Molecular Mechanisms of Estrogen Receptors' Suppression of Lipogenesis in Pancreatic β-Cells (original) (raw)
Abstract
The gonadal steroid, 17β-estradiol (E2), suppresses pancreatic islet fatty acid and glycerolipid synthesis and prevents β-cell failure in rodent models of type 2 diabetes. β-Cell estrogen receptors (ER) mediate these actions by suppressing the expression and enzymatic activity of fatty acid synthase (FAS). Here, we explored the mechanism of FAS suppression. We show that E2, and pharmacological agonists for ERα, ERβ, and the G protein-coupled ER, suppress mRNA and protein expression of the transcriptional regulators of FAS, namely, sterol regulatory element-binding protein 1c (SREBP1c) and carbohydrate response element binding protein (ChREBP) in insulin-secreting INS-1 cells. ER suppress SREBP1c and ChREBP mRNA and protein expression via an extranuclear localization. Using two mouse lines with pancreas-specific null deletion of either ERα or the signal transducer and activator of transcription 3 (STAT3), we show that ERα activation in vivo reduces SREBP1c and ChREBP mRNA expression via a direct islet action involving STAT3 activation. The master regulators of lipogenesis, liver X receptor (LXR) α and β, transcriptionally up-regulate SREBP1c and ChREBP. We find that activation of ERα, ERβ, and G protein-coupled ER suppresses LXR's mRNA expression in INS-1 cells. We also observe that activation of ERα in mouse islets in vivo suppresses LXR mRNA in a STAT3-dependent manner. Finally, we show that E2 also activates and uses AMP-activated protein kinase in INS-1 cells to suppress SREBP1c protein expression. This study identifies extranuclear ER pathways involving STAT3 and AMP-activated protein kinase in the genetic control of lipogenesis with therapeutic implications to protect β-cells in type 2 diabetes.
The gonadal steroid, 17β-estradiol (E2), is involved in reproductive, bone, cardiovascular, and neuronal physiology via estrogen receptors (ER) (1). Over the last decade, ER have also emerged as regulators of glucose and energy homeostasis (2). We and others have shown that ER protect pancreatic islet β-cells from oxidative stress- and proinflammatory cytokines-induced apoptosis, and stimulate glucose-induced insulin biosynthesis and secretion via ER present in β-cells (3–7). E2 also suppresses lipogenesis in adipose tissue, skeletal muscle (8), and liver (9). Recently, we reported that islet activation of ERα, ERβ, and G protein-coupled ER (GPER) suppresses fatty acid synthase (FAS) expression and enzymatic activity (10). This effect reduces islet lipid accumulation in rodent and human β-cells and prevents β-cell failure in rodent models of type 2 diabetes (T2D). We determined that E2 antilipogenic actions were mediated via extranuclear ER and required the activation of signal transducer and activator of transcription 3 (STAT3) in vivo (10). However, the molecular mechanism underlying FAS suppression in β-cells is still unknown. FAS is the master effector of de novo lipogenesis and the downstream target of the lipogenic transcriptional regulators, namely, the sterol regulatory element-binding protein 1c (SREBP1c), the carbohydrate response element binding protein (ChREBP), and the liver X receptor (LXR) (11–13). Here, we explored the genetic mechanisms through which extranuclear ER suppress lipogenesis in β-cells. We show that extranuclear ER are involved in the genetic control of lipogenesis in β-cells by suppressing the expression of the genes coding SREBP1c, ChREBP, and LXR. We also reveal that these actions are dependent on ER signaling outside the nucleus via STAT3 and AMP-activated protein kinase (AMPK). This study identifies extranuclear ER pathways controlling the expression of lipogenic transcription factors in β-cells with implication for T2D treatment.
Materials and Methods
Rodents
The generation of mice with a pancreas-specific deletion of ERα (PERαKO−/−) has previously been described (5). STAT3lox/lox mice were kindly provided by David E. Levy (Department of Pathology, New York University School of Medicine, New York, NY). PSTAT3KO−/− mice were generated as previously described (10). Mice were studied between 9–20 wk of age. For in vivo drug stimulation, E2 (4 μg/d or 6 μg/d), propyl-pyrazole-trizol (PPT, 200 μg/d), or vehicle (10% ethanol, 90% sesame oil) was administered twice daily by sc injection for 2 d. All animal experiments were approved by the Northwestern University Animal Care and Use Committees. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Animals.
Islet isolation
Islet isolation was performed as described elsewhere (3). Briefly, pancreases were excised, incubated (37 C, 12–15 min) in Hanks buffered saline solution (HBSS) with collagenase (2 mg/ml; Sigma Chemical Co., St. Louis, MO), filtered, rinsed with ice-cold HBSS, and separated by density gradient in Histopaque (Sigma). After an ice-cold HBSS wash, islets were hand picked under a dissection microscope or transferred en masse to culture dish.
Cell and islet culture and drug stimulation
Glucose-responsive INS-1 cells (Clone 832/13) were kindly provided by Chris Newgard (Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC). Cells/islets were cultured in phenol-red free RPMI 1640 or DMEM (5.6 mm, 11 mm or 16.0 mm glucose, 10% charcoal-stripped FBS) for the indicated amount of time before experimentation. Cells were seeded to approximately 80%, and 200–250 islets were used for experimentation. Cells/islets were incubated with either 17β-estradiol (E2, 10−8 m, Steraloids Inc., Newport, RI), propyl-pyrazole-trizol (PPT, 10−8 m), diarylpropionitrile (DPN, 10−8 m), GPER agonist (G1, 10−7m), estrogen dendrimer conjugate (EDC, 10−8 m), STAT3 PI (PpYLKTK-mts, 100 μm; Calbiochem, La Jolla, CA), SU6656 (2 μm, Sigma), Compound C (2 μm, Sigma), or vehicle (ethanol or dimethylsulfoxide) for the indicated amount of time. EDC was synthesized by J.A. Katzenellenbogen (Department of Chemistry, University of Illinois, Urbana, IL).
Western blot analysis
After treatment in media, cells were homogenized in lysis buffer as described (14). Immunoblotting primary antibodies were monoclonal mouse anti-SREBP-1 (2A4) antibody (1:150) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), polyclonal rabbit anti-ChREBP (M-300) antibody (1:1000) (Santa Cruz Biotechnology), polyclonal rabbit anti-phospho-AMPKα (Thr172) antibody (1:1000) (Cell Signaling Technology, Danvers, MA), and monoclonal mouse anti-tubulin-α Ab-2 antibody (1:10000) (Labvision Corp., Fremont, CA). Secondary antibodies were horseradish peroxidase-conjugated goat antimouse or goat antirabbit, and detection of immunoreactive bands was performed using the Pierce ECL Western Blotting Substrate (Pierce Chemical Co., Rockford, IL).
Real-time quantitative RT-PCR
After treatment or islet isolation, total RNA was extracted in TRIzol Reagent (Invitrogen, Carlsbad, CA). RNA (1 μg) was reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA) with random hexamers (iCycler, Bio-Rad). mRNA expression of target genes was normalized to β-actin expression as described elsewhere (15). Primer sequences (Sigma) are available upon request.
Statistical analysis
Results are presented as mean ± sem unless otherwise stated. Data were analyzed using the unpaired two-tailed Student's t test. A value of P < 0.05 was considered statistically significant.
Results
Activation of ERα, ERβ and GPER suppresses SREBP1c and ChREBP expression in β-cells
We showed that ER suppression of lipogenesis in β-cells involved a decrease in FAS expression and enzymatic activity (10). FAS transcriptional regulation involves two transcription factors, namely SREBP1c and ChREBP (16, 17). To investigate the effect of ER activation on SREBP1c and ChREBP expression, we used the lipogenic INS-1 insulin-secreting cell model (10). When cultured in lipogenic conditions [16 mm glucose without free fatty acid (FFA)], INS-1 cells showed a rise in SREBP1c and ChREBP protein expression (Fig. 1, A and B). Treatment with E2, the ERα-selective agonist PPT (18), the GPER/ERα36-agonist G1 (19, 20), and the ERβ-selective agonist DPN (21) prevented the increase in SREBP1c and ChREBP protein expression to similar extents (Fig. 1, A and B). Lipogenesis is also regulated posttranslationally. In nonlipogenic, low glucose concentrations, SREBP1c is sequestered in an inactive state (precursor) spanning the endoplasmic reticulum membrane, whereas ChREBP is sequestered in the cytosol by phosphorylation. In lipogenic conditions when glucose concentrations rise, the N-terminal portion of SREBP1c is cleaved (mature) and ChREBP is dephosphorylated, resulting in the translocation of both proteins to the nucleus to induce lipogenic gene expression (12, 22, 23). Using SREBP1c as a paradigm, we explored whether ER activation prevents SREBP1c activation. When cultured in lipogenic conditions, INS-1 cells showed an increase in cleaved SREBP1c (mature), and treatment with E2, PPT, G1, and DPN prevented SREBP1c cleavage into the active mature form to similar extents (Fig. 1A).
Fig. 1.

ER activation suppresses SREBP1c and ChREBP protein expression in β-cells. Effects of E2 (10−8 m), PPT (10−8 m), G1 (10−7 m), and DPN (10−8 m) treatments (4 h) on (A) SREBP1c and (B) ChREBP protein expression in INS-1 cells cultured under lipogenic conditions (16 mm glucose without FFA). Results represent the mean ± se of at least three experiments. *, **, or ***, vs. 16 mm glucose. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The islet estrogen receptor (ER)α is necessary for E2 suppression of SREBP1c and ChREBP expression in vivo
Using ERα as a paradigm of ER actions in β-cells, we investigated its role in the genetic control of islet SREBP1c and ChREBP mRNA expression in vivo in mice with a pancreas-specific deletion of ERα (PERαKO−/−). The characterization of PERαKO−/− mice has previously been described (5, 10). Consistent with the role of ERα in suppressing islet lipid accumulation (10), treatment with the ERα-selective agonist PPT decreased SREBP1c and ChREBP mRNA expression in control, but not in PERαKO−/− islets (Fig. 2A), demonstrating the importance of ERα activation in islets in vivo in this process. Of note, and as discussed previously for FAS (10), male PERαKO−/− mice showed lower islet SREBP1c and ChREBP mRNA expression than control mice (Fig. 2A), which could reflect a developmental defect. The importance of ERβ was also observed using cultured islets. E2 treatment suppressed SREBP1c and ChREBP mRNA expression in wild-type islets but had no effect in ERβKO−/− islets (Supplemental Fig. 1 published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org).
Fig. 2.

Activation of islet ER in vivo suppresses SREBP1c and ChREBP mRNA expression. A, Gene expression of lipogenic genes normalized to β-actin in islets of male control (ERαlox/lox) and PERαKO−/− mice treated with PPT (200 μg/d for 2 d) (n = 8–24 mice). Results represent the mean ± se. *, **, ***, vs. control vehicle. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Extranuclear ER suppress SREBP1c and ChREBP expression via STAT3
In the classical ER signaling pathway, E2-activated ER bind as homodimers directly to an estrogen response element in target gene promoters or indirectly to an activator protein 1 or specificity protein 1 response element through association with other transcription factors that tether the activated ER to DNA (24). This classical, genomic mechanism leads to up- or down-regulation of gene transcription. We recently showed that an extranuclear ERα suppresses lipogenesis in β-cells (10). To investigate whether an extranuclear ERα also suppresses SREBP1c and ChREBP mRNA expression, we used INS-1 β-cells treated with an estrogen dendrimer conjugate (EDC) that remains outside the nucleus and is used to study extranuclear ER actions (25). EDC was as efficient as E2 or the ERα-selective agonist PPT in suppressing SREBP1c and ChREBP mRNA and protein expression in INS-1 cells (compare Fig. 3, A–C, and Fig. 1, A and B), as well as inhibiting SREBP1c maturation (compare Fig. 3B and Fig. 1B). Thus, extranuclear ER promote the suppression of islet SREBP1c and ChREBP mRNA and protein expression. We previously showed that activation of ER suppressed islet FAS expression, activity, and triglyceride accumulation in islets in vivo after phosphorylation and nuclear translocation of STAT3 (10). Thus, we asked whether STAT3 also mediates the inhibition of SREBP1c and ChREBP mRNA expression in islets by ERs in vivo using a mouse with a pancreas-specific deletion of STAT3 (PSTAT3KO−/−) (10). In vivo treatment with E2 decreased islet SREBP1c and ChREBP mRNA expression in control, but not in PSTAT3KO−/−, islets (Fig. 4A). These data demonstrate that in vivo suppression of SREBP1c and ChREBP mRNA expression in islets by ER requires STAT3. To further examine the role of STAT3 in suppressing lipogenic gene transcription, we used the cell-permeable, selective STAT3 peptide inhibitor PpYLKTK-mts that inhibits STAT3 dimerization and activation in INS-1 cells (26). Consistent with our previous results, SREBP1c mRNA was induced in INS-1 cells cultured under high glucose, and treatment with E2 or PPT suppressed SREBP1c mRNA expression (Fig. 4B). However, inhibition of STAT3 with PpYLKTK abolished the ability of E2 and PPT to reduce SREBP1c mRNA expression (Fig. 4B). The extranuclear ERα can be associated with the tyrosine kinase Src within caveolae, a large plasma membrane signaling complex that is responsible for signal transduction compartmentalization (27). We reported that ERα complexes with Src in β-cells to enhance insulin biosynthesis (5). Thus, we explored whether Src was involved in the ERα suppression of lipogeneic genes by assessing SREBP1c mRNA expression in INS-1 cells. Exposure of INS-1 cells to the Src family kinase inhibitor SU6656 prevented the ability of E2 and PPT to reduce SREBP1c mRNA expression (Fig. 4B). Thus, in β-cells, inhibition of SREBP1c mRNA expression by ERα is dependent on STAT3 and Src.
Fig. 3.

Extranuclear ER suppress SREBP1c and ChREBP mRNA and protein expression in β-cells. A, Effects of E2 (10−8 m) and EDC (10−8 m) treatments (4 h) on lipogenic gene expression normalized to β-actin in INS-1 cells cultured under lipogenic conditions (11 mm glucose without FFA). B and C, Effects of E2 (10−8 m) and EDC (10−8 m) treatments (4 h) on (B) SREBP1c and (C) ChREBP protein expression in INS-1 cells cultured under lipogenic conditions (16 mm glucose without FFA). Results represent the mean ± se of at least three experiments. *, **, or ***, vs. 11 mm or 16 mm glucose. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Fig. 4.

ER suppress SREBP1c and ChREBP mRNA expression via STAT3. A, Gene expression of lipogenic genes normalized to β-actin in islets of male control (STAT3lox/lox) and PSTAT3KO−/− mice treated with E2 (6 μg/d for 2 d) (n = 11–20 mice). B, Effect of STAT3 and Src family kinase inhibition on E2 and PPT-induced suppression of SREBP1c mRNA expression. Cells were treated with E2 (10−8 m) or PPT (10−8 m) ± STAT3 inhibitor PpYLKTK (100 μm) or SFK inhibitor SU6656 (2 μm) for 4 h. *, **, or ***, vs. 11 mm glucose when not indicated. Results represent the mean ± se of at least three experiments. *, **, or ***, vs. control vehicle. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Activation of ER suppresses LXRα and β expression in β-cells
SREBP1c and ChREBP are regulated at the transcriptional level by liver X receptor (LXR)α and β. Whereas LXRα is the predominant stimulator of lipogenesis in liver (11, 28, 29), LXRβ predominantly stimulates lipogenesis in muscle and islets (30, 31). We thus investigated the effect of E2 treatment on LXRα and β mRNA expression in β-cells. As observed by others (30, 31), we found that LXRα is more highly expressed than LXRβ in liver and adipose tissue, whereas LXRβ is more highly expressed than LXRα in muscle, islets, and INS-1 cells (Fig. 5, A and B). In INS-1 cells, high glucose increased LXRα and -β mRNA expression, which was suppressed by treatment with E2, PPT, G1, and DPN to similar extents (Fig. 5C). Selectively activating ERα with PPT in vivo in mice decreased LXRα and β mRNA expression in control, but not in PERαKO−/−, islets (Fig. 5D), demonstrating the importance of islet ERα in this process. Furthermore, the extranuclear ER probe EDC was as efficient as E2 in suppressing LXRα and β mRNA expression in INS-1 cells (Fig. 5E). In vivo treatment with E2 decreased islet LXRα and -β mRNA expression in control, but not in PSTAT3KO−/−, islets (Fig. 6A). Lastly, because we observed that inhibition of SREBP1c mRNA expression by ERα was Src dependent (Fig. 4B), we explored whether Src was involved in the ERα suppression of LXRβ mRNA expression. Exposure of INS-1 cells to the Src family kinase inhibitor SU6656 prevented the ability of E2 and PPT to reduce LXRβ mRNA expression (Fig. 6B). Thus, in β-cells, inhibition of LXRβ mRNA expression by ERα is dependent on STAT3 and Src.
Fig. 5.

ER suppress LXR mRNA expression in β-cells. A, LXRα and -β gene expression normalized to β-actin in mouse liver, white adipose tissue (WAT), skeletal muscle, and islets (n = 12–18 mice). B, LXRα and β gene expression normalized to β-actin in INS-1 cells (n = at least 3 experiments). C, Effects of E2 (10−8 m), PPT (10−8 m), G1 (10−7 m), and DPN (10−8 m) treatments (4 h) on LXRα and -β gene expression normalized to β-actin in INS-1 cells cultured under lipogenic conditions (11 mm glucose without FFA) (n = at least 3 experiments). D, LXRα and -β gene expression normalized to β-actin in islets of male control and PERαKO−/− mice treated with PPT (200 μg/d for 2 d) (n = 8–24 mice). E, Effects of E2 (10−8 m) and EDC (10−8 m) treatments (4 h) on LXRα and -β gene expression normalized to β-actin in INS-1 cells cultured under lipogenic conditions (n = at least 3 experiments). Results represent mean ± se. *, **, or ***, vs. control vehicle or 11 mm glucose when not indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Fig. 6.

ER suppress LXR mRNA expression via STAT3. A, LXRα and -β gene expression of lipogenic genes normalized to β-actin in islets of male control and PSTAT3KO−/− mice treated with E2 (6 μg/d for 2 d) (n = 11–20 mice). B, Effect of Src inhibition on E2 and PPT-induced suppression of LXRβ mRNA expression. INS-1 cells were treated with E2 (10−8 m) or PPT (10−8 m) ± SFK inhibitor SU6656 (2 μm) for 4 h (n = at least 3 experiments). Results represent mean ± se. *, vs. control vehicle or 16 mm glucose when not indicated. *, **, or ***, P < 0.05; **, P < 0.01; ***, P < 0.001.
ERα suppresses SREBP1c protein expression via AMPK
Extranuclear ERα suppressed β-cell lipogenic gene expression via a Src-STAT3 pathway, but other parallel pathways may also be contributing to the E2 suppression of lipogenesis. For example, AMP-activated protein kinase (AMPK) is a central regulator of cellular energy homeostasis (32). AMPK inhibits lipogenesis, in part, by suppressing SREBP1c gene expression and cleavage of precursor SREBP1c into its active mature form (33, 34). Thus, we explored whether AMPK was involved in the E2 suppression of SREBP1c. In INS-1-cells, E2 transiently stimulated AMPK phosphorylation (pAMPK) to an extent close to that observed at low glucose and with maximal amplitude at 2 min (Fig. 7A). Furthermore, Exposure of INS-1 cells to the AMPK inhibitor compound C prevented the ability of E2 and PPT to suppress SREBP1c protein expression (Fig. 7B). These data suggest that ERα also suppresses SREBP1c expression through an AMPK-dependent pathway.
Fig. 7.

ER suppress SREBP1c protein expression via AMPK. A, INS-1 cells were treated with E2 (10−8 m) for up to 2 h. Phosphorylation of AMPK was determined by Western blotting with an antibody against phosphorylated AMPK. Graph represents the quantification of at least two experiments. *, **, or ***, vs. 0 min. B, Effect of AMPK inhibition on E2 and PPT-induced suppression of SREBP1c protein expression. Cells were treated with E2 (10−8 m) or PPT (10−8 m) ± compound C (2 μm) for 4 h (n = at least 3 experiments). *, **, or ***, vs. 16 mm glucose when not indicated. Results represent mean ± se. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
Previously, we reported that extranuclear ER activation suppressed islet FAS expression and enzymatic activity and prevented lipid accumulation and lipotoxic β-cell failure in rodent models of T2D (10). The induction of FAS expression is under the concerted action of the transcription factors LXRα and -β, SREBP1c, and ChREBP in response to insulin and glucose (11–13). In addition, SREBP1c and ChREBP are regulated posttranscriptionally by cleavage and phosphorylation, respectively (12, 22, 23). Here, we show that activation of ER suppresses LXR, SREBP1c, and ChREBP gene expression in pancreatic islets.
ER suppress LXR, SREBP1c, and ChREBP mRNA expression independently of the classical ligand-activated nuclear receptor actions. The extranuclear ER ligand, EDC, and the agonist for GPER and the extranuclear ERα36, lacking transcriptional activity (20), suppressed LXR, SREBP1c and ChREBP mRNA, and protein expression to a similar extent. These findings demonstrate that ER signal outside the nucleus to suppress mRNA expression. Indeed, we present in vivo evidence that ER signal though STAT3 to suppress islet LXR, SREBP1c, and ChREBP mRNA expression. This effect is consistent with the in vivo effect of the ER/STAT3 pathway in suppressing islet lipid accumulation in the same mice (10). Our data also suggest that a member of the Src family kinases is instrumental in ER suppression of lipogenesis. We previously reported that upon E2 stimulation, ERα rapidly binds Src in β-cells (5). Here, we find that pharmacological inhibition of Src family kinases abolishes ERα-induced suppression of LXRβ and SREBP1c mRNA. Because Src is clustered in caveolae and can phosphorylate STAT3 (35), the suppression of LXRβ and SREBP1c mRNA described here may be mediated via a fraction of ERα, which is associated with the plasma membrane, and activates Src leading to STAT3 activation. Interestingly, in the liver of obese ob/ob mice, in vivo E2 treatment promotes the recruitment of a nuclear ERα to the STAT3 gene promoter, which is associated with an increased expression and phosphorylation of STAT3, thus decreasing lipogenesis (9). Thus, the involvement of an extranuclear Src-ERα-STAT3 pathway in suppressing SREBP1c in β-cells provides a novel paradigm in ER suppression of lipogenesis.
In β-cells, chronic LXR activation provokes excess lipogenesis associated with lipotoxicity and apoptosis (36). SREBP1c expression is dependent upon LXR activation (11, 37), and although LXR is not required for ChREBP expression, LXRα activation is sufficient to induce ChREBP expression (13, 38). Thus, ER suppression of LXR mRNA in β-cells may account for the inhibition of SREBP1c and ChREBP gene expression observed in this study and the inhibition of lipogenesis and prevention of islet lipotoxicity we previously reported (10).
We observe that ER activation suppresses SREBP1c protein expression and inhibits the cleavage of SREBP1c into its active mature form. In vivo E2 treatment stimulates AMPK phosphorylation in mouse skeletal muscle (8), and activation of ERα increases expression of the upstream AMPK regulator liver kinase B1 in adipocytes (39). Independent from LXR regulation, AMPK is known to suppress SREBP1c activation through the inhibition of SREBP1c maturation and promoter activity in hepatoma cells (33). In hepatocytes, AMPK also directly phosphorylates SREBP1c to inhibit SREBP1c maturation and nuclear translocation (34). In accordance with these observations, we observe that in cultured insulin-secreting INS-1 cells, ligand-activated ERα also activates and uses AMPK to suppress SREBP1c protein expression. These results thus suggest that ER activation suppresses β-cell lipogenesis via at least two parallel pathways including STAT3 and AMPK. A summary of the proposed mechanism of ERα suppression of lipogenesis appears in Fig. 8.
Fig. 8.
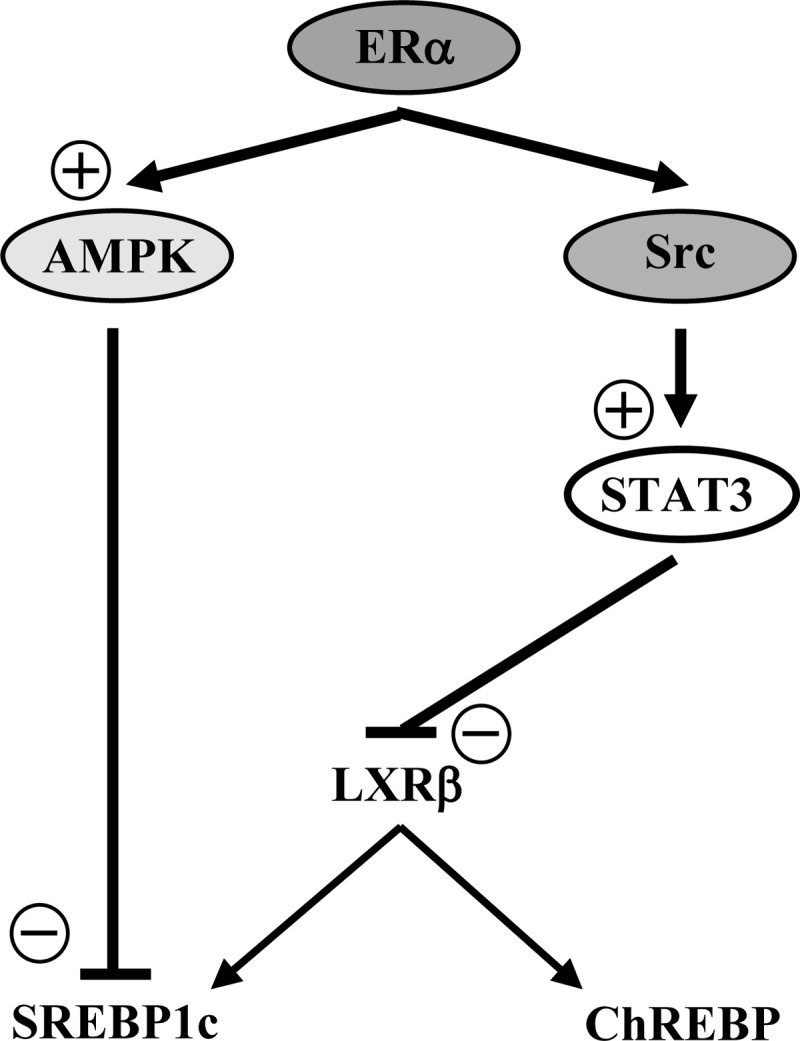
Schematic representation of the proposed mechanisms of ERα suppression of lipogenic gene expression in β-cells. The ERα suppression of lipogenic genes can occurs through 1) a Src-STAT3-dependent pathway inhibiting LXRβ mRNA expression and eventually SREBP1c and ChREBP expression; and 2) an AMPK-dependent pathway directly inhibiting SREBP1c expression.
In summary, extranuclear ER are involved in the genetic control of lipogenesis in β-cells by suppressing the expression of the genes coding SREBP1c, ChREBP, and LXR. These actions are dependent on ER signaling outside the nucleus via STAT3 and AMPK. This study identifies extranuclear ER pathways controlling the expression of lipogenic transcription factors with implication for T2D treatment.
Supplementary Material
Supplemental Data
Acknowledgments
This work was supported by grants from the National Institutes of Health (RO1 DK074970, P50 HD044405), the Juvenile Diabetes Research Foundation (1-2006-837) the March of Dimes (6-FY07-678), and the American Heart Association (11IRG5570010 to F.M.-J.). J.P.T. was supported in part by National Institutes of Health Training Grant T32 DK007169.
Disclosure Summary: The authors have declared that no conflict of interest exists
Footnotes
Abbreviations:
AMPK
AMP-activated protein kinase
ChREBP
carbohydrate response element binding protein
DPN
diarylpropionitrile
EDC
estrogen dendrimer conjugate
E2
17β-estradiol
ER
estrogen receptor
FAS
fatty acid synthase
FFA
free fatty acid
GPER
G protein-coupled ER
HBSS
Hanks buffered saline solution
LXR
liver X receptor
PPT
propyl-pyrazole-trizol
SREBP
sterol regulatory element-binding protein
STAT
signal transducer and activator of transcription
T2D
type 2 diabetes.
References
- 1.Deroo BJ, Korach KS. 2006. Estrogen receptors and human disease. J Clin Invest 116:561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mauvais-Jarvis F. 2011. Estrogen and androgen receptors: regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol Metab 22:24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, Tsai MJ, Mauvais-Jarvis F. 2006. Estrogens protect pancreatic β-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci USA 103:9232–9237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu S, Le May C, Wong WP, Ward RD, Clegg DJ, Marcelli M, Korach KS, Mauvais-Jarvis F. 2009. Importance of extranuclear estrogen receptor-α and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes 58:2292–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong WP, Tiano JP, Liu S, Hewitt SC, Le May C, Dalle S, Katzenellenbogen JA, Katzenellenbogen BS, Korach KS, Mauvais-Jarvis F. 2010. Extranuclear estrogen receptor-α stimulates NeuroD1 binding to the insulin promoter and favors insulin synthesis. Proc Natl Acad Sci USA 107:13057–13062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S, Mauvais-Jarvis F. 2010. Minireview: Estrogenic protection of β-cell failure in metabolic diseases. Endocrinology 151:859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tiano JP, Mauvais-Jarvis F. 14 February 2012. Importance of oestrogen receptors to preserve functional β-cell mass in diabetes. Nat Rev Endocrinol10.1038/nrendo.2011.242 [DOI] [PubMed] [Google Scholar]
- 8.D'Eon TM, Souza SC, Aronovitz M, Obin MS, Fried SK, Greenberg AS. 2005. Estrogen regulation of adiposity and fuel partitioning. Evidence of genomic and non-genomic regulation of lipogenic and oxidative pathways. J Biol Chem 280:35983–35991 [DOI] [PubMed] [Google Scholar]
- 9.Gao H, Bryzgalova G, Hedman E, Khan A, Efendic S, Gustafsson JA, Dahlman-Wright K. 2006. Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: a possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol Endocrinol 20:1287–1299 [DOI] [PubMed] [Google Scholar]
- 10.Tiano JP, Delghingaro-Augusto V, Le May C, Liu S, Kaw MK, Khuder SS, Latour MG, Bhatt SA, Korach KS, Najjar SM, Prentki M, Mauvais-Jarvis F. 2011. Estrogen receptor activation reduces lipid synthesis in pancreatic islets and prevents β cell failure in rodent models of type 2 diabetes. J Clin Invest 121:3331–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. 2000. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev 14:2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uyeda K, Repa JJ. 2006. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab 4:107–110 [DOI] [PubMed] [Google Scholar]
- 13.Cha JY, Repa JJ. 2007. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem 282:743–751 [DOI] [PubMed] [Google Scholar]
- 14.Mauvais-Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ, Iannacone M, Accili D, Cantley LC, Kahn CR. 2002. Reduced expression of the murine p85α subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest 109:141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cariou B, Postic C, Boudou P, Burcelin R, Kahn CR, Girard J, Burnol AF, Mauvais-Jarvis F. 2004. Cellular and molecular mechanisms of adipose tissue plasticity in muscle insulin receptor knockout mice. Endocrinology 145:1926–1932 [DOI] [PubMed] [Google Scholar]
- 16.Kim JB, Sarraf P, Wright M, Yao KM, Mueller E, Solanes G, Lowell BB, Spiegelman BM. 1998. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest 101:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishii S, Iizuka K, Miller BC, Uyeda K. 2004. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci USA 101:15597–15602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. 2000. Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-α-selective agonists. J Med Chem 43:4934–4947 [DOI] [PubMed] [Google Scholar]
- 19.Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. 2006. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol 2:207–212 [DOI] [PubMed] [Google Scholar]
- 20.Kang L, Zhang X, Xie Y, Tu Y, Wang D, Liu Z, Wang ZY. 2010. Involvement of estrogen receptor variant ER-α36, not GPR30, in nongenomic estrogen signaling. Mol Endocrinol 24:709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. 2001. Estrogen receptor-β potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem 44:4230–4251 [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Sato R, Brown MS, Hua X, Goldstein JL. 1994. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 77:53–62 [DOI] [PubMed] [Google Scholar]
- 23.Kawaguchi T, Takenoshita M, Kabashima T, Uyeda K. 2001. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc Natl Acad Sci USA 98:13710–13715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. 2001. Mechanisms of estrogen action. Physiol Rev 81:1535–1565 [DOI] [PubMed] [Google Scholar]
- 25.Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. 2006. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Mol Endocrinol 20:491–502 [DOI] [PubMed] [Google Scholar]
- 26.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R. 2001. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem 276:45443–45455 [DOI] [PubMed] [Google Scholar]
- 27.Levin ER. 2009. Plasma membrane estrogen receptors. Trends Endocrinol Metab 20:477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. 1998. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR α. Cell 93:693–704 [DOI] [PubMed] [Google Scholar]
- 29.Alberti S, Schuster G, Parini P, Feltkamp D, Diczfalusy U, Rudling M, Angelin B, Björkhem I, Pettersson S, Gustafsson JA. 2001. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRβ-deficient mice. J Clin Invest 107:565–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zitzer H, Wente W, Brenner MB, Sewing S, Buschard K, Gromada J, Efanov AM. 2006. Sterol regulatory element-binding protein 1 mediates liver X receptor-β-induced increases in insulin secretion and insulin messenger ribonucleic acid levels. Endocrinology 147:3898–3905 [DOI] [PubMed] [Google Scholar]
- 31.Hessvik NP, Boekschoten MV, Baltzersen MA, Kersten S, Xu X, Andersén H, Rustan AC, Thoresen GH. 2010. LXRβ is the dominant LXR subtype in skeletal muscle regulating lipogenesis and cholesterol efflux. Am J Physiol Endocrinol Metab 298:E602–E613 [DOI] [PubMed] [Google Scholar]
- 32.Kahn BB, Alquier T, Carling D, Hardie DG. 2005. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1:15–25 [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Craddock L, Hong S, Liu ZM. 2009. AMP-activated protein kinase suppresses LXR-dependent sterol regulatory element-binding protein-1c transcription in rat hepatoma McA-RH7777 cells. J Cell Biochem 106:414–426 [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M. 2011. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab 13:376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silva CM. 2004. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 23:8017–8023 [DOI] [PubMed] [Google Scholar]
- 36.Choe SS, Choi AH, Lee JW, Kim KH, Chung JJ, Park J, Lee KM, Park KG, Lee IK, Kim JB. 2007. Chronic activation of liver X receptor induces β-cell apoptosis through hyperactivation of lipogenesis: liver X receptor-mediated lipotoxicity in pancreatic β-cells. Diabetes 56:1534–1543 [DOI] [PubMed] [Google Scholar]
- 37.Quinet EM, Savio DA, Halpern AR, Chen L, Schuster GU, Gustafsson JA, Basso MD, Nambi P. 2006. Liver X receptor (LXR)-β regulation in LXRα-deficient mice: implications for therapeutic targeting. Mol Pharmacol 70:1340–1349 [DOI] [PubMed] [Google Scholar]
- 38.Denechaud PD, Bossard P, Lobaccaro JM, Millatt L, Staels B, Girard J, Postic C. 2008. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J Clin Invest 118:956–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McInnes KJ, Brown KA, Hunger NI, Simpson ER. 30 August 2011. Regulation of LKB1 expression by sex hormones in adipocytes. Int J Obes (Lond)10.1038/ijo.2011.172 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data