAM activities towards promoting tumor progression and metastasis by altering the function and phenotype of TAMs (Bingle et al., 2002). These cells are implicated in several key aspects of tumor progression, including tumor cell proliferation, survival, angiogenesis and metastasis (Lewis and Pollard, 2006; Yamaguchi et al., 2006). Many tumor-derived molecules, including TGF-β deactivate or suppress the cytotoxic activity of TAMs (Elgert et al., 1998). This notion that TAMs promote tumor progression is supported by the observation that a high density of TAMs correlates with a poor prognosis in patients with cancer (Balkwill et al., 2005; Bingle et al., 2002; Pollard, 2004). In addition, studies involving macrophage-depleted mice have causally linked the presence of TAMs with poor prognosis (Johnson et al., 2000).
Toll like receptors are critical components of innate immune responses in macrophages (Akira and Takeda, 2004; Akira et al., 2001; Schnare et al., 2001). Activation of TLR-family members triggers the induction of several cytokines involved in anti-tumor responses, including TNF-α, IL-12 and IFN-γ (Seya et al., 2003). In the TME, the exact trigger(s) of TLR signaling in macrophages is not known. However, the TME is rich in molecules that can potentially activate macrophage TLR signaling to trigger anti-tumor responses; including heat shock proteins, high mobility group proteins, double stranded DNA from necrotic tumor cells, and hyaluronic acid (Ben-Baruch, 2006; Elgert et al., 1998). Following recognition of pathogen associated molecular patterns, the TLR receptors bind intracellular adaptor proteins, including MyD88 and TIR domain- containing adaptor protein/Mal, which in turn recruit other serine/threonine kinases implicated in inflammation signaling (e.g., IRAK-1, IRAK-4, and TRAF6). In contrast, IRAK-M is an inactive kinase that antagonizes TLR signaling through protein-protein interactions preventing downstream activation of IRAK-1(Kobayashi et al., 2002). IRAK-M is regarded as a key negative regulator of TLR signaling in macrophages (Kobayashi et al., 2002). However, studies implicating a role for IRAK-M in other cell types are also emerging (Rosati and Martin, 2002; Wesche et al., 1999). The presence of IRAK-M is critical for inactivating innate immune signals and prevents excessive inflammatory responses (Kobayashi et al., 2002). Unfortunately, in pathologies such as sepsis and cancer, immune suppressive function of IRAK-M is exploited to evade host immune surveillance (del Fresno et al., 2005; Deng et al., 2006).
TGF-β is a multifunctional cytokine with dual and paradoxical roles in cancer biology. In early stages of carcinogenesis, this cytokine acts as a tumor suppressor, whereas in late stages TGF-β functions as a tumor promoter (Elliott and Blobe, 2005). Consistent with the tumor promoting function of TGF-β expression is frequently up-regulated in non-small cell lung cancer (Kim et al., 1999) and elevated plasma levels of TGF-β confer a poor prognosis in patients with lung cancer (Kong et al., 1999). As part of its tumor promoting effects, TGF-β has a broad influence on the immune system partly through cross-talk between TGF-β signaling intermediates and the components of both cytokine and antigen receptors, including Toll-like receptors (TLRs) (Li et al., 2006). In this study, we show TGF-β mediated induction of IRAK-M in TAMs, which serve as a key mechanism by which lung tumors may circumvent anti-tumor responses of macrophages promoting tumor immune-tolerance.
RESULTS
TAMs express significantly higher IRAK-M and produce low levels of IL-12, IFN-γ and TNF-α, as compared to peritoneal macrophages (PEMs)
It is well recognized that TAMs are defective in the production of proinflammatory and immunoregulatory cytokines such as IL-12, IFN-γ and TNF-α. Since IRAK-M is a negative regulator of pro-inflammatory TLR signaling, we assessed whether IRAK-M expression correlates with defective cytokine production by TAMs. For this analysis, TAMs were isolated from LLC tumor tissue harvested from the WT C57/B6 mice, while thioglycolate-elicited PEMs were obtained by peritoneal lavage from the same mice. As expected, we observed very low levels of IL-12, IFN–γ and TNF-α production in TAMs, compared to PEMs (Fig 1A), as assessed by real-time PCR. Interestingly, several fold greater IRAK-M expression both at RNA and protein levels was observed in TAMs, compared to PEMs (Fig 1B & C). This observation suggests that IRAK-M may mediate the immunosuppressive TAM phenotype observed in tumors.
Figure 1. IL-12, IFN-γ, TNF-α and IRAK-M expression in TAMs and PEMs.
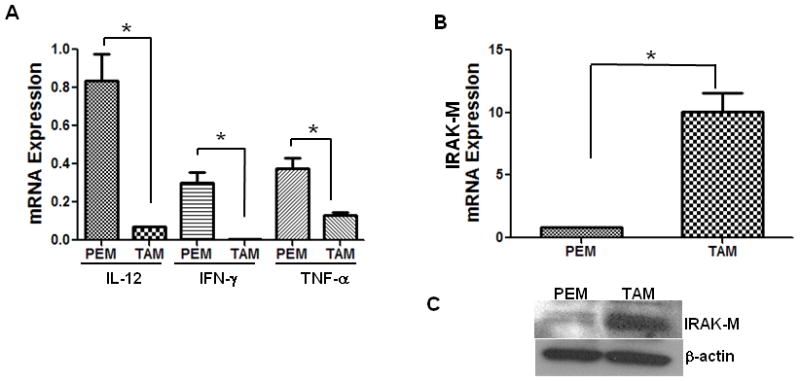
TAMs were purified from LLC tumors harvested from CB57/BL6 mice, thioglycolate-elicited PEMs were purified from peritoneal lavage fluid from the same tumor bearing mice. TAMs and PEMs were cultured for 24 h in RPMI 1640 media. Cells were lysed for RNA extraction. IL-12, IFN-γ, TNF-α (A) and IRAK-M (B) expression was assessed by real time PCR. Increase in IRAK-M expression was confirmed at protein level by western immunoblotting (C). Error bars represent SD. statistical significance is assessed by T-test and * denotes p<0.05
Inhibition of LLC tumor growth in IRAK-M−/− mice
To determine whether IRAK-M plays a role in immune surveillance against tumors, we subcutaneously implanted lewis lung carcinoma cells into IRAK-M+/+ and IRAK-M−/− mice (6 each) on both sides of the dorsal flank. Tumor growth was monitored by bi-weekly measurements for 3 weeks total. Average tumor volume for each group is plotted (Fig. 2A). As compared to wildtype (WT) mice, there was a 10 day delay in the development of palpable tumors in the IRAK-M−/− mice. Moreover, the average tumor volume was approximately 5-fold greater in the IRAK-M+/+ WT mice, as compared to IRAK-M−/− mice at day 20 (Fig. 2B). Inhibition of tumor growth in IRAK-M−/− mice implicates IRAK-M as a relevant suppressor of host immune surveillance against tumors.
Figure 2. Inhibition of syngeneic LLC tumor growth in IRAK-M−/− mice.
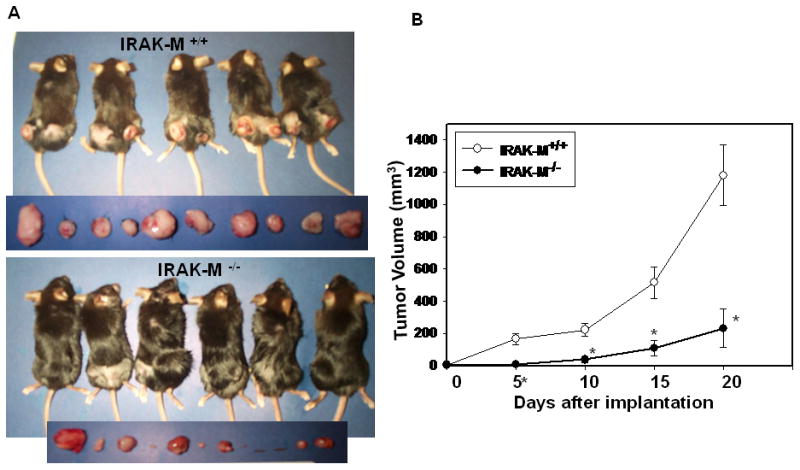
IRAK-M+/+ (n=6) and IRAK-M−/− mice (n=6) were subcutaneously implanted with 1x 106 LLC cells on either side of the dorsal flank. Tumor growth was monitored by measuring every 5th day for 21 days. (A) Mice were photographed after sacrificing. Tumors harvested from each mouse photographed before further processing. (B) Mean tumor volume for each group (6 mice x 2 tumors each =12) is plotted. Error bars represent SEM. statistical significance is assessed by two sample T-test and * denotes p<0.01
Tumors from IRAK-M−/− mice produce significantly higher levels of IL-12 compared to tumors from IRAK-M+/+ mice, but no change in TGF-β expression
Interleukin 12 is a key cytokine promoting type 1 immunity. By comparison, TGF-β is a potent immunosuppressive cytokine present in abundance within the tumor microenvironment (TME). To determine the mechanism for enhanced anti-tumor responses in IRAK-M−/− mice, we assessed the levels of TGF-β (Fig 3A) and IL-12 (Fig. 3B) in tumor lysates from IRAK-M+/+ mice and IRAK-M−/− mice by ELISA. There was no significant difference in the levels of TGF-β in the tumor tissue between IRAK-M+/+ and IRAK-M−/− mice. However, IL-12 levels in the tumors from IRAK-M−/− mice were several-fold higher than that measured in tumors from IRAK-M+/+ mice. This suggests that IRAK-M is a potential regulator of type 1 cytokine production (e.g. IL-12), but does not modulate the levels of immunosuppressive cytokine TGF-β.
Figure 3. TGF-β and IL-12 protein expression in whole tumor lysates from IRAK-M+/+ and IRAK-M−/− mice.
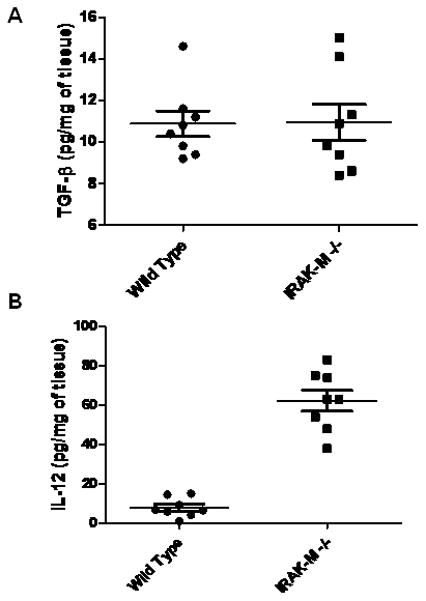
100 mg LLC tumor tissue harvested from IRAK-M+/+ and IRAK-M−/−mice was lysed by sonication in 1 ml of Pierce tissue lysis buffer supplemented with a cocktail of protease inhibitors. Tumor tissue lysates from IRAK-M+/+ (n= 8) and IRAK-M−/− mice (n=8) were assessed for TGF-β (A) and IL-12 (B) proteins by using cytokine specific ELISA. Cytokine levels were expressed as pg/mg of tumor tissue. Error bars represent SEM. Statistical significance is assessed by two sample T-test and * denotes p<0.01
Enhanced expression of IL-12 and IFN-γ in TAMs isolated from IRAK-M−/− compared to wild type TAMs
Since IRAK-M is predominantly expressed in monocytes/macrophages (Kobayashi et al., 2002; Wesche et al., 1999), the difference in cytokine production and tumor growth in IRAK-M+/+ and IRAK- M−/− mice may be due to alterations in TAM phenotypes. To test this, we assessed IL-12, IFN-γ and TNF-α production by TAMs, isolated from IRAK-M+/+ and IRAK- M−/− mice, by real-time PCR. Interestingly, there was no difference in the number of TAMs isolated per gram of tumor tissue in both WT and mutant mice (data not shown). However, we found that TAMs isolated from IRAK-M−/− mice expressed 6-fold and 4-fold greater quantities of IL-12 and IFN-γ respectively, in IRAK-M−/− TAMs, as compared to TAMs from WT mice (Fig. 4A). In contrast, no difference in TNF-α production was observed.
Figure 4. IL-12, IFN-γ TNF-α Fizz1 and iNOS expression in TAMs purified from LLC tumors of IRAK-M+/+ and IRAK-M−/−mice.
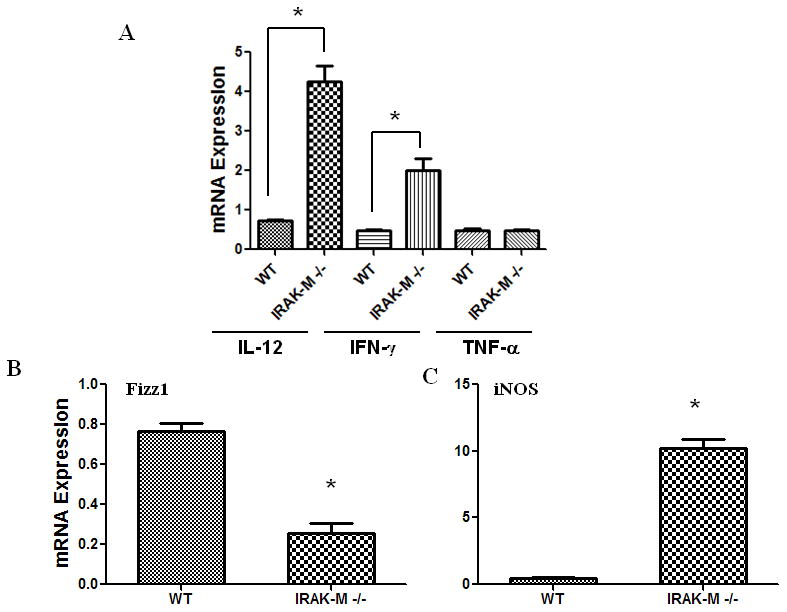
TAMs were purified from LLC tumors harvested from IRAK-M+/+ and IRAK-M−/−mice and cultured for 24 h in RPMI 1640 media. Cells were lysed for RNA extraction. IL-12, IFN-γ, TNF-α (A), Fizz1(B) and iNOS (C) expression was assessed by real time PCR. Error bars represent SD. Statistical significance is assessed by paired T-test and * denotes p<0.05
IRAK-M deficient TAMs express a classically activated rather than alternatively activated phenotype, as compared to TAMs from IRAK-M+/+ mice
It is known that TAMs exhibit an alternatively activated or M2 polarized macrophage phenotype, a response that is required to adequately regulate inflammation, tissue remodeling, and repair (Gordon, 2003). In tumors, the M2 phenotype of TAMs allows subversion of anti-tumor immunity and facilitates other tumor promoting roles of TAMs (Mantovani et al., 2002). We asked whether IRAK-M may drive the M2 phenotype in TAMs. To analyze the phenotype in purified TAMs, we assessed the expression of the M2 marker Fizz1 (RELM in mouse), and the classical activation (M1) marker iNOS in TAMs by real-time PCR. Interestingly, IRAK-M−/− TAMs displayed an approximately 3 fold decrease in Fizz1 expression (Fig. 4B) and a nearly 20-fold increase in the expression of iNOS expression (Fig. 4C), suggesting that IRAK-M may be a critical promoter of M2 phenotype in TAMs.
TGF-β induces IRAK-M expression in a time and dose dependent manner
Since TGF-β is known to have a broad suppressive influence on the immune system, partly through cross-talk between TGF-β signaling intermediates and the components of both cytokine and antigen receptors including TLRs (Jacobsen et al., 1991; Moustakas and Heldin, 2003; Xiao et al., 2003), we next investigated whether TGF-β regulates IRAK-M expression in human peripheral blood monocytes and murine macrophages (RAW264.7). We first stimulated human monocytes (1 x 106 cells) with various doses of TGF-β and assessed the time- and dose-dependent expression of IRAK-M by real time PCR. Importantly, TGF-β upregulated IRAK-M expression in a dose- and time-dependent manner in human monocytes (Fig 5A&B). Moreover, TGF-β induces IRAK-M expression even at protein level in time-dependent manner (Fig 5C). We observed much more pronounced increase in IRAK-M protein levels (approximately 3- to 5-fold increased) compared to its mRNA levels, suggesting both transcriptional and translational regulation of IRAK-M expression. In additional experiments, we assessed whether TGF-β could induce IRAK-M in a murine macrophage cell line RAW 264.7. Like human peripheral blood monocytes, TGF-β induced IRAK-M expression in RAW 264.7 cells in both a time and dose dependent manner (Fig 5D), with maximum expression observed at a concentration of 10 ng/ml dose and induction observed as early as 4h and increasing out to 24 h post stimulation.
Figure 5. TGF-β induces IRAK-M expression in human and murine cells.
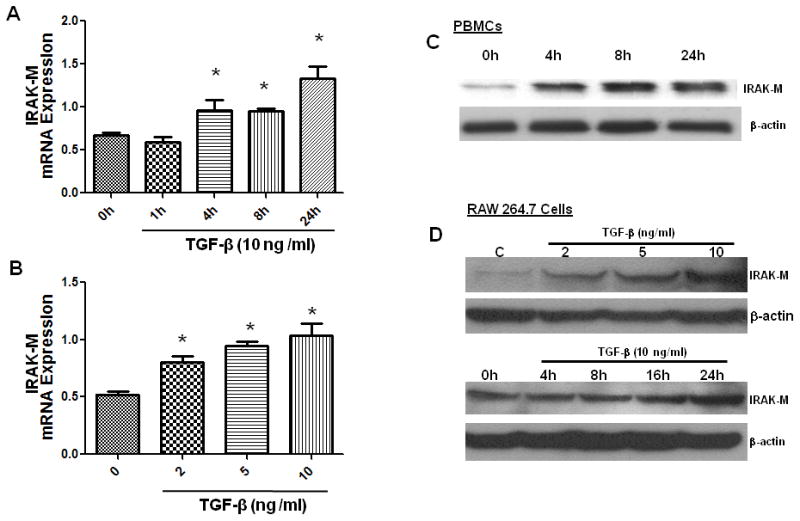
Human monocytes/macrophages isolated from the blood of a healthy donor (A, B, C) or 24 h serum starved RAW264.7 cells (D) were stimulated with various doses of TGF-β or 10 ng/ml of TGF-β was used to stimulate for different times. Cells were lysed for protein and RNA extraction. IRAK-M mRNA levels were assessed by real time PCR (A and B). Protein expression of IRAK-M was assessed by Western immunoblotting using IRAK-M specific antibody (C and D). Error bars represent SD. Statistical significance is assessed by one-way ANOVA compared to untreated control or 0 h and * denotes p<0.05
Human lung cancer cells induce IRAK-M expression in monocytes
To test whether lung cancer cells can induce IRAK-M expression, we co-cultured human peripheral blood monocyte (1 x 106 cells) with various NSCLC cells (A549, H522, and H460) either by direct contact or in transwell chambers separated by 0.4 micron porous membrane. Co-cultures of monocytes: tumor cells were performed at a ratio of 3:1. After 24 h, IRAK-M expression was assessed by real time PCR. Lung cancer cells, both by direct contact and in transwells (Fig. 6A&B) significantly stimulated IRAK-M expression in monocyte/macrophages.
Figure 6. Lung cancer cells induce IRAK-M expression in monocytes/macrophages.
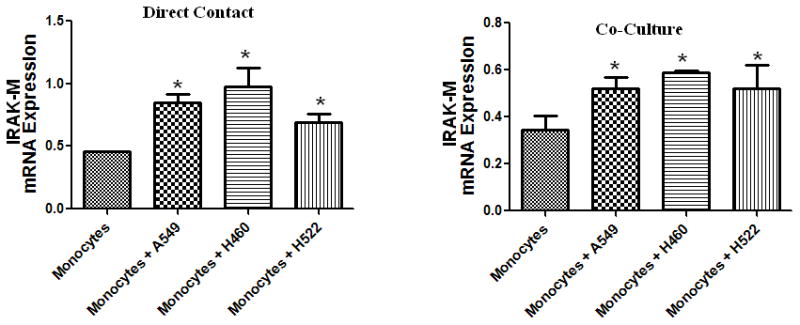
Different human lung cancer cells were used to stimulate human monocytes/macrophages isolated from the blood of a healthy donor by co-culturing them in either direct contact (A) or in transwell chambers separated by 0.4 micron porous membrane (B). A monocyte to tumor cell ratio of 3:1 was used in all experiments. Cells were lysed for RNA extraction and IRAK-M was assessed by real time PCR. Error bars represent SD. Statistical significance is assessed by one-way ANOVA compared to monocytes alone and * denotes p<0.05
TGF-β augments and TGF-β-neutralizing antibody inhibits tumor cell-induced IRAK-M expression in monocytes
Since TGF-β-induced IRAK-M expression and the lung TME is known to have significant levels of TGF-β, we tested the cooperative effect of TGF-β on tumor cell induced IRAK-M expression. In this analysis, we incubated human peripheral blood monocytes with A549 human lung adenocarcinoma cell line by co-culture in the presence or absence of TGF-β (5ng/ml) for 24 h, then IRAK-M expression assessed by real time PCR. TGF-β and A549 cells directly induced IRAK-M expression in PBM. Further, TGF-β significantly augmented tumor cell-induced IRAK-M expression (Fig. 7A). To further confirm a causal role for TGF-β in tumor cell induced IRAK-M expression, we co-cultured A549 cells with human peripheral blood monocytes that were cultured in the presence or absence of pan-TGF-β neutralizing antibody. Consistent with TGF-β-induced IRAK-M expression in human peripheral blood monocytes and murine macrophages, tumor cell-induced IRAK-M expression was significantly inhibited by pan-TGF-β neutralizing antibody (Fig. 7B). Inhibition of TGF-β-induced Smad2 phosphorylation by TGF-β neutralizing antibody, in A549 cells, demonstrates the neutralizing capacity of the antibody (Fig. 7C). These results demonstrate that tumor-derived TGF-β is present in the lung TME and may promote evasion of macrophage-mediated immune surveillance via induction of IRAK-M.
Figure 7. TGF-β augments and TGF-β-neutralizing antibody inhibits tumor cell-induced IRAK-M expression in monocyte/macrophages.
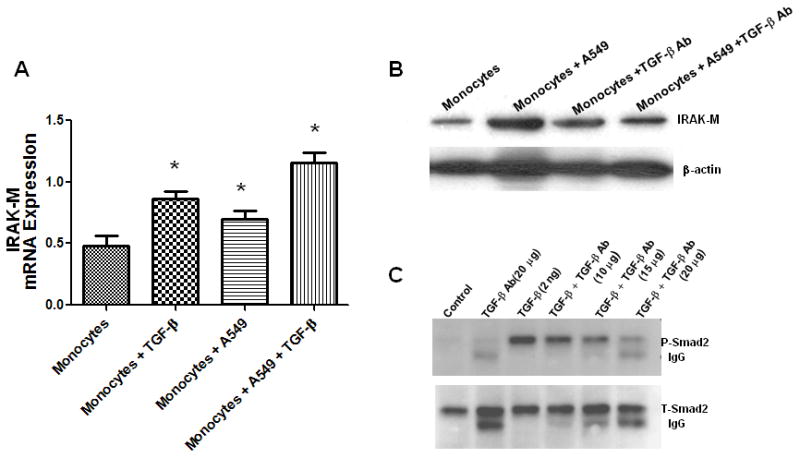
In the transwell co-culture system, A549 cells were cultured in the presence or absence of TGF-β (5 ng/ml) in the ratio of 1:3 in the upper chamber and were used to stimulate monocytes/macrophages isolated from the blood of a healthy donor in the lower chamber separated by 0.4 micron porous membrane. After 24 h Monocytes were lysed for RNA extraction and IRAK-M was assessed by real time PCR (A). Error bars represent SD. Statistical significance is assessed by one-way ANOVA compared to monocytes alone and * denotes p<0.05. To assess the effect of TGF-β neutralizing antibody, monocytes/macrophages were cultured in the lower chamber in the presence or absence of TGF-β-neutralizing antibody (50 μg/ml), and stimulated with A549 cells (1:3) in the upper chamber of transwell system. After 24 h Monocytes/macrophages were lysed for protein extraction. IRAK-M protein levels were assessed by Western immunoblotting (B). To demonstrate the efficacy of TGF-β neutralizing antibody, A549 cells were cultured in the absence or presence of indicated concentrations of TGF-β neutralizing antibody for 1 hr. Cells were lysed for protein extraction and TGF-b-induced Smad2 phosphorylation as well as total Smad2 levels were assessed by western immunoblotting (C).
Gene expression of IRAK-M in human lung cancer correlates with poor survival
Given the observed tumor promoting activity of IRAK-M, we tested for an association between IRAK-M mRNA levels and survival in a previously described cohort of 439 patients with lung adenocarcinoma using Affymetrix GeneChip array analysis (Shedden et al., 2008). We fit Cox Proportional-Hazards Regression models that included age, gender, and stage (I, II, or III), which were reported to be significant clinical variables in the original publication (Shedden et al., 2008). All co-variables were found to be independently correlated with survival (Table I). IRAK-M minimum and maximum values differed by 11-fold, and base-2 logarithms had standard deviation of 0.54. Importantly, the magnitude of IRAK-M expression was associated with a relative risk of 1.33 for each doublingof the expression values (p =0.023). The mean expression of IRAK-M for stages I, II, and III were −0.006, 0.018, and 0.024. Even though these means were not significantly different (p = 0.68 comparing stage I vs. III by 1-way ANOVA model), their magnitude increased with the stage of the tumor. The similar increased relative risk was observed for male sex and for everyadditional 10 years of age at time of diagnosis increased risk by a similar amount, as did being male. Stage of disease was the most significant risk factor, with a2.4-foldincreased relative riskfor stage II and a nearly 5-fold increased risk for stage IIIdisease. Collectively, this data demonstrates that in addition to age, gender, and stage, the magnitude of IRAK-M expression in tumors is an independent predictor of mortality in patients with adenocarcinoma of the lung, even when corrected for other co-variables.
Table I.
Factors correlating with relative risk of death in patients with adenocarcinoma of the lung.
| Effect | P-value (Wald test) | Relative Risk (95% CI) |
|---|---|---|
| IRAK-M (base-2 log) | .021 | 1.33 (1.05 – 1.70) |
| Age, decades | .000011 | 1.36 (1.18 – 1.55) |
| Male gender | .030 | 1.33 (1.03 – 1.73) |
| Stage II vs. Stage I | 3 × 10−8 | 2.40 (1.76 – 3.27) |
| Stage III vs. Stage I | < 10−15 | 4.77 (3.44 – 6.60) |
DISCUSSION
Lung cancer is the single most deadly cancer in the world, with 150,000 new cases diagnosed every year in US alone (Jemal et al., 2005). General cytotoxic therapies such as chemotherapy and radiation are standard of care for both resectable and non-resectable non-small cell lung cancers (Scagliotti and Novello, 2003; Souquet and Geriniere, 2001), although the 5-year survival rate for lung cancer patients remains less than 15 percent (Edwards et al., 2005). This highlights the urgent need for novel strategies to improve treatment outcomes. Despite the well recognized role of dynamic interactions between various cell types present within the TME in fostering tumor progression, investigations to define these interactions or to elucidate underlying molecular mechanisms in lung cancer are limited. In this study we define one such interaction between lung cancer cells and TAMs, including mechanisms involved and clinical relevance to human lung cancer.
Macrophages present in healthy tissues can exhibit cytolytic activity against tumor cells, present tumor-associated antigens, and stimulate anti-tumor functions of T-cells (Letterio and Roberts, 1998; Li et al., 2006). However, TAMs present in the TME often display reduced effector functions, rendering the host incapable of mounting an effective immune response. Instead, cells/factors present within the TME skews TAMs towards a distinct tumor permissive phenotype (Lewis and Pollard, 2006; Mantovani et al., 1992). The molecular basis for such a distinct tumor promoting phenotype of TAMs and their role in lung cancer progression is yet undefined. It is worth noting that TAMs play distinct roles in different tumor microenvironments (Lewis and Pollard, 2006). For that reason, it is essential to investigate TAM functions in lung tumor models to derive inferences regarding lung cancer-specific roles of TAMs. A blunted inflammatory cytokine response was observed, as manifested by reduced production of IL-12 and IFN-γ by TAMs isolated from LLC tumors, similar to the phenotype displayed by TAMs in other malignancies (Mytar et al., 2003). Conversely, TAMs expressed higher levels of Fizz1 (RELM) and reduced levels of iNOS, reflecting the M2-phenotype of TAMs. Previously it has been shown that LPS tolerance in human endotoxemia models is associated with IRAK-M up-regulation (de Vos et al., 2009; van 't Veer et al., 2007). Analogous to endotoxin tolerance, similar increase in IRAK-M expression was reported in alveolar macrophages during sepsis-induced immune suppression (Deng et al., 2006) and in PBMCs isolated from patients with chronic myeloid leukemia (del Fresno et al., 2005). Correlating with blunted cytokine responses, we observed significantly elevated levels of IRAK-M in TAMs relative to other macrophage populations (PEMs). This is the first report demonstrating increased IRAK-M expression in macrophages isolated from solid tumors.
It has been shown that IRAK-M is a negative regulator of TLR signaling, as mice deficient in IRAK-M demonstrate enhanced macrophage-mediated innate immunity (Kobayashi et al., 2002). In addition, IRAK-M−/− mice exhibit striking resistance to development of immune tolerance. In this study, mice lacking IRAK-M demonstrated greater suppression of syngeneic lung cancer tumor growth, as compared to wild type mice. Analysis of TAMs isolated from tumors in IRAK-M−/− mice showed a dramatic increase in the production of type 1 cytokines, indicating resistance to the tolerance phenotype of TAMs. However, lack of IRAK-M did not rescue TNF-α production in TAMs, suggesting a potential IRAK-M independent mechanism. Interestingly, we also noted inhibition of the M2-like phenotype in TAMs isolated from IRAK-M−/− mice, as indicated by reduced Fizz1 and increased iNOS expression. This suggests that IRAK-M, at least in part, is responsible for promoting alternative activation in TAMs. The observed resistance to tumor growth in IRAK-M−/− mice is consistent with an earlier study using a melanoma tumor model, which attributed increased resistance to elevated populations of CD4+ and CD8+ T-cells and a decrease in Foxp3+ regulatory T-cells in IRAK-M−/− splenocytes (Xie et al., 2007). The aforementioned study did not assess the phenotype of TAMs, and it is quite plausible that the changes in T-cell populations are reflective of altered TAM phenotype in IRAK-M−/− mice.
Tumors are known to evade host immune surveillance by multiple mechanisms, one of which being the production of TGF-β. For that reason, we explored potential regulation of IRAK-M in TAMs by TGF-β. Analysis of tumor lysates showed a dramatic increase in the levels of IL-12 protein in IRAK-M−/− tumors, compared to tumors from wild type mice. However, there was no difference in the levels of TGF-β protein between the tumors of wild type and IRAK-M−/− mice, demonstrating that IRAK-M had no effect on the production of TGF-β in the TME. Interestingly, stimulation of either human PBMCs or murine macrophage cell line RAW 264.7 with TGF-β induced IRAK-M expression in both a dose- and time-dependent manner. Our findings are consistent with a recent study demonstrating a role for TGF-β/Smad4 in LPS-induced IRAK-M expression in THP-1 cells (Pan et al.). As reported previously for other tumor cell types (del Fresno et al., 2005), we found that human lung cancer cells also induced IRAK-M expression in human PBMCs when co-cultured either in direct contact or separated by a porous membrane, implicating a soluble factor in the induction of IRAK-M. Co-culturing human PBMCs with lung cancer cells in the presence of a pan-TGF-β neutralizing antibody significantly abrogated tumor cell induced IRAK-M expression in PBMCs, confirming a role for TGF-β in the regulation of IRAK-M expression. Given the pleiotropic biological functions of TGF-β, it is not surprising to observe cross-regulation between this cytokine and TLR signaling. For example, investigations in mice lacking a functional TGF-β signaling pathway have revealed aberrantly increased TLR4 expression (McCartney-Francis et al., 2004). TGF-β has also been shown to inhibit LPS-induced TLR4 signaling by inducing degradation of MyD88, a critical downstream adaptor molecule required for the signaling of most TLRs (Naiki et al., 2005). Consistent with these observations, our findings demonstrate yet another novel mechanism by which TGF-β regulates TAM function in lung tumors.
Analysis of IRAK-M gene expression in human lung adenocarcinoma tumors revealed that high IRAK-M expression in tumors correlated with poor survival in patients with this disease. Given the regulation of IRAK-M by TGF-β, this observation is consistent with earlier reports that TGF-β expression is frequently up-regulated in patients with lung cancer (Hasegawa et al., 2001) and elevated plasma levels of TGF-β confer a poor prognosis (Hasegawa et al., 2001; Toonkel et al.). Together, our data demonstrates that TGF-β-dependent induction of IRAK-M expression is an important and clinically relevant mechanism by which tumors may circumvent anti-tumor responses of macrophages. Thus, we propose that the targeting IRAK-M may represent a viable immunotherapy in the treatment of patients with lung cancer.
MATERIALS AND METHODS
Cell culture
The A549, H460, H522 human non-small cell cancer (NSCLC) cell lines, murine lung cancer cell line, LLC (Lewis lung carcinoma) which are syngeneic to C57/BL6 mice and RAW 264.7 murine macrophage cell line were obtained from the American Type Culture Collection. NSCLC, LLC and RAW 264.7 cells were maintained in RPMI-1640 medium with glutamine, supplemented with 10% FBS and antibiotics penicillin and streptomycin.
Isolation of TAMs from tumors
To isolate TAMs, tumor tissue was chopped into small pieces before incubation with an enzyme-mixture (400 U/ml collagenase type IV, 0.05 mg/ml collagenase type I, 0.025 mg/ml hyaluronidase (Sigma-Aldrich), 0.01 mg/ml DNAse I, 0.2 U/ml soybean trypsin inhibitor (BoehringerMannheim) dissolved in RPMI 1640 for 2 h’ at 37°C. Single cell suspensions were prepared in MACS buffer (degassed PBS, 0.5% BSA, 2 mM EDTA), and incubated (15 min, 8°C) with anti-CD11b-microbeads (Miltenyi Biotec) (5 μl beads/107 cells). CD11b+ cells were purified on LS columns (Miltenyi Biotec), using the MidiMACS system according to the manufacturer’s instructions. Cells were plated in Petri dishes, after 1 h of incubation, non-adherent cells were washed off thoroughly and the purity of the prep was confirmed by morphology and staining with macrophage specific marker F4/80.
Isolation of human peripheral blood monocytes
Whole blood (anticoagulated with 1000 U of heparin per 60 ml) was obtained by venipuncture from healthy donors, and monocytes were obtained by Ficoll-plaque (Amersham, Uppsala, Sweden) density centrifugation and adherence purification. Cells were cultured in the absence of serum and were stimulated as indicated.
Isolation of peritoneal macrophages (PEMs)
Cells were obtained by peritoneal lavage from mice injected 4 days previously with 0.5 ml of 3% thioglycolate. Cells were suspended in MACS buffer (degassed PBS, 0.5% BSA, 2 mM EDTA), and incubated (15 min, 8°C) with anti-CD11b-microbeads (Miltenyi Biotec) (5 μl beads/107 cells). CD11b+ cells were purified on LS columns (Miltenyi Biotec), using the MidiMACS system according to the manufacturer’s instructions. Cells were plated in petridishes, after 1 h of incubation, non-adherent cells were washed off thoroughly and the purity of the prep was confirmed by morphology and staining with macrophage specific marker F4/80.
Western blotting
Crude protein extracts were obtained by lysing 5 x 106 cells in a buffer containing 50 mM Tris-HCl (pH 7.6), 1% Nonidet P-40, 2 mM EDTA, 0.5% sodium deoxycholate, 150 mM NaCl, 1 mM sodium orthovanadate, 2 mM EGTA, 4 mM sodium p-nitrophenyl phosphate, 100 mM sodium fluoride and supplemented with protease inhibitors (leupeptin (0.5%), aprotinin (0.5%) and phenylmethylsulfonyl fluoride (0.02%)). Samples containing 20 μg of total protein were electrophoresed on SDS-polyacrylamide gels and transferred on to nitrocellulose membrane by electro blotting. Membranes were probed with antibodies as indicated followed by horseradish peroxidase-conjugated mouse or rabbit secondary antibodies and visualized by West Pico chemiluminescence detection reagents (Pierce, Rockford, IL).
Heterotopic LLC tumor model
0.5 x 106 LLC cells suspended in 100 ml of serum-free RPMI 1640 media were implanted subcutaneously on either side of the dorsal flank of C57/BL-6 mice. Tumor growth was monitored by weekly measurements of tumors with calipers. After 21 days, animals were sacrificed and tumors were dissected from the mice, measured for three-dimensional size and photographed. A portion of the tumor was snap-frozen in liquid nitrogen for protein analysis.
Real time quantitative PCR for IRAK-M: (TaqMan)
Relative expression of IRAK-M was determined by using TaqMan real time quantitative PCR as described before (Reddy et al., 2004). Briefly, Gene-specific primers and probes were designed using Primer Express software (Perkin-Elmer/PE Applied Biosystems, Foster City, CA). Oligonucleotide primers and TaqMan probe for b-actin internal control was purchased from Perkin-Elmer/PE Applied Biosystems. The real-time quantitative RT-PCR was performed following the manufacturer’s protocol. Briefly, the reaction mixture contained 5.5 mM MgCl2, 500 M dNTP, 2.5 M random hexamers, 200 nM FAM probe, and forward and reverse primers at 600 nM in a final volume of 25 μl and was analyzed in an ABI PRISM 7700 sequence detection system. Relative expression of IRAK-M mRNA levels was plotted after normalization against β-actin mRNA. The sequences used for IRAK-M and other cytokines are as follows:
| HuIRAK-M: | TTG-GTC-CTG-GGC-ACA-GAA-A-3’ (forward) |
|---|---|
| GCT-CGA-CGA-TGT-CCC-ATC-TC-3’ (reverse) | |
| CCA-TCG-GTG-ACC-TTT-TAC-AGG-TCC-TCC (probe) | |
| mIRAK-M: | TGA GCA ACG GGA CGC TTT (forward) |
| GAT TCG AAC GTG CCA GGA A (reverse) | |
| TTA CAG TGC ACA AAT GGC ACA ACC CC (probe) | |
| mTNF-α: | CAG CCG ATG GGT TGT ACC TT (forward) |
| TGT GGG TGA GGA GCA CGT AGT (reverse) | |
| TCC CAG GTT CTC TTC AAG GGA CAA GGC (probe) | |
| mIL-12P40 | AGA CCC TGC CCA TTG AAC TG (forward) |
| GAA GCT GGT GCT GTA GTT CTC ATA TT (reverse) | |
| CGT TGG AAG CAC GGC AGC AGA A (probe) | |
| mIFN-γ: | CTG CGG CCT AGC TCT GAG A (forward) |
| CAG CCA GAA ACA GCC ATG AG (reverse) | |
| CAC ACT GCA TCT TGG CTT TGC AGC TCT (probe) | |
| mFizz1(RELM): | CCC TGC TGG GAT GAC TGC TA (forward) |
| TCC ACT CTG GAT CTC CCA AGA (reverse) | |
| TGG GTG TGC TTG TGG CTT TGC C (probe) | |
| miNOS: | CCC TCC TGA TCT TGT GTT GGA (forward) |
| CAA CCC GAG CTC CTG GAA (reverse) | |
| TGACCATGGAGCATCCCAAGTACGAGT (probe) |
Cytokine specific ELISAs
Levels of murine TNF-α, IL-12, TGF-β and IFN-γ were assessed using a modification of double-ligand method, as previously described (Reddy et al., 2004). Briefly, flat-bottomed 96-well microtiter plates were coated with 50 μl/well of rabbit antibody against specific cytokines for 16 h at 4°C . The plates were rinsed four times with wash buffer, diluted cell-free supernatants (50 μl) in duplicate were added, and the samples were incubated for 1 h at 37°C. The plates were washed again four times before adding biotinylated rabbit antibodies against the specific cytokines (50 μl/well) were and incubated for 30 min at 37°C. After washing the plates four more times, streptavidin-peroxidase conjugate (Bio-Rad Laboratories, Richmond, CA) was added, and incubated for 30 min at 37°C. The chromogen substrate (Bio-Rad Laboratories) was added after four more washes. The plates were further incubated at room temperature to the desired extinction, and the reaction was terminated with 3 M H2SO4 solution (50 μl/well) and OD was measured at 490 nm with an ELISA reader.
Association between IRAK-M mRNA expression and survival in patients with adenocarcinoma of the lung
To determine if the IRAK-M expression was predictive of patient survival, we analyzed IRAK-M mRNA levels in a cohort of 442 human lung adenocarcinomas assayed for mRNA by Affymetrix HG-U133A arrays. This is the largest available set of microarray data with extensive pathological and clinical annotation for lung adenocarcinomas (Shedden et al., 2008). The data for IRAK-M was available for 439 out of 442 samples. For this analysis, we log-transformed mRNA expression levels using Y = log2 (max(X,0)+1) as in the original analysis. The data were generated at four institutions (University of Michigan Cancer Center, Moffitt Cancer Center, Memorial Sloan-Kettering Cancer Center and the Dana-Farber Cancer Institute) and we normalized values by subtracting the mean for each institution. Patients with lung adenocarcinoma were sequentially enrolled from the four institutions. Patient characteristics and the treatments they received were described previously (Shedden et al., 2008). Consent was obtained and the project was approved by the local Institutional Review Boards. Primary tumors and adjacent non-neoplastic lung tissue were obtained at the time of surgery. Peripheral portions of resected lung carcinomas were sectioned, evaluated by a study pathologist and compared with routine H&E sections of the same tumors, and utilized for mRNA isolation. Regions chosen for analysis contained a tumor cellularity greater than 70%, with no mixed histology, potential metastatic origin, extensive lymphocytic infiltration or fibrosis.
Acknowledgments
This research is funded by NIH/NCI (R01 CA132571-01), and American Cancer Society (RSG -CSM-116801) grants to V.G.K., and NIH/NHLBI HL25243 and HL097564 to T.J.S.
References
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Ben-Baruch A. Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Semin Cancer Biol. 2006;16:38–52. doi: 10.1016/j.semcancer.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- de Vos AF, Pater JM, van den Pangaart PS, de Kruif MD, van 't Veer C, van der Poll T. In vivo lipopolysaccharide exposure of human blood leukocytes induces cross-toleranceto multiple TLR ligands. J Immunol. 2009;183:533–42. doi: 10.4049/jimmunol.0802189. [DOI] [PubMed] [Google Scholar]
- del Fresno C, Otero K, Gomez-Garcia L, Gonzalez-Leon MC, Soler-Ranger L, Fuentes-Prior P, et al. Tumor cells deactivate human monocytes by up-regulating IL-1 receptor associated kinase-M expression via CD44 and TLR4. J Immunol. 2005;174:3032–40. doi: 10.4049/jimmunol.174.5.3032. [DOI] [PubMed] [Google Scholar]
- Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, et al. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest. 2006;116:2532–42. doi: 10.1172/JCI28054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BK, Brown ML, Wingo PA, Howe HL, Ward E, Ries LA, et al. Annual report to the nation on the status of cancer, 1975–2002, featuring population-based trends in cancer treatment. J Natl Cancer Inst. 2005;97:1407–27. doi: 10.1093/jnci/dji289. [DOI] [PubMed] [Google Scholar]
- Elgert KD, Alleva DG, Mullins DW. Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol. 1998;64:275–90. doi: 10.1002/jlb.64.3.275. [DOI] [PubMed] [Google Scholar]
- Elliott RL, Blobe GC. Role of transforming growth factor Beta in human cancer. J Clin Oncol. 2005;23:2078–93. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Takanashi S, Kanehira Y, Tsushima T, Imai T, Okumura K. Transforming growth factor-beta1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer. 2001;91:964–71. [PubMed] [Google Scholar]
- Jacobsen SE, Keller JR, Ruscetti FW, Kondaiah P, Roberts AB, Falk LA. Bidirectional effects of transforming growth factor beta (TGF-beta) on colony-stimulating factor-induced human myelopoiesis in vitro: differential effects of distinct TGF-beta isoforms. Blood. 1991;78:2239–47. [PubMed] [Google Scholar]
- Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- Johnson SK, Kerr KM, Chapman AD, Kennedy MM, King G, Cockburn JS, et al. Immune cell infiltrates and prognosis in primary carcinoma of the lung. Lung Cancer. 2000;27:27–35. doi: 10.1016/s0169-5002(99)00095-1. [DOI] [PubMed] [Google Scholar]
- Kim WS, Park C, Jung YS, Kim HS, Han J, Park CH, et al. Reduced transforming growth factor-beta type II receptor (TGF-beta RII) expression in adenocarcinoma of the lung. Anticancer Res. 1999;19:301–6. [PubMed] [Google Scholar]
- Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- Kong F, Jirtle RL, Huang DH, Clough RW, Anscher MS. Plasma transforming growth factor-beta1 level before radiotherapy correlates with long term outcome of patients with lung carcinoma. Cancer. 1999;86:1712–9. [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–61. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. TRANSFORMING GROWTH FACTOR-beta REGULATION OF IMMUNE RESPONSES. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today. 1992;13:265–70. doi: 10.1016/0167-5699(92)90008-U. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- McCartney-Francis N, Jin W, Wahl SM. Aberrant Toll receptor expression and endotoxin hypersensitivity in mice lacking a functional TGF-beta 1 signaling pathway. J Immunol. 2004;172:3814–21. doi: 10.4049/jimmunol.172.6.3814. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Ecsit-ement on the crossroads of Toll and BMP signal transduction. Genes Dev. 2003;17:2855–9. doi: 10.1101/gad.1161403. [DOI] [PubMed] [Google Scholar]
- Mytar B, Woloszyn M, Szatanek R, Baj-Krzyworzeka M, Siedlar M, Ruggiero I, et al. Tumor cell-induced deactivation of human monocytes. J Leukoc Biol. 2003;74:1094–101. doi: 10.1189/jlb.0403140. [DOI] [PubMed] [Google Scholar]
- Naiki Y, Michelsen KS, Zhang W, Chen S, Doherty TM, Arditi M. Transforming growth factor-beta differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling. J Biol Chem. 2005;280:5491–5. doi: 10.1074/jbc.C400503200. [DOI] [PubMed] [Google Scholar]
- Pan H, Ding E, Hu M, Lagoo AS, Datto MB. Lagoo-Deenadayalan SA SMAD4 is required for development of maximal endotoxin tolerance. J Immunol. 184:5502–9. doi: 10.4049/jimmunol.0901601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807–39. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Reddy RC, Keshamouni VG, Jaigirdar SH, Zeng X, Leff T, Thannickal VJ, et al. Deactivation of murine alveolar macrophages by peroxisome proliferator-activated receptor-gamma ligands. Am J Physiol Lung Cell Mol Physiol. 2004;286:L613–9. doi: 10.1152/ajplung.00206.2003. [DOI] [PubMed] [Google Scholar]
- Rosati O, Martin MU. Identification and characterization of murine IRAK-M. Biochem Biophys Res Commun. 2002;293:1472–7. doi: 10.1016/S0006-291X(02)00411-4. [DOI] [PubMed] [Google Scholar]
- Scagliotti G, Novello S. Adjuvant chemotherapy after complete resection for early stage NSCLC. Lung Cancer. 2003;42(Suppl 1):S47–51. doi: 10.1016/s0169-5002(03)00304-0. [DOI] [PubMed] [Google Scholar]
- Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activationof adaptive immune responses. Nat Immunol. 2001;2:947–50. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- Seya T, Akazawa T, Uehori J, Matsumoto M, Azuma I, Toyoshima K. Role of toll-like receptors and their adaptors in adjuvant immunotherapy for cancer. Anticancer Res. 2003;23:4369–76. [PubMed] [Google Scholar]
- Shedden K, Taylor JM, Enkemann SA, Tsao MS, Yeatman TJ, Gerald WL, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med. 2008;14:822–7. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souquet PJ, Geriniere L. The role of chemotherapy in early stage of non-small cell lung cancer. Lung Cancer. 2001;34(Suppl 2):S155–8. doi: 10.1016/s0169-5002(01)00361-0. [DOI] [PubMed] [Google Scholar]
- Toonkel RL, Borczuk AC, Powell CA. Tgf-beta signaling pathway in lung adenocarcinoma invasion. J Thorac Oncol. 5:153–7. doi: 10.1097/JTO.0b013e3181c8cc0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van 't Veer C, van den Pangaart PS, van Zoelen MA, de Kruif M, Birjmohun RS, Stroes ES, et al. Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J Immunol. 2007;179:7110–20. doi: 10.4049/jimmunol.179.10.7110. [DOI] [PubMed] [Google Scholar]
- Wesche H, Gao X, Li X, Kirschning CJ, Stark GR, Cao Z. IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J Biol Chem. 1999;274:19403–10. doi: 10.1074/jbc.274.27.19403. [DOI] [PubMed] [Google Scholar]
- Xiao C, Shim JH, Kluppel M, Zhang SS, Dong C, Flavell RA, et al. Ecsit is required for Bmp signaling and mesoderm formation during mouse embryogenesis. Genes Dev. 2003;17:2933–49. doi: 10.1101/gad.1145603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Gan L, Wang J, Wilson I, Li L. Loss of the innate immunity negative regulator IRAK-M leads to enhanced host immune defense against tumor growth. Mol Immunol. 2007;44:3453–61. doi: 10.1016/j.molimm.2007.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Pixley F, Condeelis J. Invadopodia and podosomes in tumor invasion. Eur J Cell Biol. 2006;85:213–8. doi: 10.1016/j.ejcb.2005.10.004. [DOI] [PubMed] [Google Scholar]