Integration of β-Catenin, Sirtuin, and FOXO Signaling Protects from Mutant Huntingtin Toxicity (original) (raw)
Abstract
One of the current challenges of neurodegenerative disease research is to determine whether signaling pathways that are essential to cellular homeostasis might contribute to neuronal survival and modulate the pathogenic process in human disease. In Caenorhabditis elegans, sir-2.1/SIRT1 overexpression protects neurons from the early phases of expanded polyglutamine (polyQ) toxicity, and this protection requires the longevity-promoting factor _daf-16/_FOXO. Here, we show that this neuroprotective effect also requires the DAF-16/FOXO partner bar-1/β-catenin and putative DAF-16-regulated gene ucp-4, the sole mitochondrial uncoupling protein (UCP) in nematodes. These results fit with a previously proposed mechanism in which the β-catenin FOXO and SIRT1 proteins may together regulate gene expression and cell survival. Knockdown of β-catenin enhanced the vulnerability to cell death of mutant-huntingtin striatal cells derived from the HdhQ111 knock-in mice. In addition, this effect was compensated by SIRT1 overexpression and accompanied by the modulation of neuronal UCP expression levels, further highlighting a cross-talk between β-catenin and SIRT1 in the modulation of mutant polyQ cytoxicity. Taken together, these results suggest that integration of β-catenin, sirtuin and FOXO signaling protects from the early phases of mutant huntingtin toxicity.
Introduction
Huntington's disease (HD) is a dominantly inherited CAG repeat disorder with expanded polyglutamine (polyQ) tracts in huntingtin (htt), causing striatal and cortical degeneration (Walker, 2007). While a person carrying more than 39 CAG repeats will invariably develop HD, the age at onset of HD varies considerably. The CAG expansions account for about 60% of the variability of the age at onset of HD, and the residual variability may be heritable, suggesting that other genes influence age at onset (Li et al., 2003; Gayán et al., 2008; Metzger et al., 2008; Taherzadeh-Fard et al., 2009; Xu et al., 2009). Given that HD is an age-related disorder, candidate gene approaches based on longevity modulators have been examined to identify disease modifiers. Studies of TCERG1, an evolutionary conserved transcriptional regulator that modulates longevity in Caenorhabditis elegans (Ghazi et al., 2009), showed that this protein is neuroprotective in models of HD (Arango et al., 2006) and that its polymorphic Gln-Ala repeat modifies HD in American patients (Holbert et al., 2001). Another longevity modulator with neuroprotective activities is the transcription factor daf-16/FOXO. FOXO proteins are essential for stress resistance (Brunet et al., 2004), and daf-16 is required for neuroprotection by increased dosage of sirtuin _sir-2.1/_SIRT1 in expanded-polyQ nematodes (Parker et al., 2005). Several cofactors regulate FOXO activity, and FOXOs have many targets (Greer and Brunet, 2008; Landis and Murphy, 2010; Yen et al., 2011), suggesting that a network of genes centered onto FOXO might regulate mutant polyglutamine neuron survival and have a role in HD variability. Interestingly, in this regard FOXO activity may be conserved from C. elegans to humans, as human FOXO3A was associated with the ability to be long lived in several populations of centenarians (Willcox et al., 2008; Anselmi et al., 2009; Li et al., 2009; Soerensen et al., 2010). Here, we investigated the neuroprotective role of the Wnt effector BAR-1/β-catenin, a protein that also binds to DAF-16/FOXO3a to regulate its transcriptional activity in response to oxidative stress in both C. elegans and mammalian cells (Essers et al., 2005), and that of ucp-4, the sole mitochondrial uncoupling protein (UCP) in C. elegans and putative DAF-16 transcriptional target. To this end, we used C. elegans transgenics expressing exon-1 like htt in touch receptor neurons (Parker et al., 2005). Animals expressing expanded polyQs show a strong neuronal dysfunction phenotype (defective response to touch) in the absence of cell death (Parker et al., 2005), recapitulating an early phase of mutant htt neurotoxicity. Additionally, we tested for the effects of SIRT1, β-catenin, and neuronal UCPs (UCP2, UCP4) on the survival of striatal cells derived from htt knock-in mice (Trettel et al., 2000). As detailed below, our results suggest that integration of β-catenin, sirtuin, and FOXO signaling protects against the early phases of mutant htt toxicity.
Materials and Methods
Nematode experiments.
The wild-type strain of C. elegans used was Bristol N2. Standard methods of culturing and handling worms, either hermaphrodites or males, were used. All strains were scored at 20°C. Touch tests, scoring of PLM cell processes, drug response assays, and quantitative real-time PCR were performed as described previously (Parker et al., 2005). Western analysis was performed using standard protocols and htt fusion proteins detected with the GFP antibody ab6556 (Abcam). Mutations and transgenes used in this study are listed in Table 1. All strains were obtained from the C. elegans Genetics Center (University of Minnesota, Minneapolis, MN), except for those generated in the laboratory. For strain construction with polyQ transgenes, mutants were verified by visible phenotypes, deletion mutants by PCR analysis, point mutations by sequencing, or a combination thereof. Deletion mutants were outcrossed a minimum of three times to wild type, and the _geIs3_[sir-2.1(+)] strain was outcrossed 10 times to wild type before use. The _geIs3_[sir-2.1(+)] animals were tested for dye-filling defect as described previously (Burnett et al., 2011) and showed no defect in this regard.
Table 1.
Names and genotypes of the C. elegans strains used in this study
| Name | Genotypea | Origin |
|---|---|---|
| Bristol N2 | Standard wild type | CGCb |
| TJ356 | _zIs356_[pdaf-16::daf-16-gfp; _rol-6_]IV | CGC |
| CB1370 | daf-2(e1370)III | CGC |
| GR1307 | daf-16(mgDf50)I | CGC |
| HT1890 | daf-16(mgDf50)I;daf-2(e1370)III | CGC |
| CF1038 | daf-16(mu86)I | CGC |
| EW15 | bar-1(ga80)X | CGC |
| CY121 | ucp-4(ok195)V | CGC |
| LG100 | geIn3_[sir-2.1; rol-6_]; dyf-(?) | L. Guarente (MIT, Boston, MA) |
| ID1280c | _geIn3_[sir-2.1; _rol-6_] | This study |
| ID245d | _igIs245_[mec-3p::htt57–19Q::CFP; lin-15(+); mec-7p::_YFP_] | See Parker et al., 2005 |
| ID1d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_] | Parker et al., 2005 |
| ID191d | _igIs245_[mec-3p::htt57–19Q::CFP; lin-15(+); mec-7p::_YFP_]; bar-1(ga80)X | This study |
| ID1281d | _igIs1_[mec-3p::htt57–128Q:CFP; lin-15(+); mec-7p::_YFP_]; bar-1(ga80)X | This study |
| ID192d | _igIs245_[mec-3p::htt57–19Q::CFP; lin-15(+); mec-7p::_YFP_]; ucp-4(ok195)V | This study |
| ID1282d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; ucp-4(ok195)V | This study |
| ID1283d | _igIs245_[mec-3p::htt57–19Q::CFP; lin-15(+); mec-7p::_YFP_]; sir-2.1(ok434)X | This study |
| ID1285d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; sir-2.1(ok434)X | This study |
| ID1286d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; daf-16(mgDf50)I | This study |
| ID1287d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; _geIn3_[sir-2.1; _rol-6_] | This study |
| ID1288d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; _geIn3_[sir-2.1; _rol-6_]; bar-1(ga80)X | This study |
| ID1289d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; _geIn3_[sir-2.1; _rol-6_]; ucp-4(ok195)V | This study |
| ID1290d | _igIs245_[mec-3p::htt57–19Q::CFP; lin-15(+); mec-7p::_YFP_]; sir-2.1(ok434)X; _igEx500_[mec-3p::_sir-2.1-biGFP_] | This study |
| ID1291d | _igIs1_[mec-3p::htt57–19Q::CFP; lin-15(+); mec-7p::_YFP_]; sir-2.1(ok434)X; _igEx501_[mec-3p::_sir-2.1-biGFP_] | This study |
| ID1292d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; sir-2.1(ok434)X; _igEx502_[mec-3p::_sir-2.1-biGFP_] | This study |
| ID1293d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; sir-2.1(ok434)X; _igEx503_[mec-3p::_sir-2.1-biGFP_] | This study |
| ID1296d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; sir-2.1(ok434)X; _igEx506_[mec-3p::_GFP_] | This study |
| ID1297d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; sir-2.1(ok434)X; _igEx507_[mec-3p::_GFP_] | This study |
| ID1298 | _igEx508_[ucp-4p::ucp-4::mCherry; myo-2p::_GFP_] | This study |
| ID1299 | _igEx509_[ucp-4p::ucp-4::mCherry; myo-2p::_GFP_] | This study |
| ID1300 | _igEx510_[ucp-4p::ucp-4::mCherry; myo-2p::_GFP_]; zIs356 | This study |
| ID1301 | _igEx511_[ucp-4p::ucp-4::mCherry; myo-2p::_GFP_]; zIs356 | This study |
| ID1302e | _igEx512_[ucp-4pWT::mCherry; myo-2p::_GFP_] | This study |
| ID1303e | _igEx513_[ucp-4pWT::mCherry; myo-2p::_GFP_] | This study |
| ID1304e | _igEx514_[ucp-4pWT::mCherry; myo-2p::_GFP_]; daf-16(mu86)I | This study |
| ID1305e | _igEx515_[ucp-4pWT::mCherry; myo-2p::_GFP_]; daf-16(mu86)I | This study |
| ID1306e | _igEx516_[ucp-4pWT::mCherry; myo-2p::_GFP_]; zIs356 | This study |
| ID1307e | _igEx517_[ucp-4pWT::mCherry; myo-2p::_GFP_]; zIs356 | This study |
| ID1308e | _igEx518_[ucp-4pSCR::mCherry; myo-2p::_GFP_] | This study |
| ID1309e | _igEx519_[ucp-4pSCR::mCherry; myo-2p::_GFP_] | This study |
| ID1310e | _igEx520_[ucp-4pSCR::mCherry; myo-2p::_GFP_]; daf-16(mu86)I | This study |
| ID1311e | _igEx521_[ucp-4pSCR::mCherry; myo-2p::_GFP_]; daf-16(mu86)I | This study |
| ID1312e | _igEx522_[ucp-4pSCR::mCherry; myo-2p::_GFP_]; zIs356 | This study |
| ID1313e | _igEx523_[ucp-4pSCR::mCherry; myo-2p::_GFP_]; zIs356 | This study |
| ID1314d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; bar-1(ga80)X; _igEx524_[mec-3p::bar-1-bi-mCherry; myo-2p::_GFP_] | This study |
| ID1315d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; bar-1(ga80)X; _igEx525_[mec-3p::bar-1-bi-mCherry; myo-2p::_GFP_] | This study |
| ID1316d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; bar-1(ga80)X; _igEx526_[mec-3p::mCherry; myo-2p::_GFP_] | This study |
| ID1317d | _igIs1_[mec-3p::htt57–128Q::CFP; lin-15(+); mec-7p::_YFP_]; bar-1(ga80)X; _igEx527_[mec-3p::mCherry; myo-2p::_GFP_] | This study |
To test for rescue of the effects of sir-2.1 loss of function (LOF) in 128Q animals, constructs encoding SIR-2.1 were generated as follows. We assembled the sir-2.1 cDNA with a bicistronic GFP (biGFP) by PCR fusion. We obtained the sir-2.1 cDNA from wild-type animals by RT-PCR, using RV197 (5′-GGGGACAACTTTGTATACAAAAGTTGATGTCACGTGATAGTGGCAAC-3′) and RV198 (5′-GTGAAAGTAGGATGAGACAGCTCAGATACGCATTTCTTCAC-3′) primers. RV198 contains a sequence complementary to the 5′ region of biGFP. We amplified biGFP from pAN51 using RV192 (5′-GCTGTCTCATCCTACTTTCAC) and RV178 (5′-GGGGACCACTTTGTA CAAGAAAGCTGGGTATTATAGTTCATCCATGCCATGTGTA-3′). Then, we fused both PCR products by nested PCR using primers RV197 and RV178. These primers contain the sequences of attB5 and attB2, respectively, for recombination in the pDONR221-P5-P2 vector using the Gateway system (Invitrogen). In parallel, we produced a clone in pDONR221-P1-P5r containing the promoter of mec-3, mec-3p using primers RV3 (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCCTGCAGGTACCCGGAGTAGTTG-3′) and RV4 (5′-GGGGACAACTTTTGTATA CAAAGTTGTGGCGCGCCAATGCGCGAAATTGTGGCTACTC-3′). Both clones were used to assemble mec-3p and sir-2.1-biGFP using Gateway technology in the destination vector pDEST-AN, which is suitable for C. elegans transgenesis.
To test for rescue of the effects of bar-1 LOF in 128Q animals, constructs encoding BAR-1 were generated as follows. We assembled the bar-1 cDNA with a bicistronic mCherry (bi-mCherry) by PCR fusion. We obtained the bar-1 cDNA from wild-type animals by RT-PCR using RV229 (5′-GGGGACAACTTTGTATACAAAAGTTGATGGAC CTGGATCCGAACCTAG-3′) and RV230 (5′-GTGAAAGTAGGATGAGACAGCTTAAAATCGACTATTCCTAGAAG-3′) primers. RV230 contains a sequence complementary to the 5′ region of bi-mCherry. We amplified bi-mCherry from the pGEM-T-bicistronic mCherry construct using RV192 (5′-GCTGTCTCATCCTACTTTCAC-3′) and the universal primer SP6. Then, we fused both PCR products by nested PCR using primers RV229 and RV8 (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTATTATTTGTATAGTTCATCCATGCCACC-3′). These primers contain respectively attB5 and attB2 sequences for recombination in the pDONR221-P5-P2 vector using the Gateway system (Invitrogen). We combined the resulting construct with the plasmid pDONR221-P1-P5r (which contains mec-3p) to generate the bar-1 overexpression construct by means of the Gateway technology in the destination vector pDEST-AN, which is suitable for C. elegans transgenesis.
Constructs for mCherry expression under the control of the UCP-4 promoter (1768 bp) were generated as follows. We fused the promoter of UCP-4 (from position 1768 bp upstream of the ATG of ucp-4) and the ucp-4 gene to mCherry::unc-54Terminator. Primers used to amplify ucp-4 were Forward 5′-TTTTGCGTTTGCTCGTCGCAC-3′ and Reverse 5′-AGTCGACCTGCAGGCATGCAAGCT-3′. We amplified mCherry from a plasmid generated by replacing GFP from pPD95.75 by mCherry and by using the primers Forward 5′-AGCTTGCATGCCTGCAGGTCGACT-e′ and Reverse 5′-GGAAACAGTTATGTTTGGTATATTGGG-3′. Constructs encoding mCherry under the control of the ucp-4 promoter (1768 bp), either wild-type or scrambled at binding site 2, were generated as follows. We amplified the promoter of ucp-4 (from position 1768 bp upstream of the ATG of ucp-4) and cloned PCR fragments into pRV10 (a derivative of pPD95.75, in which GFP has been substituted by mCherry). Primers used to amplify the ucp-4 promoter sequence were Forward 5′-GGGGAAGCTTTTTTGCGTTTGCTCGTCGCAC-3′ and Reverse 5′-GGGG CCCGGGCATCTGAATAAAAGTTATAATTCC-3′. Primers used to mutagenize DAF-16 binding site 2 were Forward 5′-AAATACATGTTTGATTCTTTTTGAATACAGCTGTGACGTAA-3′ and Reverse 5′-GCTGTATTCAAAAAGAATCAAACATGTATTTAAATACATTTG-3′ (the scrambled sequence is underlined). All of these constructs were verified for sequence integrity.
For transgenesis, 5–20 ng/μl DNA of the constructs was injected into 19Q and 128Q nematodes, together with pPD118.33 (a plasmid containing myo-2p::GFP), at a final total DNA concentration of 100–150 ng/μl using standard methods. We isolated at least two independent strains from each construct to perform the touch tests. Strains stably expressing the UCP-4-mCherry reporter were crossed to the wild-type N2 strain or to a strain overexpressing a translational construct encoding DAF-16::GFP (TJ356, insertion zIs356IV) (Henderson and Johnson, 2001), and animals homozygous for each allele (w/w, daf-16::GFP/daf-16::GFP) were isolated. Similar crosses were performed using strains stably expressing mCherry under the control of the ucp-4 promoter, which carried a wild-type or scrambled DAF-16 binding site 2. The mCherry signals were scored blindly using a Zeiss fluorescence microscope (10×), and quantification was performed using NIH ImageJ.
Touch tests involved scoring for the response to a light touch at the tail by using a fine hair. Touch tests were performed blindly by scoring 10 touches at the tail of the young adult animal for a minimum of 200 animals per genotype (50–60 animals per independent experiment for a total of at least 4 independent experiments). Touch tests in strains expressing extrachromosomal arrays involved a minimum of 60 animals per array (20–30 animals per independent experiment for a total of at least 3 independent experiments). Ordinarily, wild-type animals will respond by backing away from the touch. The responses were recorded for every animal such that, for example, 3 responses out of 10 at the tail is given as 30% responsiveness, and the mean values for responsiveness were retained for comparison of nematode groups. Although nematodes become habituated after ∼30 touch stimuli, restricting analysis to 10 touch challenges ensures that no significant habituation occurs within this range (Giles and Rankin, 2009). Axonal aggregation and axonal swelling were scored as described previously (Parker et al., 2005) by testing a minimum of 100 animals per treatment. The primers used for quantitative (q)RT-PCR analysis of htt transgene expression were Forward 5′-CACTTGTCACTACTTTCTCAT-3′ and Reverse 5′-GTAGTTCCCGTCATCTTTG-3′, with the primers Forward 5′-CTTCTGCAAGGGATCTGCTAAG-3′ and Reverse 5′-GATTTGGAGCTGGGTTGACTTC-3′ used for amplification of the housekeeping gene col-1 as a control. Proteins were extracted from whole animals as described previously (Parker et al., 2005), separated by SDS-PAGE, and analyzed by Western blotting using mouse anti-htt (GenScript A00089, 1:500) and mouse anti-actin (MP Biomedicals, 1:10,000). Secondary antibodies were goat anti-mouse IgG HRP conjugated (Bio-Rad). Proteins were detected using ECL+ and evaluated by densitometry using a GS-800 densitometer (Bio-Rad), and quantification was performed using ImageJ.
Chromatin immunoprecipitation (ChIP) was performed as described previously (Oh et al., 2006; Mukhopadhyay et al., 2008) using a DAF-16 antibody generated against the C-terminal portion of DAF-16a2 (Oh et al., 2006) and wild-type (N2), daf-2(e1370), daf-16(mgDf50), and daf-16(mgDf50);daf-2(e1370) worms. Binding on ChIP samples was determined by real-time PCR using the following primers: binding site 1, tctgtgtcagcagcttttcg and acggccgtccgtaatagata; binding site 2, tgagcaagttgtaatggggtta and catctggcacttatggggtta; and 3′ region, aaccaggagcaccattcaac and gcaacttcttcctgctgacc.
Mammalian experiments.
Cell death assays for HdhQ111 mouse striatal cells expressing full-length htt with either wild-type (7Q/7Q) or mutant (109Q/109Q) polyglutamines were performed as described previously, with cell death induced by one-day serum deprivation (Arango et al., 2006). We used jetPEI for transfection with cDNA and jetSI-ENDO for transfection with RNA molecules as indicated by the manufacturer (PolyPlus Transfection). The siRNAs (si-htt, si-β-catenin, si-SIRT1, si-SIRT2, si-SIRT3, si-UCP2, si-UCP4) and scramble RNAs were obtained from QIAGEN. Except for htt, for which siRNAs have been described (DiFiglia et al., 2007), mixes of 3–4 different siRNA sequences per gene (25 or 33 nm) were systematically tested for modulation of cell survival and siRNA target gene expression, followed by the evaluation of individual siRNA sequences at optimal concentration (25–100 nm). Effects on cell survival were considered to be reliable if two different siRNAs showed similar effects on target mRNA/protein expression and cell survival and if scrambled RNAs (25–100 nm) did not show any effect. The active siRNA sequences shown in figures are as follows: htt siRNA, 5′- TTCATCAGCTTTTCCAGGGTC-3′ (100 nm); β-catenin siRNA, 5′-GGCTTTTCCCAGTCCTTCATT-3′ (100 nm); SIRT1 siRNA, 5′-GATTGTTATTAATATCCTTTT-3′ (25 nm); SIRT2 siRNA 5′-TTGGGTGAAGTTCTATTTGTT-3′ (25 nm); SIRT3 siRNA, 5′-TTACCGATCAACATGCTAGTT-3′ (25 nm); UCP2 siRNA, 5′–GAAGAACGAGACACCTTTA-3′ (33 nm); and UCP4 siRNA 5′-CGCCATTTACAGACAGGTA-3′ (33 nm). The corresponding scramble RNAs were as it follows: htt, 5′-GTCCAGATTTCCCTGTCGTAT-3′ (100 nm); β-catenin, 5′-GCTCTCGCCATATGAATCTCT-3′ (100 nm); SIRT1, 5′-GTTTATGCTCATTATATAT-3′ (25 nm); SIRT2 5′-GTTAGTTAGTTCGTGTAGT-3′ (25 nm); SIRT3, 5′-GCTATGTATCAGCACACTA-3′ (25 nm); UCP2, 5′-GCATAAGACCGACAATAGT-3′ (33 nm); and UCP4, 5′-GAGCCATCAACTGACTAGT-3′ (33 nm). The construct encoding an active variant of SIRT1 (2 kb cDNA) lacking an internal segment in the N terminus was pCDNA3.1-sirt1-Flag. Cells were subjected to DAPI staining, and cell death was scored 48 h after cell transfection by counting pyknotic versus normal nuclei in DAPI-and JetSI-ENDO-positive cells. Cytoplasmic and nuclear proteins were extracted as described previously (Arango et al., 2006), separated by SDS-PAGE, and analyzed by Western blotting using the following primary antibodies: mouse anti-SIRT1 (Millipore, 1:1000), mouse anti-SIRT2 (Santa Cruz Biotechnology, 1:100), rabbit anti-SIRT-3 (Abgent, 1:100), rabbit anti-β-catenin (Cell Signaling Technology, 1:5000), goat anti-UCP2 (Santa Cruz Biotechnology, dilution), rabbit anti-UCP4 (Abcam, 1:25), mouse anti-htt (4C8, Millipore Bioscience Research Reagents, 1:5000 or GenScript A00089, 1:500), mouse anti-actin (Invitrogen, 1:2000), rabbit anti-NCAM (Millipore Bioscience Research Reagents, 1:10,000). Secondary antibodies were goat anti-mouse IgG HRP-conjugated and goat anti-rabbit IgG HRP-conjugated (Biorad). Proteins were detected using ECL+ and evaluated by densitometry using a GS-800 densitometer (Bio-Rad), and quantification was performed using ImageJ. Chemicals were purchased from Calbiochem [6-bromoindirubin-3′-oxime (BIO) and LiCl].
RNA Isolation and quantitative RT-PCR in striatal cells.
Quantitative RT-PCR was performed on RNA extracted from 109Q/109Q mouse striatal cells transfected with the appropriate siRNAs and scramble RNAs to assess gene silencing efficiency. Two days after cell transfection, RNA was extracted with a Qiagen RNeasy kit and Dnase I (Sigma) treatment (as per the manufacturer's protocol). Single-strand cDNA synthesis was done using oligo(dT), random hexamer priming, and 100 ng of total RNA with ABsolute 2-Step QRT-PCR SYBR ROX kit (ABgene). Quantitative PCR was performed using SYBR Green with the ABI PRISM 7700 Sequence Detection System (Applied Biosystems) and oligonucleotides as follows: ucp2-f (5′-TCCTGCTACCTCCCAGAAGA-3′), ucp2-r (5′-TGAGACCTCAAAGCAGCCTC-3′), ucp4-f (5′-GTGACGCCCGCCATTTACAG-3′), ucp4-r (5′-CATCCCTCCAATGACCGATTTCC-3′), sirt2-f (5′-CCCTTCGCCTCCCTCATC-3′), sirt2-r (5′-GTCCCTGTAAGCCTTCTTGG-3′), sirt3-f (5′-TGGCTGACTTCGCTTTGG-3′), sirt3-r (5′-TCCACACCATGAACTACATCC-3′), rpl13a-f (5′-CTGCTGCTCTCAAGGTTGTTC-3′), rpl13a-r (5′-CTGCTTCTTCTTCCGATAGTGC-3′), hprt-f (5′-TTTGCCGCGAGCCG-3′), hprt-r (5′-TAACCTGGTTCATCATCGCTAATC-3′). Assays and data analysis were performed according to the manufacturer's protocol (User Bulletin #2, ABI PRISM 7700 Sequence Detection System, PerkinElmer). All samples were run at least in triplicate using rpl13a or hprt as the calibrator gene with a dilution of 1/100 of cDNA. The amount of target, normalized to an endogenous reference (ucp2 or ucp4) and relative to the calibrator (rpl13a or hprt), was calculated using the 2−ΔΔCT method, and statistical significance was determined using paired t tests.
Statistics.
Statistics of nematode data were performed using one-way ANOVA, with correction for multiple testing by Tukey's Multiple Comparison Test. Data were expressed as mean ± SEM for >60–200 nematodes in each group, depending on the experiments performed (see figure legends). Student's _t test_s were used for striatal cell data. Data were expressed as mean ± SD for >150 cells in each group. Student's t test was used for gene and protein expression data. All experiments were repeated at least three times. p < 0.05 was considered significant.
Results
β-Catenin and ucp-4 are required for neuroprotection by increased sir-2.1 dosage in C. elegans
Nematodes expressing normal (19Q) polyQs in touch receptor neurons show a moderate loss of response to touch, whereas nematodes bearing an expanded (128Q) polyQ show a strong loss of response to touch (85%). This touch phenotype is accompanied by axonal swelling and, importantly, occurs in the absence of cell death (Parker et al., 2005), providing an in vivo model to manipulate the early phases of expanded polyQ neurotoxicity.
We previously reported that the sir-2.1/SIRT1-daf-16/FOXO pathway is neuroprotective in expanded polyQ nematodes (Parker et al., 2005). Importantly, we showed that the neuroprotective effects of sir-2.1 overexpression in these animals is unrelated to background mutations that may produce dye-filling defects and influence lifespan in C. elegans (Burnett et al., 2011), and the sir-2.1 overexpresser strain used herein does not show dye-filling defects. Recent work in C. elegans has suggested that for DAF-16 to gain specificity in the regulation of lifespan and other biological processes, DAF-16 may interact with different cofactors (Wolff et al., 2006; Landis and Murphy, 2010; Yen et al., 2011). Therefore, to further investigate the regulation of neuronal survival by sir-2.1/SIRT1-daf-16/FOXO, we first examined the evolutionary conserved FOXO cofactor BAR-1/β-catenin. BAR-1/β-catenin directly interacts with both DAF-16 and FOXO3a and has been shown to have a role in oxidative stress signaling (Essers et al., 2005). Loss of function of bar-1 enhanced the loss of response to touch in 128Q nematodes with no effect detected in 19Q nematodes (Fig. 1A) and no change detected in 128Q transgene expression (Fig. 1C), indicating that bar-1 protects from neuron dysfunction induced by expanded polyQs.
Figure 1.

β-Catenin and ucp-4 are required for neuroprotection by sir-2.1 in 128Q nematodes. A, bar-1/β-catenin null mutation enhanced touch insensitivity at the tail of 128Q nematodes. No change was detected in 19Q animals. ***p < 0.001 compared to 128Q alone. B**, ucp-4 deletion enhanced 128Q neuronal dysfunction. No change was detected in 19Q animals. ***p < 0.001 compared to 128Q alone. **_C_**, 128Q transgene expression is unchanged at the protein and mRNA levels in 128Q nematodes bearing _bar-1_ or _ucp-4_ LOF. **_D_**, Aggravation of neuron dysfunction by _sir-2.1_ LOF (_sir-2.1_(_ok434_)) was suppressed in animals specifically overexpressing (O/E) wild-type SIR-2.1 in touch receptor neurons under the control of the promoter of the _mec-3_ gene with no effect detected in animals overexpressing empty vector (ID1296 and ID1297; see Table 1). Aggravation of neuron dysfunction by _bar-1_ LOF (_bar-1_(_ga80_)) was suppressed in animals specifically overexpressing wild-type BAR-1 in touch receptor neurons under the control of the promoter of the _mec-3_ gene, with no effect detected in detected in animals overexpressing empty vector (ID1316 and ID1317; see Table 1). Shown are data compiled from two independent extrachromosomal arrays per genotype. ***_p_ < 0.001 versus empty vector. n.s., Not significant. **_E_**, Neuroprotection by increased Sir2 dosage (_sir-2.1_(_O/E_)) against 128Q toxicity was lost in animals mutant for _bar-1_ or _ucp-4_. *_p_ < 0.001 versus 128Q; **_p_ < 0.001 versus _sir-2.1_(_O/E_) alone. Data in **_A_**, **_B_**, and **_E_** are means ± SEM with >200 animals per genotype (50–60 animals per independent experiment for a total of at least 4 independent experiments). Data for touch tests in strains expressing extrachromosomal arrays (D**) are means ± SEM with >60 animals per array (20–30 animals per independent experiment for a total of at least 3 independent experiments).
Next, we examined a putative DAF-16 target gene, ucp-4, to expand on how daf-16 might affect neuronal survival. The ucp-4 gene encodes for the sole mitochondrial uncoupling protein, UCP, in C. elegans (Iser et al., 2005). We tested ucp-4 for two reasons. First, UCPs might be transcriptional targets of FOXO as suggested by a bioinformatics survey of DAF-16 binding sites in the C. elegans genome (Lee et al., 2003) and a transcriptome analysis of Foxo3a-null mouse neural stem cells (Paik et al., 2009). Second, UCPs are thought to be essential to neuron survival and neurodegenerative disease pathogenesis (Andrews et al., 2005b). Deletion of ucp-4 resulted in enhanced neuronal dysfunction in 128Q animals with no effect in 19Q animals (Fig. 1B) and no change detected in 128Q transgene expression (Fig. 1C), indicating that ucp-4 protects from neuron dysfunction induced by expanded polyQs.
In the 128Q nematodes, neuronal dysfunction is intrinsically connected to the dosage of sir-2.1 (Parker et al., 2005; Burnett et al., 2011). Neuron dysfunction is aggravated by sir-2.1 LOF (Parker et al., 2005), and this effect is rescued by overexpression of wild-type sir-2.1 in mechanosensory neurons with no effect of empty vector control, suggesting that this effect of sir-2.1 LOF is cell-autonomous (Fig. 1D). The aggravation of neuron dysfunction by LOF of bar-1 in 128Q nematodes (Fig. 1A) is also cell autonomous, as suggested by the rescue of this effect by overexpression of wild-type bar-1 in mechanosensory neurons with no effect of empty vector control (Fig. 1D). In contrast to aggravation by sir-2.1 LOF, increased sir-2.1 dosage is neuroprotective and requires daf-16 (Parker et al., 2005). Therefore we tested how the DAF-16 cofactor bar-1 and the putative DAF-16 target ucp-4 may also be required for this neuroprotective activity of sir-2.1. The increase in touch response induced by sir-2.1 overexpression was lost in 128Q animals bearing either a bar-1 or a ucp-4 LOF mutation, and the detrimental effects of bar-1 LOF and ucp-4 LOF on touch response were similar in 128Q nematodes and 128Q nematodes bearing increased sir-2.1 dosage (Fig. 1E). Together, these results indicated that the neuroprotective effect of sir-2.1 in addition to requiring daf-16 is also dependent on bar-1 and ucp-4.
The ucp-4 promoter is regulated by DAF-16
Having shown that ucp-4 is required for sir-2.1 to regulate expanded polyQ toxicity in C. elegans neurons, we next tested whether the ucp-4 gene, a putative DAF-16 target (Lee et al., 2003; Paik et al., 2009), may be directly regulated by DAF-16. We scanned the promoter of ucp-4 and identified 3 consensus DAF-16 binding sites within 5 kb upstream of the translation start site (Fig. 2A). We then isolated whole worm extracts from mix staged populations of wild-type (N2), as well as animals mutant for _daf-2/_IR-IGF1R or daf-16/FOXO or both, and we performed ChIP with an anti-DAF-16 antibody to test for DAF-16 direct binding to the ucp-4 promoter (Oh et al., 2006; Mukhopadhyay et al., 2008). There was no statistically significant difference in binding signals for the binding site 1 and the 3′ region in daf-16 mutant animals, signifying DAF-16 independent recruitment (Fig. 2A). In contrast, binding signals were strongly decreased for binding site 2 when DAF-16 was absent, signifying DAF-16 dependence (Fig. 2A). To test for the functional role of binding site 2, we generated C. elegans transgenic strains stably expressing mCherry under the control of the ucp-4 promoter (1768 bp fragment), which carried a wild-type or scrambled DAF-16 binding site 2, and we crossed these strains into a DAF-16 protein overexpression or daf-16 knock-out background. First, constructs encoding UCP-4::mCherry under the control of the ucp-4 promoter were stably expressed in a wild-type N2 strain or a strain overexpressing DAF-16::GFP (translational construct TJ356), and the intensity of mCherry signals was quantified in late L4 animals in the pharynx area, an area where DAF-16 isoforms and UCP-4 are known to be expressed (Lee et al., 2001; Lin et al., 2001) using two independent arrays per genotype and >60 animals per array (20–30 animals per independent experiment for a total of at least 3 independent experiments). The expression of mCherry was increased (p < 0.005) in animals bearing DAF-16 overexpression (mean ± SEM: 40 ± 2.7, arbitrary unit) compared to wild-type (mean ± SEM: 57 ± 2.8, arbitrary unit) (see also Fig. 2B). Second, mCherry expression required DAF-16 binding site 2, as shown by the loss of variation of mCherry expression in animals bearing either daf-16 LOF or DAF-16 overexpression when the ucp-4 promoter carried a scrambled sequence for this binding site (Fig. 2C). Together, these experiments suggested that the ucp-4 promoter may be regulated by DAF-16.
Figure 2.
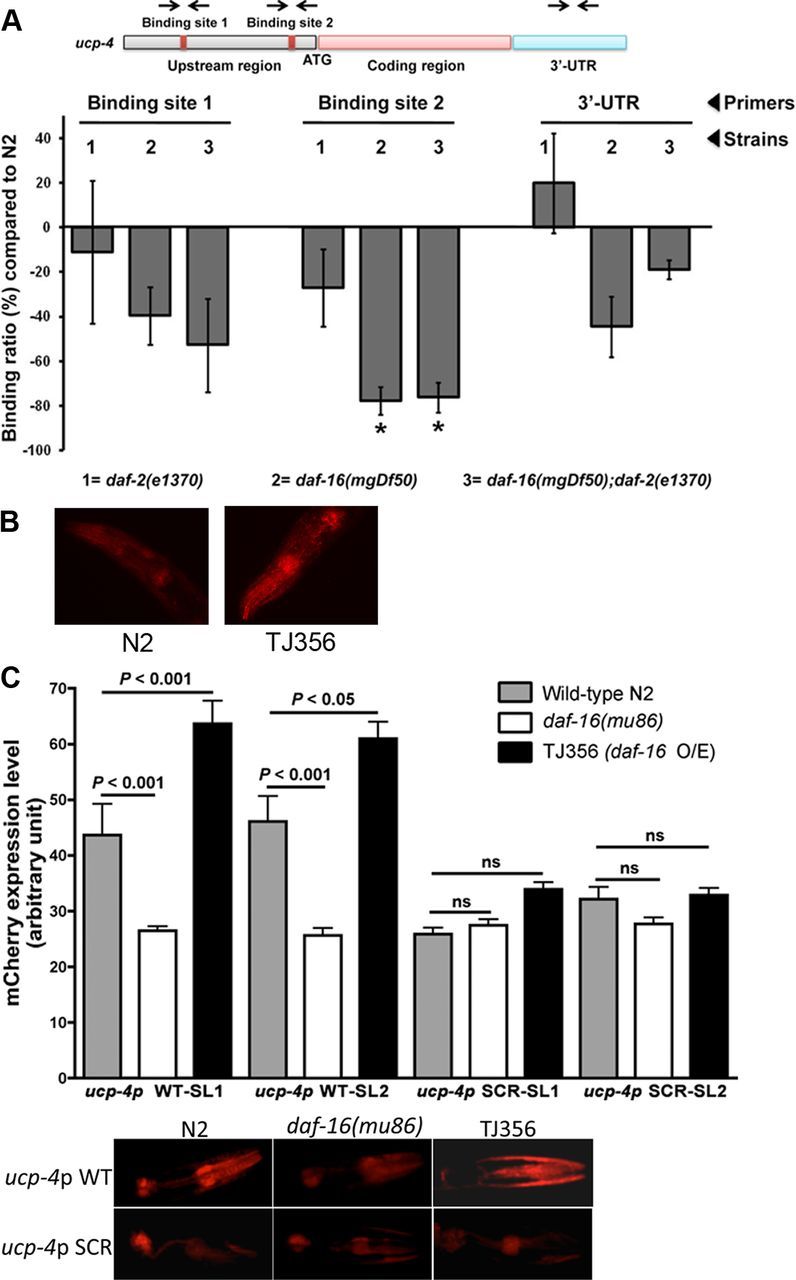
The UCP-4 promoter is regulated by DAF-16. A, DAF-16 binds to the UCP-4 promoter. The 5′ region of UCP-4 has three consensus DAF-16 binding sites (top; only 2 sites are shown; binding site 1 has two closely located consensus sites) that are separated by 3.7 kb. One of the binding sites is located 894 bp upstream of the ATG (binding site 2), while two others are located 4.655 and 4.964 kb upstream of ATG. Since the latter sites are closely situated, primers were designed for the site at 4.655 kb (binding site 1). Primers were also designed for region 654 bp downstream of the stop codon as a control site in the 3′-region. Chromatin immunoprecipitation (bottom) using anti-DAF-16 antibody (bottom) using N2, daf-2(e1370), daf-16(mgDf50), and daf-16(mgDf50);daf-2(e1370). The binding was normalized to that of N2, and effects for which p < 0.01 were considered significant. *_p_ < 0.005 compared to N2. **_B_**, Representative images for DAF-16 overexpression to increase the activity of the UCP-4 promoter (1768 bp) in late L4 _C. elegans_ nematodes as inferred from quantifying the intensity of _mCherry_ signals in the pharynx (see Results). **_C_**, The effects of DAF-16 on the activity of the _ucp-4_ promoter requires binding site 2. Constructs encoding _mCherry_ under the control of the _ucp-4_ promoter (1768 bp), which carried a wild-type (_ucp-4p_ WT) or scrambled (_ucp-4p_ SCR) binding site 2, were stably expressed in N2, _daf-16_(_mu86_) or TJ356 strain, and _mCherry_ signals were quantified as indicated in **_B_**. Data are mean ± SEM as compiled from two independent arrays (SL1, SL2) per genotype, and >60 animals per array (20–30 animals per independent experiment for a total of at least 3 independent experiments).
GSK-3β inhibitors require bar-1, sir-2.1, daf-16, and ucp-4 for neuroprotection
To further expand the mechanistic basis for neuroprotection by sir-2.1 in connection with daf-16 and bar-1 activities, we sought to look at the regulation of β-catenin, a protein that is part of the Wnt signaling pathway and is primarily targeted for degradation by direct phosphorylation by the GSK-3β kinase. Interestingly, previous studies on a simple model system have shown GSK-3β inhibitors such as lithium may protect from expanded polyQs (Berger et al., 2005; Voisine et al., 2007), suggesting that GSK-3β inhibitors may be useful for HD therapy. Therefore, we tested whether sir-2.1, daf-16, bar-1, and ucp-4 may be required for neuroprotection by GSK-3β inhibitors. We first tested BIO, a selective inhibitor of GSK-3β, and observed that at 100–30 μm BIO significantly reduced neuron dysfunction in 128Q animals with no effect detected in 19Q animals (Fig. 3A) and no change detected in 128Q transgene expression (Fig. 3C). However, neuroprotection by BIO was lost in 128Q nematode mutant for any of the four genes including sir-2.1, daf-16, bar-1 and ucp-4 (Fig. 3A), indicating these genes are required for BIO activity. BIO also reduced axonal swelling (Fig. 3B), which may reflect increased neuron health and is consistent with the ability of BIO to reduce the loss of touch response in 128Q nematodes. We also tested lithium chloride, a drug that has GSK-3β inhibition properties, and observed neuroprotective effects similar to those of BIO (data not shown). While BIO and lithium may have secondary targets other than GSK-3β, a genome-scale functional RNAi screen indicated that gsk-3 inactivation is neuroprotective in expanded-polyQ nematodes (Lejeune et al., 2012), suggesting that the neuroprotective effects of BIO and lithium may involve the GSK-3β target. Together, these results suggested that the neuroprotective properties of GSK-3β inhibitors require sir-2.1, daf-16, bar-1, and ucp-4, which may be useful for protecting HD neurons from the dysfunction induced by mutant polyQs.
Figure 3.

The GSK-3β inhibitor BIO is neuroprotective via FOXO signaling in 128Q nematodes. A, Neuron dysfunction in 128Q nematodes is higher compared to 19Q nematodes, with about 85% of the 128Q animals having a defective response to touch (Parker et al., 2005). BIO strongly rescues expanded polyQ neurotoxicity at 100–33.3 μm (*p < 0.001 and **_p_ < 0.01 versus DMSO controls) with no effect in 19Q animals. BIO rescuing activity was lost in mutants for _daf-16_, _sir-2.1_, _bar-1_, and _ucp-4_. Dilution factor is 3×. Percent rescue was calculated from percentages of touch response as ((test − control)/(100 − control) * 100). Data are means ± SEM with >200 animals per point for all genotypes (50–60 animals per independent experiment for a total of at least 4 independent experiments). The percentages of touch response (means ± SEM) were 52 ± 3% in 19Q animals treated with vehicle, 23 ± 2% in 128Q animals treated with vehicle, 46 ± 3% in 128Q animals treated with 100 μm BIO, and 34.6 ± 4% in 128Q animals treated with 33.3 μm BIO. B, BIO reduced axonal swelling in PLM cells of 128Q animals (*p < 0.002 versus DMSO controls). Data are means ± SEM with >100 animals per treatment (25–30 animals per independent experiment for a total of at least 4 independent experiments). C, BIO treatment (50 μm, 100 μm) does not modify transgene expression in 128Q nematodes. n.s., Not significant.
β-Catenin, SIRT1 overexpression, and UCPs modulate the survival of mouse striatal cells
Having shown that sir-2.1, bar-1, and ucp-4 modulate the neurotoxicity of a N-terminal htt fragment in nematodes, we tested whether their mammalian counterparts may have an effect in a full-length cellular model of HD. To this end, we used striatal cells derived from the htt knock-in mice HdhQ111 (Trettel et al., 2000). Mutant htt (109Q/109Q) striatal cells have a higher rate of cell death induced by serum deprivation compared to wild-type (7Q/7Q) cells (Trettel et al., 2000), thus providing an assay to test for cell vulnerability to mutant htt expressed at endogenous levels (Fig. 4A). To test for the implication of htt in cell death induced by serum deprivation, we subjected these cells to htt siRNA treatment using a previously described siRNA (DiFiglia et al., 2007) and corresponding scrambled RNA. Reducing htt levels in 109Q/109Q cells strongly decreased cell mortality upon serum deprivation, with no change detected in 7Q/7Q cells (Fig. 4B), suggesting that a significant percent of cell mortality is associated to mutant htt expression. As reported previously (Gines et al., 2003), mutant htt cells showed increased β-catenin levels (data not shown) that may correspond to a prosurvival response (Gines et al., 2003). To test for the role of β-catenin in striatal cell survival, we subjected these cells to β-catenin siRNA treatment (see Materials and Methods for details of siRNA experiments). Reducing β-catenin levels decreased the survival of 109Q/109Q cells, an effect accompanied by reduction of β-catenin expression, and no change in htt expression was detected (Fig. 4C,D). Consistent with the effect of reducing β-catenin levels, the GSK-3β inhibitor BIO enhanced the survival of 109Q/109Q cells (Fig. 4C) with no detectable change in htt expression (Fig. 4D). This amelioration of cell survival was accompanied by increased β-catenin expression (Fig. 4D) and, consistently, was lost when 109Q/109Q cells were incubated with BIO and β-catenin siRNA together (Fig. 4C). Therefore, in both mouse and C. elegans models for HD, β-catenin has neuroprotective effects, and altering its dosage is detrimental to neuronal cell survival.
Figure 4.

β-Catenin and UCP2/UCP4 modulate the survival of mutant htt striatal cells from HdhQ111 mice. In these assays, cells were subjected to serum deprivation. A, Representative graph showing that 109Q/109Q cells were more susceptible to cell death compared to 7Q/7Q cells (N = 3 with SD; *p < 0.01 compared to 7Q/7Q). B, Effects of reducing htt levels. Htt siRNA reduced 109Q/109Q cell mortality (N = 3 with SD; **p < 0.001 versus untreated). Scramble RNA showed no effect. ns, Not significant. C, Representative Western blot showing that Htt siRNA treatment reduces Htt expression levels in 7Q/7Q and 109Q/109Q striatal cells. In the following panels, data are normalized due to the variability of striatal cell survival after serum deprivation across experiments. D, Effects of reducing β-catenin levels and of BIO (0.5 μm) treatment. Left, β-Catenin siRNA enhanced 109Q/109Q cell mortality (N = 3 with SD; *p < 0.01 versus untreated). Scramble RNA showed no effect. BIO treatment reduced cell mortality (N = 3 with SD; **p < 0.05 versus DMSO controls). When β-catenin siRNA and BIO were combined, no change in cell mortality was detected compared to cells treated with scramble RNA and DMSO (N = 3 with SD). Right, β-catenin siRNA and BIO showed no effect in 7Q/7Q cells. E, Mutant htt expression was unchanged by treatment with BIO or β-catenin siRNA. β-Catenin levels are increased by BIO (N = 3, p < 0.02) and reduced by β-catenin siRNA (n = 3, p < 0.05). Scramble RNA had no effect. E, Effects of reducing SIRT1, UCP2, and UCP4 (N = 4 with SD). UCP2 siRNA enhanced 109Q/109Q cell mortality (*p < 0.01 versus untreated) with no effect detected in 7Q/7Q cells. UCP4 siRNA reduced 109Q/109Q cell mortality (*p < 0.01 versus untreated) with no effect detected in 7Q/7Q cells. Scramble RNAs had no effect. G, Mutant htt expression was unchanged by siRNAs against SIRT1, UCP2, and UCP4. Shown are representative Western blots. Scramble RNAs showed no effect. H, At left is a representative Western blot image showing that 109Q/109Q cells have lower SIRT1 levels upon SIRT1 siRNA treatment (N = 3; p < 0.05). Quantitative RT-PCR experiments indicated that UCP2 and UCP4 siRNA decreased mRNA levels of UCP2 and UCP4, respectively (N = 5 with SD; p < 0.05). The effect of UCP2/4 siRNAs on target protein expression could not be evaluated, as the antisera were repeatedly unable to detect any protein in Western blot experiments, and scramble RNAs showed no effect. For all panels, N indicates the number of independent experiments performed.
Next, we explored the role of SIRTs and UCPs on mouse striatal cell survival. Reducing SIRT1 levels showed no effect on cell survival (Fig. 4E). We tested whether reducing levels of either SIRT2 and SIRT3, two additional sirtuin family members, may modulate striatal cell survival, and observed no effect as well. Therefore, lowering these sirtuins did not affect neuronal survival. Second, we examined the two neuronal UCPs (UCP2, UCP4). UCP2 siRNAs decreased the survival of 109Q/109Q cells (Fig. 4E) with no change in htt expression (Fig. 4F), and they reduced UCP2 mRNA levels (Fig. 4G). In contrast, UCP4 siRNAs increased the survival of 109Q/109Q cells (Fig. 4E) with no change in htt expression (Fig. 4F), and they reduced UCP4 mRNA levels (Fig. 4G), suggesting evolutionary diversity for mouse UCP activity compared to the sole UCP in C. elegans.
Next, we tested whether SIRT1 and β-catenin might cooperate in the modulation of mouse striatal cell survival since sir-2.1/SIRT1 and bar-1/β-catenin are linked to modulate neuron dysfunction in 128Q nematodes. To this end, we overexpressed SIRT1 either alone or in combination with β-catenin siRNA. The overexpression of SIRT1 slightly enhanced the survival of 109Q/109Q cells subjected to serum deprivation with no change in htt levels (Fig. 5A,B). As observed previously, reducing β-catenin levels was strongly detrimental to 109Q/109Q cell survival (Fig. 5A), and this effect was suppressed by SIRT1 overexpression, suggesting that raising SIRT1 levels can compensate for the detrimental effect of β-catenin reduction, which further connected the SIRT1 and β-catenin pathways. We then examined further the individual effects of SIRT1 and β-catenin in mouse striatal cells. Given that UCP gene expression may be regulated by FOXO proteins as suggested by previous studies (Lee et al., 2003; Paik et al., 2009), we tested for the effects of β-catenin reduction and SIRT1 overexpression on the expression levels of UCP2 and UCP4. In mutant htt striatal cells, reducing β-catenin, which aggravates cell death vulnerability, increased mRNA levels of UCP4 (detrimental to cell survival), whereas SIRT1 overexpression, which is slightly neuroprotective, had the opposite effect (Fig. 5C). Conversely, reducing β-catenin decreased mRNA levels of UCP2 (a neuroprotective gene) in mutant htt cells, with however no significant effect of SIRT1 overexpression (Fig. 5C). These results indicated that while SIRT1 and β-catenin may have common effects such as the repression of UCP4 levels, they may differ in their ability to promote UCP2 levels.
Figure 5.

Effects of β-catenin and SIRT1 in mutant htt striatal cells from HdhQ111 mice. A, Mutant htt cells showed increased susceptibility to cell death induced by serum deprivation (***p < 0.001 compared to 7Q/7Q). SIRT1 overexpression slightly enhanced the survival of 109Q/109Q striatal cells with no effects in 7Q/7Q cells (**p < 0.01 compared to untreated 109Q/109Q). Reducing β-catenin enhanced the mortality of 109Q/109Q striatal cells with no effects in 7Q/7Q cells (***p < 0.001 compared to untreated 109Q/109Q). When SIRT1 overexpression was combined with β-catenin reduction in 109Q/109Q striatal cells, the detrimental effect of reducing β-catenin was compensated by SIRT1 overexpression (N (number of experiments performed) = 3 with SD; ***p < 0.001 compared to β-catenin reduction). n.s., Not significant. B, Representative Western blots showing that striatal cells transfected with the SIRT1 construct (an active variant lacking an internal segment in the N terminus) have increased SIRT1 levels. n.a., Not applicable. C, Effects of reducing β-catenin or overexpressing SIRT1 on the expression levels of UCP2. The expression of the UCP4 gene is downregulated by β-catenin siRNA with no effect by SIRT1 overexpression (O/E) in 109Q/109Q cells (N = 4 with SD; *p < 0.05). No effect was detected in 7Q/7Q cells. ns, Not significant. Scramble RNA (β-catenin) showed no effect on the UCP2/UCP4 gene expression levels (C, D).
Collectively, these results were consistent with a well documented model in which the FOXO, SIRT1, and β-catenin proteins may form a complex in the nucleus to regulate gene expression in nematodes and other species (Brunet et al., 2004). Further, these results highlighted a cross-talk between β-catenin and SIRT1 in the regulation of vulnerability to cell death caused by mutant htt expression, which may be primarily mediated by modulating the levels of UCP4, a UCP detrimental to mutant htt cell survival.
Discussion
The Sir2/SIRT1 gene and its target daf-16/FOXO have emerged as potent protective factors for HD and other degenerative diseases (Morley et al., 2002; Parker et al., 2005; Cohen et al., 2006; Kim et al., 2007; Jeong et al., 2011; Jiang et al., 2011). Since FOXO integrates numerous signals and regulates the expression of many genes (Greer and Brunet, 2008; Landis and Murphy, 2010; Yen et al., 2011), an essential question is how the FOXO network protects HD neurons. Does it play a role in the early phases of mutant htt toxicity such as neuronal dysfunction and vulnerability to cell death?
Our data show that β-catenin, SIRT1, and UCPs may participate in the regulation of vulnerability to cell death of mutant htt striatal cells, which is consistent with the protective effect of increased sir-2.1 dosage against neuron dysfunction in 128Q nematodes, an effect that requires daf-16/FOXO (Parker et al., 2005), the FOXO cofactor bar-1/β-catenin (Essers et al., 2005), and ucp-4, which we suggest to be a DAF-16 regulated gene. As summarized in Figure 6, these results are consistent with a previously proposed model in which the SIRT1, FOXO, and β-catenin proteins may form a complex in the nucleus to regulate gene expression in nematodes and other species (Tissenbaum and Guarente, 2001; Brunet et al., 2004; Essers et al., 2005; Berdichevsky et al., 2006; Wang et al., 2006). Importantly, neuroprotective effects of SIRT1 activation were recently reported in several mouse models of HD (Jeong et al., 2011; Jiang et al., 2011), and FOXO3a acetylation may be involved in SIRT1 protection (Jiang et al., 2011), which corroborates the C. elegans data. In addition, neuroprotective effects of β-catenin were consistently observed in expanded-polyQ nematodes and mutant htt striatal cells. However, we observed differences between C. elegans and mouse striatal cells for UCP activity. This is expected, as evolutionary diversity in gene family function is a widely observed phenomenon. The UCPs can have profound effects on neurons with chronic uncoupling, promoting mitochondrial biogenesis and elevated ATP levels (Andrews et al., 2005b). The enhancement of 128Q toxicity in nematode ucp-4 mutants suggests that ucp-4 normally protect neurons from the dysfunction induced by expanded polyQ expression. However, for mouse striatal cells that express mutant htt, while UCP4 may normally contribute to their vulnerability, UCP2 may protect from it. This is consistent with the neuroprotective effect of UCP2 overexpression in mouse models of Parkinson's disease (Andrews et al., 2005a). While the C. elegans and mouse striatal cell model both emphasized the neuroprotective effects of β-catenin, they showed differences in their ability to account for the cross-talk between Sir2/SIRT1 and β-catenin. Expanded polyQ nematodes indicated that bar-1/β-catenin is required for sir-2.1/SIRT1 overexpression to be neuroprotective. However, mutant htt striatal cells poorly recapitulated the neuroprotective effects of SIRT1 overexpression and primarily indicated that SIRT1 overexpression compensates for the detrimental effect of β-catenin reduction.
Figure 6.

Working model for the role of bar-1/β-catenin, daf-16/FOXO and sir-2.1/SIRT1 in the regulation of mutant polyglutamine neuron homeostasis. The effects observed in C. elegans and mouse striatal cells, two models of the early cytotoxicity/cell-vulnerability induced by mutant htt, are summarized in the context of prior knowledge on the binding of BAR-1/β-catenin to DAF-16/FOXO (Essers et al., 2005) and the role of GSK-3/β-catenin and SIRT1 in the modulation of FOXO activity in the nucleus and its ability to regulate gene expression (Greer and Brunet, 2008; Landis and Murphy, 2010; Yen et al., 2011). Genes required for neuroprotection by sir-2.1 in C. elegans as previously reported (Parker et al., 2005) or indicated herein are underlined. Bold indicates gene activity in mouse striatal cells as suggested by the ability of SIRT1 overexpression to compensate for the detrimental effect of reducing β-catenin on cell survival, and by the effects on UCP2 expression levels.
These results highlight the complexity of the regulation around FOXO factors (Greer and Brunet, 2008; Landis and Murphy, 2010; Yen et al., 2011), and they illustrate how the conclusions about the role of cell survival genes may be influenced by the methodological and cellular context in which their activity is assessed. For example, SIRT1 was reported to deacetylate β-catenin and suppress its ability to activate transcription in mammalian cells (Firestein et al., 2008), which appears to contrast with our findings. However, this study was performed in human colon cancer cell lines, where SIRT1-β-catenin homeostasis may be different compared to neurons and cells expressing expanded polyQs. In addition, several studies have indicated that β-catenin is an important survival factor in normal (Essers et al., 2005) and diseased (Inestrosa and Arenas, 2010) cells through its ability to coactivate either TCF (T-cell factor) or FOXO transcription factors. However, a study of neuronal cell death in HD has suggested that abnormal accumulation of β-catenin may cause neurotoxicity (Godin et al., 2010). Since β-catenin is neuroprotective against the early phases of mutant polyQ cytotoxicity (neuron dysfunction, cell vulnerability), this suggests that β-catenin homeostasis and activity may change along the pathogenic process in HD.
Finally, the activation of cell survival mechanisms controlled by FOXO factors may contribute to delaying HD pathogenesis. Our data indeed suggest that GSK-3β inhibitors require the FOXO network for protecting neurons from the dysfunction induced by expanded polyQs, suggesting that GSK-3β may have potential for the treatment of HD. In conclusion, our data reveal that neuroprotection by sirtuin sir-2.1 from expanded polyQs requires β-catenin and ucp-4 in C. elegans, suggesting that FOXO interactors modulate the early phases of mutant polyQ cytotoxicity. Our findings raise the possibility that FOXO interactors might have a role in the modification of the pathogenic process in HD, and this will be addressed in future studies.
Footnotes
This project was funded in part by an endowment from the William Randolph Hearst Foundation and a grant (AG025891) from the National Institute of Aging to H.A.T. This work is supported by INSERM, AP-HP, ANR, FRM, and AFM, Paris, France, and by the CHDI Foundation and the Hereditary Disease Foundation. J.A.P. was supported by an Inserm Young Researcher award. H.A.T. is a William Randolph Hearst Young Investigator. We thank Cheryl Wellington and Michael Hayden for constructs encoding htt with 128 Glns, Andrew Fire for C. elegans plasmids, Leonard Guarente for the LG100 strain, Elena Cattaneo for cells from HdhQ111 mice, Robert E. Hughes for constructs encoding SIRT1, and Maryline Mage for technical assistance. Some of the strains were kindly provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources.
References
- Andrews ZB, Horvath B, Barnstable CJ, Elsworth J, Yang L, Beal MF, Roth RH, Matthews RT, Horvath TL. Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson's disease. J Neurosci. 2005a;25:184–191. doi: 10.1523/JNEUROSCI.4269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Diano S, Horvath TL. Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nat Rev Neurosci. 2005b;6:829–840. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- Anselmi CV, Malovini A, Roncarati R, Novelli V, Villa F, Condorelli G, Bellazzi R, Puca AA. Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 2009;12:95–104. doi: 10.1089/rej.2008.0827. [DOI] [PubMed] [Google Scholar]
- Arango M, Holbert S, Zala D, Brouillet E, Pearson J, Régulier E, Thakur AK, Aebischer P, Wetzel R, Déglon N, Néri C. CA150 expression delays striatal cell death in overexpression and knock-in conditions for mutant huntingtin neurotoxicity. J Neurosci. 2006;26:4649–4659. doi: 10.1523/JNEUROSCI.5409-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell. 2006;125:1165–1177. doi: 10.1016/j.cell.2006.04.036. [DOI] [PubMed] [Google Scholar]
- Berger Z, Ttofi EK, Michel CH, Pasco M, Tenant S, Rubinsztein DC, O'Kane C J. Lithium rescues toxicity of aggregate-prone proteins in Drosophila by perturbing Wnt pathway. Hum Mol Genet. 2005;14:3003–3011. doi: 10.1093/hmg/ddi331. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvári M, Piper MD, Hoddinott M, Sutphin GL, Leko V, McElwee JJ, Vazquez-Manrique RP, Orfila AM, Ackerman D, Au C, Vinti G, Riesen M, Howard K, Neri C, Bedalov A, Kaeberlein M, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011;477:482–485. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sena-Esteves M, Chase K, Sapp E, Pfister E, Sass M, Yoder J, Reeves P, Pandey RK, Rajeev KG, Manoharan M, Sah DW, Zamore PD, Aronin N. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A. 2007;104:17204–17209. doi: 10.1073/pnas.0708285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, Hahn WC, Guarente LP, Sinclair DA. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS One. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayán J, Brocklebank D, Andresen JM, Alkorta-Aranburu G, Zameel Cader M, Roberts SA, Cherny SS, Wexler NS, Cardon LR, Housman DE. Genomewide linkage scan reveals novel loci modifying age of onset of Huntington's disease in the Venezuelan HD kindreds. Genet Epidemiol. 2008;32:445–453. doi: 10.1002/gepi.20317. [DOI] [PubMed] [Google Scholar]
- Ghazi A, Henis-Korenblit S, Kenyon C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 2009;5:e1000639. doi: 10.1371/journal.pgen.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles AC, Rankin CH. Behavioral and genetic characterization of habituation using Caenorhabditis elegans. Neurobiol Learn Mem. 2009;92:139–146. doi: 10.1016/j.nlm.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Gines S, Ivanova E, Seong IS, Saura CA, MacDonald ME. Enhanced Akt signaling is an early pro-survival response that reflects N-methyl-d-aspartate receptor activation in Huntington's disease knock-in striatal cells. J Biol Chem. 2003;278:50514–50522. doi: 10.1074/jbc.M309348200. [DOI] [PubMed] [Google Scholar]
- Godin JD, Poizat G, Hickey MA, Maschat F, Humbert S. Mutant huntingtin-impaired degradation of beta-catenin causes neurotoxicity in Huntington's disease. EMBO J. 2010;29:2433–2445. doi: 10.1038/emboj.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Brunet A. Signaling networks in aging. J Cell Sci. 2008;121:407–412. doi: 10.1242/jcs.021519. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Holbert S, Denghien I, Kiechle T, Rosenblatt A, Wellington C, Hayden MR, Margolis RL, Ross CA, Dausset J, Ferrante RJ, Nér C. The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proc Natl Acad Sci U S A. 2001;98:1811–1816. doi: 10.1073/pnas.041566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- Iser WB, Kim D, Bachman E, Wolkow C. Examination of the requirement for ucp-4, a putative homolog of mammalian uncoupling proteins, for stress tolerance and longevity in C. elegans. Mech Ageing Dev. 2005;126:1090–1096. doi: 10.1016/j.mad.2005.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Cohen DE, Cui L, Supinski A, Savas JN, Mazzulli JR, Yates JR, 3rd, Bordone L, Guarente L, Krainc D. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med. 2011;18:159–165. doi: 10.1038/nm.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wang J, Du L, Jeong H, West T, Xiang L, Peng Q, Hou Z, Cai H, Seredenina T, Arbez N, Zhu S, Sommers K, Qian J, Zhang J, Mori S, Yang XW, Tamashiro KL, Aja S, Moran TH, et al. Neuroprotective role of Sirt1 in mammalian models of Huntington's disease through activation of multiple Sirt1 targets. Nat Med. 2011;18:153–158. doi: 10.1038/nm.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis JN, Murphy CT. Integration of diverse inputs in the regulation of Caenorhabditis elegans DAF-16/FOXO. Dev Dyn. 2010;239:1405–1412. doi: 10.1002/dvdy.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RY, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol. 2001;11:1950–1957. doi: 10.1016/s0960-9822(01)00595-4. [DOI] [PubMed] [Google Scholar]
- Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- Lejeune FX, Mesrob L, Parmentier F, Bicep C, Vazquez-Manrique RP, Parker JA, Vert JP, Tourette C, Neri C. Large-scale functional RNAi screen in C. elegans identifies genes that regulate the dysfunction of mutant polyglutamine neurons. BMC Genomics. 2012;13:91. doi: 10.1186/1471-2164-13-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JL, Hayden MR, Almqvist EW, Brinkman RR, Durr A, Dodé C, Morrison PJ, Suchowersky O, Ross CA, Margolis RL, Rosenblatt A, Gómez-Tortosa E, Cabrero DM, Novelletto A, Frontali M, Nance M, Trent RJ, McCusker E, Jones R, Paulsen JS, et al. A genome scan for modifiers of age at onset in Huntington disease: the HD MAPS study. Am J Hum Genet. 2003;73:682–687. doi: 10.1086/378133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang WJ, Cao H, Lu J, Wu C, Hu FY, Guo J, Zhao L, Yang F, Zhang YX, Li W, Zheng GY, Cui H, Chen X, Zhu Z, He H, Dong B, Mo X, Zeng Y, Tian XL. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum Mol Genet. 2009;18:4897–4904. doi: 10.1093/hmg/ddp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet. 2001;28:139–145. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- Metzger S, Rong J, Nguyen HP, Cape A, Tomiuk J, Soehn AS, Propping P, Freudenberg-Hua Y, Freudenberg J, Tong L, Li SH, Li XJ, Riess O. Huntingtin-associated protein-1 is a modifier of the age-at-onset of Huntington's disease. Hum Mol Genet. 2008;17:1137–1146. doi: 10.1093/hmg/ddn003. [DOI] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat Protoc. 2008;3:698–709. doi: 10.1038/nprot.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Mukhopadhyay A, Dixit BL, Raha T, Green MR, Tissenbaum HA. Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat Genet. 2006;38:251–257. doi: 10.1038/ng1723. [DOI] [PubMed] [Google Scholar]
- Paik JH, Ding Z, Narurkar R, Ramkissoon S, Muller F, Kamoun WS, Chae SS, Zheng H, Ying H, Mahoney J, Hiller D, Jiang S, Protopopov A, Wong WH, Chin L, Ligon KL, DePinho RA. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell. 2009;5:540–553. doi: 10.1016/j.stem.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JA, Arango M, Abderrahmane S, Lambert E, Tourette C, Catoire H, Néri C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet. 2005;37:349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- Soerensen M, Dato S, Christensen K, McGue M, Stevnsner T, Bohr VA, Christiansen L. Replication of an association of variation in the FOXO3A gene with human longevity using both case-control and longitudinal data. Aging Cell. 2010;9:1010–1017. doi: 10.1111/j.1474-9726.2010.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taherzadeh-Fard E, Saft C, Andrich J, Wieczorek S, Arning L. PGC-1alpha as modifier of onset age in Huntington disease. Mol Neurodegener. 2009;4:10. doi: 10.1186/1750-1326-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Voisine C, Varma H, Walker N, Bates EA, Stockwell BR, Hart AC. Identification of potential therapeutic drugs for Huntington's disease using Caenorhabditis elegans. PLoS ONE. 2007;2:e504. doi: 10.1371/journal.pone.0000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FO. Huntington's disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Wang Y, Oh SW, Deplancke B, Luo J, Walhout AJ, Tissenbaum HA. C. elegans 14-3-3 proteins regulate life span and interact with SIR-2.1 and DAF-16/FOXO. Mech Ageing Dev. 2006;127:741–747. doi: 10.1016/j.mad.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A. 2008;105:13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff S, Ma H, Burch D, Maciel GA, Hunter T, Dillin A. SMK-1, an essential regulator of DAF-16-mediated longevity. Cell. 2006;124:1039–1053. doi: 10.1016/j.cell.2005.12.042. [DOI] [PubMed] [Google Scholar]
- Xu EH, Tang Y, Li D, Jia JP. Polymorphism of HD and UCHL-1 genes in Huntington's disease. J Clin Neurosci. 2009;16:1473–1477. doi: 10.1016/j.jocn.2009.03.027. [DOI] [PubMed] [Google Scholar]
- Yen K, Narasimhan SD, Tissenbaum HA. DAF-16/Forkhead box O transcription factor: many paths to a single Fork(head) in the road. Antioxid Redox Signal. 2011;14:623–634. doi: 10.1089/ars.2010.3490. [DOI] [PMC free article] [PubMed] [Google Scholar]