Nitric oxide formation versus scavenging: the red blood cell balancing act (original) (raw)
Abstract
Nitric oxide (NO) is a key modulator of vascular homeostasis controlling critical functions related to blood flow, respiration, cell death and proliferation, and protecting the vasculature from pro-inflammatory and coagulative stresses. Inhibition of NO formation, and/or diversion of NO away from its physiological signalling targets lead to dysregulated NO bioavailability, a hallmark of numerous vascular and pulmonary diseases. Current concepts suggest that the balance between NO formation and NO scavenging is critical in disease development, with the corollary being that redressing the balance offers a target for therapeutic intervention. Evidence presented over the last two decades has seen red blood cells (RBCs) and haemoglobin specifically emerge as prominent effectors in this paradigm. In this symposium review article, we discuss recent insights into the mechanisms by which RBCs may modulate the balance between NO-formation and inhibition. We discuss how these mechanisms may become dysfunctional to cause disease, highlight key questions that remain, and discuss the potential impact of these insights on therapeutic opportunities.

Benjamin Owusu received his bachelor's degree from University of Ghana and is currently pursuing his PhD in Biochemistry and Structural Biology at the University of Alabama at Birmingham (UAB). His research interests focus on the role of erythrocytes in vascular and pulmonary nitric oxide signaling. Rakesh Patel received his PhD from the University of Essex, UK in 1996. He moved to pursue post-doctoral studies at UAB, where he is currently a Professor in the Department of Pathology. His research interests have centred on understanding the role of erythrocytes and oxidative/nitrosative intermediates in modulating acute and chronic inflammatory diseases.
Physiological role of erythrocyte haemoglobin in the regulation of NO bioavailability
Several lines of evidence implicate red blood cells (RBCs) as modulators of NO signalling by effecting both NO formation and inhibition of NO signalling (illustrated in Fig. 1). In the context of mediating NO signalling, the primary investigated function is stimulation of hypoxic dilatation (Singel & Stamler, 2005; Gladwin et al. 2006; Sprague et al. 2011), although as discussed below, modulation of coagulation and inflammation may also need to be considered (Crawford et al. 2004_a_; Srihirun et al. 2012). In the context of blood flow, the requirement for hypoxia, or more specifically hypoxaemia (haemoglobin deoxygenation) is key for understanding biological functions and molecular mechanisms. Hypoxic dilatation is a critical physiological process that ensures blood flow increases to tissue beds under hypoxic stress to provide nutrients and oxygen to support respiration, exemplified by increased blood flow to exercising skeletal muscle. Physiology studies in animals and humans have shown that hypoxic blood flow especially in the context of exercise does not track with dissolved oxygen tensions, but is directly correlated with the haemoglobin oxygen fractional saturation (Singel & Stamler, 2005; Gladwin et al. 2006), i.e. the amount of oxygen bound by haemoglobin, which in turn is controlled by several allosteric effectors. Within this framework three distinct, haemoglobin deoxygenation-dependent mechanisms have been investigated: _S_-nitrosohaemoglobin (SNOHb)-dependent bioactivity (Singel & Stamler, 2005), adenosine 5′-triphosphate (ATP) release (Sprague & Ellsworth, 2012) and deoxyhaemoglobin-mediated nitrite reduction to NO (Patel et al. 2011). We focus our discussion here on nitrite-dependent mechanisms, and limit discussion of ATP and SNOHb to the context of potential overlap between mechanisms and roles in disease and therapeutics. We also note that other mechanisms for RBC-dependent stimulation of NO signalling have been proposed and include effects of haematocrit on shear stress-dependent activation of endothelial nitric oxide synthase (eNOS) as well as the presence of an active eNOS within the RBC (Kleinbongard et al. 2006; Salazar Vazquez et al. 2008). However, to our knowledge, a haemoglobin oxygenation dependence to these eNOS-dependent pathways has not been established and is thus not discussed further here.
Figure 1. Current models for how RBCs can inhibit and stimulate NO signalling.
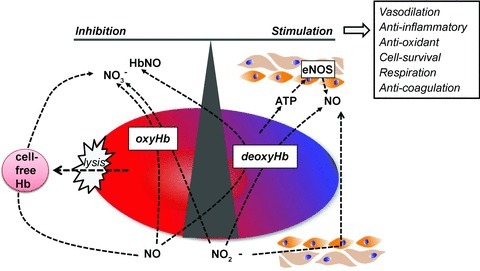
Shown is how haemoglobin oxygen sensing may be coupled to NO scavenging (by intact or cell-free haemoglobin), or formation (from deoxyhaemoglobin-mediated nitrite reduction or ATP release and subsequent eNOS activation) to control NO bioavailability in the vasculature. Also shown is the concept that nitrite reduction to elicit NO signalling during hypoxia may occur by tissues independent of RBCs, but RBCs can affect this process by controlling nitrite concentration via oxidation to nitrate.
Deoxyhaemoglobin-mediated nitrite reduction to NO
Deoxygenated RBCs and haemoglobin reduce nitrite by one electron to produce NO (eqn (1)), which should be contrasted with nitrite oxidation to nitrate upon reaction with oxyhaemoglobin (eqn (2); note this is an autocatalytic reaction and hydrogen peroxide (H2O2) can react further with methaemoglobin (Hb3+)).
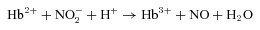 |
(1) |
|---|
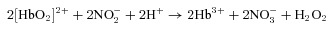 |
(2) |
|---|
In this model, as haemoglobin becomes deoxygenated its nitrite reductase function is activated, resulting in NO production in ischaemic environments. NO formation from eNOS will decrease at very low limiting oxygen tensions, and thus the nitrite reductase activity of haemoglobin has been discussed as a mechanism to ensure sufficient NO bioavailability to sustain the multiple signalling roles associated with this free radical in ischaemic tissues (van Faassen et al. 2009). Biochemical studies demonstrate that haemoglobin oxygen sensing is key in controlling nitrite reduction with maximal rates of this reaction occurring at the haemoglobin p50 (oxygen tension at which haemoglobin is 50% bound with oxygen) (Huang et al. 2005; Crawford et al. 2006).In other words the nitrite reductase activity of haemoglobin is integrated with allosteric mechanisms that control oxygen delivery. Support for a RBC-dependent nitrite reduction mechanism to stimulate hypoxic NO signalling comes from in vitro data showing that a combination of deoxygenated RBCs or cell-free haemoglobin and nitrite produce detectable free NO, which can promote vasodilatation of isolated aortic rings, and inhibit platelet aggregation and mitochondrial respiration (Cosby et al. 2003; Nagababu et al. 2003; Crawford et al. 2006; Roche & Friedman, 2010; Cantu-Medellin et al. 2011; Shiva et al. 2011; Srihirun et al. 2012). Moreover, these effects are reversed by an NO scavenger. Several in vivo studies have complemented insights gained from biochemical studies supporting a role for nitrite-reduction by deoxyhemoglobin in NO homeostasis and include (i) positive arterial–venous nitrite concentration gradients in humans (which is associated with concomitant formation of markers of nitrite–deoxyhaemoglobin reactions in RBCs, the magnitude of which increases with exercise stress), which is associated with increased blood flow (Cosby et al. 2003; Rogers et al. 2007; Maher et al. 2008; Dufour et al. 2010), (ii) nitrite-dependent stimulation of blood flow only in ischaemic, but not in normoxic, murine hind limbs (Kumar et al. 2008), (iii) nitrite-dependent increases in cerebral, pulmonary, intestinal blood flow (Kozlov et al. 2005; Blood & Power, 2007; Rifkind et al. 2007), and (iv) data suggesting that nitrite derived from nitrate therapy improves human exercise performance and decreases blood pressure in normo- and hypertensive individuals (Larsen et al. 2006; Kapil et al. 2010; Kenjale et al. 2011; Lansley et al. 2011). Definitively proving a functional role for the RBCs in vivo remains challenging, however, largely due to the fact that disrupting nitrite reduction by RBCs (through modulating oxygen affinity for example) will also affect oxygen transport (Vitturi & Patel, 2011). This is an area that requires further research.
It should also be stated that some studies have failed to observe free NO formation by haemoglobin-mediated nitrite reduction (Mikulski et al. 2010). Central to this issue is the question: how could RBCs stimulate NO-dependent signalling via a mechanism that involves NO formation by erythrocyte haemoglobin? This question should be considered in the context that both deoxy- and oxyhaemoglobin react and inhibit NO bioavailability very rapidly (bimolecular rate constants = 3–6 × 107 m−1 s−1) (Kim-Shapiro et al. 2011), which when coupled with high millimolar haem concentrations implies that any nitrite-derived NO formed at the haem should not be able to escape the RBC. Thus a critical question in this area remains to resolve the paradox of stimulation of NO signalling by deoxygenated RBCs and nitrite, versus rapid NO scavenging by haem. Proposed solutions include nitrite–deoxyhaem reactions proceeding via formation of intermediate species that avoid rapid inhibitory reactions by the haem, allowing these to diffuse out of the RBCs and then produce NO to elicit endocrine effects (Robinson & Lancaster, 2005; Basu et al. 2007; Salgado et al. 2011). Regulation of this process may occur by controlling transport of nitrite or intermediates in and out of the RBC as recently suggested (Vitturi et al. 2009; Jensen & Rohde, 2010; de Almeida et al. 2011). One candidate is dinitrogen trioxide (N2O3), which may be formed by a reaction between NO and a nitrite–ferric haem intermediate (Basu et al. 2007; Roche & Friedman, 2010). N2O3 does not react with haem, and is small and uncharged and could therefore diffuse out of the RBC where its homolysis can generate NO (and the nitrogen dioxide radical). Further support for this mechanism is that N2O3 is a nitrosating agent, and indeed, nitrosated products (e.g. _S_-nitrosothiols) are also products of nitrite–deoxyhaemoglobin reactions (Cosby et al. 2003; Vitturi et al. 2009). That said, this remains a working model since theoretical concerns still need to be resolved. For example, N2O3 also readily hydrolyses in water and its rapid reactions with amines and thiols (which are present in the RBC and circulation) provide effective competitive pathways for homolysis and NO formation. Thus the search for mechanism(s) that can explain how NO is produced from nitrite and RBCs remains intriguing and its elucidation will not only inform on physiological linkages between hypoxia and vascular NO signalling, but also provide insights that may be utilized to develop targeted therapies to improve NO bioavailability in vascular inflammatory diseases. We also note that other RBC-dependent mechanisms for nitrite bioactivity have been proposed including acidic pH-dependent reduction by RBC associated xanthine oxidoreductase (Webb et al. 2008), nitrite-dependent stimulation of ATP release (Cao et al. 2009) and carbonic anhydrase-dependent nitrite reduction (Aamand et al. 2009).
Dysfunction in RBC-dependent NO signalling and disease
Beyond physiological functions, emerging data are beginning to highlight how disruption in the mechanisms by which RBCs affect NO signalling can contribute to haematologic, vascular, and pulmonary inflammatory diseases. This can occur by physical disruption of the RBC itself leading to spillage of cell-free haemoglobin into the circulation, as well as by biochemical and molecular defects in the pathways that couple RBC oxygen sensing to stimulation of NO signalling. In this section we provide a brief overview of these processes and the diseases involved, and highlight potential and novel targets for therapeutic intervention.
Haemolysis and acute loss of NO bioavailability
The longest standing insights into NO–haemoglobin reactions are that they are rapid, which combined with the high concentration of haemoglobin and high diffusability of NO, should lead to inactivation of the NO signal (and see above). The question why all NO generated in the vascular compartment is not immediately scavenged by RBCs, but is able to elicit many signalling events despite the presence of millimolar haem in the vicinity, was initially raised by Lancaster and colleagues (Lancaster, 1994). A series of seminal studies by multiple groups have established that physical encapsulation of haemoglobin within the RBC is critical in slowing down haemoglobin–NO reactions by ∼1000-fold (Liao et al. 1999; Liu et al. 2002; Azarov et al. 2011). The biophysical and molecular mechanisms underlying this decrease in rate remain under investigation, but effectively increase NO diffusion barriers into the RBCs. Intriguingly, oxygen fractional saturation of RBC modulates NO diffusion barriers (Azarov et al. 2005), suggesting that preventing inhibition of NO signalling should also be considered as a mechanism in the same context as fractional saturation-dependent formation of NO and stimulation of NO signalling. An additional diffusion barrier is an RBC-free zone, generated adjacent to the endothelial monolayer and created according to the Fahraeus effect (Liao et al. 1999). The focus of most recent studies in this area has been to understand factors intrinsic to the RBC that slow NO diffusion, but little attention has been paid to factors that modulate the RBC free zone. The latter is regulated by the glycocalyx, which is rapidly emerging to be an extracellular structure that is dynamically regulated with respect to composition and size, especially during inflammation (Becker et al. 2010). How RBC free zone-dependent modulation of NO bioavailability changes under different inflammatory stress conditions remains to be elucidated.
The importance of haemoglobin encapsulation is best evidenced by observations that haemolysis leads to potent inhibition of NO bioavailability. Without compartmentalization-dependent diffusion barriers, haemoglobin increases blood pressure, pro-coagulation and inflammatory stress – all effects implicated in the pathogenesis of sickle cell disease, sub-arachnoid haemorrhage, pulmonary hypertension, toxicity associated with transfusion of older RBC units (so called RBC storage lesion) and failure of acellular haemoglobins as blood substitutes (Rother et al. 2005; Buehler et al. 2011). How haemolysis occurs and how it could be prevented remains to be determined together with a better understanding of endogenous mechanisms that protect against cell-free haemoglobin will provide much needed insights (Baek et al. 2012). Clearly many questions still remain, but it is evident that haemolysis dramatically disrupts NO homeostasis tilting the scales towards the scavenging side of the balance. Haemolysis is the dramatic conclusion to perturbations in RBC structure. However, it is important not to exclude potential effects of more subtle changes in the biophysical properties of RBCs and NO-scavenging kinetics. For example during RBC storage in the blood bank, or in chronic obstructive pulmonary disease, RBC morphology is dramatically altered, being smaller less deformable and more echinocytic in shape. Preliminary data (Stapley et al. submitted) suggest that these changes lead to an intact RBC that also scavenges NO at faster rates. Combined with other recent studies showing that haemoglobin containing microparticles emanating from RBC scavenge NO at similar rates compared to cell-free haemoglobin (Donadee et al. 2011), the emerging paradigm is that a spectrum of changes to the RBCs from the intact discoidal ‘healthy’ shape to haemolysis may disrupt mechanisms that slow NO scavenging. More and more studies are beginning to document potential links to RBC alterations and disease and further insights into this question will provide novel therapeutic targets to replete NO signalling in diseases characterized by deficits in vascular NO bioavailability.
Uncoupling of intact RBC-dependent activation of NO signalling
Separate studies have implicated dysfunction in either ATP or SNOHb pathways in various diseases. For example experimental evidence for cause–effect relationships between dysfunctional ATP release from RBCs and pulmonary arterial hypertension (PAH), microvascular flow deficits in diabetes and inflammatory stress during transfusion toxicity has been provided (Sprague et al. 2001, 2006; Zhu et al. 2011). Moreover dysfunctional ATP release has been demonstrated in RBCs isolated from cystic fibrosis patients (Liang et al. 2005). Elegant insights into signalling processes that regulate ATP release from hypoxic RBCs are providing the precise proteins whose function is altered during diabetes that then leads to diminished ATP release. These studies are exciting as they provide therapeutic targets for potentially improving microcirculatory blood flow. Similarly, deficits in SNOHb-dependent signalling have been reported in PAH, diabetes, transfusion toxicity, congestive heart failure and sickle cell disease (Datta et al. 2004; James et al. 2004; McMahon et al. 2005; Pawloski et al. 2005; Bennett-Guerrero et al. 2007; Reynolds et al. 2007). The overlap between diseases implicated by dysfunctional ATP release and SNOHb bioactivity should be noted and underlies in part, we feel, the debate in the literature regarding the SNOHb hypothesis. Our previous data using transgenic mice that express either wild-type human haemoglobin or haemoglobin in which the site of _S_-nitrosothiol formation, β93cys, has been replaced with an alanine showed no deficit in isolated RBC hypoxic vasodilatation responses (Isbell et al. 2008). Using a combination of approaches including apyrase-dependent ATP degradation, our data suggested that addition of isolated RBCs to hypoxic vessel rings induce vasodilatation via ATP release (Crawford et al. 2006; Isbell et al. 2008). Moreover, the underlying mechanism of dilatation depended on the type of vessel and the species from which it was isolated; whereas rabbit vessels did (Crawford et al. 2006). We note that other data using eNOS knockout aorta, which preclude ATP-dependent effects, show no deficit in isolated RBC-dependent hypoxic vasodilatation supporting a role for SNOHb (Diesen et al. 2008). However, ATP can still promote dilatation of de-endothelialized vessels (Isbell et al. 2008). These data underscore that the experimental system employed plays a critical role in insights gained and suggest that broad conclusions supporting or dismissing hypotheses must be made with all factors considered. Notwithstanding mechanistic questions, strategies that restore controlled ATP release and/or promote nitrosylation of RBC proteins are intriguing possibilities for all the diseases mentioned above.
Less is known about if, and if so how, altered nitrite reactions with RBCs could play a role in disease. A clinical study in critically ill patients with sepsis, demonstrated that arterial–venous nitrite gradients were approximately half (∼25 nm) in non-survivors compared to survivors (∼45 nm) or healthy volunteers (∼39 nm) with the association with higher arterial–venous gradients and survival being significant (Morgan et al. 2010). Artery–vein (A-V) nitrite consumption may reflect reactions to produce NO-based dilatory signals from deoxygenated RBCs, although differential consumption by tissues present in the arterial vs. venous circuits cannot be excluded. That said, diminished A-V gradients in sepsis is suggestive of a defect in RBC–nitrite reactions and other studies have shown that RBC are targets for oxidative damage in sepsis indicated by altered antioxidant levels and shape (Crawford et al. 2004_b_; Spolarics et al. 2004). Another perspective is that since nitrite serves as a source for NO in ischaemia to limit ischaemia–reperfusion injury (higher endogenous or exogenously applied nitrite protects against numerous ischaemia–reperfusion injury insults; Kevil et al. 2011), RBCs may affect inflammatory disease by modulating the concentration of circulating nitrite. Indeed, antioxidants in the RBCs are thought to be important in limiting nitrite oxidation to nitrate. Oxidative damage to RBCs has been documented in several pathologies; it remains to be determined if this leads to increased nitrite oxidation, and if so, whether this predisposes tissues to inflammatory tissue injury. This remains speculation, although our recent studies with stored RBCs, a treatment that decreases cellular antioxidant levels, are demonstrating increased rates of nitrite oxidation and decreased levels of circulating nitrite in vivo (Stapley et al. submitted).
Summary
RBCs should no longer be considered as only inhibitors of NO bioavailability, but as hubs that regulate NO signalling in the vasculature to affect numerous physiological processes. Emerging paradigms suggest that understanding the balance between how RBCs stimulate NO signalling and how they inhibit NO signalling, especially during hypoxemia, will provide novel insights into diseases characterized by inflammation and microcirculatory dysfunction.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL092624 and HL095468 to R.P.P.).
Conflicts of interest
R.P.P. is a coinventor on a patent for use of nitrite salts for the treatment of cardiovascular conditions.
References
- Aamand R, Dalsgaard T, Jensen FB, Simonsen U, Roepstorff A, Fago A. Generation of nitric oxide from nitrite by carbonic anhydrase: a possible link between metabolic activity and vasodilation. Am J Physiol Heart Circ Physiol. 2009;297:H2068–2074. doi: 10.1152/ajpheart.00525.2009. [DOI] [PubMed] [Google Scholar]
- Azarov I, Huang KT, Basu S, Gladwin MT, Hogg N, Kim-Shapiro DB. Nitric oxide scavenging by red blood cells as a function of hematocrit and oxygenation. J Biol Chem. 2005;280:39024–39032. doi: 10.1074/jbc.M509045200. [DOI] [PubMed] [Google Scholar]
- Azarov I, Liu C, Reynolds H, Tsekouras Z, Lee JS, Gladwin MT, Kim-Shapiro DB. Mechanisms of slower nitric oxide uptake by red blood cells and other hemoglobin-containing vesicles. J Biol Chem. 2011;286:33567–33579. doi: 10.1074/jbc.M111.228650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek JH, D’Agnillo F, Vallelian F, Pereira CP, Williams MC, Jia Y, Schaer DJ, Buehler PW. Hemoglobin-driven pathophysiology is an in vivo consequence of the red blood cell storage lesion that can be attenuated in guinea pigs by haptoglobin therapy. J Clin Invest. 2012;122:1444–1458. doi: 10.1172/JCI59770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Grubina R, Huang J, Conradie J, Huang Z, Jeffers A, Jiang A, He X, Azarov I, Seibert R, Mehta A, Patel R, King SB, Hogg N, Ghosh A, Gladwin MT, Kim-Shapiro DB. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat Chem Biol. 2007;3:785–794. doi: 10.1038/nchembio.2007.46. [DOI] [PubMed] [Google Scholar]
- Becker BF, Chappell D, Jacob M. Endothelial glycocalyx and coronary vascular permeability: the fringe benefit. Basic Res Cardiol. 2010;105:687–701. doi: 10.1007/s00395-010-0118-z. [DOI] [PubMed] [Google Scholar]
- Bennett-Guerrero E, Veldman TH, Doctor A, Telen MJ, Ortel TL, Reid TS, Mulherin MA, Zhu H, Buck RD, Califf RM, McMahon TJ. Evolution of adverse changes in stored RBCs. Proc Natl Acad Sci U S A. 2007;104:17063–17068. doi: 10.1073/pnas.0708160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood AB, Power GG. In vitro and in vivo kinetic handling of nitrite in blood: effects of varying hemoglobin oxygen saturation. Am J Physiol Heart Circ Physiol. 2007;293:H1508–1517. doi: 10.1152/ajpheart.01259.2006. [DOI] [PubMed] [Google Scholar]
- Buehler PW, Karnaukhova E, Gelderman MP, Alayash AI. Blood aging, safety, and transfusion: capturing the “radical” menace. Antioxid Redox Signal. 2011;14:1713–1728. doi: 10.1089/ars.2010.3447. [DOI] [PubMed] [Google Scholar]
- Cantu-Medellin N, Vitturi DA, Rodriguez C, Murphy S, Dorman S, Shiva S, Zhou Y, Jia Y, Palmer AF, Patel RP. Effects of T- and R-state stabilization on deoxyhemoglobin-nitrite reactions and stimulation of nitric oxide signaling. Nitric Oxide. 2011;25:59–69. doi: 10.1016/j.niox.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Bell JB, Mohanty JG, Nagababu E, Rifkind JM. Nitrite enhances RBC hypoxic ATP synthesis and the release of ATP into the vasculature: a new mechanism for nitrite-induced vasodilation. Am J Physiol Heart Circ Physiol. 2009;297:H1494–1503. doi: 10.1152/ajpheart.01233.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, 3rd, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- Crawford JH, Chacko BK, Kevil CG, Patel RP. The red blood cell and vascular function in health and disease. Antioxid Redox Signal. 2004a;6:992–999. doi: 10.1089/ars.2004.6.992. [DOI] [PubMed] [Google Scholar]
- Crawford JH, Chacko BK, Pruitt HM, Piknova B, Hogg N, Patel RP. Transduction of NO-bioactivity by the red blood cell in sepsis: novel mechanisms of vasodilation during acute inflammatory disease. Blood. 2004b;104:1375–1382. doi: 10.1182/blood-2004-03-0880. [DOI] [PubMed] [Google Scholar]
- Crawford JH, Isbell TS, Huang Z, Shiva S, Chacko BK, Schechter AN, Darley-Usmar VM, Kerby JD, Lang JD, Jr, Kraus D, Ho C, Gladwin MT, Patel RP. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;107:566–574. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta B, Tufnell-Barrett T, Bleasdale RA, Jones CJ, Beeton I, Paul V, Frenneaux M, James P. Red blood cell nitric oxide as an endocrine vasoregulator: a potential role in congestive heart failure. Circulation. 2004;109:1339–1342. doi: 10.1161/01.CIR.0000124450.07016.1D. [DOI] [PubMed] [Google Scholar]
- de Almeida JP, Freitas-Santos T, Saldanha C. Evidence that the degree of band 3 phosphorylation modulates human erythrocytes nitric oxide efflux – in vitro model of hyperfibrinogenemia. Clin Hemorheol Microcirc. 2011;49:407–416. doi: 10.3233/CH-2011-1490. [DOI] [PubMed] [Google Scholar]
- Diesen DL, Hess DT, Stamler JS. Hypoxic vasodilation by red blood cells: evidence for an S-nitrosothiol-based signal. Circ Res. 2008;103:545–553. doi: 10.1161/CIRCRESAHA.108.176867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadee C, Raat NJ, Kanias T, Tejero J, Lee JS, Kelley EE, Zhao X, Liu C, Reynolds H, Azarov I, Frizzell S, Meyer EM, Donnenberg AD, Qu L, Triulzi D, Kim-Shapiro DB, Gladwin MT. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation. 2011;124:465–476. doi: 10.1161/CIRCULATIONAHA.110.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour SP, Patel RP, Brandon A, Teng X, Pearson J, Barker H, Ali L, Yuen AH, Smolenski RT, Gonzalez-Alonso J. Erythrocyte-dependent regulation of human skeletal muscle blood flow: role of varied oxyhemoglobin and exercise on nitrite, S-nitrosohemoglobin, and ATP. Am J Physiol Heart Circ Physiol. 2010;299:H1936–1946. doi: 10.1152/ajpheart.00389.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladwin MT, Raat NJ, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, Patel RP. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol. 2006;291:H2026–2035. doi: 10.1152/ajpheart.00407.2006. [DOI] [PubMed] [Google Scholar]
- Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, Gladwin MT. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isbell TS, Sun CW, Wu LC, Teng X, Vitturi DA, Branch BG, Kevil CG, Peng N, Wyss JM, Ambalavanan N, Schwiebert L, Ren J, Pawlik KM, Renfrow MB, Patel RP, Townes TM. SNO-hemoglobin is not essential for red blood cell-dependent hypoxic vasodilation. Nat Med. 2008;14:773–777. doi: 10.1038/nm1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James PE, Lang D, Tufnell-Barret T, Milsom AB, Frenneaux MP. Vasorelaxation by red blood cells and impairment in diabetes: reduced nitric oxide and oxygen delivery by glycated hemoglobin. Circ Res. 2004;94:976–983. doi: 10.1161/01.RES.0000122044.21787.01. [DOI] [PubMed] [Google Scholar]
- Jensen FB, Rohde S. Comparative analysis of nitrite uptake and hemoglobin-nitrite reactions in erythrocytes: sorting out uptake mechanisms and oxygenation dependencies. Am J Physiol Regul Integr Comp Physiol. 2010;298:R972–982. doi: 10.1152/ajpregu.00813.2009. [DOI] [PubMed] [Google Scholar]
- Kapil V, Milsom AB, Okorie M, Maleki-Toyserkani S, Akram F, Rehman F, Arghandawi S, Pearl V, Benjamin N, Loukogeorgakis S, Macallister R, Hobbs AJ, Webb AJ, Ahluwalia A. Inorganic nitrate supplementation lowers blood pressure in humans: role for nitrite-derived NO. Hypertension. 2010;56:274–281. doi: 10.1161/HYPERTENSIONAHA.110.153536. [DOI] [PubMed] [Google Scholar]
- Kenjale AA, Ham KL, Stabler T, Robbins JL, Johnson JL, Vanbruggen M, Privette G, Yim E, Kraus WE, Allen JD. Dietary nitrate supplementation enhances exercise performance in peripheral arterial disease. J Appl Physiol. 2011;110:1582–1591. doi: 10.1152/japplphysiol.00071.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevil CG, Kolluru GK, Pattillo CB, Giordano T. Inorganic nitrite therapy: historical perspective and future directions. Free Radic Biol Med. 2011;51:576–593. doi: 10.1016/j.freeradbiomed.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Shapiro DB, Lee J, Gladwin MT. Storage lesion: role of red blood cell breakdown. Transfusion. 2011;51:844–851. doi: 10.1111/j.1537-2995.2011.03100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinbongard P, Schulz R, Rassaf T, Lauer T, Dejam A, Jax T, Kumara I, Gharini P, Kabanova S, Ozuyaman B, Schnurch HG, Godecke A, Weber AA, Robenek M, Robenek H, Bloch W, Rosen P, Kelm M. Red blood cells express a functional endothelial nitric oxide synthase. Blood. 2006;107:2943–2951. doi: 10.1182/blood-2005-10-3992. [DOI] [PubMed] [Google Scholar]
- Kozlov AV, Costantino G, Sobhian B, Szalay L, Umar F, Nohl H, Bahrami S, Redl H. Mechanisms of vasodilatation induced by nitrite instillation in intestinal lumen: possible role of hemoglobin. Antioxid Redox Signal. 2005;7:515–521. doi: 10.1089/ars.2005.7.515. [DOI] [PubMed] [Google Scholar]
- Kumar D, Branch BG, Pattillo CB, Hood J, Thoma S, Simpson S, Illum S, Arora N, Chidlow JH, Jr, Langston W, Teng X, Lefer DJ, Patel RP, Kevil CG. Chronic sodium nitrite therapy augments ischemia-induced angiogenesis and arteriogenesis. Proc Natl Acad Sci U S A. 2008;105:7540–7545. doi: 10.1073/pnas.0711480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster JR., Jr Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proc Natl Acad Sci U S A. 1994;91:8137–8141. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansley KE, Winyard PG, Bailey SJ, Vanhatalo A, Wilkerson DP, Blackwell JR, Gilchrist M, Benjamin N, Jones AM. Acute dietary nitrate supplementation improves cycling time trial performance. Med Sci Sports Exerc. 2011;43:1125–1131. doi: 10.1249/MSS.0b013e31821597b4. [DOI] [PubMed] [Google Scholar]
- Larsen FJ, Ekblom B, Sahlin K, Lundberg JO, Weitzberg E. Effects of dietary nitrate on blood pressure in healthy volunteers. N Engl J Med. 2006;355:2792–2793. doi: 10.1056/NEJMc062800. [DOI] [PubMed] [Google Scholar]
- Liang G, Stephenson AH, Lonigro AJ, Sprague RS. Erythrocytes of humans with cystic fibrosis fail to stimulate nitric oxide synthesis in isolated rabbit lungs. Am J Physiol Heart Circ Physiol. 2005;288:H1580–1585. doi: 10.1152/ajpheart.00807.2004. [DOI] [PubMed] [Google Scholar]
- Liao JC, Hein TW, Vaughn MW, Huang KT, Kuo L. Intravascular flow decreases erythrocyte consumption of nitric oxide. Proc Natl Acad Sci U S A. 1999;96:8757–8761. doi: 10.1073/pnas.96.15.8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Samouilov A, Lancaster JR, Jr, Zweier JL. Nitric oxide uptake by erythrocytes is primarily limited by extracellular diffusion not membrane resistance. J Biol Chem. 2002;277:26194–26199. doi: 10.1074/jbc.M201939200. [DOI] [PubMed] [Google Scholar]
- Maher AR, Milsom AB, Gunaruwan P, Abozguia K, Ahmed I, Weaver RA, Thomas P, Ashrafian H, Born GV, James PE, Frenneaux MP. Hypoxic modulation of exogenous nitrite-induced vasodilation in humans. Circulation. 2008;117:670–677. doi: 10.1161/CIRCULATIONAHA.107.719591. [DOI] [PubMed] [Google Scholar]
- McMahon TJ, Ahearn GS, Moya MP, Gow AJ, Huang YC, Luchsinger BP, Nudelman R, Yan Y, Krichman AD, Bashore TM, Califf RM, Singel DJ, Piantadosi CA, Tapson VF, Stamler JS. A nitric oxide processing defect of red blood cells created by hypoxia: deficiency of S-nitrosohemoglobin in pulmonary hypertension. Proc Natl Acad Sci U S A. 2005;102:14801–14806. doi: 10.1073/pnas.0506957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikulski R, Tu C, Swenson ER, Silverman DN. Reactions of nitrite in erythrocyte suspensions measured by membrane inlet mass spectrometry. Free Radic Biol Med. 2010;48:325–331. doi: 10.1016/j.freeradbiomed.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MA, Frasier LM, Stewart JC, Mack CM, Gough MS, Graves BT, Apostolakos MJ, Doolin KP, Darling DC, Frampton MW, Pietropaoli AP. Artery-to-vein differences in nitric oxide metabolites are diminished in sepsis. Crit Care Med. 2010;38:1069–1077. doi: 10.1097/CCM.0b013e3181d16a3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagababu E, Ramasamy S, Abernethy DR, Rifkind JM. Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin-mediated nitrite reduction. J Biol Chem. 2003;278:46349–46356. doi: 10.1074/jbc.M307572200. [DOI] [PubMed] [Google Scholar]
- Patel RP, Hogg N, Kim-Shapiro DB. The potential role of the red blood cell in nitrite-dependent regulation of blood flow. Cardiovasc Res. 2011;89:507–515. doi: 10.1093/cvr/cvq323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawloski JR, Hess DT, Stamler JS. Impaired vasodilation by red blood cells in sickle cell disease. Proc Natl Acad Sci U S A. 2005;102:2531–2536. doi: 10.1073/pnas.0409876102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JD, Ahearn GS, Angelo M, Zhang J, Cobb F, Stamler JS. S-Nitrosohemoglobin deficiency: a mechanism for loss of physiological activity in banked blood. Proc Natl Acad Sci U S A. 2007;104:17058–17062. doi: 10.1073/pnas.0707958104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkind JM, Nagababu E, Barbiro-Michaely E, Ramasamy S, Pluta RM, Mayevsky A. Nitrite infusion increases cerebral blood flow and decreases mean arterial blood pressure in rats: a role for red cell NO. Nitric Oxide. 2007;16:448–456. doi: 10.1016/j.niox.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Robinson JM, Lancaster JR., Jr Hemoglobin-mediated, hypoxia-induced vasodilation via nitric oxide: mechanism(s) and physiologic versus pathophysiologic relevance. Am J Respir Cell Mol Biol. 2005;32:257–261. doi: 10.1165/rcmb.F292. [DOI] [PubMed] [Google Scholar]
- Roche CJ, Friedman JM. NO reactions with sol-gel and solution phase samples of the ferric nitrite derivative of HbA. Nitric Oxide. 2010;22:180–190. doi: 10.1016/j.niox.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SC, Khalatbari A, Datta BN, Ellery S, Paul V, Frenneaux MP, James PE. NO metabolite flux across the human coronary circulation. Cardiovasc Res. 2007;75:434–441. doi: 10.1016/j.cardiores.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293:1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- Salazar Vazquez BY, Cabrales P, Tsai AG, Johnson PC, Intaglietta M. Lowering of blood pressure by increasing hematocrit with non nitric oxide scavenging red blood cells. Am J Respir Cell Mol Biol. 2008;38:135–142. doi: 10.1165/rcmb.2007-0081OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado MT, Ramasamy S, Tsuneshige A, Manoharan PT, Rifkind JM. A new paramagnetic intermediate formed during the reaction of nitrite with deoxyhemoglobin. J Am Chem Soc. 2011;133:13010–13022. doi: 10.1021/ja1115088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S, Rassaf T, Patel RP, Gladwin MT. The detection of the nitrite reductase and NO-generating properties of haemoglobin by mitochondrial inhibition. Cardiovasc Res. 2011;89:566–573. doi: 10.1093/cvr/cvq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol. 2005;67:99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- Spolarics Z, Condon MR, Siddiqi M, Machiedo GW, Deitch EA. Red blood cell dysfunction in septic glucose-6-phosphate dehydrogenase-deficient mice. Am J Physiol Heart Circ Physiol. 2004;286:H2118–2126. doi: 10.1152/ajpheart.01085.2003. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Bowles EA, Achilleus D, Ellsworth ML. Erythrocytes as controllers of perfusion distribution in the microvasculature of skeletal muscle. Acta Physiol (Oxf) 2011;202:285–292. doi: 10.1111/j.1748-1716.2010.02182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML. Erythrocyte-derived ATP and perfusion distribution: Role of intracellular and intercellular communication. Microcirculation. 2012;19:430–439. doi: 10.1111/j.1549-8719.2011.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Stephenson AH, Bowles EA, Stumpf MS, Lonigro AJ. Reduced expression of G(i) in erythrocytes of humans with type 2 diabetes is associated with impairment of both cAMP generation and ATP release. Diabetes. 2006;55:3588–3593. doi: 10.2337/db06-0555. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Stephenson AH, Ellsworth ML, Keller C, Lonigro AJ. Impaired release of ATP from red blood cells of humans with primary pulmonary hypertension. Exp Biol Med (Maywood) 2001;226:434–439. doi: 10.1177/153537020122600507. [DOI] [PubMed] [Google Scholar]
- Srihirun S, Sriwantana T, Unchern S, Kittikool D, Noulsri E, Pattanapanyasat K, Fucharoen S, Piknova B, Schechter AN, Sibmooh N. Platelet inhibition by nitrite is dependent on erythrocytes and deoxygenation. PLoS One. 2012;7:e30380. doi: 10.1371/journal.pone.0030380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Faassen EE, Bahrami S, Feelisch M, Hogg N, Kelm M, Kim-Shapiro DB, Kozlov AV, Li H, Lundberg JO, Mason R, Nohl H, Rassaf T, Samouilov A, Slama-Schwok A, Shiva S, Vanin AF, Weitzberg E, Zweier J, Gladwin MT. Nitrite as regulator of hypoxic signaling in mammalian physiology. Med Res Rev. 2009;29:683–741. doi: 10.1002/med.20151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitturi DA, Patel RP. Current perspectives and challenges in understanding the role of nitrite as an integral player in nitric oxide biology and therapy. Free Radic Biol Med. 2011;51:805–812. doi: 10.1016/j.freeradbiomed.2011.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitturi DA, Teng X, Toledo JC, Matalon S, Lancaster JR, Jr, Patel RP. Regulation of nitrite transport in red blood cells by hemoglobin oxygen fractional saturation. Am J Physiol Heart Circ Physiol. 2009;296:H1398–1407. doi: 10.1152/ajpheart.01303.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AJ, Milsom AB, Rathod KS, Chu WL, Qureshi S, Lovell MJ, Lecomte FM, Perrett D, Raimondo C, Khoshbin E, Ahmed Z, Uppal R, Benjamin N, Hobbs AJ, Ahluwalia A. Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia: role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ Res. 2008;103:957–964. doi: 10.1161/CIRCRESAHA.108.175810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Zennadi R, Xu BX, Eu JP, Torok JA, Telen MJ, McMahon TJ. Impaired adenosine-5′-triphosphate release from red blood cells promotes their adhesion to endothelial cells: a mechanism of hypoxemia after transfusion. Crit Care Med. 2011;39:2478–2486. doi: 10.1097/CCM.0b013e318225754f. [DOI] [PMC free article] [PubMed] [Google Scholar]