Brain Infiltration of Leukocytes Contributes to the Pathophysiology of Temporal Lobe Epilepsy (original) (raw)
Abstract
Clinical and experimental evidence indicates that inflammatory processes contribute to the pathophysiology of epilepsy, but underlying mechanisms remain mostly unknown. Using immunohistochemistry for CD45 (common leukocyte antigen) and CD3 (T-lymphocytes), we show here microglial activation and infiltration of leukocytes in sclerotic tissue from patients with mesial temporal lobe epilepsy (TLE), as well as in a model of TLE (intrahippocampal kainic acid injection), characterized by spontaneous, nonconvulsive focal seizures. Using specific markers of lymphocytes, microglia, macrophages, and neutrophils in kainate-treated mice, we investigated with pharmacological and genetic approaches the contribution of innate and adaptive immunity to kainate-induced inflammation and neurodegeneration. Furthermore, we used EEG analysis in mutant mice lacking specific subsets of lymphocytes to explore the significance of inflammatory processes for epileptogenesis. Blood–brain barrier disruption and neurodegeneration in the kainate-lesioned hippocampus were accompanied by sustained ICAM-1 upregulation, microglial cell activation, and infiltration of CD3+ T-cells. Moreover, macrophage infiltration was observed, selectively in the dentate gyrus where prominent granule cell dispersion was evident. Unexpectedly, depletion of peripheral macrophages by systemic clodronate liposome administration affected granule cell survival. Neurodegeneration was aggravated in kainate-lesioned mice lacking T- and B-cells (RAG1-knock-out), because of delayed invasion by Gr-1+ neutrophils. Most strikingly, these mutant mice exhibited early onset of spontaneous recurrent seizures, suggesting a strong impact of immune-mediated responses on network excitability. Together, the concerted action of adaptive and innate immunity triggered locally by intrahippocampal kainate injection contributes seizure-suppressant and neuroprotective effects, shedding new light on neuroimmune interactions in temporal lobe epilepsy.
Introduction
Experimental and clinical evidence indicates that inflammatory processes contribute to the pathogenesis of several forms of epilepsy (Turrin and Rivest, 2004; Vezzani and Granata, 2005; Schultzberg et al., 2007; Fabene et al., 2010). Thus, interactions between leukocytes and vascular endothelial cells modulate spontaneous recurrent seizures (SRSs) in a rodent model of temporal lobe epilepsy (TLE) (Fabene et al., 2008). Furthermore, altered blood–brain barrier (BBB) function and increased expression of proinflammatory cytokines (Oby and Janigro, 2006; van Vliet et al., 2007; Vezzani et al., 2008; Friedman et al., 2009) underscore the activation of inflammatory responses during epileptogenesis. Thus, interleukin-1β (IL-1β) and IL-1R are rapidly upregulated in the brain after seizures (Jankowsky and Patterson, 2001; Ravizza et al., 2006, 2008), whereas IL-1β, IL-6, and TNFα (tumor necrosis factor-α) are increased in microglia and astrocytes in epileptic tissue and may lead to recruitment of adaptive immune system cells (Nguyen et al., 2002; Vezzani and Granata, 2005; Riazi et al., 2008). Conversely, experimental strategies to block either CNS or systemic inflammation reduce status epilepticus duration and seizure frequency (Marchi et al., 2009; Maroso et al., 2010). However, the respective contribution of brain-resident and peripheral immune cells to epileptogenesis is poorly understood (Fabene et al., 2008).
In this study, we investigated how interactions between innate and adaptive immunity contribute to functional and histopathological reorganization of the hippocampal formation in an experimental model of TLE with hippocampal sclerosis (HS), well characterized for morpho-functional adaptations underlying seizures (Bouilleret et al., 2000; Straessle et al., 2003; Arabadzisz et al., 2005; Fedele et al., 2005; Le Duigou et al., 2005, 2008; Heinrich et al., 2006; Ledergerber et al., 2006; Dugladze et al., 2007; Antonucci et al., 2008). In this model, a single intrahippocampal kainic acid (KA) injection causes gradual development of an epileptic focus and onset of SRSs after ∼2 weeks (Bouilleret et al., 1999). The lesion, characterized by neurodegeneration in the ipsilateral hilus and CA1/CA3c regions, extensive astrogliosis, and pronounced dispersion of dentate gyrus granule cells, is restricted to the dorsal hippocampus, no damage being evident contralaterally or in any extrahippocampal region, including entorhinal cortex.
Using immunohistochemistry, we investigated the presence of immune cells in brain tissue resected from mesial temporal lobe epilepsy (MTLE) patients with HS and from KA-injected mice. Next, we characterized with specific markers the time course of inflammation and activation of major components of adaptive and innate immunity in KA-treated mice and investigated with pharmacological and genetic approaches their contribution to KA-induced tissue damage and epileptogenesis. In particular, we analyzed the significance of T-lymphocyte infiltration for regulating SRS onset, using EEG analysis in various lines of mutant mice, including RAG1- and RAG2-knock-out (KO) mice (lacking B- and T-cells) (Mombaerts et al., 1992; Shinkai et al., 1992), β2-microglobulin-KO (β2M-KO) mice (lacking CD8+ T-cells) (Zijlstra et al., 1990), and major histocompatibility class II-KO (MHCII-KO) mice (lacking CD4+ cells) (Madsen et al., 1999). The results point to complex interactions between innate and adaptive immune responses in KA-lesioned hippocampus, modulating neuronal damage and SRS onset.
Materials and Methods
An overview of all animal experiments performed is given in Table 1.
Table 1.
Design of experiments performed

Animals.
Experiments were performed on 12- to 14-week-old male wild-type and KO mice weighing 25–35 g (Table 2) bred and housed in small groups in the animal facility of the Institute of Pharmacology and Toxicology on a 12 h light/dark cycle with food and water provided ad libitum. During experiments, they were housed in individual cages. Animal procedures were approved by the Cantonal Veterinary Office of Zurich (Switzerland). All efforts were made to minimize animal suffering and the number of animals used.
Table 2.
List of mice used for experiments
| Short name | Genetic background | Phenotype | Supplier |
|---|---|---|---|
| B6, wild type | C57BL/6J | Control mice for all experiments | Harlan (RCC Switzerland) |
| RAG1-KO | C57BL/6-CD45.1/T × B6.129S7-Rag1tmMom/J | Lack of B- and T-cells [inability to initiate V(D)J rearrangement] | The Jackson Laboratory |
| B6-RAG2-KO | B6.129S6-Rag2tm1Fwa N12 | Lack of B- and T-cells (B6 background) | Taconic Farms |
| BALB/c-RAG2-KO | BALB/c-IL2rgtm1 B6.129S6-Rag2tm1Fwa | Lack of B- and T-cells (BALB/c background) | Prof. A. Aguzzi (University of Zurich, Zurich, Switzerland) |
| β2M-KO | B6;129P2-B2mtm1Jae | Lack of CD8+ T-cells | Prof. B. Becher (University of Zurich) |
| MHCII-KO | B6-I-Atm1Blt | Lack of CD4+ T-cells | Prof. B. Becher (University of Zurich) |
Kainic acid injection and electrodes implantation.
Mice anesthetized by inhalation of 2.5–3% isoflurane (Baxter) in oxygen received a unilateral stereotaxic injection of KA (70 nl, 5 mm; Tocris) or vehicle into the right CA1 area of the dorsal hippocampus [coordinates with bregma as reference: anteroposterior, −1.8 mm; mediolateral, −1.6 mm; dorsoventral, −1.9 mm] as described (Bouilleret et al., 1999). Control mice were treated identically but were injected with vehicle only (0.9% NaCl).
For EEG recordings, mice were implanted immediately after KA injection with bipolar electrodes aimed the same coordinates (for details, see Arabadzisz et al., 2005). A monopolar reference electrode was inserted over the cerebellum. Electrodes were fixed to the skull with cyanoacrylate and dental acrylic cement. After surgery, the mice were allowed to recover from anesthesia on a warm pad, injected with 1 mg/kg buprenorphine (Temgesic; Essex Chemicals) and then returned to their home cage with daily monitoring of well-being.
EEG recordings.
Intrahippocampal EEG activity was recorded in freely moving mice connected via a swivel to a digital acquisition system (AcqKnowledge MP100; Biopac Systems). After 30 min habituation to the Faraday cage, recordings were taken during 3–4 h at a sampling rate of 200 Hz. Mice were recorded every other day starting from day 2–6 after KA injection until maximum day 20 (Table 1). Recordings were taken around midday to minimize potential circadian variations. Data were analyzed off-line and artifacts attributable to animal movements were discarded. Paroxysmal events were defined as seizures when they lasted >20 s and were separated by intervals of at least 3 s. For each animal, the average frequency and duration SRS was calculated.
Neutrophil depletion.
Repeated systemic administration of functional rat anti-granulocyte receptor-1 (Gr-1) monoclonal antibodies (clone RB6-8C5; catalog #16-5931; eBioscience) or control IgGs (catalog #16-4031-85; eBioscience) to KA-lesioned mice was used to deplete neutrophils (Stirling et al., 2009). In B6 mice, treatment was started 1 d before KA injection and repeated three times at 3–4 d intervals until killing at 10 d after KA for histological analysis. In RAG1-KO mice, to avoid infections after surgery, anti-Gr-1 treatment was started at day 6 after KA and repeated three times before perfusion/fixation at day 14 after KA. Depletion was verified by Gr-1 staining of neutrophils in the spleen.
Macrophage depletion.
Depletion of peripheral macrophages was performed by intraperitoneal administration of clodronate liposomes (Clodrolip) (75 μg/g body weight in B6 and 25 μg/g in RAG1-KO mice). Empty liposomes were used as control. Clodrolip contains ∼18 ± 2 mg of clodronate per milliliter (0.5 μmol of clodronic acid disodium salt tetrahydrate, CH2Cl2Na2O6P2·4H2O). After systemic injection, liposomes are phagocytosed and digested by macrophages, followed by intracellular release and accumulation of clodronate, causing >90% depletion of macrophages within 24–36 h (Seiler et al., 1997). Treatment was started 1 d before KA injection and repeated twice at 3–4 d interval until killing at day 10 after KA for histological analysis (Table 1). Efficacy of the treatment was verified by F4/80 immunostaining of mononuclear phagocytes in the spleen.
Tissue preparation.
Human tissue preparation and storage were described previously (Loup et al., 2000). All samples used here were taken from the same patients and postmortem controls.
Mice were deeply anesthetized with pentobarbital (Nembutal; 60 mg/kg, i.p.) and perfused transcardially with ice-cold fixative (0.15 m sodium phosphate buffer, 4% paraformaldehyde, 15% saturated picric acid solution, pH 7.4) as described (Fritschy and Mohler, 1995). Brains were removed immediately after perfusion, postfixed for 3 h at 4°C, and impregnated overnight with 30% sucrose in PBS for cryoprotection. Transverse 40-μm-thick sections were cut from frozen blocks with a sliding microtome and collected in PBS. Sections were then stored in antifreeze solution (50 mm phosphate buffer, 15% glucose, 30% ethylene glycol, sodium azide; pH 7.4) at −20°C until use.
Immunoperoxidase staining.
Primary antibodies used in this study are listed in Table 3. Immunoperoxidase staining was performed to visualize the regional distribution of cellular markers in human tissue (CD45 and CD3) and KA-treated mice (CD45, CD3, B220, CD68, Gr-1, F4/80), as well as intercellular cell adhesion molecule (ICAM-1) and BBB leakage (fibrinogen). Sections were incubated overnight at 4°C with primary antibodies diluted in Tris buffer, pH 7.4, containing 2% normal goat serum and 0.2% Triton X-100 (ICAM-1, 1:500; CD3, CD4, CD8, CD45, B220, Gr-1, F4/80, 1:1000; CD68, 1:2000; Iba-1, 1:3000; fibrinogen, 1:10,000). The presence of IgGs in the tissue was probed by incubation with biotinylated anti-mouse IgGs, skipping the primary antibody incubation. Sections were washed and incubated for 30 min in biotinylated secondary antibodies (1:300; Jackson ImmunoResearch), followed by ABC complex for 30 min (Vectastain Elite kit; Vector Laboratories) and reaction with diaminobenzidine tetrahydrochloride (Sigma-Aldrich) for 5–15 min in Tris, pH 7.7. Finally, sections were washed thoroughly, mounted onto gelatin-coated slides, air-dried overnight, dehydrated, and coverslipped with Eukitt (Erne Chemie).
Table 3.
List of primary antibodies
| Epitope | Host species | Catalog # | Company | Marker of: |
|---|---|---|---|---|
| Human CD3ϵ | Mouse | MCA1477 | Serotec | CD3+ T-cells |
| Mouse CD3ϵ | Hamster | 553058 | BD Biosciences Pharmingen | |
| Mouse CD4 | Rat | 553727 | BD Biosciences Pharmingen | CD4+ T helper cells |
| Mouse CD8a | Rat | 553027 | BD Biosciences Pharmingen | Cytotoxic CD8+ T-cells |
| Mouse CD45R/B220 | Rat | 550286 | BD Biosciences Pharmingen | B-cells |
| Mouse Gr-1 | Rat | 553123 | BD Biosciences Pharmingen | Neutrophils, some monocytes |
| Human CD45 (common leukocyte antigen) | Mouse | M0701 | Dako | Leukocytes, microglia |
| Mouse CD45 | Rat | 0111D | BD Biosciences Pharmingen | |
| Mouse CD68 | Rat | MCA1957GA | Serotec | (Activated) microglia |
| Mouse Iba-1 | Rabbit | 019-19741 | Wako Chemicals | Microglia |
| Mouse F4/80 | Rat | b6640 | Abcam | Monocytic lineage (tissue macrophages, microglia) |
| NeuN | Mouse | MAB377 | Millipore Bioscience Research Reagents | Neurons |
| Mouse ICAM-1 (CD54) | Hamster | 553250 | BD Biosciences | Activated endothelial cells |
| Human fibrinogen | Rabbit | A0080 | Dako | Blood–brain barrier leakage |
Immunofluorescence staining.
Immunofluorescence staining was performed to compare the cellular distribution of two (or three) markers in the same tissue section. CD3+ T-cells were differentiated by double staining for either CD4 or CD8. The interactions between microglial cells and neutrophils were determined by triple labeling for Gr-1, Iba-1, and the nucleic acid stain Sytox Green (Invitrogen). Comparative morphological analysis of microglia and macrophage-like cells in the dentate gyrus was determined by triple labeling for Iba-1, F4/80, and the neuronal marker NeuN. Free-floating sections were incubated overnight in a mixture of primary antibodies, followed by washes and incubation in corresponding goat secondary antibodies coupled to Alexa 488 (1:1000), Alexa 647 (1:1000), or Cy3 (1:500) for 30 min. Sections were mounted and coverslipped with fluorescent mounting medium (Dako).
Nissl staining.
A 1:6 series of sections was Nissl-stained with cresyl violet to verify pattern of degeneration induced by KA and the placement of EEG electrodes.
Image acquisition and processing.
Histological analysis of cresyl violet and immunoperoxidase staining was performed by bright-field microscopy (Zeiss AxioCam) and images acquired with an 8 bit digital color camera controlled by AxioVision 4.5 (Zeiss). Exposure times were held constant during acquisition of all images for each experiment.
Sections processed for double and triple immunofluorescence staining were visualized by confocal laser-scanning microscopy (LSM 710 Zen; Zeiss) using a 63× objective [numerical aperture (NA), 1.4] and sequential acquisition of separate channels. Pinhole was set to 1 Airy unit and the acquisition parameters set to use the full dynamic range of photomultipliers. Stacks of images (1024 × 1024 pixels; pixel size, 70–100 nm) spaced by 0.35–0.5 μm were acquired and processed using Imaris software (Bitplane). For display, maximal intensity projection images (10–35 layers) were prepared with either merged or color-separated channels.
Quantification and densitometry analysis.
T-cells immunoreactive for CD3 (CD3+) were counted in a one-in-six series of sections (three to four sections per mouse) through the dorsal hippocampal formation using stereological methods (optical fractionator method). For each animal, the volume of the granule cell layer was determined by using Mercator software (Explora Nova). The region of interest was outlined in each section and the total volume was estimated by summing the surface area enclosed by all the traces and multiplying by the section thickness and the sampling interval. CD3+ T-cells were not homogeneously distributed precluding random sampling using a counting frame. Therefore, all immunopositive cells were counted at high magnification (63×; NA, 1.3) in the sampled region of interest. Data were analyzed statistically by one-way ANOVA followed by post hoc Bonferroni's multiple-comparison test (GraphPad Prism 5).
Extravasation of CD3+ T-cells and neutrophils was visualized by differential interference contrast microscopy, distinguishing between intravascular/perivascular and intraparenchymal cells. In each case investigated, the proportion of each subpopulation was determined on a sample of 50 randomly selected immunopositive cells.
Densitometry measurements of CD68 immunoreactivity were performed to assess degree of microglia activation in the hippocampal formation. Digital images were acquired using a 12 bit digital camera. Relative optical density measurements were calibrated with a gray-value density scale. Background was measured in the thalamus and subtracted from measurements made in the same section. Data were averaged per region of interest and animal and were analyzed by two-way ANOVA followed by Bonferroni's post hoc tests. Densitometry analysis of ICAM-1 immunoreactivity was performed in 12 bit digital images from bright microscopy were captured with a 10× (NA, 0.3) lens. The proportional surface area occupied by immunopositive pixels selected by threshold segmentation was determined in the contralateral and in KA-injected hippocampal formation and compared statistically (paired t test).
Correlation between number of F4/80+ cells and integrity of the dentate gyrus granule cell layer.
The analysis was performed to evaluate the impact of macrophage depletion on granule cell dispersion. In each section analyzed (four per mouse), the granule cell layer was subdivided into six segments, in which the average thickness of the granule cell layer and number of F4/80+ macrophage-like cells in the molecular and granule cell layers were determined. Data were analyzed statistically using linear regression analysis to correlate the thickness of the granule cell layer with the number of F4/80+ cells.
Results
Activation of immune cells in human MTLE and in the KA mouse model of TLE
Immune cell activation in hippocampal tissue resected from patients with intractable MTLE and HS was investigated immunohistochemically with antibodies against CD45 (common leukocyte antigen) (Fig. 1A,B). The patients (n = 9) and postmortem controls (n = 4) have been described previously (Loup et al., 2000). In every surgical specimen examined (Fig. 1A′,B′), we observed prominent activation of microglial cells, along with the presence of immunoreactive leukocytes, either associated with blood vessels or distributed intraparenchymally in CA1-CA3, hilus, and, to a lesser extent, dentate gyrus. In comparison, in tissue from postmortem controls, resting microglial cells were observed throughout the hippocampal formation, whereas intravascular leukocytes were detected only sporadically (Fig. 1A,B).
Figure 1.
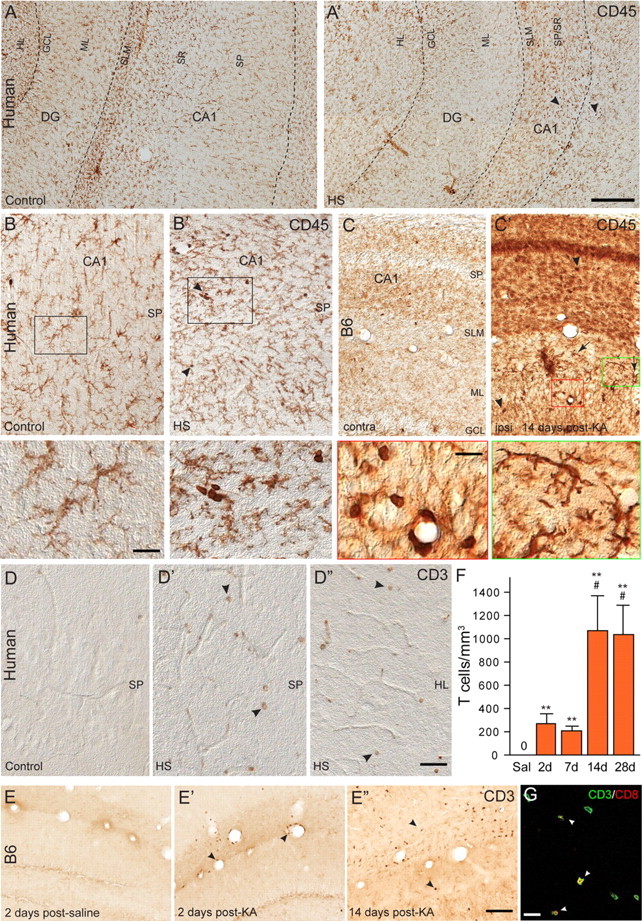
Leukocyte infiltration in human (A, C) and C57BL/6J (B6) mouse (B, D, E, F) epileptic tissue. A, A′, Low-magnification view of CD45 immunoreactivity in hippocampal tissue from a postmortem control (A) and resected from a patient with intractable MTLE (A′) depicting the presence of resting and activated microglial cells, respectively, and of blood vessel-associated and intraparenchymal leukocytes (A′, arrowheads). The marked sclerosis of the epileptic tissue is denoted by the shrinking of the CA1 area. B, B′, CD45 immunoreactivity in control and sclerotic CA1 area, with arrowheads pointing to leukocytes; the boxed areas are enlarged at the bottom. C, C′, For comparison, CD45 immunoreactivity in a mouse with a unilateral intrahippocampal KA injection; the contralateral side (C) is depicted as control. Ipsilaterally (C′), a mixture of activated microglial cells, leukocytes (arrowheads), and large macrophage-like cells (arrows) are evident. The green and red boxed areas are enlarged at the bottom. D–D″, Detection of intraparenchymal CD3+ lymphocytes (arrowheads) in sclerotic tissue from CA1 (D′) and hilus (D″) in MTLE patients, but not postmortem control (D). E–E″, Selective infiltration by CD3+ lymphocytes in mouse epileptic tissue; in control (saline) (E), no CD3 immunoreactivity was seen. In KA-treated tissue, CD3+ cells were seen first in blood vessels (E′) (2 d time point) and later intraparenchymally (E″) (14 d time point). F, Unbiased quantification of CD3+ lymphocytes in the KA-lesioned dorsal hippocampal formation, demonstrating their absence in control mice (Sal) and a significant biphasic increase after KA injection (mean ± SEM; #p < 0.05 compared with 7 d; **p < 0.01 compared with Sal; n = 6 per group; Bonferroni's posttest analysis). G, Example of double immunofluorescence staining for CD3 (green) and CD8 (red) depicting a subset of double-labeled cells in the CA1 region (arrowheads). Abbreviations: GCL, Granule cell layer; HL, hilus; HS, hippocampal sclerosis; ML, molecular layer; SLM, stratum lacunosum-moleculare; SP, stratum pyramidale; SR, stratum radiatum. Scale bars: A, 500 μm; B, C, 20 μm; D, E, 50 μm; G, 20 μm.
These observations in human tissue were addressed in KA-treated mice to determine the validity of this TLE model for investigating inflammatory responses in epilepsy. KA injection induces severe neurodegeneration in the hilus, CA1, and CA3c along with delayed, but prominent granule cell dispersion (see Introduction). In lesioned hippocampal tissue from these mice, CD45 immunoreactivity revealed strong microgliosis, along with the presence of large cells with short ramified branches in the dentate gyrus molecular layer and of leukocytes in blood vessels and in the neuropil (Fig. 1C′), like those seen in human tissue. In the contralateral, nonlesioned side, a weak staining of resting microglia was observed (Fig. 1C).
To further characterize these CD45+ leukocytes, we performed immunoperoxidase staining for CD3+ T-cells and B220+ B-cells, both in human and B6 mouse tissue (Fig. 1D,E). Although CD3+ T-lymphocytes readily were detected in epileptic tissue, including blood vessels and brain parenchyma, no B-cells were observed in our analysis. In human sclerotic hippocampal tissue resected from surgery, CD3+ cells were seen mostly within blood vessels. Intraparenchymal infiltration was evident in seven of the nine samples examined, restricted to the hippocampal sclerosis, and involved between 9 and 31% of CD3+ cells (Fig. 1D–D″). In KA-treated mice, unbiased quantitative analysis revealed a gradual accumulation of CD3+ T-cells (one-way ANOVA, F(3,17) = 6.18, p < 0.0049), reaching a maximum at 2 weeks after injection and remaining present at 4 weeks (Fig. 1F). Initially, CD3+ T-cells were detected within blood vessels (Fig. 1E′); at 2–4 weeks, they were found mainly within the neuropil (Fig. 1E″), reaching up to 74% of CD3+ cells, as seen by differential interference contrast microscopy (data not shown). Remarkably, no CD3+ T-cells were detected in saline-treated control mice (Fig. 1E), as well as extrahippocampally in KA-treated mice. Double-immunofluorescence staining performed at 14 d after KA to distinguish CD4+ and CD8+ cells (Fig. 1G) (data not shown) revealed that 60–75% T-cells (n = 4 mice) present in the hippocampus were CD8+, suggesting a preferential infiltration of cytotoxic T-cells in this model of TLE.
Microglial activation mirrors neurodegeneration in KA-lesioned hippocampus
CD45 immunoreactivity in KA-treated mice suggested widespread activation of innate immunity, in addition to T-cell infiltration. Therefore, we investigated with specific markers the distribution of cells of the monocytic lineage (microglia and mononuclear phagocytes). Preliminary experiments (data not shown) with antibodies to Iba1, CD11b, and CD68 revealed that CD68 allows best to discriminate between resting and activated microglia. Systematic analysis of CD68 immunoreactivity revealed a prominent and widespread activation of microglial cells, which reproduced precisely the pattern of neurodegeneration seen in adjacent Nissl-stained sections (Fig. 2). Thus, the strongest CD68 immunoreactivity occurred in the pyramidal cell layer of CA1 and CA3c, followed by stratum radiatum and oriens of CA1. Little microglia activation was detected in the dentate granule cell layer and molecular layer. Compared with saline-injected controls, no microglia activation was observed in the contralateral hippocampal formation. Neuronal death progressed further between 2 and 4 weeks after KA (Fig. 2A′,A″), paralleled by increased CD68 staining intensity (Fig. 2B′,B″). Therefore, we used CD68 immunoreactivity to quantify the severity of neurodegeneration in KA-treated mice by densitometry analysis at day 14 after KA (Fig. 2C). Two-way ANOVA revealed a significant difference in CD68 immunoreactivity between KA- and saline-treated mice (F(1,120) = 133.3; p < 0.0001), as well as a significant difference among the regions analyzed (F(7,120) = 5.151; p < 0.0001), notably in CA1, CA3, and hilus compared with saline-treated animals (Fig. 2C), confirming our visual impressions that the KA-induced lesion is most severe in CA1 and CA3c, but mostly spares the dentate gyrus and, to a lesser extent, the CA3a,b region.
Figure 2.
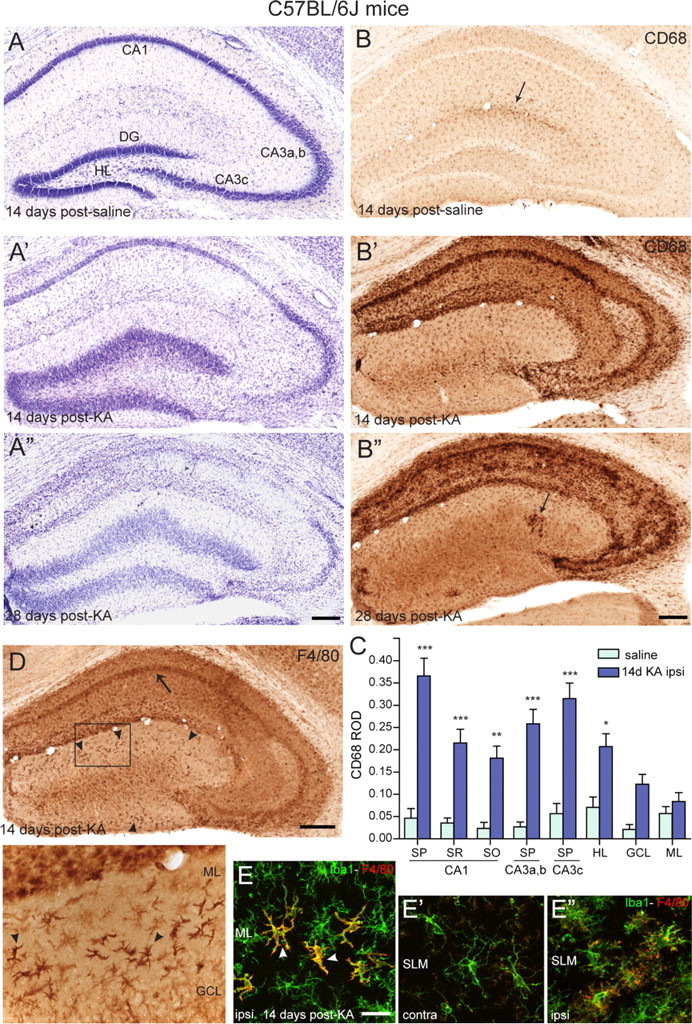
Activation of innate immunity in KA-lesioned hippocampal formation of B6 mice. A–A″, Gradual degeneration of CA1, CA3c, and hilar neurons, along with dentate gyrus granule cell dispersion, as seen in KA-injected hippocampus by cresyl violet staining compared with control (saline). B–B″, CD68 immunoreactivity in sections from the same mice, depicting the regular distribution of resting microglia in control (B), with a slight increase in staining around the saline injection site (arrow), and the strong increase in all regions where neurodegeneration was evident in KA-treated tissue (B′, B″). Note that microglia activation does not occur in the dentate gyrus, except for a few isolated cells at late time points (B″, arrow). C, Densitometric analysis of CD68 immunoreactivity, showing selective increase in areas of neuronal cell loss compared with saline control. The increase was significant in the pyramidal layer of CA1, CA3a,b, and CA3c, in the stratum radiatum and oriens of CA1 and in the hilus. The relative optical density (ROD) are given as mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001, Bonferroni's posttest; n = 11). D, Detection of activated microglia (arrow) and macrophage-like cells (arrowheads) by F4/80 immunostaining in KA-injected hippocampus. The boxed area is enlarged below the panel. E–E″, Double-immunofluorescence staining for Iba1 (green) and F4/80 (red) showing the distinctive morphology of microglial cells and macrophage-like cells in the molecular layer 14 d after KA injection; for comparison, the contralateral and ipsilateral stratum-lacunosum-moleculare is also depicted. CA, Cornu ammonis; DG, dentate gyrus; GCL, granule cell layer; ML, molecular layer; SLM, stratum lacunosum-moleculare; SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum; HL, hilus. Scale bars: A, B, D, 250 μm; E, 20 μm.
Detection of macrophage-like cells in the dentate gyrus
Unexpectedly, CD68 staining did not label macrophage-like cells exhibiting prominent CD45 immunoractivity in the dentate gyrus (Fig. 1B). Therefore, we tested antibodies against F4/80, a marker of a wide variety of mature tissue macrophages. This marker labeled activated microglial cells in KA-lesioned hippocampus, albeit less distinctly than CD68 (Fig. 2D). Interestingly, however, it strongly stained macrophage-like cells around the dentate granule cell layer, as seen with CD45 (Fig. 2D, inset). Double immunofluorescence using antibodies to Iba1 confirmed that prominent F4/80 immunoreactivity was restricted to this subset of cells in the dentate gyrus, whereas weak F4/80 staining was seen also in activated microglia (Fig. 2E). Together, these observations suggest that dentate gyrus granule cells, which undergo profound morphological changes and become dispersed in this TLE model, are selectively associated with a specific subpopulation of F4/80+ cells, possibly representing macrophages derived from peripheral mononuclear phagocytes.
Exacerbation of neurodegeneration in RAG1-knock-out mice
Since RAG1-KO mice lack adaptive immune cells (Mombaerts et al., 1992), we used them to understand the significance of T-cell infiltration with regard to activation of innate immunity in the KA mouse model of TLE. RAG1-KO mice treated with KA were analyzed at various time points after treatment (Table 1) and compared with age-matched B6 mice. As seen by cresyl violet staining and CD68 immunoreactivity (Fig. 3A,B), saline-treated RAG1-KO mice exhibited no abnormality. Despite the lack of lymphocytes, stereotaxic surgery did not cause infections in RAG1-KO mice, suggesting effective protection by innate immunity. Nevertheless, on KA injection, RAG1-KO mice showed the typical lesion pattern and activation of microglial cells induced by KA, but overall with increased severity relative to wild-type mice (compare Figs. 3A,B, 2A,B). Thus, quantification of CD68 staining at 14 d after KA injection revealed a significant increase in RAG1-KO mice, notably in CA1 and CA3 (two-way ANOVA; effect of genotype: F(1,158) = 53.3, p < 0.0001; effect of region: F(7,158) = 14.57, p < 0.0001) (Fig. 3C). Remarkably, extensive hippocampal neurodegeneration, including loss of dentate granule cells was observed 28 d after KA injection (Fig. 3A″). At this time point, the cytoarchitecture of the ipsilateral hippocampus was characterized by strong gliosis and became almost unrecognizable in some animals. These changes were restricted to the ipsilateral side, extending into the ventral hippocampus, whereas no changes were apparent contralaterally. Although these alterations were present in all six mice examined after 28 d, they were already evident in a subset of mice (4 of 11) at 14 d after KA, in particular in the CA3 area, which was extensively degenerated (Fig. 3D). Together, these findings indicated that KA-induced lesion eventually led to a complete destruction of the ipsilateral hippocampus in RAG1-KO mice, a feature never seen in wild-type mice.
Figure 3.
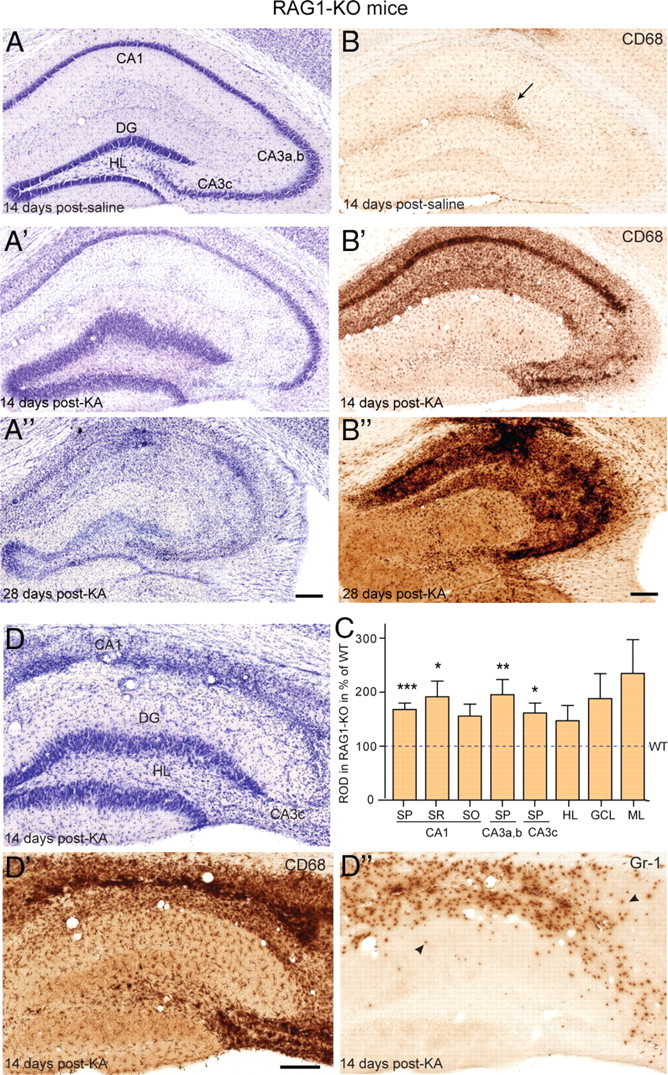
Enhanced KA-induced neurodegeneration in RAG1-KO mice. A–A″, The general pattern of neurodegeneration induced by KA was globally similar to wild type, but was most severe and led to complete destruction by 28 d after KA, as seen by cresyl violet staining. B–B″, CD68 immunoreactivity again mirrored precisely the neurodegeneration. In control (B), a slight enhancement was seen at the injection site (arrow). C, Densitometric analysis of CD68 immunoreactivity, with ROD being normalized to wild type (indicated by a dashed line; two-way ANOVA, Bonferroni's posttest; *p < 0.05, **p < 0.01, ***p < 0.001). Error bars indicate SEM. D–D″, Example of a mouse with severe degeneration seen at 14 d after KA by Nissl staining (D) and CD68 immunoreactivity (D′); in such case, staining for Gr-1 (D″) revealed the presence of numerous Gr-1-positive neutrophils (arrowheads) in zones of extensive degeneration. For abbreviations, see Figure 2. Scale bars: All panels, 250 μm.
Neutrophil infiltration in lesioned RAG1-KO mouse hippocampus underlies increased neurodegeneration
Tissue vacuolization in KA-treated RAG1-KO mice with extensive neurodegeneration suggested the active participation of immune cells to the destruction of neurons. Staining for Gr-1 was performed to determine whether neutrophils were involved in the innate response to KA lesion (Fig. 3). The Gr-1 epitope is a marker of early myeloid lineage commitment expressed on mature neutrophils, as well as some monocytes (Fleming et al., 1993). No Gr-1 immunoreactivity was detectable in saline control wild-type or RAG1-KO mice; on KA injection, it weakly labeled activated microglial cells in wild-type mice, but not macrophage-like cells (data not shown). Remarkably, in RAG1-KO mice, a population of strongly labeled, round cells with a lobulated nucleus (Fig. 4) was detected in sections from all animals with extensive neurodegeneration at 14 and 28 d after KA, suggesting infiltration of the lesioned tissue by Gr-1-positive neutrophils (Fig. 3D″). Examination of earlier time points revealed isolated Gr-1-positive cells within blood vessels up to 10 d after KA, followed by tissue infiltration on day 14, selectively in the lesioned tissue. Differential interference contrast microscopy showed that 100% Gr-1-positive cells were intravascular at 10 d after KA, and 50–70% extravascular at 14 d. Importantly, increased neurodegeneration affecting a subset of RAG1-KO mice coincided with the presence of Gr-1+ neutrophils within the lesioned tissue (Fig. 3D–D″). Conversely, neutrophils (identified by their lobulated nucleus and strong Gr-1 staining) were never detected in wild-type mice, neither in brain parenchyma nor in blood vessels, suggesting activation of chemoattractant signals specifically in the absence of T-lymphocytes.
Figure 4.

Engulfment of Gr-1+ neutrophils by activated microglial cells in RAG1-KO mice, as visualized by double immunofluorescence for Gr-1 (red) and Iba1 (green). Nuclei were counterstained with Sytox Green (blue). Images were acquired by confocal laser-scanning microscopy and depicted as maximal intensity projection of 10–15 layers spaced by 0.5 μm. A, Representative image from a B6 mouse, stratum radiatum of CA1, 14 d after KA injection, depicting the bushy appearance of activated microglial cells. The weak Gr-1 staining observed in B6 animals was not detected here because of the laser setting used for these images. B, C, Images from a RAG1-KO mice 14 d after KA injection. Gr-1+ cells (red) were identified as neutrophils based on their lobulated nucleus, as shown in color-separated images (B′, C′); the asterisk (*) in B and B′ indicates a transversally cut capillary; note that most nuclei belong to microglial cells, emphasizing their predominance in the lesioned tissue. Gr-1+ neutrophils were surrounded by one or several activated microglial cells, often strongly labeled for Iba1 (B, arrows), or even engulfed by them (B, arrowheads). These features are illustrated at higher magnification in C and C′, depicting two microglial cells establishing direct contacts with a Gr-1+ neutrophil, covering most of its surface. Scale bars: A, B, 20 μm; C, 10 μm.
Additional insight into the role of neutrophils was gained by double staining for Gr-1 and Iba1, to label both neutrophils and microglial cells (Fig. 4). Combined with Sytox Green fluorescence to visualize cell nuclei, these experiments confirmed that Gr-1+ cells had lobulated nuclei, identifying them as neutrophils, and showed that they systematically were surrounded, and even engulfed, by reactive microglial cells (Fig. 4B,C).
The contribution of Gr-1+ neutrophils to the exacerbated neurodegeneration seen in KA-treated RAG1-KO mice was tested experimentally by inactivation of circulating neutrophils on systemic treatment with functional anti-Gr-1 antibody (clone RB6-8C5) or nonimmune IgGs as control (Fig. 5). Wild-type mice were treated repeatedly every 3–4 d by intravenous infusion of 100 μg of antibodies, starting 1 d before KA injection (Table 1) and were examined histologically after 10 d to determine whether neutrophil neutralization during the acute phase of KA lesion had an influence on neurodegeneration. Because of concerns that neutrophil depletion might lead to postoperative infections in RAG1-KO mice, the anti-Gr-1 treatment was started 6 d after KA treatment with the aim to prevent the delayed neutrophil invasion of the lesioned hippocampus. Mice were treated three more times with inactivating antibodies and examined 14 d after KA (Table 1).
Figure 5.
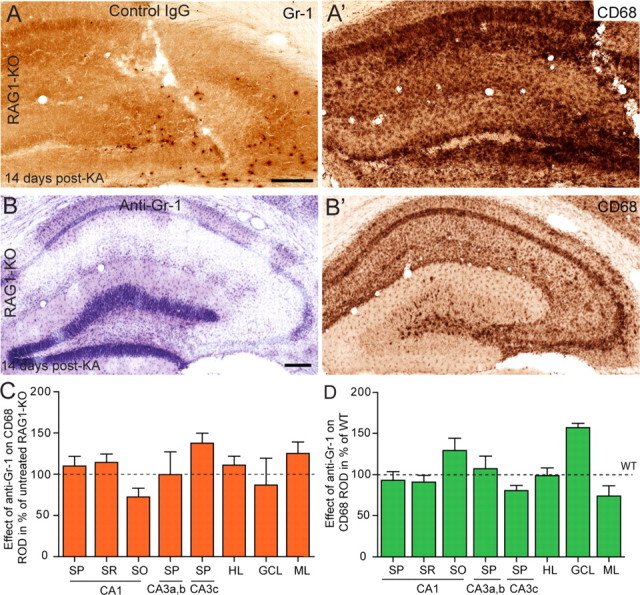
Depletion of Gr-1+ neutrophils by systemic anti-Gr-1 treatment prevents exacerbated neurodegeneration in RAG1-KO mice. A, A′, Representative example of a control RAG1-KO mouse, administered with control IgGs. A depicts infiltrating Gr1+ neutrophils into the KA-lesioned hippocampus and A′ depicts the corresponding CD68 staining. B, B′, Example of a RAG1-KO mouse treated with anti-Gr-1 antibodies. Nissl staining (B) and CD68 immunoreactivity (B′) illustrate a lesion similar to those seen in B6 mice (Fig. 2). C, Densitometric analysis of CD68 staining at 10 d after KA in RAG1-KO mice treated with anti-Gr-1 antibodies compared with control, untreated mice (dashed line). D, Densitometric analysis of CD68 staining in KA-injected RAG1-KO mice treated with anti-Gr-1 antibodies compared with KA-injected B6 mice (dashed lines). In both cases, no single region of interest had significantly different intensity of CD68 immunoreactivity in the two groups. Error bars indicate SEM. Scale bar, 250 μm.
As detected in Nissl-stained sections, anti-Gr-1 treatment did not influence the KA-induced lesion in wild-type mice (data not shown), whereas in RAG1-KO mice, it reduced lesion severity to wild-type levels (Fig. 5B,B′). Infusion of control IgG did not prevent neutrophil invasion in two of four mice, leading to extensive degeneration (Fig. 5A,A′). These conclusions were confirmed by quantification of CD68 immunoreactivity, as above. In KA-injected wild-type mice, there was no effect of anti-Gr-1 treatment (two-way ANOVA; F(1,112) = 1.24; p = 0.2672) despite significant main effect of the regions (F(7,112) = 17.59; p < 0.0001). However, post hoc analysis showed that no single region analyzed differed in CD68 staining between Gr-1-treated versus control mice (Fig. 5C). Likewise, comparison of Gr-1-treated RAG1-KO and wild-type mice revealed no significant difference in CD68 immunoreactivity across lesioned areas (F(1,64) = 2.72; p = 0.1041) (Fig. 5D), confirming that neutralization of neutrophils was sufficient to prevent exacerbation of the KA lesion. Effective depletion of neutrophils was confirmed by examination of spleen tissue stained for Gr-1, as well as brain sections, in which no Gr-1+ neutrophils were detectable (data not shown).
F4/80+ macrophages in dentate gyrus protect granule cells from degeneration
To determine whether additional parameters of innate immune response are activated by KA treatment in RAG1-KO mice, the distribution of F4/80+ macrophage-like cells in the dentate gyrus was analyzed (Fig. 6). As expected from the prominent granule cell dispersion seen at day 14 after KA, numerous strongly stained macrophage-like cells were readily detected in lesioned dentate gyrus of RAG1-KO mice, like in wild-type (Fig. 6A,B). Unexpectedly, these macrophages were missing in sections from mice with severe degeneration, affecting the integrity of the granule cell layer.
Figure 6.
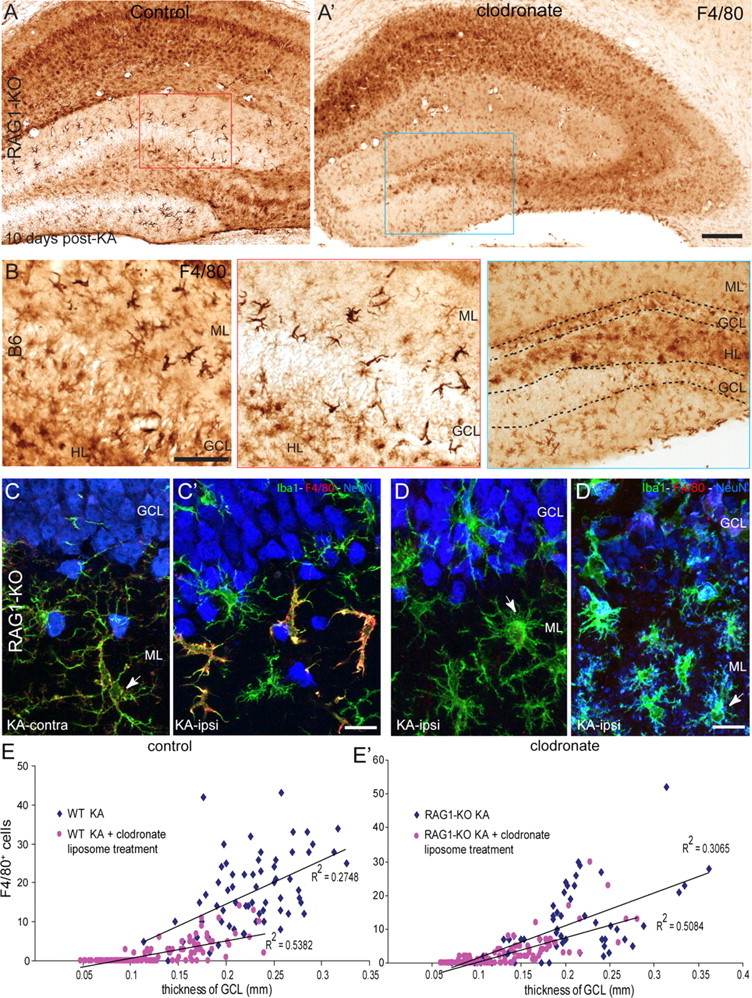
Depletion of peripheral mononuclear phagocytes affects dispersion and survival of dentate gyrus granule cells. A, A′, Effect of clodronate liposome treatment on the distribution of F4/80+ macrophage like-cells in the dentate gyrus of representative RAG1-KO mice. The red and blue boxed areas are enlarged below the main panels. B, For comparison, F4/80+ macrophages seen in B6 mice are depicted. C, D, Images of triple-immunofluorescence staining in the dentate gyrus against Iba1 (green), F4/80 (red), and NeuN (blue) of KA-treated RAG1-KO mice. Images were acquired by confocal laser-scanning microscopy and depicted as maximal intensity projection of 10–15 layers spaced by 0.5 μm. C, C′, In control mice, 10 d after KA, Iba1+ resting microglial cells were recognized contralaterally, by their long and slender processes and faint F4/80-IR (C; arrow). “Alerted” microglial cells were seen ipsilaterally (arrow), along with strongly labeled F4/80+ macrophage-like cells characterized by their larger cell body and short and thick processes (C′). Note the preservation of granule cells, which were enlarged and dispersed. D, D′, Representative images from RAG1-KO KA-injected mice treated with clodronate liposomes. Strongly labeled F4/80+ macrophages were conspicuously absent; microglial cells were alerted in zones where the granule cell layer was intact (D), whereas numerous activated microglial cells, with a “bushy” appearance, were seen where granule cell degeneration was evident (D′, arrow). E, E′, Correlation between granule cell dispersion and density of F4/80+ macrophages in B6 (E) and RAG1-KO (E′) mice. Each scatter plot depicts data from KA-injected mice treated with clodronate liposomes and untreated control mice. Regression analysis (thickness of granule cell layer versus number of F4/80+ cells) revealed in each genotype a significant correlation in both groups of mice. In addition, effect of clodronate liposome treatment was reflected by significantly reduced thickness of the granule cell layer and significantly reduced number of F4/80+ macrophages, occurring in both genotypes. Scale bars: A, 250 μm; B, 50 μm; C, D, 20 μm.
To determine the origin of these macrophage-like cells and investigate whether their presence is causally related to granule cell dispersion, peripheral mononuclear phagocytes were depleted using repeated systemic treatment with clodronate liposomes, first administered 1 d before KA injection and continuing every 3 d until day 10 after KA (Table 1). These liposomes do not pass the BBB, resulting in selective depletion of peripheral mononuclear phagocytes. Longer treatment was not feasible because of systemic toxicity, and the dose had to be reduced in RAG1-KO mice (25 μg/g body weight, instead of 75 μg/g in B6 mice). As control, both untreated mice (injected with KA only) and mice treated with “empty” liposomes were used; effectiveness of the treatment was verified by F4/80 immunostaining in the spleen, where a 60–85% reduction of F4/80+ cells was observed (data not shown).
Essentially identical results were obtained in wild-type and RAG1-KO mice, as evaluated 10 d after KA by cresyl violet (data not shown) and F4/80 immunoperoxidase staining (Fig. 6A,B). By depleting peripheral macrophages, clodronate liposome treatment caused a partial reduction of macrophage-like cells in the dentate gyrus, whereas the KA-induced lesion pattern in CA1 and CA3 was comparable with that seen in control mice. Overall, granule cell dispersion was reduced, and in five of eight clodronate-treated mice, zones of the granule cell layer containing pyknotic granule cells were observed (Fig. 6A′). The reduction of F4/80+ cells was most profound where granule cell dispersion was absent (granule cell layer width, ∼0.15–0.2 mm) and where granule cell degeneration occurred (granule cell layer width, <0.15 mm) (Fig. 6E,E′), suggesting that macrophage-like cells are required for survival of granule cells in KA-lesioned hippocampal formation. No effect of clodronate liposome treatment was seen on activated microglial cells, as verified by CD68 immunoreactivity (data not shown), suggesting a selective action on peripheral phagocytic mononuclear cells.
These results from immunoperoxidase staining were substantiated by triple immunofluorescence for NeuN (to visualize granule cells), Iba1, and F4/80 (Fig. 6C,D). In these images, presence of macrophage-like cells, recognized by strong F4/80 staining and by their distinctive morphology, was restricted to zones of marked granule cell dispersion, in which microglial cells were not activated (Fig. 6C′). In contrast, absence of macrophage-like cells coincided with microglia activation and, in part, with granule cell degeneration (Fig. 6D,D′).
These various conditions typically could be seen within a single section. Therefore, to quantify the relationship between F4/80+ cells and granule cell dispersion, we subdivided the granule cell layer into six segments of equal length, and determined the mean width of each segment (as an index of granule cell dispersion), as well as the number of F4/80+ macrophage-like cells associated with the granule cell layer. Linear regression analysis with data from four sections per mouse revealed a highly significant correlation between density of macrophage-like cells and width of the granule cell layer in both wild-type and RAG1-KO mice (Fig. 6E,E′). After clodronate liposome treatment, a significant reduction of both parameters (number of macrophages and granule cell layer thickness) was obtained. Although the correlation coefficient was reduced, it was still statistically significant (Fig. 6E,E′).
Together, these data suggest that F4/80+ macrophage-like cells are of peripheral origin and infiltrate the KA-lesioned hippocampus to selectively reside within the dentate gyrus. The presence of these cells correlates with granule cell hypertrophy and dispersion, and they appear to be required for long-term survival of granule cells in this model of TLE.
Early seizure onset in mutant mice with impaired adaptive immunity
A distinctive feature of the KA mouse model of TLE is the delayed onset of SRSs, which typically arise ∼2 weeks after KA injection, as shown previously in Swiss and NMRI mice (Riban et al., 2002; Arabadzisz et al., 2005). Here, to confirm that this delay also occurs in B6 mice, intrahippocampal EEG activity was recorded in six mice between days 2 and 20 after KA (Table 1). Each mouse was recorded every second day during 3–4 h after 30 min habituation to the recording chamber. The lesion pattern caused by KA injection and the placement of intrahippocampal electrodes were verified histologically after the last recording session. Some mice were discarded when either parameter was not satisfactory (Table 1). Finally, recording sessions had to be ended prematurely in some mice because of loss of connector.
No SRSs (defined as paroxysmal hippocampal discharges lasting for at least 20 s) were detectable before day 12 after KA in any of the six B6 mice recorded, which exhibited only isolated spikes-and-waves and occasional short paroxysmal events, lasting a few seconds. Thereafter, SRSs were detected in every recording session, with single events lasting up to 100 s (Fig. 7A,B). Their frequency was variable within and between animals with a maximum of ∼10 seizures per hour, although sometimes no seizures occurred for up to 1 h. Typically, long seizures coincided with behavioral arrest and head nodding.
Figure 7.
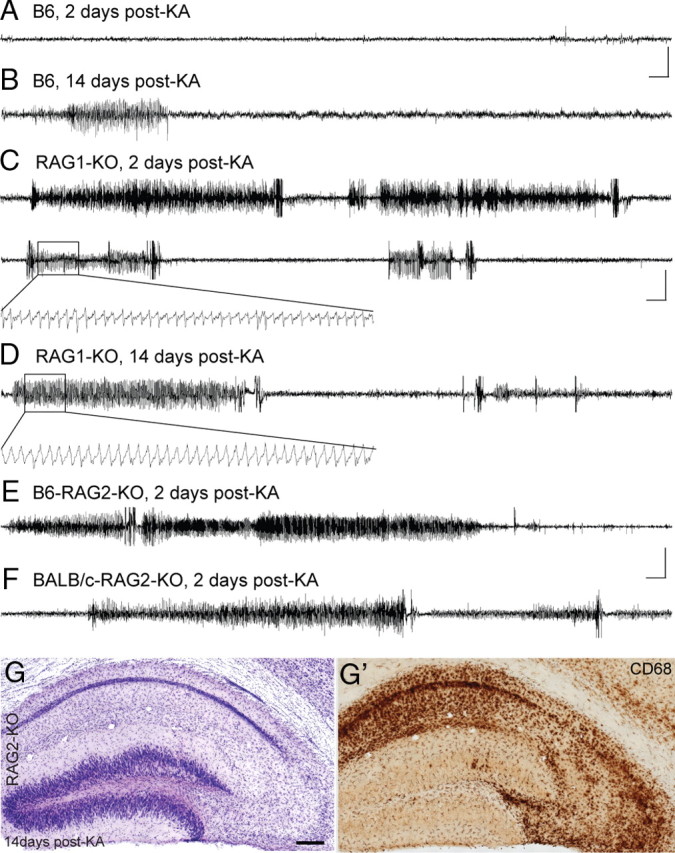
Representative hippocampal EEG recordings during the latent and chronic phases of TLE. A, B, B6 mice during the latent phase (2 d) and at the onset of the chronic phase (14 d) after KA injection; only isolated spikes were detected during the latent phase. C, D, KA-treated RAG1-KO mice at 2 and 14 d after KA; in both cases, SRSs with similar discharge frequency and duration were detected. The boxed areas in the upper trace (10 s duration) are depicted with better time resolution. E, F, Recordings in KA-treated RAG2-KO (B6 and BALB/c strains) at 2 d after KA, showing prolonged seizures. Calibration: horizontal, 5 s; vertical, 20 mV. G, G′, Histological analysis of RAG2-KO mice by Nissl staining (G) and CD68 immunoreactivity (G′) revealed a neurodegeneration similar to wild type, with increased severity, notably in CA3. Scale bar, 250 μm.
Next, we investigated whether impaired adaptive immunity was influencing seizure onset. Unexpectedly, intrahippocampal EEG recordings in RAG1-KO mice revealed the occurrence of characteristic SRSs already during the first session in all animals analyzed (starting at day 6 after KA in a first group of mice and at day 2 in the second group) (Table 1, Fig. 7C,D). These seizures lasted up to 300 s and their frequency was variable, reaching a maximum of 12 per hour. The seizures observed at 2–3 d after KA in RAG1-KO mice were indistinguishable from the ones recorded in B6 mice at 2–3 weeks after KA.
Based on these results, we wondered whether early seizure onset was correlated with severity of the KA lesion. Therefore, histological changes in KA-injected hippocampus were analyzed by Nissl staining at the end of the experiments. These observations revealed that 3 of the 10 recorded RAG1-KO mice had a rather mild lesion, with substantial preservation of the CA3a,b area and moderate granule cell dispersion, whereas the other 7 mice had a severe lesion, affecting most of the CA1-CA3 pyramidal layer and leading to prominent dispersion of the granule cell layer. No mice showed the complete destruction of the hippocampal formation associated with neutrophil invasion (Fig. 2). Nevertheless, frequency of SRSs did not correlate with lesion severity (Fig. 8A,B).
Figure 8.
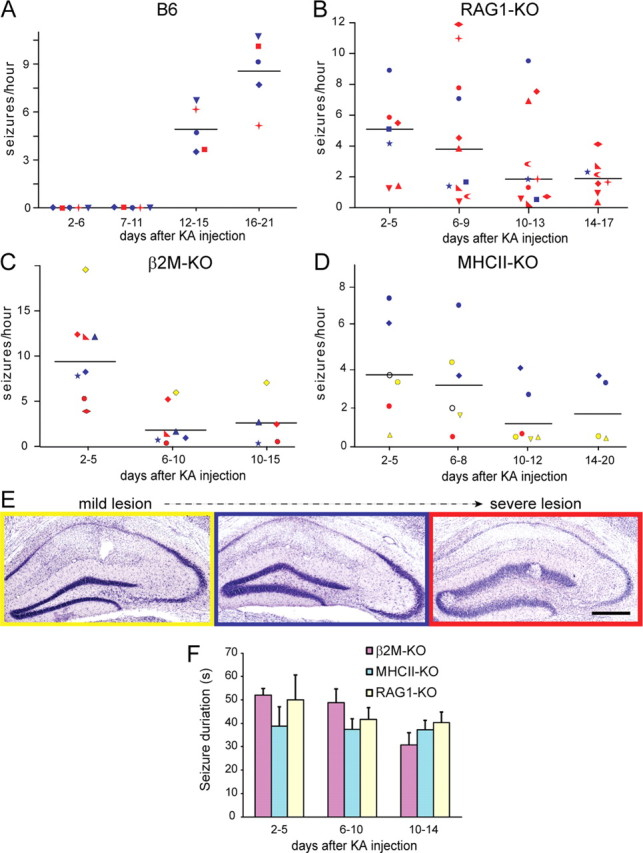
Scatter plot graphs showing the frequency of intrahippocampal SRSs in B6 (A), RAG1-KO (B), β2M-KO (C), and MHCII-KO (D) mice over time. Each mouse is represented by a different symbol. The color denotes the severity of the lesion, as depicted with a representative image in E (yellow, mild lesion; blue, moderate lesion; red, severe lesion). In D, the mouse depicted with an empty symbol died before tissue collection. At each time point on the _x_-axis, the median seizure frequency over two to three consecutive recording sessions is given for every mouse. The horizontal lines denote the overall median. A, Note the absence of SRSs up to 12–14 d after KA in B6 mice. B, In contrast, SRSs were detected already during the first recording session in all RAG1-KO mice. The frequency was variable between animals but declined at 14–17 d after KA. C, β2M-KO mice also showed seizure activity during the first recording session; thereafter, seizure frequency declined regardless of lesion severity, with the exception of a single mouse with a mild lesion. D, In MHCII-KO mice, SRSs occurred early on, with an apparent correlation between lesion severity and SRS frequency. E, Cresyl violet staining of the ipsilateral KA-injected hippocampus depicting representative examples of lesion severity seen after EEG recordings in KA-treated mice. F, Histogram of average seizure duration (mean ± SD) in the three lines of mutant mice, for three time points after KA injection. Two-way ANOVA analysis revealed no significant difference. Scale bar, 200 μm.
Together, the occurrence of a latent phase of epileptogenesis in this TLE model appears related to the integrity of the adaptive immune system. Deletion of the RAG1 gene is sufficient to enable early SRS onset even in mice with a mild lesion, although the mutation selectively affects maturation of lymphocytes. However, a potential pitfall of RAG1-KO mice is that this expression of this gene has also been reported in hippocampus and cerebellum (Mombaerts et al., 1992). To discard the possibility that early seizure onset was attributable to the absence of RAG1 in the CNS, we performed a series of EEG recordings in KA-treated RAG2-KO mice. This gene is functionally homologous to RAG1 but is not expressed in the CNS (Chun et al., 1991), thereby excluding confounding effects potentially present in RAG1-KO mice.
RAG2-KO mice were available in two different strains (B6 and BALB/c) (Table 2), allowing us to test for possible strain-related effects. KA-treated B6-RAG2-KO and BALB/c-RAG2-KO mice were recorded every second day, starting at days 2–3 after KA injection and continuing until day 16 (Table 1). As seen in RAG1-KO mice, SRSs occurred already at days 2–3 after KA injection in all RAG2-KO mice analyzed (Fig. 7E,F), regardless of strain, reproducing the shortening of the latent phase seen in RAG1-KO mice. Likewise, the pattern of hippocampal damage caused by the KA injection was comparable in extent and variability in RAG2-KO mice with that occurring in RAG1-KO mice (Fig. 7G,G′). These results demonstrate that lack of RAG1 in the brain was not responsible for early onset of SRSs after KA treatment.
To determine whether CD4+ or CD8+ T-cells are differentially implicated in the abolition of the latent phase observed in RAG1-KO mice, we performed intrahippocampal EEG recordings in β2M-KO and in MHCII-KO mice, lacking CD8+ and CD4+ T-cells, respectively (Fig. 8C,D, Table 2). Mice were recorded first at days 2–3 after KA and successively every second day until days 14–15 in β2M-KO and days 16–20 in MHCII-KO mice (Table 1). Similar to RAG1- and RAG2-KO mice, SRSs were detected already during the first recording session in β2M-KO mice. Their frequency was variable, reaching up to 20 seizures per hour. Thereafter, however, we observed a marked reduction of seizure frequency, with the majority of mice having less than two seizures per hour during the second week of recordings (Fig. 8C). In these mutants, conspicuous interictal paroxysmal activity was evident, ranging from isolated spikes-and-wave to short “seizure-like” episodes typically lasting 5–10 s. Histological analysis revealed that severity of the lesion in these β2M-KO mice was mostly similar to that seen in B6 mice (Fig. 8C). However, one mouse had a moderate lesion and two mice a severe lesion, which did not correlate with time of onset or frequency of SRSs (Fig. 8C).
The analysis of intrahippocampal EEG recording in MHCII-KO mice reproduced the abolition of the latent phase observed in RAG1-KO mice. Median seizure frequency was low, between 2 and 4 SRSs per hour, although two mice with a moderate lesion had up to 10 SRSs per hour (Fig. 8D). Like in β2M-KO mice, short interictal paroxysmal discharges were conspicuous. Overall lesion severity in this specific cohort of mice was less than in wild-type mice, which might have contributed to reduce the incidence of SRSs. Finally, comparison of SRS duration among the three lines of mutant mice revealed no significant differences and no change over time after KA injection (Fig. 8F).
Intrahippocampal KA injection causes local inflammation and BBB disruption
To determine whether early seizure occurrence in RAG1-KO mice compared with B6 is attributable to a difference in acute immune-mediated responses triggered by KA injection, possibly on B-cell activation in B6 mice, we examined the expression of ICAM-1, BBB integrity, and microglial cell activation at 24 h after KA (Fig. 9). In both genotypes, ICAM-1 immunoreactivity in brain blood vessels was increased selectively in lesioned hippocampus relative to the contralateral side (Fig. 9A,B). Quantification by densitometry using the same intensity threshold across all samples showed that the proportional area occupied on the ipsilateral side by pixels positive for ICAM-1 was on average 240% higher than contralaterally in B6 mice (contralateral, 6.5 ± 0.3%; ipsilateral, 15.4 ± 0.5%; p < 0.01) and 241% in RAG1-KO mice (contralateral, 7.8 ± 0.3%; ipsilateral, 17.3 ± 0.3%; mean ± SD; n = 3–4 mice). At later time points, ICAM-1 was most strongly upregulated in glial cells (Fig. 9C,C′), pointing to persistent activation in line with a role in mediating leukocyte–endothelial cell adhesion required for brain infiltration of T-lymphocytes. Evidence for acute focal BBB leakage was evidenced by fibrinogen immunostaining, revealing accumulation notably in the lesioned CA1 and CA3c areas in both B6 (data not shown) and RAG1-KO mice (Fig. 9D,D′). Finally, as seen by CD68 immunoreactivity, microglial cells were activated ipsilaterally in the hippocampus and dentate gyrus, without any evidence for a difference between B6 and RAG1-KO mice (Fig. 9E,E′). These experiments also revealed that the “background” staining typically produced by IgGs in tissue from lesioned mice was abolished in RAG1-KO tissue (Fig. 9F,F′), reflecting the lack of IgG production in the absence of T- and B-lymphocytes. Collectively, these results suggest that the acute effects of KA injection are very similar in B6 and RAG1-KO mice, inducing a local inflammatory reaction, presumably by triggering cytokine and chemokine release from activated glial cells.
Figure 9.
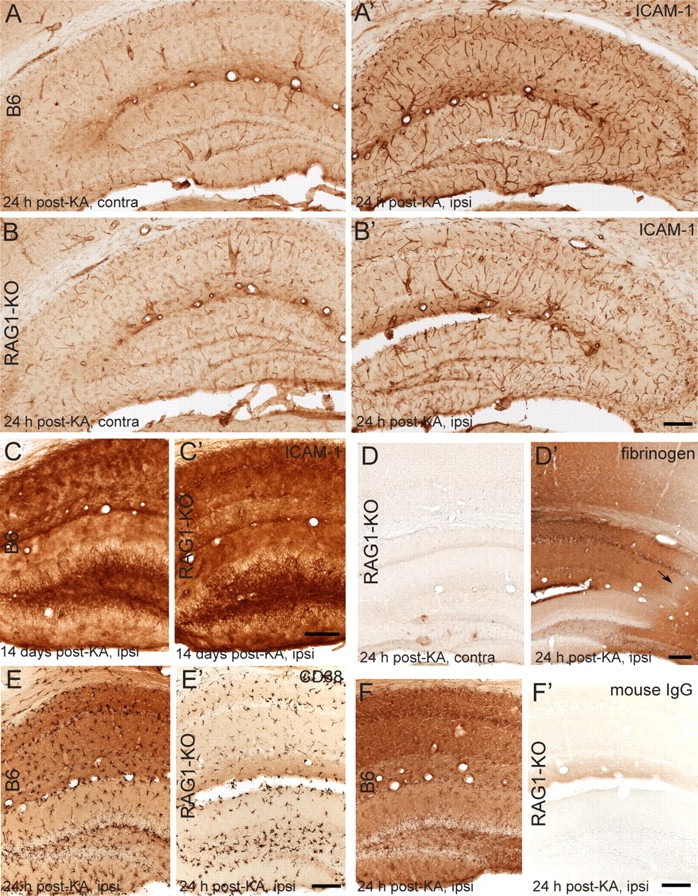
KA injection causes a local inflammatory response in both B6 and RAG1-KO mice, with overexpression of ICAM-1 in vascular endothelium, focal BBB disruption, and microglia activation. A–C, Comparison of ICAM-1 immunoreactivity in B6 and RAG1-KO mice in the ipsilateral (ipsi) (KA-injected) and contralateral (contra) hippocampus, as seen after 1 d (A, A′; B, B′) and 14 d (C, C′) after KA injection. Pronounced increase in staining intensity in vascular endothelium of the lesioned hippocampus is evident acutely in both genotypes. At 14 d after KA (C, C′), ICAM-1 immunoreactivity is increased further, reflecting strong expression in glial cells. D, D′, Staining for fibrinogen reveals focal extravasation in the ispilateral hippocampus and along the needle track in the overlaying cortex, whereas the contralateral side of the same section shows very little immunoreactivity, as illustrated for a RAG1-KO mice. Note the relative sparing of the CA2 sector (arrow) and of the dentate gyrus. E, E′, Similar acute activation of microglial cells in the lesioned hippocampal formation, as seen in both genotypes by CD68 staining at 24 h after KA injection. The background in the section from B6 mice is attributable to IgGs in the damaged tissue, as confirmed by incubation of sections with secondary anti-mouse IgG antibodies (F, F′). The absence of IgGs in RAG1-KO mice (devoid of B- and T-lymphocytes) completely prevents this nonspecific staining. Scale bars: All panels, 100 μm.
Discussion
This study demonstrates activation of multiple components of adaptive and innate immunity in the KA mouse model of TLE, leading to local inflammation, BBB disruption, and infiltration of leukocytes, as well as sustained endothelial and microglial cell activation in areas of neurodegeneration. Genetic and pharmacological interventions to prevent T-cell infiltration and deplete peripheral macrophages markedly worsened the KA-induced lesion. Thus, neurodegeneration was worsened in RAG1-KO mice on Gr-1+ neutrophil infiltration, whereas macrophage depletion induced granule cell degeneration. These findings point to a neuroprotective role of lymphocytes and macrophages infiltrating the epileptic focus. Functionally, onset of SRSs shortly after status epilepticus in mice lacking CD4+ and/or CD8+ T-cells revealed strong impact of immune-derived factors on neuronal networks sustaining seizure activity. Together, concerted action of adaptive and innate immunity provides neuroprotective and seizure-suppressant effects, shedding new light on the role of neuroimmune interactions in TLE with HS.
Validation of the KA mouse model
A distinctive feature of KA-mouse model of TLE is the focal nature of seizures, which corresponds to the strictly unilateral lesion (Arabadzisz et al., 2005; Duveau and Fritschy, 2010). Furthermore, the temporal gap between KA-induced status epilepticus and onset of SRSs (Riban et al., 2002) allows separating epileptogenic mechanisms from excitotoxic action of KA. Thus, activation of innate immunity and leukocyte infiltration in KA-treated mice and in tissue from patients with intractable MTLE (Fabene et al., 2008; Ravizza et al., 2008) indicate that this mouse model is relevant for studying the role of neuroimmune interactions in MTLE with HS.
The importance for epileptogenesis of proinflammatory signaling cascades, as well as leukocyte–endothelium interactions and BBB leakage, has been demonstrated in the pilocarpine model of TLE (Marchi et al., 2007; Ravizza et al., 2008; Fabene et al., 2010). Neurodegeneration induced by pilocarpine is more discrete than in KA-injected mice but extends to extrahippocampal regions (Curia et al., 2008). However, comparison of in vitro and in vivo effects of pilocarpine pointed to a role of systemic inflammation in ictogenesis (Marchi et al., 2007), possibly because of activation of muscarinic receptors on leukocytes. Furthermore, neutrophils are also involved, as shown by antibody-mediated suppression (Fabene et al., 2008), underscoring the importance of systemic inflammation for ictogenesis. Finally, extravasation of plasma proteins and K+ ions contributes to SRSs in pilocarpine-treated rodents (Marchi et al., 2007, 2010), in line with proepileptogenic effects of BBB disruption (Friedman et al., 2009).
By analogy, since lymphocytes express ionotropic glutamate receptors (Ganor et al., 2003), one might hypothesize that intrahippocampal KA injection induces a systemic inflammation leading to intraparenchymal CD3+ T-cell diapedesis. However, this idea implies that such B-cell-mediated systemic inflammation would not occur in KA-treated RAG1-KO mice, unlike our present results. Rather, KA injection causes similar acute effects in B6 and RAG1-KO mice, with local upregulation of ICAM-1, focal BBB disruption, and activation of microglial cells occurring selectively in the lesioned hippocampus.
Together, comparison of the two models shows that a systemic inflammation is not a precondition for onset of SRSs in rodents. Furthermore, its absence in KA-treated mice might explain their focal nature, whereas local inflammation might contribute to model-specific histopathological features, notably granule cell dispersion linked to macrophage invasion in the dentate gyrus.
Significance of microglial cell activation
Although microglial cell activation reflects CNS damage and can be deleterious (Rodgers et al., 2009; Haselkorn et al., 2010, Maroso et al., 2010), increasing evidence indicates that it can also be beneficial by increasing neurotrophic and antiinflammatory factors, clearing debris, and possibly promoting repair (Batchelor et al., 1999; Boscia et al., 2009; Rivest, 2009). Here, in line with the study by Siao et al. (2003), demonstrating that microglia activation after intrahippocampal KA injection depends on tPA (tissue plasminogen activator) release from injured neurons, several indications suggest that microglia activation occurs as a consequence rather than a cause of KA-induced neurodegeneration. First, the distribution of activated microglial cells overlapped perfectly with that of pyknotic neurons in the chronic, but not acute phase after KA injection (Fig. 9E,E′). Second, aggravated neurodegeneration in RAG1-KO mice, mirrored by increased CD68 immunoreactivity, was dependent on neutrophil infiltration (Fig. 5); particularly striking was the observation of Gr1+ neutrophils engulfed by activated microglial cells, suggesting an attempt to protect brain tissue from neutrophil-mediated destruction (Fig. 4). Third, in the dentate gyrus, macrophage depletion induced a clear transition from resting to “alerted” microglia (Hanisch and Kettenmann, 2007) in the absence of granule cell death, whereas activated microglia were seen on granule cell degeneration (Fig. 6D′); of note, denervation of granule cells on lesion of the perforant pathway induces microglial cell activation without causing cell loss in the dentate gyrus (Nielsen et al., 2009). Therefore, microglial cell activation likely is triggered by neurodegeneration in this TLE model. However, the lack of correlation between SRS frequency/duration and lesion severity suggests that microglial cells do not contribute directly to SRS generation.
Blood-borne macrophages infiltrate the molecular layer of the lesioned hippocampus
The presence of strongly labeled CD45+ and F4/80+ macrophage-like cells in the dentate gyrus raised questions about their identity, origin, and function. They were distinct from microglial cells based on differential CD68 staining and morphology, but belong to the same lineage and therefore likely represent mononuclear phagocytic cells. Previous studies have shown macrophage infiltration into the dentate gyrus after denervation (Babcock et al., 2003; Fux et al., 2008). Here, the reduction of macrophage-like cells, but not microglia, after systemic clodronate liposome treatment is the strongest argument in favor of their peripheral origin. Finally, decreased dispersion and even partial degeneration of dentate granule cells on decreased macrophage brain penetration strongly suggested a neuroprotective role of macrophages for granule cells.
This beneficial effect of macrophages suggests release of factors supporting neuronal survival and migration, but precise mechanism of action is unclear. Alternatively, since granule cell dispersion after KA injection depends on TrkB receptor activation after status epilepticus (Inoue et al., 1998) and on disrupted Reelin signaling (Duveau et al., 2010; Haas and Frotscher, 2010), it is conceivable that macrophages are involved in these processes.
Infiltration of CD3+ T-cells in the KA-injected hippocampus is neuroprotective
In KA-treated mice, absence of T-cells curtails the latent phase of epileptogenesis, underscoring the relevance of immune system integrity for protecting against premature seizure activity. Silverberg et al. (2010) showed that brief episodes of tonic-clonic seizure activity are sufficient to trigger CD3+ lymphocyte infiltration into the neocortex and hippocampus. Here, the likely driving force behind lymphocyte recruitment in the lesioned hippocampus is the local inflammation evidenced by focal fibrinogen infiltration and persistent upregulation of ICAM-1. As discussed above, leukocyte infiltration implicates functional changes at the BBB, along with increased expression of cytokines that facilitate diapedesis into the brain (Engelhardt, 2008). IL-1β, which induces ICAM-1 and MCP-1, is credited primarily for recruitment of macrophages but also acting as T-cell chemoattractant (Gibson et al., 2004). Accordingly, blockade of IL-1β reduces pilocarpine-induced status epilepticus and BBB leakage (Marchi et al., 2009) and has been proposed as therapeutic venue to prevent development of epilepsy (Vezzani and Baram, 2007). Therefore, recruitment of CD3+ T-cells in KA-lesioned hippocampus might represent a secondary outcome of the inflammatory response serving to limit activation of innate immunity. The infiltration of Gr-1+ neutrophils in RAG1-KO mice gives additional credit to this idea.
Onset of SRSs in KA-treated mice depends on integrity of CD3+ T-cell-mediated adaptive immunity
Prolonged bilateral stimulation of the perforant pathway in mice is sufficient for reproducing key features of MTLE with HS, including hippocampal atrophy, mossy fiber sprouting, and SRSs (Kienzler et al., 2009). Therefore, seizure-induced hippocampal damage might contribute significantly to the pathophysiology of MTLE, allowing to disregard potential confounding factors linked to systemic administration of convulsants such as pilocarpine. In contrast, seizure activity in the pilocarpine model of TLE is associated with inflammatory changes in the CNS vasculature (Fabene et al., 2008; Marchi et al., 2009). Therefore, to reconcile these outcomes, it is likely that specific factors from the immune system influence neuronal mechanisms of epileptogenesis. This possibility is strongly supported by early SRS onset in mice lacking subsets of lymphocytes, despite having a similar local inflammation in the lesioned hippocampus. Furthermore, a major implication of our results is that synaptic reorganization in the lesioned hippocampus, notably mossy fiber sprouting, is not necessary for triggering SRSs. Rather, acute damage caused by KA injection, including destruction of hilar cells and parvalbumin-positive interneurons in CA1 and dentate gyrus (Bouilleret et al., 2000), is sufficient for eliciting SRSs, but their occurrence is prevented by mechanisms dependent on CD3+ T-cells.
This interpretation is complicated by the fact that none of the mutant mice analyzed here can be considered simply as lacking a subset of T-cells. Rather, multiple cytokines and chemokines produced by CD4+ and CD8+ cells to influence the maturation and production of other cells types are lacking, as well. Furthermore, the role of IgGs, produced by B-cells and present in lesioned tissue of B6 mice (Fig. 9F,F′), and of MHCII molecules, expressed for instance by dendritic cells, microglial cells, and macrophages, is not known in the context of epileptogenesis. Determining which cytokines and chemokines are produced in wild type, but not in mutant mice after KA injection will be required to understand the role of local inflammatory mechanisms for seizure occurrence in TLE.
Footnotes
This work was supported by the Swiss National Science Foundation (National Center of Competence in Research–Neuro), a grant to J.-M.F. and M.Z. from the Swiss League against Epilepsy, a fellowship to M.Z. from the Center for Neuroscience Zurich, and a fellowship to M.L.M. from the region of Sardinia, Italy. We are grateful to Prof. B. Becher and Prof. A. Aguzzi (University of Zurich, Zurich, Switzerland) for providing us with mutant mice lacking subsets of lymphocytes.
References
- Antonucci F, Di Garbo A, Novelli E, Manno I, Sartucci F, Bozzi Y, Caleo M. Botulinum neurotoxin E (BoNT/E) reduces CA1 neuron loss and granule cell dispersion, with no effects on chronic seizures, in a mouse model of temporal lobe epilepsy. Exp Neurol. 2008;210:388–401. doi: 10.1016/j.expneurol.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Arabadzisz D, Antal K, Parpan F, Emri Z, Fritschy JM. Epileptogenesis and chronic seizures in a mouse model of temporal lobe epilepsy are associated with distinct EEG patterns and selective neurochemical alterations in the contralateral hippocampus. Exp Neurol. 2005;194:76–90. doi: 10.1016/j.expneurol.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23:7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, Donnan GA, Howells DW. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci. 1999;19:1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscia F, Esposito CL, Di Crisci A, de Franciscis V, Annunziato L, Cerchia L. GDNF selectively induces microglial activation and neuronal survival in CA1/CA3 hippocampal regions exposed to NMDA insult through Ret/ERK signalling. PLoS One. 2009;4:e6486. doi: 10.1371/journal.pone.0006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouilleret V, Ridoux V, Depaulis A, Marescaux C, Nehlig A, Le Gal La Salle G. Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience. 1999;89:717–729. doi: 10.1016/s0306-4522(98)00401-1. [DOI] [PubMed] [Google Scholar]
- Bouilleret V, Loup F, Kiener T, Marescaux C, Fritschy JM. Early loss of interneurons and delayed subunit-specific changes in GABAA-receptor expression in a mouse model of mesial temporal lobe epilepsy. Hippocampus. 2000;10:305–324. doi: 10.1002/1098-1063(2000)10:3<305::AID-HIPO11>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Chun JJ, Schatz DG, Oettinger MA, Jaenisch R, Baltimore D. The recombination activating gene-1 (RAG-1) transcript is present in the murine central nervous system. Cell. 1991;64:189–200. doi: 10.1016/0092-8674(91)90220-s. [DOI] [PubMed] [Google Scholar]
- Curia G, Longo D, Biagini G, Jones RS, Avoli M. The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods. 2008;172:143–157. doi: 10.1016/j.jneumeth.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugladze T, Vida I, Tort AB, Gross A, Otahal J, Heinemann U, Kopell NJ, Gloveli T. Impaired hippocampal rhythmogenesis in a mouse model of mesial temporal lobe epilepsy. Proc Natl Acad Sci U S A. 2007;104:17530–17535. doi: 10.1073/pnas.0708301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duveau V, Fritschy JM. PSA-NCAM-dependent GDNF signaling limits neurodegeneration and epileptogenesis in temporal lobe epilepsy. Eur J Neurosci. 2010;32:89–98. doi: 10.1111/j.1460-9568.2010.07272.x. [DOI] [PubMed] [Google Scholar]
- Duveau V, Madhusudan A, Caleo M, Knuesel I, Fritschy JM. Impaired reelin processing and secretion by Cajal-Retzius cells contributes to granule cell dispersion in a mouse model of temporal lobe epilepsy. Hippocampus. 2010 doi: 10.1002/hipo.20793. [DOI] [PubMed] [Google Scholar]
- Engelhardt B. Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci. 2008;274:23–26. doi: 10.1016/j.jns.2008.05.019. [DOI] [PubMed] [Google Scholar]
- Fabene PF, Navarro Mora G, Martinello M, Rossi B, Merigo F, Ottoboni L, Bach S, Angiari S, Benati D, Chakir A, Zanetti L, Schio F, Osculati A, Marzola P, Nicolato E, Homeister JW, Xia L, Lowe JB, McEver RP, Osculati F, et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14:1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabene PF, Bramanti P, Constantin G. The emerging role for chemokines in epilepsy. J Neuroimmunol. 2010;224:22–27. doi: 10.1016/j.jneuroim.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Gouder N, Güttinger M, Gabernet L, Scheurer L, Rülicke T, Crestani F, Boison D. Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain. 2005;128:2383–2395. doi: 10.1093/brain/awh555. [DOI] [PubMed] [Google Scholar]
- Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol. 1993;151:2399–2408. [PubMed] [Google Scholar]
- Friedman A, Kaufer D, Heinemann U. Blood-brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res. 2009;85:142–149. doi: 10.1016/j.eplepsyres.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Mohler H. GABAA-receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J Comp Neurol. 1995;359:154–194. doi: 10.1002/cne.903590111. [DOI] [PubMed] [Google Scholar]
- Fux M, van Rooijen N, Owens T. Macrophage-independent T cell infiltration to the site of injury-induced brain inflammation. J Neuroimmunol. 2008;203:64–72. doi: 10.1016/j.jneuroim.2008.06.025. [DOI] [PubMed] [Google Scholar]
- Ganor Y, Besser M, Ben-Zakay N, Unger T, Levite M. Human T cells express a functional ionotropic glutamate receptor GluR3, and glutamate by itself triggers integrin-mediated adhesion to laminin and fibronectin and chemotactic migration. J Immunol. 2003;170:4362–4372. doi: 10.4049/jimmunol.170.8.4362. [DOI] [PubMed] [Google Scholar]
- Gibson RM, Rothwell NJ, Le Feuvre RA. CNS injury: the role of the cytokine IL-1. Vet J. 2004;168:230–237. doi: 10.1016/j.tvjl.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Haas CA, Frotscher M. Reelin deficiency causes granule cell dispersion in epilepsy. Exp Brain Res. 2010;200:141–149. doi: 10.1007/s00221-009-1948-5. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Haselkorn ML, Shellington DK, Jackson EK, Vagni VA, Janesko-Feldman K, Dubey RK, Gillespie DG, Cheng D, Bell MJ, Jenkins LW, Homanics GE, Schnermann J, Kochanek PM. Adenosine A1 receptor activation as a brake on the microglial response after experimental traumatic brain injury in mice. J Neurotrauma. 2010;27:901–910. doi: 10.1089/neu.2009.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich C, Nitta N, Flubacher A, Müller M, Fahrner A, Kirsch M, Freiman T, Suzuki F, Depaulis A, Frotscher M, Haas CA. Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus. J Neurosci. 2006;26:4701–4713. doi: 10.1523/JNEUROSCI.5516-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Hirai H, Onteniente B, Suzuki F. Correlated long-term increase of brain-derived neurotrophic factor and Trk B proteins in enlarged granule cells of mouse hippocampus after kainic acid injection. Neuroscience. 1998;86:723–728. doi: 10.1016/s0306-4522(98)00112-2. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Patterson PH. The role of cytokines and growth factors in seizures and their sequelae. Prog Neurobiol. 2001;63:125–149. doi: 10.1016/s0301-0082(00)00022-8. [DOI] [PubMed] [Google Scholar]
- Kienzler F, Norwood BA, Sloviter RS. Hippocampal injury, atrophy, synaptic reorganization, and epileptogenesis after perforant pathway stimulation-induced status epilepticus in the mouse. J Comp Neurol. 2009;515:181–196. doi: 10.1002/cne.22059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledergerber D, Fritschy JM, Kralic JE. Impairment of dentate gyrus neuronal progenitor cell differentiation in a mouse model of temporal lobe epilepsy. Exp Neurol. 2006;199:130–142. doi: 10.1016/j.expneurol.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Le Duigou C, Wittner L, Danglot L, Miles R. Effects of focal injection of kainic acid into the mouse hippocampus in vitro and ex vivo. J Physiol. 2005;569:833–847. doi: 10.1113/jphysiol.2005.094599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Duigou C, Bouilleret V, Miles R. Epileptiform activities in slices of hippocampus from mice after intra-hippocampal injection of kainic acid. J Physiol. 2008;586:4891–4904. doi: 10.1113/jphysiol.2008.156281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loup F, Wieser HG, Yonekawa Y, Aguzzi A, Fritschy JM. Selective alterations in GABAA receptor subtypes in human temporal lobe epilepsy. J Neurosci. 2000;20:5401–5419. doi: 10.1523/JNEUROSCI.20-14-05401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen L, Labrecque N, Engberg J, Dierich A, Svejgaard A, Benoist C, Mathis D, Fugger L. Mice lacking all conventional MHC class II genes. Proc Natl Acad Sci U S A. 1999;96:10338–10343. doi: 10.1073/pnas.96.18.10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Oby E, Batra A, Uva L, De Curtis M, Hernandez N, Van Boxel-Dezaire A, Najm I, Janigro D. In vivo and in vitro effects of pilocarpine: relevance to ictogenesis. Epilepsia. 2007;48:1934–1946. doi: 10.1111/j.1528-1167.2007.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Fan Q, Ghosh C, Fazio V, Bertolini F, Betto G, Batra A, Carlton E, Najm I, Granata T, Janigro D. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. 2009;33:171–181. doi: 10.1016/j.nbd.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Teng Q, Ghosh C, Fan Q, Nguyen MT, Desai NK, Bawa H, Rasmussen P, Masaryk TK, Janigro D. Blood–brain barrier damage, but not parenchymal white blood cells, is a hallmark of seizure activity. Brain Res. 2010;1353:176–186. doi: 10.1016/j.brainres.2010.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- Nielsen HH, Ladeby R, Fenger C, Toft-Hansen H, Babcock AA, Owens T, Finsen B. Enhanced microglial clearance of myelin debris in T cell-infiltrated central nervous system. J Neuropathol Exp Neurol. 2009;68:845–856. doi: 10.1097/NEN.0b013e3181ae0236. [DOI] [PubMed] [Google Scholar]
- Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia. 2006;47:1761–1774. doi: 10.1111/j.1528-1167.2006.00817.x. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Boer K, Redeker S, Spliet WG, van Rijen PC, Troost D, Vezzani A, Aronica E. The IL-1b system in epilepsy-associated malformations of cortical development. Neurobiol Dis. 2006;24:128–143. doi: 10.1016/j.nbd.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Gagliardi B, Noé F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29:142–160. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Riazi K, Galic MA, Kuzmiski JB, Ho W, Sharkey KA, Pittman QJ. Microglial activation and TNFα production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci U S A. 2008;105:17151–17156. doi: 10.1073/pnas.0806682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riban V, Bouilleret V, Pham-Lê BT, Fritschy JM, Marescaux C, Depaulis A. Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy. Neuroscience. 2002;112:101–111. doi: 10.1016/s0306-4522(02)00064-7. [DOI] [PubMed] [Google Scholar]
- Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- Rodgers KM, Hutchinson MR, Northcutt A, Maier SF, Watkins LR, Barth DS. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain. 2009;132:2478–2486. doi: 10.1093/brain/awp177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultzberg M, Lindberg C, Aronsson AF, Hjorth E, Spulber SD, Oprica M. Inflammation in the nervous system—physiological and pathophysiological aspects. Physiol Behav. 2007;92:121–128. doi: 10.1016/j.physbeh.2007.05.050. [DOI] [PubMed] [Google Scholar]
- Seiler P, Aichele P, Odermatt B, Hengartner H, Zinkernagel RM, Schwendener RA. Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur J Immunol. 1997;27:2626–2633. doi: 10.1002/eji.1830271023. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- Siao CJ, Fernandez SR, Tsirka SE. Cell type-specific roles for tissue plasminogen activator released by neurons or microglia after excitotoxic injury. J Neurosci. 2003;23:3234–3242. doi: 10.1523/JNEUROSCI.23-08-03234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverberg J, Ginsburg D, Orman R, Amassian V, Durkin HG, Stewart M. Lymphocyte infiltration of neocortex and hippocampus after a single brief seizure in mice. Brain Behav Immun. 2010;24:263–272. doi: 10.1016/j.bbi.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Liu S, Kubes P, Yong VW. Depletion of Ly6G/Gr-1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J Neurosci. 2009;29:753–764. doi: 10.1523/JNEUROSCI.4918-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straessle A, Loup F, Arabadzisz D, Ohning GV, Fritschy JM. Rapid and long-term alterations of hippocampal GABAB receptors in a mouse model of temporal lobe epilepsy. Eur J Neurosci. 2003;18:2213–2226. doi: 10.1046/j.1460-9568.2003.02964.x. [DOI] [PubMed] [Google Scholar]
- Turrin NP, Rivest S. Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol Dis. 2004;16:321–334. doi: 10.1016/j.nbd.2004.03.010. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, da Costa Araújo S, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130:521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Baram TZ. New roles for interleukin-1b in the mechanisms of epilepsy. Epilepsy Curr. 2007;7:45–50. doi: 10.1111/j.1535-7511.2007.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Balosso S, Ravizza T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun. 2008;22:797–803. doi: 10.1016/j.bbi.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R. β2-microglobulin deficient mice lack CD4−8+ cytolytic T cells. Nature. 1990;344:742–746. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]