Characterization of the RpoN regulon reveals differential regulation of T6SS and new flagellar operons in Vibrio cholerae O37 strain V52 (original) (raw)
Abstract
The alternative sigma factor RpoN is an essential colonization factor of Vibrio cholerae and controls important cellular functions including motility and type VI secretion (T6SS). The RpoN regulon has yet to be clearly defined in T6SS-active V. cholerae isolates, which use T6SS to target both bacterial competitors and eukaryotic cells. We hypothesize that T6SS-dependent secreted effectors are co-regulated by RpoN. To systemically identify RpoN-controlled genes, we used chromatin immunoprecipitation coupled with sequencing (ChIP-Seq) and transcriptome analysis (RNA-Seq) to determine RpoN-binding sites and RpoN-controlled gene expression. There were 68 RpoN-binding sites and 82 operons positively controlled by RpoN, among which 37 operons had ChIP-identified binding sites. A consensus RpoN-binding motif was identified with a highly conserved thymine (−14) and an AT-rich region in the middle between the hallmark RpoN-recognized motif GG(−24)/GC(−12). There were seven new RpoN-dependent promoters in the flagellar regions. We identified a small RNA, flaX, downstream of the major flagellin gene flaA. Mutation of flaX substantially reduced motility. In contrast to previous results, we report that RpoN positively regulates the expression of hcp operons and vgrG3 that encode T6SS secreted proteins but has no effect on the expression of the main T6SS cluster encoding sheath and other structural components.
INTRODUCTION
Cholera is a serious threat to public health in the developing countries. The cholera outbreak in Haiti has resulted in >6000 deaths and about a half-million confirmed cases (1). As the causative pathogen for cholera, Vibrio cholerae has many virulence factors including cholera toxin, the critical colonization factor TCP pili, and virulence regulators ToxR and ToxT [reviewed in (2)]. A newly identified protein secretion system, Type VI secretion system (T6SS), has also been implicated in the virulence of V. cholerae by causing cytotoxicity in macrophage and inducing intestinal inflammation (3–5).
T6SS is a conserved protein delivery system present in >100 Gram-negative bacterial species including many important pathogens, V. cholerae (5), Pseudomonas aeruginosa (6), Burkholderia thailandensis (7), Serratia marcescens (8) and avian pathogenic Escherichia coli (9). In V. cholerae, T6SS genes are organized in one large locus consisting of 17 genes encoding mostly basal components and two hcp-vgrG operons encoding secreted proteins (5) (Supplementary Figure S1). Structural analysis shows that T6SS components are highly similar to phage tail proteins (10,11). For example, the T6SS-secreted Hcp and VgrG proteins are homologous to the tail tube protein and spike-like protein of T4 phage, respectively (11). Expression of T6SS genes is regulated by quorum sensing, TsrA and RpoN (5,12,13).
RpoN is an alternative sigma factor that binds to the core RNA polymerase and directs gene expression from RpoN-recognized promoters (14). RpoN positively regulates the expression of 70 genes in E. coli K-12, including genes for motility and nitrogen assimilation (15). However, the RpoN regulon has not been clearly defined in V. cholerae. RpoN-mediated transcription requires an enhancer that activates transcription by binding to upstream regions (16). The T6SS locus encodes an RpoN-enhancer VasH that is required for Hcp secretion and T6SS function (17). RpoN is essential for colonization in V. cholerae O1 classical strain O395, and it controls motility and expression of glutamine synthetase GlnA (18). However, the colonization defect of the rpoN mutant is not because of loss of motility or GlnA expression, suggesting the involvement of other RpoN-controlled traits (18). Microarray profiling studies of the effect of rpoN mutation on rugosity (19) and motility (20) show the expression of a large number of genes is altered in rpoN mutants of V. cholerae O1 El Tor strain and serotype O1 classical strain O395, respectively. Interestingly, T6SS genes are up-regulated in the rpoN mutants of these strains (19,20). This difference in RpoN control of T6SS is likely because of strain variation, as T6SS is not active in the seventh pandemic V. cholerae O1 El Tor strain and O395 strain under laboratory conditions (5). In addition, heterologous expression of T6SS-promoter reporter fusions was shown to be RpoN-dependent (21), suggesting that the T6SS cluster and hcp operons are co-regulated by RpoN.
T6SS targets both eukaryotic hosts and prokaryotic competitors, although the mechanism is not fully understood. Pseudomonas aeruginosa T6SS secretes effector proteins that are toxic to E. coli by degrading peptidoglycan (22,23). T6SS of V. cholerae is also required for killing E. coli (24), but the effectors have not been clearly defined. The actin-crosslinking domain of VgrG1 is translocated into macrophage cells resulting in cytotoxicity (4,25), and a newly identified secreted factor VasX is important for T6SS-mediated virulence in the model amoeba Dictyostelium discoideum (26).
In this study, our primary interest is to fully understand the regulatory role of RpoN in T6SS secretion and other cellular functions in V. cholerae. Chromatin-immunoprecipitation coupled with next-generation sequencing, ChIP-Seq, provides a powerful tool to identify protein–DNA interactions. ChIP-Seq has been mostly used in eukaryotic systems and there are few reports in bacteria. Our lab previously demonstrated the effectiveness of ChIP-Seq in bacterial studies and identified the Fur regulon in V. cholerae (27). Here we used the same approach combined with RNA-Seq to characterize the RpoN regulon in a T6SS-active, serotype O37 clinical isolate V52. There were 37 operons that show RpoN-dependent expression and RpoN-binding sites, indicative of direct control by RpoN. We uncovered new RpoN-controlled promoters in the flagellar gene clusters, and a small RNA that is important for motility. In addition, RpoN was found to selectively control the expression of T6SS secreted proteins but not the structural components.
MATERIALS AND METHODS
Bacterial strains and growth conditions
Strains and plasmids are listed in Table 1. A serotype O37 clinical isolate of V. cholerae strain V52 was used in this study. Escherichia coli DH5α and SM10 λpir were used for cloning and conjugation, respectively. Antibiotics were used at the following concentrations: ampicillin (100 µg/ml), streptomycin (100 µg/ml), kanamycin (50 µg/ml) and chloramphenicol (2.5 µg/ml for V. cholerae and 25 µg/ml for E. coli). Arabinose (0.1%) was used for induction, unless otherwise specified. Motility was tested on Luria Bertani (LB) medium with 0.3% agar.
Table 1.
Strains and plasmids used in this study
| Strain and plasmid | Genotype or phenotype | Reference |
|---|---|---|
| V. cholerae | ||
| V52 | Serotype 37 clinical isolate from Sudan | Pukatzki et al., 2006 (5) |
| V52 rpoN | Knock-out rpoN mutant of V52 | This study |
| T6SS mutants | Nonpolar deletion mutants of V52 | Zheng et al., 2010 (12) |
| SM10 | thi thr leu tonA lac Y supE recA::RP4-2-Tc::Mu | Miller and Mekalanos, 1988 (28) |
| Plasmid | ||
| pWM91 | Suicidal conjugation vector | Metcalf et al., 1996 (29) |
| pBAD18V5 | Expression vector with 3xV5 tag | Davies et al., 2011 (27) |
| pRpoN | The rpoN gene cloned into pBAD18V5 | This study |
| pNM12 | sRNA expression vector derivative of pBAD24 | Majdalani et al., 1998 (30) |
| pflaX | flaX inserted between NheI and KpnI sites on pNM12 | This study |
All strains were routinely grown at 37°C in LB medium containing 5 g/l sodium chloride. For ChIP- and RNA-Seq analyses, cultures were grown in LB medium to exponential phase (OD600 = 0.5) and induced with 0.1% arabinose for 30 minutes at 37°C.
DNA manipulation
In-frame gene deletion in V. cholerae was performed as described (28,29). Briefly, the flanking region of the respective gene was amplified by crossover PCR, cloned into the pWM91 suicide vector, and transferred into recipient strains through conjugation. Independent transconjugants were purified and incubated on 5% sucrose to select for sucrose-resistant segregants. Deletion was confirmed by sequencing. For gene induction, the respective gene was PCR-amplified, digested with KpnI and SalI, and cloned into the pBAD18 plasmid (31) carrying a C-terminal 3xV5 epitope tag (32). The small RNA flaX was cloned into a plasmid pNM12, a pBAD24-derivative vector that lacks ribosomal binding sites upstream of the cloning site and is commonly used for expressing sRNAs (30). All constructs were verified by sequencing. Primers used in this study are listed in Supplementary Table S1.
Western blot
Proteins were resolved in a precast 10% SDS PAGE gel (Life Technologies) and transferred to a PVDF membrane (Millipore) by electrophoresis. The membrane was then blocked in 5% non-fat milk for 1 hour at room temperature, and incubated with respective monoclonal antibodies at 4°C overnight. The monoclonal antibodies to V5-tag, RpoB and T6SS components were from Sigma Aldrich, NeoClone and laboratory stock, respectively. The membrane was then washed three times in TBST buffer (50 mM Tris, 150 mM NaCl, 0.05% Tween-20, pH 7.6) and incubated with a HRP-conjugated secondary antibody (Pierce) for 1 hour at room temperature. Signals were detected using the ECL solution and ECL films (Amersham). Western blotting was performed at least twice for each experiment.
Protein secretion assay
Cultures were grown in LB medium to exponential phase (OD600 = 0.5). Expression was induced by 0.1% L-arabinose for 1 hour. One millilitre culture was centrifuged twice at 20 000 × g for 2 minutes, and the supernatant was then filtered through a 0.2 -µm filter. A mixture of 900 µl of supernatant and 100 µl of 100% TCA solution was placed on ice for 2 hours and centrifuged at 15 000 × g for 20 minutes at 4°C. The supernatant was discarded and the pellet was washed twice with 1 ml of 100% acetone by centrifugation at 20 000 × g for 5 minutes. The resultant pellet was mixed with 30 µl of SDS-loading dye and proteins analyzed by western blot analysis.
Chromatin immunoprecipitation paired with next-generation sequencing ChIP-Seq
ChIP assay was performed as described previously (27). Briefly, 50 ml culture was mixed with formaldehyde (1% final concentration) for cross-linking at room temperature for 20 minutes, and the reaction was quenched by glycine (0.5 M). Cells were collected by centrifugation, washed twice with TBS buffer, and resuspended in 1 ml lysis buffer (10 mM Tris, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% deoxycholate, 0.5% N-lauroylsarcosine, pH 8.0) with protease inhibitor cocktail (Sigma). Cells were then lysed with 1 mg/ml lysozyme at 37°C for 30 minutes and sonicated for 10 minutes using a cup horn sonicator at 60% power setting (Misonix). Cell debris was removed by centrifugation at 16 000 × g for 15 minutes at 4°C, and the supernatant containing DNA fragments with an average size of 200 bp was incubated with pretreated Dynal-Protein G beads coated with anti-V5 monoclonal antibody (Sigma). A control sample (100 µl) was taken before Dynal-bead treatment. ChIP reaction was carried out at 4°C overnight on a rotary shaker, and samples were washed five times with RIPA buffer (50 mM HEPES buffer, 500 mM LiCl, 1 mM EDTA, 1% Nonidet P-40, 0.7% deoxycholate, pH 7.5) and once with TBS buffer. ChIP DNA was eluted in 100 μl elution buffer (50 mM TE buffer, 1% EDTA, pH 7.5) at 65°C for 30 minutes and de-cross-linked by incubation at 65°C overnight. Samples were then treated with RNase A and proteinase K, and purified with a ChIP DNA clean kit (Zymo). Experiments were performed in triplicate.
RNA extraction
RNA was extracted from three independent replicates of exponential phase cultures using acidic phenol (pH 4.3, Sigma) and purified with a Direct-zol RNA kit (Zymo). The quality of RNA was examined with Bioanalyzer (Agilent). Ten micrograms of total RNA was treated with an Ambion rRNA kit to remove ribosomal RNA, and the efficiency was confirmed by Bioanalyzer analysis.
Library construction and Illumina HiSeq sequencing
Libraries for ChIP-Seq and RNA-Seq were prepared using NEBNext® ChIP-Seq Sample Prep Master Mix Set 1 (NEB) and NEBNext® mRNA Sample Prep Master Mix kit (NEB), respectively. Briefly, ChIP DNA was end-repaired ligated to Illumina adaptors, and selected for a fragment size of around 200 bp by gel extraction. RNA samples were treated similarly, except for two additional steps of fragmentation and cDNA synthesis before end-repair. Multiplex Illumina primers were used to PCR-amplify gel-extracted products, and products were purified with a DNA clean-up kit (Zymo).
Sequencing was performed using an Illumina HiSeq2000 platform in the Biopolymer core facility at Harvard Medical School. ChIP-Seq and RNA-Seq reads were mapped to V. cholerae N16961 genome using the software CLC Genomics Workbench as previously described (27). ChIP-Seq and RNA-Seq data generated average genome coverage of 4-fold and 40-fold, respectively. Three independent ChIP samples were compared with the reference, and enriched peaks were detected by scanning the genome with a 120-bp sliding window. Identified peaks are ranked by the false discovery rate (FDR), which is estimated as previously describe (33). Genuine RpoN-binding sites are peaks identified in all three ChIP samples with a cut-off FDR value (1E-26) that corresponds to the highest FDR for previously known binding sites. Consensus RpoN-binding motif was generated by searching ChIP peak regions using the MEME Suite program (34). For RNA-Seq, gene expression level is represented as the RPKM value (reads per kilobase per million mapped reads) (35). We use a two-fold change in average gene expression and Student’s t-test P value < 0.05 as criteria to identify RpoN-controlled genes. The average RPKM value for the rpoN gene was 345-fold higher in the _rpoN_-positive strain than in the rpoN mutant that had only background values, indicating a good quality of RNA-Seq data.
Quantitative PCR (qPCR)
To validate ChIP-Seq and RNA-Seq results, qPCR was performed using the Fast SYBR green mix (Kapa Biosystems). For transcriptional expression analysis, one-step qPCR was performed with the RNA-Ct one-step system (Applied Systems). The rrsA gene, encoding 16 S RNA, was used as a control sample for normalization of difference in sample quantities.
Northern blot analysis
Total RNA (10 μg) and ssRNA ladder (2 μg) were mixed with ssRNA ladder loading buffer (NEB), respectively, and incubated at 75°C for 5 minutes and 4°C for 2 minutes. Samples were resolved on a 6% TBE-Urea gel (Life Technologies) at 150 V for 1 hour and transferred to a Hybond N membrane at 70 V for 2 hours. After UV-cross-linking twice at 1200 J, the membrane was pre-hybridized with ULTRAhyb®-Oligo buffer (Life Technologies) at 45°C for 30 minutes and then hybridized with 100 pmol of 32P labeled probe overnight. The membrane was washed twice with 2 × SSC (saline-sodium citrate) buffer and exposed to film for 24 hours before development.
RESULTS
RpoN-recognized promoter regions identified by ChIP-Seq
As a transcription factor, the importance of RpoN in virulence and other functions results from the effects of its regulated genes. To characterize these genes and identify RpoN-binding promoters, we performed ChIP experiments in an rpoN knockout mutant expressing an epitope-tagged RpoN or the empty vector as a control subject. The C-terminus of RpoN was fused with a 3xV5 tag that has been shown with high ChIP efficiency in V. cholerae (27). To test whether this construct is functional, we examined two known RpoN-dependent traits, motility and Hcp expression. The rpoN mutant was complemented by the RpoN-3V5 construct for motility and expression of Hcp (Figure 1), indicative of a functional RpoN.
Figure 1.
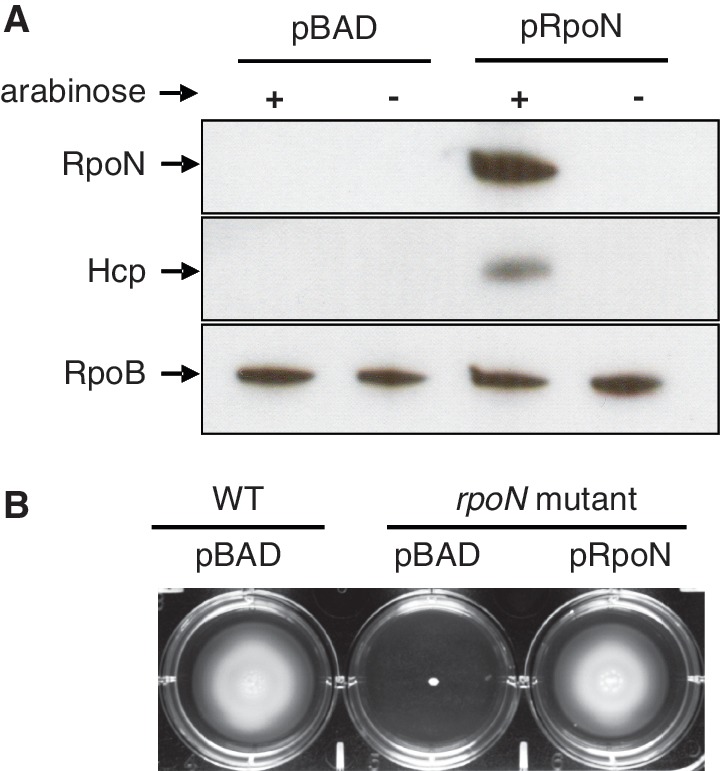
RpoN-3V5 complements the rpoN deletion mutant. The pBAD-derivative plasmid pRpoN carrying arabinose-inducible RpoN-3V5 complements two known RpoN-dependent traits in the rpoN mutant, Hcp expression (A) and motility (B).
Using ChIP-Seq analysis, we found 68 RpoN-binding peaks common in three replicate samples (Figure 2, Supplementary Table S2). These peak-associated genes are involved in motility, T6SS secretion, nitrogen utilization, lipoprotein and membrane and unknown functions. By comparing all peak sequences, we identified a consensus 15 bp RpoN-binding motif, tGG(−24)cacnnttttTGC(−12), capital letter indicating highly conserved sequences (Figure 2). Although it closely resembles the canonical RpoN-recognized sequence identified in E. coli (15), the RpoN-binding motif in V. cholerae differs from E. coli motif with a more AT-rich middle region and a conserved thymine (−14). All 12 previously shown RpoN-controlled promoters were confirmed, including hcp and flaA (Supplementary Table S2). Forty-eight (70%) RpoN-biding peaks were found within 250-bp upstream of an open reading frame (ORF). There were seven RpoN-binding sites located inside genes and distant from the 5’ end of the nearest downstream gene (Supplementary Table S2). To validate ChIP-Seq results, we tested the ChIP enrichment by qPCR. Results show at least 10-fold enrichment for all binding sites except for VCA0925, which had a 2.9- ± 0.6-fold enrichment (Supplementary Figure S2).
Figure 2.

A sample of ChIP-Seq and RNA-Seq data covering the flagellar region and consensus RpoN-binding motif. (A) Genomic structures are shown above the peak panel. The height of each peak corresponds to the number of reads bound to the corresponding ChIP-site. (B) Motif was generated based on all ChIP-peaks using MEME program. The height of each letter represents the occurrence frequency at each location. (C) RNA-Seq data show difference in regulation of flagellar genes by RpoN. The RpoN-dependence (expression ratio) is shown at the bottom with the left axis. The absolute expression for each gene in the rpoN mutant is shown on the top with the right axis. Genes with high-RpoN-dependence and low-expression in the rpoN mutant require RpoN for expression, whereas genes with high-expression in the rpoN mutant likely have other promoters independent of RpoN.
Transcriptome profiling of RpoN-controlled genes by RNA-Seq
The identified RpoN-binding sites may include both silent sites not involved in transcription and active promoters that drive gene expression. To differentiate these, we compared the effect of RpoN on transcriptome expression in RpoN-expressed and RpoN-deleted conditions using RNA-Seq analysis. The expression of 144 genes in 82 operons was positively controlled by RpoN (≥two-fold, P < 0.05) (Supplementary Table S3). These genes include >50 genes for motility and chemotaxis, all genes in the T6SS hcp1 and hcp2 operons, nitrogen utilization genes glnB1, glnA and ntrB, genes coding for formate dehydrogenase and phage shock proteins and >40 genes encoding unknown functions. Of RpoN-controlled operons, 37 possess upstream ChIP-identified RpoN-binding sites (Table 2). Thirty-five of these promoter sites are within 250 bp from the 5’end of downstream genes. It is important to note that the activity of RpoN-binding promoters requires the presence of an enhancer, and our analysis would not detect RpoN-dependent transcripts if their cognate enhancers were not expressed under the conditions tested. A large number of genes were expressed higher in the rpoN mutant, including many genes for metabolism (Supplementary Table S4). This is likely an indirect effect because of competition of sigma factors for binding to the limited number of RNA core polymerase (36,37). Alteration in the expression of one sigma factor results in changes in gene expression controlled by other sigma factors (37,38). For example, deletion of rpoN increases the expression of genes controlled by another sigma factor RpoS in E. coli (39).
Table 2.
RpoN-regulated genes identified by RNA-Seq and ChIP-Seq
| Operon | Gene | Function | Fold change (pRpoN/pBAD) | _P_-value | ChIP-peak |
|---|---|---|---|---|---|
| T6SS | |||||
| VC1415-20 | hcp1/vgrG1 | T6SS secretion | 511.1/61.9/14.8/6.4/7.0/4.7 | 0.001 | TGGCATCCCACTTGC |
| VCA0017-23 | hcp2/vgrG2 | T6SS secretion | 532.5/94.1/19.0/14.5/12.7/2.8/3.8 | 0.001 | TGGCATCCCACTTGC |
| VCA0123 | vgrG3 | T6SS secretion | 2.4 | 0.001 | TGGCATTGAGTTTGC |
| Motiliy and Chemotaxis | |||||
| VC1008 | motY | Sodium-type flagellar protein | 16.9 | 0.001 | TGGCTAGATTTTTGC |
| VC1384 | Putative outer memberane protein | 7.6 | 0.001 | AGGTACGAAATTTGC | |
| VC2068-66 | flhFG-fliA | Flagellar biosynthesis/sigma factor FliA | 16.2/6.8/3.3 | 0.001 | TGGAACAAATTTTGC |
| VC2069 | flhA | Flagellar biosynthesis | 7.4 | 0.001 | TGGACTGAAAATTGC |
| VC2128 | fliK | Flagellar hook-length control | 36.2 | 0.001 | TGGCTTACTTCTTGC |
| VC2134-29 | fliEFGHIJ | Flagellar assembly | 5.6/5.5/5.3/4.7/6.0/6.6 | 0.001 | TGGCATACAAATTGC |
| VC2136-35 | flrBC | Flagellar regulator | 3.2/2.3 | 0.001 | TGGCATGACTCTTGC |
| VC2140-38 | fliD/flaI/fliS | Flagellar rod/hook-associated protein | 2.1/3.4/5.2/5.4 | 0.001 | TGGCACTAAAATTGC |
| VC2188 | flaA | Flagellin core protein | 251.0 | 0.001 | TGGCACACTAATTGA |
| VC2189 | Hypothetical protein | 176.4 | 0.001 | TGGCACGGAAGTTGC | |
| VC2191-90 | flgKL | Flagellar hook-associated protein | 79.0/46.2 | 0.001 | TGGCATACATATTGC |
| VC2193-92 | flgIJ | Flagellar P-ring/flagellar protien | 67.6/47.6 | 0.001 | TGGCACGATTTTTTA |
| VC2196-94 | flgFGH | Flagellar basal rod/L-ring | 183.9/84.0/93.1 | 0.001 | TGGCATGCTGCTTGC |
| VC2200-2197 | flgBCDE | Basal rod protein/hook protein E | 155.2/36.9/51.0/47.3 | 0.001 | TGGTACGCTAATTGC |
| VC2207-06 | flgOP | Outer membrane proteins | 79.5/23.5 | 0.001 | GGGTATAAATTTTGC |
| VC2208 | flgT | Flagellar assembly protein | 2.4 | 0.001 | TGGAACGCTCCTTGC |
| Regulator | |||||
| VC0606-07 | glnB-1 | Nitrogen regulatory protein P-II | 32.7/14.8 | 0.005 | TGGCACGCCCCTTGG |
| VC2748 | ntrB | Nitrogen regulation protein | 2.8 | 0.001 | CGGCAAGATTATTGC |
| Enzyme and biosynthesis | |||||
| VC1516-10 | Formate dehydrogenase | 63.9/44.6/40.7/69.4/48.1/51.6/22.3 | 0.001 | TGGAACGCTATTTGC | |
| VC1519 | fdhD | Formate dehydrogenase accessory | 3.0 | 0.001 | CGGCACCCTTTTTGC |
| VC1523-27 | ABC transporter and molybdopterin synthesis | 5.4/5.7/3.9/2.3/2.1 | 0.001 | TGGCATCCCATTTGC | |
| VC2746 | glnA | Glutamate–ammonia ligase | 3.4 | 0.004 | TGGCACGCTTTTCGC |
| Unknown function | |||||
| VC1154 | Hypothetical protein | 3.8 | 0.001 | TGGCACTCTAATTGC | |
| VC1518-17 | Hypothetical protein | 7.5/10.9 | 0.001 | TGGCGCAATTATTGC | |
| VC1678-76 | pspABC | Phage shock proteins | 5.9/2.5/3.2 | 0.001 | TGGATTTATCTTTGC |
| VC1699 | Hypothetical protein | 3.8 | 0.012 | TGGCATCGGTTTTGC | |
| VC2005 | Hypothetical protein | 28.5 | 0.002 | AGGCACAGCATTTGC | |
| VCA0051-48 | Hypothetical/GGDEF family protein | 2.0/2.0/2.6/2.3 | 0.001 | TGGCACAATTTATGC | |
| VCA0105-06 | Hypothetical protein | 17.5/14.4 | 0.001 | TGGAACATTAATTGC | |
| VCA0144 | immunogenic protein | 4.9 | 0.012 | TGGCATCTTCTTTGC | |
| VCA0195 | Hypothetical protein | 4.6 | 0.002 | CGGCGCATTTATTGC | |
| VCA0284-86 | Hypothetical protein | 47.3/34.9/19.4 | 0.001 | CGGCACCGATCATGA | |
| VCA0734 | Hypothetical protein | 3.1 | 0.003 | TGGCCTGTAATTTGC | |
| VCA1016 | Putative lipoprotein | 4.3 | 0.010 | TGGCACGCACTATGC |
New RpoN-dependent promoters in flagellar gene clusters
Consistent with ChIP-Seq results, all genes for motility and chemotaxis, except for flrA, flgA, cheV-3 and cheR-2, showed significant difference in expression when RpoN was expressed (P < 0.05) (Supplementary Table S3). This is as expected because RpoN is required for motility in V. cholerae (18). However, the RpoN-dependence of these flagellar genes varies greatly. Flagellar genes are located in three major clusters and are regulated by RpoN, FlrA, FlrC and FliA, which form a four-tiered hierarchical regulation (20). Expression of flrA was independent of RpoN, whereas flrC and fliA were positively controlled by RpoN. We expect genes that require RpoN for expression to have basal expression levels in the rpoN mutant and high RpoN-dependence. These include most genes in cluster 3 (flgBCDEFGHIJKL, flaA and flgOP), fliK and motY (Figure 2). In contrast, most genes in the other clusters exhibited little RpoN-dependence (<4 fold) and were expressed at relatively high levels in the rpoN mutant, suggesting the existence of other promoters. For example, the fliA gene was expressed at considerable levels in the rpoN mutant, and the fliA gene in E. coli is known to possess both RpoD- and FliA- recognized promoters (40). In addition, cheR-2, a previously identified RpoN-dependent gene (20), was expressed independent of RpoN in V52.
Using ChIP-Seq and RNA-Seq, we identified new operon structures for flagellar genes (Figure 2). In addition to the previously characterized flagellar gene regulation (41), we found seven new RpoN-binding promoters for flgF, flgI, flhF, fliD, fliK, motY and flgT (Table 2). Two RpoN-binding peaks were identified upstream of the major flagellin gene VC2188 flaA: one previously known as FlrC-bound RpoN-controlled promoter upstream of VC2189 (42) and a new RpoN promoter located 144 bp upstream of VC2188 (Table 2) (Figure 2). Notably, previously identified RpoN-controlled genes fliLMNOPQR, flhB, cheABWYZ and motX were lack of RpoN-recognized promoters and transcribed from other promoters (Supplementary Table S3).
Identification of a small RNA (sRNA), flaX, important for motility
RNA-Seq data revealed a highly expressed transcript that is downstream of flaA (Figure 3). We performed sequence analysis to scan the transcript region and did not identify any potential ORF with >20 codons, suggesting it is likely a small RNA. To test whether it is a true transcript, we performed northern blot analysis using sequence-specific oligo probes. Results confirmed the existence of this sRNA (named flaX), with an approximate size of 150 nt. We also used reverse-transcription PCR with primers specifically targeting flaX and its adjacent 3’end of flaA and found 164.2- ± 13.6-fold higher expression for flaX, consistent with the RNA-Seq results. We constructed a knock-out mutant of flaX that lacks 43 nt in the middle and found that its motility was impaired in comparison with wild type, indicating an important role of flaX in motility. In addition, the impaired motility of the flaX mutant was complemented by expressing flaX in trans on a sRNA-expression vector pNM12 (30), which has no ribosomal-binding site upstream of the cloning site. This sRNA is likely conserved since it was also expressed in the V. cholerae El Tor biotype strain C6706 (data not shown). It is also important to note that transcription of flaX was also found in the rpoN mutant, suggesting its expression is independent of RpoN.
Figure 3.

The new sRNA flaX downstream of VC2188. (A) RNA-Seq data show a substantial increase of coverage in the downstream region of flaA. (B) Northern blot analysis confirmed the existence of flaX. (C) Mutation in flaX attenuated motility. (D) Motility of the flaX mutant was complemented by expressing flaX in trans on a sRNA expression vector pNM12. Arabinose (0.02%) was used for inducing the expression of flaX.
RpoN controls the expression of hcp but not genes on the T6SS cluster
Data from RNA-Seq and ChIP-Seq show that none of the T6SS genes on the main cluster was differentially expressed on RpoN induction and there was no RpoN-binding site. This is surprising, as previous data examining Vibrio T6SS promoter activity in E. coli suggest that T6SS genes on the main cluster and hcp were co-regulated by RpoN (21). To confirm our ChIP- and RNA-Seq data, we first compared the effect of RpoN on the expression of hcp and the first two genes of T6SS cluster, vipA (VCA0107) and vipB (VCA0108) (Figure 4). As expected, induction of RpoN increased Hcp levels substantially (Figure 4). Surprisingly, both VipA and VipB levels were independent of RpoN expression. In addition, the transcription levels of hcp and vgrG2 were greatly induced on RpoN induction, whereas vipA and vipB levels were unaffected (Figure 4). We then tested whether RpoN binds to the promoter regions of hcp and vipAB by ChIP. There was a 90-fold enrichment for the hcp promoter region, whereas no difference was observed for the vipAB promoters and for the control gene vgrG2 that is downstream of hcp, indicating that RpoN binds to the promoter of hcp but not vipAB in vivo. These results validate the RNA-Seq and ChIP-Seq data and confirm that the hcp operons and the major T6SS cluster are regulated differently by RpoN.
Figure 4.

RpoN is required for the expression of Hcp but not VipA or VipB. (A) Protein levels of Hcp, VipA (VCA0107) and VipB (VCA0108) by western blot analysis. The rpoN mutant was transformed with the plasmid pRpoN or the empty vector pBAD18. Cultures were grown aerobically in LB medium at 37°C to exponential phase (OD600 = 0.5), and RpoN was induced by the addition of arabinose (0.1%) for 30 minutes. Samples were taken at different time points and protein expression was detected by western blot analysis. (B) Transcriptional levels of hcp and and vipAB and ChIP-binding of their promoter regions. The rrsA gene encoding 16 S RNA was used as an internal control sample to normalize difference in total RNA quantity among samples for qPCR. VgrG2 is located downstream of hcp2 and used as a control sample for ChIP assay. Monoclonal antibody to RpoN-3V5 was used for ChIP assay.
RpoN enhancer-binding protein VasH is required for the expression of hcp and not vipA or vipB
Because VasH is an enhancer-binding protein that activates RpoN-controlled transcription, we expect that VasH and RpoN have similar effect on the expression of T6SS genes. To test this, we compared the transcriptional levels of hcp, vipA and vipB, in the wild type V52 and a vasH deletion mutant. The expression of hcp was 1000-fold higher in the wild type than in the vasH mutant, whereas no significant change in expression was observed for vipA/B in the vasH mutant (Figure 5). This result confirms that RpoN-VasH regulates only hcp operons but not the major cluster.
Figure 5.

VasH, the enhancer-binding protein for RpoN, controls the expression of hcp but not the major cluster genes vipA and vipB. Relative gene expression was compared in wild type V52 and the vasH mutant by qPCR. The 16 S ribosomal RNA gene rrsA was used as a control sample. RNA was extracted in exponential phase cultures OD600 = 0.5.
DISCUSSION
Development of next-generation sequencing technologies provides a revolutionary tool for studying the interaction between transcriptional factors and target DNA. In comparison with ChIP-chip and microarray, ChIP-Seq and RNA-Seq analyses provide much increased sensitivity to identify binding sites and reveal transcriptional units. However, in comparison with the large number of ChIP-Seq studies done in eukaryotes, much fewer have been performed in bacteria. Here, we present the first example of combining ChIP and RNA-Seq data to study the transcriptome and regulon structure controlled by an important transcription factor RpoN. We identified 68 RpoN-binding sites and 82 RpoN-positively regulated operons containing 144 genes. This result is comparable with previous microarray studies in which RpoN was found to control the expression of a large number of genes in the classical V. cholerae strain O395 (20) and El Tor O1 strain (19). Although the effect of RpoN on expression of motility genes seems to be conserved, its control on T6SS varies substantially among strains. In El Tor O1 strain, the rpoN mutation resulted in significant up-regulation of the T6SS locus, whereas it had little effect on hcp expression (19). In the classical O1 strain, the rpoN mutation also led to increased expression of genes on the main T6SS cluster (20). Here, we report that RpoN controls only the two hcp operons but not the main T6SS cluster. This adds to the notion that there is considerable genomic and genetic variation in V. cholerae isolates, especially in the context of virulence genes. For example, the seventh pandemic El Tor O1 strain has two unique islands, ‘Vibrio seventh pandemic island’ (VSP-I and VSP-II), and the RTX toxin that are not present in classical strains (43). In the case of T6SS, it is constitutively active in some Vibrio isolates (e.g. the O37-serotype V52) but is repressed in the seventh pandemic strain under laboratory conditions.
Of the RpoN-regulated operons, 37 operons have upstream RpoN-binding motifs indicating direct control by RpoN. The other operons lacking an RpoN-recognized promoter are likely controlled through intermediate regulators. For example, RpoN directly regulates the flagellar sigma factor FliA, which in turn regulates Class IV flagellar genes including flaB and flaC (41). In addition, many genes downstream of RpoN-binding sites were not affected by rpoN induction. This might be because of the presence of other promoters or the inactivity of RpoN-binding sites. The transcriptional activity of RpoN-directed promoter depends completely on cognate enhancer proteins, including their expression levels and the presence of enhancer-binding sites close to the promoter. Notably, binding of RpoN to promoter does not require enhancer binding proteins (44). Therefore, ChIP-Seq is able to detect RpoN-DNA interaction sites independent of active transcription. Indeed, we found seven binding sites that are not in close vicinity to a downstream gene. The physiological relevance of these binding sites is not clear, but it is consistent with previous results that sigma factors and RNA polymerase bind to chromosomal locations not involved in transcription. For example, RNA polymerase binds to 300 inactive transcription units (45) and a quarter of sigma 70 bound-promoters are inactive (46). Interestingly, when using the identified RpoN-binding motif to scan the genome, we found >300 potential RpoN-binding sites (data not shown), a number much larger than the reported RpoN-controlled promoter here. One predicted RpoN-binding motif is located upstream of the major T6SS cluster VCA0107, which was previously shown in E. coli that E. coli RpoN controls the activity of a plasmid-borne VCA0107 promoter and binds to the promoter region in vitro (21). However, this RpoN-binding motif is unlikely to be active and transcribe VCA0107 in V. cholerae, as the predicted transcription start site maps to the third nucleotide of the start codon of VCA0107, which would result in an out of frame translation. Indeed, our results of ChIP and expression analysis of RNA and protein levels show that RpoN does not control VCA0107 expression. How RpoN selectively binds to some but not other similar sites is still not clear. This selectivity may result from the effect of DNA topology and/or exclusive binding of other global regulators, such as IHF, Fis and H-NS. Nonetheless, this difference underscores the importance of experimental verification for in silico prediction of transcriptional factor binding sites.
The T6SS-encoded RpoN enhancer-binding protein, VasH, binds to the promoter of the major cluster in vitro (21) and is required for T6SS functions (17). However, we show that VasH, like RpoN, controls only the expression of hcp but not vipAB. This uncoupled regulation of T6SS major cluster and hcp operons by RpoN provides a plausible explanation for the interesting results that overexpression of vasH increases Hcp cellular levels but not secretion in the T6SS-inactive pandemic El Tor strain N16961 (17), as the major T6SS cluster is not subject to RpoN regulation and thus, remains non-induced. We postulate that regulation of the major T6SS cluster is the key determinant for T6SS activity. Expression of the major T6SS cluster results in not only the expression of T6SS structural genes for Hcp secretion but also the cognate RpoN enhancer protein VasH for hcp expression.
The flagellar gene regulatory network has been well-characterized in previous studies (20,41). Our data modify the previous model and identify seven new RpoN-controlled promoters within the large clusters of flagellar genes. Some previously characterized class II flagellar genes controlled by RpoN likely have separate promoters, as their expression does not require RpoN. We identified an sRNA flaX downstream of flaA that is required for full motility. The in silico analysis failed to predict a clear target for flaX. We are further investigating the target using an exponential enrichment pull down approach (47).
T6SS is under tight and complex control by regulators LuxO, TsrA and HapR (12), and it is recently found to be activated under high-osmolarity and low-temperature conditions (48). Although RpoN has been known as the critical regulator for T6SS function since T6SS was originally discovered, our report differentiates clearly its effect on the secreted hcp promoter and the main T6SS promoter. Our recent model shows that the T6SS sheath proteins VipA and VipB are in a dynamic stepwise process of forming tubular structures, contraction, disassembly and reassembly, whereas Hcp and other T6SS-dependent molecules are secreted (49). Our findings are consistent with this model that the regulation of secretion molecules differs from the components that are recycled.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4 and Supplementary Figures 1 and 2.
FUNDING
National Institute of Allergy and Infectious Diseases (NIAID) [AI-026289 to J.J.M]; T.G.D. is a recipient of the Banting Postdoctoral Fellowship. Funding for open access charge: NIAID [AI-026289].
Conflict of interest statement. None declared.
Supplementary Material
Supplementary Data
ACKNOWLEDGEMENTS
The authors thank B.D. Davies for critical reading of this manuscript. They also thank R. Bogard, W.P. Robins and M. Basler for technical assistance and the Mekalanos group for helpful discussion.
REFERENCES
- 1.Weil AA, Ivers LC, Harris JB. Cholera: lessons from haiti and beyond. Curr. Infect. Dis. Rep. 2012;14:1–8. doi: 10.1007/s11908-011-0221-9. [DOI] [PubMed] [Google Scholar]
- 2.Ritchie JM, Waldor MK. Vibrio cholerae interactions with the gastrointestinal tract: lessons from animal studies. Curr. Top. Microbiol. Immunol. 2009;337:37–59. doi: 10.1007/978-3-642-01846-6_2. [DOI] [PubMed] [Google Scholar]
- 3.Ma AT, Mekalanos JJ. In vivo actin cross-linking induced by Vibrio cholerae type VI secretion system is associated with intestinal inflammation. Proc. Natl. Acad. Sci. U.S.A. 2010;107:4365–4370. doi: 10.1073/pnas.0915156107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl. Acad. Sci. U.S.A. 2007;104:15508–15513. doi: 10.1073/pnas.0706532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl. Acad. Sci. USA. 2006;103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordonez CL, Lory S, et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312:1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwarz S, West TE, Boyer F, Chiang WC, Carl MA, Hood RD, Rohmer L, Tolker-Nielsen T, Skerrett SJ, Mougous JD. Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 2010;6:e1001068. doi: 10.1371/journal.ppat.1001068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murdoch SL, Trunk K, English G, Fritsch MJ, Pourkarimi E, Coulthurst SJ. The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J. Bacteriol. 2011;193:6057–6069. doi: 10.1128/JB.05671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Pace F, Nakazato G, Pacheco A, de Paiva JB, Sperandio V, da Silveira WD. The type VI secretion system plays a role in type 1 fimbria expression and pathogenesis of an avian pathogenic Escherichia coli strain. Infect. Immun. 2010;78:4990–4998. doi: 10.1128/IAI.00531-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pell LG, Kanelis V, Donaldson LW, Howell PL, Davidson AR. The phage lambda major tail protein structure reveals a common evolution for long-tailed phages and the type VI bacterial secretion system. Proc. Natl. Acad. Sci. USA. 2009;106:4160–4165. doi: 10.1073/pnas.0900044106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leiman PG, Basler M, Ramagopal UA, Bonanno JB, Sauder JM, Pukatzki S, Burley SK, Almo SC, Mekalanos JJ. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl. Acad. Sci. USA. 2009;106:4154–4159. doi: 10.1073/pnas.0813360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng J, Shin OS, Cameron DE, Mekalanos JJ. Quorum sensing and a global regulator TsrA control expression of type VI secretion and virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. USA. 2010;107:21128–21133. doi: 10.1073/pnas.1014998107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa T, Rompikuntal PK, Lindmark B, Milton DL, Wai SN. Quorum sensing regulation of the two hcp alleles in Vibrio cholerae O1 strains. PLoS One. 2009;4:e6734. doi: 10.1371/journal.pone.0006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunt TP, Magasanik B. Transcription of glnA by purified Escherichia coli components: core RNA polymerase and the products of glnF, glnG, and glnL. Proc. Natl. Acad. Sci. USA. 1985;82:8453–8457. doi: 10.1073/pnas.82.24.8453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao K, Liu M, Burgess RR. Promoter and regulon analysis of nitrogen assimilation factor, sigma54, reveal alternative strategy for E. coli MG1655 flagellar biosynthesis. Nucleic Acids Res. 2010;38:1273–1283. doi: 10.1093/nar/gkp1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buck M, Gallegos MT, Studholme DJ, Guo Y, Gralla JD. The bacterial enhancer-dependent sigma(54) (sigma(N)) transcription factor. J. Bacteriol. 2000;182:4129–4136. doi: 10.1128/jb.182.15.4129-4136.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitaoka M, Miyata ST, Brooks TM, Unterweger D, Pukatzki S. VasH is a transcriptional regulator of the type VI secretion system functional in endemic and pandemic Vibrio cholerae. J. Bacteriol. 2011;193:6471–6482. doi: 10.1128/JB.05414-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klose KE, Mekalanos JJ. Distinct roles of an alternative sigma factor during both free-swimming and colonizing phases of the Vibrio cholerae pathogenic cycle. Mol. Microbiol. 1998;28:501–520. doi: 10.1046/j.1365-2958.1998.00809.x. [DOI] [PubMed] [Google Scholar]
- 19.Yildiz FH, Liu XS, Heydorn A, Schoolnik GK. Molecular analysis of rugosity in a Vibrio cholerae O1 El Tor phase variant. Mol. Microbiol. 2004;53:497–515. doi: 10.1111/j.1365-2958.2004.04154.x. [DOI] [PubMed] [Google Scholar]
- 20.Syed KA, Beyhan S, Correa N, Queen J, Liu J, Peng F, Satchell KJ, Yildiz F, Klose KE. The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J. Bacteriol. 2009;191:6555–6570. doi: 10.1128/JB.00949-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernard CS, Brunet YR, Gavioli M, Lloubes R, Cascales E. Regulation of type VI secretion gene clusters by sigma54 and cognate enhancer binding proteins. J. Bacteriol. 2011;193:2158–2167. doi: 10.1128/JB.00029-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hood RD, Singh P, Hsu F, Guvener T, Carl MA, Trinidad RR, Silverman JM, Ohlson BB, Hicks KG, Plemel RL, et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host. Microbe. 2010;7:25–37. doi: 10.1016/j.chom.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD. Type VI secretion delivers bacteriolytic effectors to target cells. Nature. 2011;475:343–347. doi: 10.1038/nature10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacIntyre DL, Miyata ST, Kitaoka M, Pukatzki S. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc. Natl. Acad. Sci. USA. 2010;107:19520–19524. doi: 10.1073/pnas.1012931107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma AT, McAuley S, Pukatzki S, Mekalanos JJ. Translocation of a Vibrio cholerae type VI secretion effector requires bacterial endocytosis by host cells. Cell Host. Microbe. 2009;5:234–243. doi: 10.1016/j.chom.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyata ST, Kitaoka M, Brooks TM, McAuley SB, Pukatzki S. Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect. Immun. 2011;79:2941–2949. doi: 10.1128/IAI.01266-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies BW, Bogard RW, Mekalanos JJ. Mapping the regulon of Vibrio cholerae ferric uptake regulator expands its known network of gene regulation. Proc. Natl. Acad. Sci. USA. 2011;108:12467–12472. doi: 10.1073/pnas.1107894108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller VL, Mekalanos JJ. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 1988;170:2575–2583. doi: 10.1128/jb.170.6.2575-2583.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid. 1996;35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 30.Majdalani N, Cunning C, Sledjeski D, Elliott T, Gottesman S. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc. Natl. Acad. Sci. USA. 1998;95:12462–12467. doi: 10.1073/pnas.95.21.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies BW, Bogard RW, Dupes NM, Gerstenfeld TA, Simmons LA, Mekalanos JJ. DNA damage and reactive nitrogen species are barriers to Vibrio cholerae colonization of the infant mouse intestine. PLoS Pathog. 2011;7:e1001295. doi: 10.1371/journal.ppat.1001295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat. Biotechnol. 2008;26:1293–1300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 35.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 36.Grigorova IL, Phleger NJ, Mutalik VK, Gross CA. Insights into transcriptional regulation and sigma competition from an equilibrium model of RNA polymerase binding to DNA. Proc. Natl. Acad. Sci. USA. 2006;103:5332–5337. doi: 10.1073/pnas.0600828103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farewell A, Kvint K, Nystrom T. Negative regulation by RpoS: a case of sigma factor competition. Mol. Microbiol. 1998;29:1039–1051. doi: 10.1046/j.1365-2958.1998.00990.x. [DOI] [PubMed] [Google Scholar]
- 38.Jishage M, Kvint K, Shingler V, Nystrom T. Regulation of sigma factor competition by the alarmone ppGpp. Genes Dev. 2002;16:1260–1270. doi: 10.1101/gad.227902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong T, Yu R, Schellhorn H. Antagonistic regulation of motility and transcriptome expression by RpoN and RpoS in Escherichia coli. Mol. Microbiol. 2011;79:375–386. doi: 10.1111/j.1365-2958.2010.07449.x. [DOI] [PubMed] [Google Scholar]
- 40.Mytelka DS, Chamberlin MJ. Escherichia coli fliAZY operon. J. Bacteriol. 1996;178:24–34. doi: 10.1128/jb.178.1.24-34.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prouty MG, Correa NE, Klose KE. The novel sigma54- and sigma28-dependent flagellar gene transcription hierarchy of Vibrio cholerae. Mol. Microbiol. 2001;39:1595–1609. doi: 10.1046/j.1365-2958.2001.02348.x. [DOI] [PubMed] [Google Scholar]
- 42.Correa NE, Klose KE. Characterization of enhancer binding by the Vibrio cholerae flagellar regulatory protein FlrC. J. Bacteriol. 2005;187:3158–3170. doi: 10.1128/JB.187.9.3158-3170.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dziejman M, Balon E, Boyd D, Fraser CM, Heidelberg JF, Mekalanos JJ. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc. Natl. Acad. Sci. USA. 2002;99:1556–1561. doi: 10.1073/pnas.042667999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buck M, Cannon W. Specific binding of the transcription factor sigma-54 to promoter DNA. Nature. 1992;358:422–424. doi: 10.1038/358422a0. [DOI] [PubMed] [Google Scholar]
- 45.Herring CD, Raffaelle M, Allen TE, Kanin EI, Landick R, Ansari AZ, Palsson BO. Immobilization of Escherichia coli RNA polymerase and location of binding sites by use of chromatin immunoprecipitation and microarrays. J. Bacteriol. 2005;187:6166–6174. doi: 10.1128/JB.187.17.6166-6174.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reppas NB, Wade JT, Church GM, Struhl K. The transition between transcriptional initiation and elongation in E. coli is highly variable and often rate limiting. Mol. Cell. 2006;24:747–757. doi: 10.1016/j.molcel.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 47.Lorenz C, von Pelchrzim F, Schroeder R. Genomic systematic evolution of ligands by exponential enrichment (Genomic SELEX) for the identification of protein-binding RNAs independent of their expression levels. Nat. Protoc. 2006;1:2204–2212. doi: 10.1038/nprot.2006.372. [DOI] [PubMed] [Google Scholar]
- 48.Ishikawa T, Sabharwal D, Broms J, Milton DL, Sjostedt A, Uhlin BE, Wai SN. Pathoadaptive conditional regulation of the type VI secretion system in Vibrio cholerae O1 strains. Infect. Immun. 2012;80:575–584. doi: 10.1128/IAI.05510-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature. 2012;483:182–186. doi: 10.1038/nature10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data