Chemoselective Ligation of Sulfinic Acids with Aryl-Nitroso Compounds (original) (raw)
. Author manuscript; available in PMC: 2013 Jun 25.
Published in final edited form as: Angew Chem Int Ed Engl. 2012 May 29;51(26):6502–6505. doi: 10.1002/anie.201201812
Hydrogen peroxide (H2O2) acts as a second messenger during cell signaling and, at low levels, regulates an array of physiological functions.[1] Conversely, excessive H2O2 can lead to oxidative stress, which is a chronic state implicated in the etiology or progression of human diseases, including cancer.[2] Owing to the high nucleophilicity of the thiol group, reactive cysteine residues in proteins can be modified by H2O2 to form sulfenic acid (RSOH).[3] This cysteine oxoform can be reduced back to the thiol group or be further oxidized to sulfinic (RSO2H) and sulfonic acid (RSO3H) (see Figure S1 in the Supporting Information). Each of these species exhibits unique chemical properties and affords a versatile mechanism to alter protein function.[4]
Although the regulatory function of protein sulfenic acids is now established,[5] little is known about the role of sulfinic acids. Indeed, this modification was long dismissed solely as an artifact of protein isolation. However, mounting evidence indicates that cysteine is oxidized to sulfinic acid in cells to a greater extent, and is more controlled, than first thought. For example, quantitative amino acid analysis of soluble proteins from normal rat liver indicates that approximately 5%of cysteine residues exist in this oxidation state.[6] Sulfinic acid modification (with concomitant regulation) is also associated with a growing list of proteins, including nitrile hydratase,[7] matrilysin,[8] and the Parkinson’s disease protein, DJ-1.[9] Peroxiredoxins are also highly susceptible to sulfinic acid formation at their catalytic cysteine and leads to a loss in peroxidase activity.[10] Cysteine sulfinic acid is not reduced by typical cellular reductants such as glutathione and thus, this derivative was considered to be biologically irreversible. Recently, this viewpoint was revised when an enzyme called sulfiredoxin was found to reduce the sulfinic form of certain peroxiredoxins.[11] The discovery of a sulfinic acid reductase suggests a more fundamental role for this modification, thereby leading Jacob and colleagues to propose a new paradigm for protein regulation by H2O2 known as the “sulfinic acid switch”.[12]
Robust methods for detecting sulfinic acid are required to understand the physiological and pathological function of this modification. Sulfinic acid derivatives can be detected by an increase in cysteine residue mass of 32 Da,[13] however, there is increasing concern with this potential indicator given that modification of proteins by hydrogen sulfide (H2S) leads to a persulfide species (RSSH) with the same nominal mass shift. Antibodies directed against the sulfinic acid form of specific proteins are known,[14] but are not suited for global profiling studies. Aryl diazonium salts have been used for the quantitation of methanesulfinic acid.[15] Nevertheless, this system suffers from several significant limitations inherent to the instability of diazonium salts in aqueous solution[16] and formation of stable adducts with tyrosine[17] and cysteine.[18] Herein, we describe a novel selective ligation reaction of sulfinic acids with potential utility for detection of protein sulfinylation in biological systems.
With a p_K_a of approximately 2, sulfinic acids are fully deprotonated at physiological pH (Figure 1; 1a–e). The ambident sulfinate anion behaves primarily as a soft nucleophile. Sulfur attack is favored and proceeds toward the more thermodynamically stable sulfone.[19] The key challenge is to develop a ligation method for sulfinic acid that is orthogonal to cysteine, related oxyacids, and other common biological functionalities. With these considerations, we focused on the reaction of C-nitroso compounds (2) with aryl sulfinic acids to provide an N-sulfonyl hydroxylamine (3; Scheme 1).
Figure 1.

Resonance and structures of sulfinate anions in this study. Boc = _tert_-butoxycarbonyl.
Scheme 1.

Condensation reaction between a C-nitroso compound and aryl sulfinic acid.
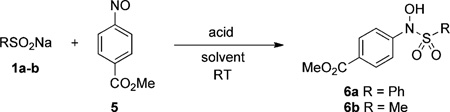
Reports of this condensation date back to the end of the 19th century,[20] however this topic remains surprisingly understudied. For instance, reactions with aromatic sulfinic acids and C-nitroso compounds have been reported,[21] but the reactivity of alkyl sulfinic acids have not been explored. With respect to our goal for sulfinic acid ligation, it is also important to note that the resulting adduct is unstable in basic solutions. In fact, the N-sulfonyl hydroxylamine 3 may be deprotonated (4) and readily dissociate back into the starting materials.[21] In this context, we investigated the reactivity of _p_-nitroso methyl benzoate (5) with sulfinic acids (Table 1).
Table 1.
N-sulfonyl hydroxylamine formation from RNO and RSO2Na.
 |
|||||
|---|---|---|---|---|---|
| Entry | RSO2Na | Solvent | Acid | Product | Yield [%][a] |
| 1 | 1a | DMSO | CF3CO2H | 6a | > 98 |
| 2 | 1a | DMSO | HCO2H | 6a | 98 |
| 3 | 1a | DMSO | CH3CO2H | 6a | 95 |
| 4 | 1b | DMSO | CH3CO2H | 6b | 97 |
| 5 | 1b | DMF | CH3CO2H | 6b | > 98 |
| 6 | 1b | MeOH | CH3CO2H | 6b | 85 |
| 7 | 1b | CH3CN | CH3CO2H | 6b | 89 |
Encouragingly, in the presence of weak acids, we found that both aryl (1a) and alkyl sulfinates (1b) reacted rapidly with high yields in a wide variety of organic solvents. Given that these condensation reactions only proceed in aqueous media of very low pH (0–3),[21] we next needed to develop a strategy to convert the sulfonyl hydroxylamine adduct into a more stable product. We hypothesized that the deprotonated form of 4 is a potential nucleophile, which in the presence of an electrophilic center on the aromatic group could trap the oxyanion by intramolecular rearrangement. In support of this proposal, Moinet et al. have described the intramolecular cyclization reaction of N-sulfonyl aromatic hydroxylamines with an ester group in the ortho position (e.g., 8; Scheme 2).[22] Since cyclization proceeds through acid-catalyzed transesterification, long reaction times (24 h) are required to obtain moderate yields. However, we reasoned that the reaction kinetics could be improved by performing the ligation in neutral or slightly basic conditions.
Scheme 2.

Proposed mechanism of N-sulfonylbenzisoxazolone formation.
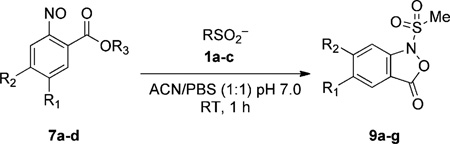
To evaluate this hypothesis, we synthesized _ortho_-nitroso benzoic esters (7a–b) and tested their reactivity with methane sulfinic acid (1b) in a solvent system containing pH 7.0 PBS buffer (50%; PBS = phosphate buffered saline) and CH3CN (50%; Table 2). Interestingly, the methyl ester 7a reacted with 1b, but the intermediate species was not converted into the N-sulfonylbenzisoxazolone 9a (Table 2, entry 1). Considering that the analogous result was obtained with ethyl ester 7b (Table 2, entry 2), we speculated that low conversion reflects the relatively weak acidity of the sulfonyl hydroxylamine 10 (see Scheme 2). Thus, to increase the acidity of this group, a nitro substitutent was introduced on the phenyl ring (7c). Compound 7c was converted in good yield into the sulfonyl adducts (Table 2, entries 3 and 4), consistent with our hypothesis. To improve solubility in water, while preserving reactivity, we then synthesized 2-nitroso terephthalic acid methyl ester (7d), in which the scaffold is elaborated with a carboxylic acid group. Compound 7d showed excellent solubility under neutral pH conditions and robust reactivity toward a variety of sulfinic acids (Table 2, entries 5–7), as envisioned. Importantly, the nitroso 7d proved stable in neutral aqueous conditions (_t_1/2 ≥ 24 h; Figure S2). Together, these studies highlight the potential compatibility of this reaction with biological systems.
Table 2.
Reactivity of 2-nitroso benzoic acid derivatives towards sulfinic acids.
 |
||||||
|---|---|---|---|---|---|---|
| Entry | C-Nitroso compound | R3 | RSO2− | Yield [%][a] | ||
| R1 | R2 | |||||
| 1 | 7a | H | H | Me | 1b | − (9a) |
| 2 | 7b | H | H | Et | 1b | − (9b) |
| 3 | 7c | NO2 | H | Me | 1b | 84 (9c)] |
| 4 | 7c | NO2 | H | Me | 1a | 88 (9d) |
| 5 | 7d | H | CO2H | Me | 1b | 84 (9e) |
| 6 | 7d | H | CO2H | Me | 1a | 89 (9f) |
| 7 | 7d | H | CO2H | Me | 1c | 81 (9g) |
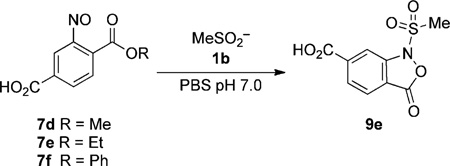
Next, we investigated the kinetics for this conversion by 1H NMR spectroscopy and LC-MS. The rate of ligation was measured using a large excess of 1b over the nitroso compound, so that pseudo-first-order rate constants could be obtained (Table 3 and see Figures S3–S8 in the Supporting Information). In accord with the proposed mechanism (Scheme 2), the rate of reaction between 7d and 1b accelerated with increasing pH (Table 3, entries 1–3). To further optimize this reaction, we evaluated the rate constants for 2-nitroso terephthalic acid ester derivatives (7e–f). Although the ethyl ester 7e gave similar reactivity to that of 7d (Table 3, entry 4), the phenyl ester 7f exhibited excellent conversion rates, even at lower pH (Table 3, entries 5–7).
Table 3.
Rate constants for the reaction of 2-nitroso benzoic ester derivatives with methane sulfinic acid.
 |
|||
|---|---|---|---|
| Entry | C-Nitrosocompound | pH | k (× 10−3 s−1) |
| 1 | 7d | 6 | 0.2 |
| 2 | 7d | 7 | 1.1 |
| 3 | 7d | 8 | 2.3 |
| 4 | 7e | 7 | 0.6 |
| 5 | 7f | 5 | 16 |
| 6 | 7f | 6 | 54 |
| 7 | 7f | 7 | 131 |
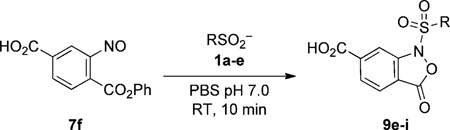
With the improved compound 7f in hand, we then explored its reactivity with sulfinic acids of increasing complexity in PBS buffer (Table 4). We were delighted to observe complete ligation of 7f within 10 minutes, with simple (1a–b) as well as more elaborate sulfinic acids (1c–e). In all cases, LC-MS analysis verified formation of a single product with an m/z corresponding to the N-sulfonylbenzisoxazolone adduct (see Figure S9 in the Supporting Information). As compared to the methyl ester 7d, phenyl ester 7f was slightly less stable under aqueous conditions (_t_1/2 ≥ 8 h; Figure S10). Nonetheless, the higher chemical reactivity of 7f with sulfinic acids assures short reaction times. Furthermore, the resulting sulfonyl adducts (9e–i) were stable in buffer (Figure S11) and unreactive, even with nucleophiles, such as lysine and cysteine (Figure S12).
Table 4.
Reactivity of 2-nitroso terephthalic acid phenyl ester in buffer.[a]
 |
|||
|---|---|---|---|
| Entry | RSO2− | Product | Yield [%] |
| 1 | 1a | 9 f | quantitative |
| 2 | 1b | 9e | quantitative |
| 3 | 1c | 9g | quantitative |
| 4 | 1d | 9h | quantitative |
| 5 | 1e | 9i | quantitative |
To further test the suitability of our sulfinic acid ligation approach, we examined the potential cross-reactivity between 7f and a variety of biological functional groups (Table 5). The phenyl ester 7f did not react with the lysine ε-amino group (11); alcohol-containing amino acids, such as serine (12) and tyrosine (13), were also inert (Table 5, entries 1–3 and see Figure S13 in the Supporting Information). Thiol compounds are known to reduce aromatic C-nitroso compounds.[23] In this reaction, the thiol attacks the nitroso to yield an N-hydroxylsulfenamide which condenses with a second thiol to yield N-hydroxylamine and disulfide products (Scheme S1). Consistent with this mechanism, treatment of cysteine (14) with 7f generated cystine and reduced nitroso products (Table 5, entry 4; Scheme S2 and Figure S13). Importantly, however, no stable adduct was formed between 7f and cysteine, and reduced nitroso species did not react with biological moieties (Figures S14 and S15).
Table 5.
Reactivity of 7f toward biological functional groups in buffer.
If we apply this method to biological systems, glutathione (GSH) is likely to be present in many-fold excess, relative to protein sulfinic acids, even under oxidative stress conditions. Thus, a major concern is that excess thiol could reduce 7f and preclude ligation to sulfinic acids. To address this question, the sulfinic acid 1b (1 equiv) was treated with 7f (100 equiv) in the presence of a 50-fold excess of cysteine or GSH [Eq. (1); Scheme 3 and see Table S1 in the Supporting Information]. Under these reaction conditions, 1b was converted into the ligation product 9e almost quantitatively (98%) after 30 minutes. An alternative strategy, commonly employed in analysis of related redox modifications, is to alkylate free thiols with _N_-ethylmaleimide (NEM).[24] In this manner, a mixture of sulfinic acid 1b and GSH (1:50) was treated with NEM for 15 minutes prior to the addition of 7f (1 equiv). Notably, this reaction sequence also led to the desired ligation product 9e in excellent yield (96%) [Eq. (2); Scheme 3 and see Table S2].
Scheme 3.

Control experiments for the ligation of sulfinic acid by 7f in the presence of excess thiols.
Finally, we investigated the reactivity of 7f with oxidized thiol species, including sulfenic acid, sulfenamide, disulfide, sulfonic acid, and nitrosothiol (Table 5, entries 5–9). In all cases, LC-MS analysis showed no evidence for the ligation product (see Figure S16 in the Supporting Information). Even sulfenic acid, which is known for its ambiphilic reactivity, did not react with 7f. Rather, thiosulfinate 15c (obtained by self-condensation of sulfenic acid; Scheme S3) was detected as the exclusive product in this reaction.
To summarize, we have developed a robust ligation reaction that selectively converts sulfinic acid moieties into stable conjugates. To the best of our knowledge, this is the first report of a one-step method to selectively convert alkyl sulfinic acids into stable conjugates in aqueous media under physiological pH. In view of data obtained in several control experiments, we expect that the selective and facile reaction presented herein can serve as the foundation for the future development of methods to detect sulfinic acid formation in biological systems. Current studies focus on this area and the details of these ongoing efforts will be reported in due course.
Experimental Section
See the Supporting Information for experimental details.
Supplementary Material
SI
Footnotes
**
The authors acknowledge funding from the Camile Henry Dreyfus Teacher Scholar Award (to K.S.C.) and the American Heart Association Scientist Development Award (0835419N to K.S.C.).
References
- 1.Dickinson BC, Chang CJK. Nat. Chem. Biol. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 4th ed. New York: Oxford University Press; 2007. [Google Scholar]
- 3.Reddie KG, Carroll KS. Curr. Opin. Chem. Biol. 2008;12:746–754. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 4.Jacob C, Giles GI, Giles NM, Sies H. Angew. Chem. 2003;115:4890–4907. doi: 10.1002/anie.200300573. Angew. Chem. Int. Ed.2003, 42, 4742 – 4758. [DOI] [PubMed] [Google Scholar]
- 5.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Nat. Chem. Biol. 2011;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamann M, Zhang T, Hendrich S, Thomas JA. Methods Enzymol. 2002;348:146–156. doi: 10.1016/s0076-6879(02)48634-x. [DOI] [PubMed] [Google Scholar]
- 7.Murakami T, Nojiri M, Nakayama H, Odaka M, Yohda M, Dohmae N, Takio K, Nagamune T, Endo I. Protein Sci. 2000;9:1024–1030. doi: 10.1110/ps.9.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu X, Kassim SY, Parks WC, Heinecke JW. J. Biol. Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 9.Blackinton J, Lakshminarasimhan M, Ahmad KJT, Greggio E, Raza AS, Cookson MR, Wilson MA. J. Biol. Chem. 2009;284:6476–6485. doi: 10.1074/jbc.M806599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowther WT, Haynes AC. Antioxid. Redox Signaling. 2011;15:99–109. doi: 10.1089/ars.2010.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biteau B, Labarre J, Toledano MB. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 12.Jacob C, Holme AL, Fry FH. Org. Biomol. Chem. 2004;2:1953–1956. doi: 10.1039/b406180b. [DOI] [PubMed] [Google Scholar]
- 13.Witze ES, Old WM, Resing KA, Ahn NG. Nat. Methods. 2007;4:798–806. doi: 10.1038/nmeth1100. [DOI] [PubMed] [Google Scholar]
- 14.Woo HA, Kang SW, Kim HK, Yang KS, Chae HZ, Rhee SG. J. Biol. Chem. 2003;278:47361–47364. doi: 10.1074/jbc.C300428200. [DOI] [PubMed] [Google Scholar]
- 15.Babbs CF, Gale MJ. Anal. Biochem. 1987;163:67–73. doi: 10.1016/0003-2697(87)90093-5. [DOI] [PubMed] [Google Scholar]
- 16.Detar DF. J. Am. Chem. Soc. 1956;78:3911. [Google Scholar]
- 17.Hooker JM, Kovacs EW, Francis MB. J. Am. Chem. Soc. 2004;126:3718–3719. doi: 10.1021/ja031790q. [DOI] [PubMed] [Google Scholar]
- 18.Patt J, Patt M. J. Labelled Compd. Radiopharm. 2002;45:1229–1238. [Google Scholar]
- 19.Baidya M, Kobayashi S, Mayr H. J. Am. Chem. Soc. 2010;132:4796–4805. doi: 10.1021/ja9102056. [DOI] [PubMed] [Google Scholar]
- 20.Bamberger E, Buesdorf H, Szolayski B. Chem. Ber. 1899;32:210–221. [Google Scholar]
- 21.Darchen A, Moinet C. J. Chem. Soc. Chem. Commun. 1976:820a. [Google Scholar]
- 22.Guilbaud-Criqui A, Moinet C. Bull. Soc. Chim. Fr. 1992;129:295–300. [Google Scholar]
- 23.Montanari S, Paradisi C, Scorrano G. J. Org. Chem. 1999;64:3422–3428. doi: 10.1021/jo981889t. [DOI] [PubMed] [Google Scholar]
- 24.Leonard SE, Carroll KS. Curr. Opin. Chem. Biol. 2011;15:88–102. doi: 10.1016/j.cbpa.2010.11.012. and references therein. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SI