Cancer-Stromal Cell Interactions Mediated by Hypoxia-Inducible Factors Promote Angiogenesis, Lymphangiogenesis, and Metastasis (original) (raw)
. Author manuscript; available in PMC: 2015 Apr 30.
Published in final edited form as: Oncogene. 2012 Dec 10;32(35):4057–4063. doi: 10.1038/onc.2012.578
Abstract
Interactions between cancer cells and stromal cells, including blood vessel endothelial cells (BECs), lymphatic vessel endothelial cells (LECs), bone marrow-derived angiogenic cells (BMDACs) and other bone marrow-derived cells (BMDCs) play important roles in cancer progression. Intratumoral hypoxia, which affects both cancer and stromal cells, is associated with a significantly increased risk of metastasis and mortality in many human cancers. Recent studies have begun to delineate the molecular mechanisms underlying the effect of intratumoral hypoxia on cancer progression. Reduced O2 availability induces the activity of hypoxia-inducible factors (HIFs), which activate the transcription of target genes encoding proteins that play important roles in many critical aspects of cancer biology. Included among these are secreted factors, including angiopoietin 2, angiopoietin-like 4, placental growth factor, platelet-derived growth factor B, stem cell factor (kit ligand), stromal-derived factor 1, and vascular endothelial growth factor. These factors are produced by hypoxic cancer cells and directly mediate functional interactions with BECs, LECs, BMDACs and other BMDCs that promote angiogenesis, lymphangiogenesis, and metastasis. In addition, lysyl oxidase (LOX) and LOX-like proteins, which are secreted by hypoxic breast cancer cells, remodel extracellular matrix in the lungs, which leads to BMDC recruitment and metastatic niche formation.
Keywords: breast cancer, extravasation, intravasation, lung metastasis, lymph node, lymphatic metastasis, metastatic niche formation
Introduction
Although considerable effort has been devoted to the identification of genetic alterations in cancer cells, it is clear that knowledge of changes at the DNA level is necessary but not sufficient to understand cancer biology or to develop effective cancer therapies. Complementing the analysis of DNA alterations are studies of changes at the RNA level, which reflect both alterations in the cancer cell genome and responses to the tumor microenvironment. The microenvironment within the primary tumor differs from that of the normal tissue from which the tumor arose in many critical aspects,1 only two of which will be highlighted here. First, cancer cells recruit and interact with stromal cells2,3 and these interactions play critical roles in (a) the establishment and growth of the primary tumor; and (b) the metastasis of cancer cells via lymphatic vessels and blood vessels to local lymph nodes and distant tissues throughout the body. Included among the tumor stromal cells are blood vessel endothelial cells (BECs), lymphatic vessel endothelial cells (LECs), bone marrow-derived angiogenic cells (BMDACs) and other bone marrow-derived cells (BMDCs), cancer-associated fibroblasts, and inflammatory cells. A second critical difference between the tumor microenvironment and that of the surrounding normal tissue is the presence of intratumoral hypoxia. In cancers in which it is possible to directly measure tumor oxygenation by Eppendorf microelectrode, such as cervical cancer4 and soft tissue sarcoma,5 the presence of severe intratumoral hypoxia (_P_O2 < 10 mm Hg [~1.5% O2]) is associated with an increased risk of metastasis and decreased disease-free and overall survival. This review will summarize recent studies that have delineated molecular mechanisms by which hypoxic cancer cells functionally interact with BECs, LECs, BMDACs and other BMDCs, and thereby promote angiogenesis, lymphangiogenesis, and metastasis.
Reduced O2 availability is a stimulus for the activation of hypoxia-inducible factor 1 (HIF-1), which is a heterodimer protein that is composed of an O2-regulated HIF-1α subunit and a constitutively expressed HIF-1β subunit.6,7 HIF-1 activates the transcription of hundreds of target genes in hypoxic stromal cells and cancer cells.8,9 The presence of increased HIF-1α protein levels in the primary tumor biopsy is associated with increased mortality in bladder, breast, cervical, colorectal, endometrial, esophageal, gastric, laryngeal, lung, oropharyngeal, ovarian, and pancreatic cancer.10 In breast cancer, in which the median _P_O2 is 10 mm Hg as compared to 65 mm Hg in normal breast tissue,11 increased HIF-1α levels in the diagnostic biopsy are associated with increased metastasis and mortality even in patients with lymph node-negative breast cancer, who would otherwise have a good prognosis.12,13 High HIF-1α levels in the primary tumor are also predictive of disease recurrence in breast and prostate cancer.13,14
HIF-2α (also known as EPAS1), which shares 48% amino acid identity with HIF-1α,15 is also induced by hypoxia, dimerizes with HIF-1β, and activates transcription of target genes, some of which are shared with HIF-1α and some of which are distinct.16 In some breast cancer cell lines, such as MDA-MB-231, both HIF-1α and HIF-2α contribute to HIF target gene transactivation, primary tumor growth and metastasis.17 However, in MDA-MB-435 breast cancer cells, although both HIF-1α and HIF-2α are expressed, only HIF-1α contributes to HIF target gene transactivation, tumor growth and metastasis.18 In contrast, HIF-2α plays a critical role in the progression of renal carcinoma and neuroblastoma,16 while in colon cancer HIF-2α expression is lost with advanced tumor stage.19
Several drugs have been shown to block HIF transcriptional activity and inhibit tumor growth, angiogenesis, and/or metastasis in mouse models, including (among others): acriflavine,20,21 cetuximab and other EGFR inhibitors;22 digoxin and other cardiac glycosides;17,23 doxorubicin and other anthracyclines;24 geldanamycin and other HSP90 inhibitors;25,26 HDAC inhibitors;27 rapamycin and other mTOR inhibitors;28-30 selenium compounds;31 and topotecan and other topoisomerase I inhibitors.32,33 Translation to the clinic has begun: effective inhibition of HIF-1α and angiogenesis in tumors of patients treated with topotecan has been demonstrated34 and a clinical trial of digoxin in patients with prostate cancer is ongoing (NCT01162135, ClinicalTrials.gov).
Recent studies have delineated molecular mechanisms by which intratumoral hypoxia promotes angiogenesis, lymphangiogenesis, and metastasis through the HIF-dependent production by cancer cells of secreted factors that mediate functional interactions with BECs, LECs, BMDACs, and other BMDCs. These functional interactions are summarized in Figure 1 and described in detail below. HIF target genes encode proteins that play critical roles in many aspects of cancer biology that will not be addressed in this review, including immortalization; metabolic reprogramming, epithelial-mesenchymal transition, stem cell self-renewal, and tissue invasion.35
Figure 1.
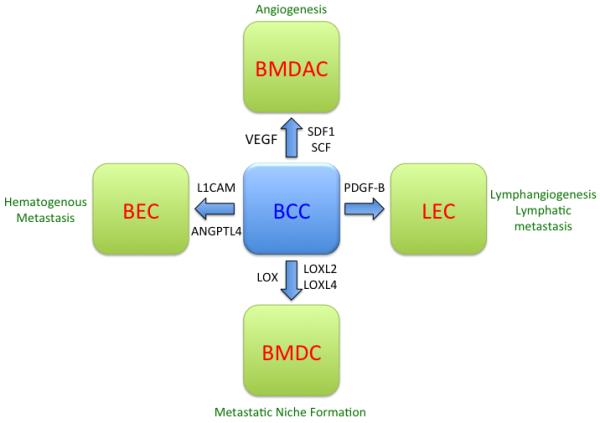
Functional interactions between breast cancer cells and stromal cells that are mediated by the HIF-dependent expression of secreted factors. In response to hypoxia, HIFs are induced and activate transcription of genes encoding angiopoietin-like 4 (ANGPTL4), L1 cell adhesion molecule (L1CAM), lysyl oxidase (LOX), LOX-like 2 (LOXL2), LOXL4, platelet-derived growth factor B (PDGF-B), stem cell factor (SCF), stromal-derived factor 1 (SDF1), and vascular endothelial growth factor (VEGF). These factors promote functional interactions with blood endothelial cells (BECs), lymphatic endothelial cells (LECs) bone marrow-derived angiogenic cells (BMDACs), and other bone marrow-derived cells (BMDCs). These interactions promote angiogenesis, lymphangiogenesis, lymphatic metastasis, metastatic niche formation, and blood vessel (hematogenous) metastasis.
HIF-mediated BEC activation and BMDAC recruitment promotes angiogenesis and primary tumor growth
HIFs have been shown to activate the transcription of multiple angiogenic growth factors and cytokines, including vascular endothelial growth factor (VEGF; also known as VEGF-A), stromal-derived factor 1 (SDF1; also known as CXCL12), angiopoietin 2 (ANGPT2), and stem cell factor (SCF; also known as kit ligand).36-40 VEGF and ANGPT2 play direct roles in angiogenesis by binding to their cognate receptors (VEGFR2 and TIE2, respectively) on BECs, which activates pro-angiogenic signaling programs.41 In addition to the local effects of these factors, angiogenic cytokines such as VEGF, SDF1, and SCF produced by hypoxic cells bind to their cognate receptors (VEGFR2, CXCR4, and CKIT [also known as CD117], respectively) located on the surface of bone marrow cells, which are released into the circulation; these mobilized BMDACs home to the tumor and stimulate vascularization.20,24,42 When immunodeficient mice bearing human prostate cancer xenografts were treated with doxorubicin or acriflavine, the consequences of reduced HIF activity included: decreased VEGF, SDF1, and SCF mRNA levels within the tumor; impaired mobilization of CXCR4+Sca1+, VEGFR2+CD34+, and VEGFR2+CD117+ BMDACs into the circulation; and inhibition of tumor vascularization, thereby impairing tumor growth.20,24
Whereas treatment with VEGF inhibitors selects for cancer cells that induce angiogenesis by VEGF-independent mechanisms,43 treatment with HIF inhibitors blocks all of the major pro-angiogenic signaling pathways. Furthermore, VEGF receptor tyrosine kinase inhibitors induce tumor hypoxia, increase the number of cancer stem cells,44 and promote metastasis,45 which may involve activation of many of the HIF-dependent pathways that are described below.
HIF-mediated LEC proliferation and migration promotes lymphangiogenesis and lymphatic metastasis
Most cancer-related deaths are due to metastasis,46 the systemic dissemination of cancer cells from the site of the primary tumor to one or more distant sites. Cancer cells disseminate systemically by invading blood vessels or lymphatic vessels. In the latter case, cancer cells access blood vessels either within lymph nodes or through the lymphatic ducts, which enter the blood circulation at the junction of the jugular and subclavian veins. Just as cancer cells produce angiogenic factors that bind to BECs stimulate angiogenesis, they also produce lymphangiogenic factors that bind to LECs and stimulate lymphangiogenesis. The greatest attention has been focused on VEGF-C and VEGF-D, which bind to VEGFR3 on the surface of LECs.47 Other secreted factors that have been implicated in lymphangiogenesis include VEGF-A and members of the fibroblast growth factor (FGF), insulin-like growth factor (IGF), and platelet-derived growth factor (PDGF) families.47-49
In the case of breast cancer, most if not all patients with distant metastases have lymph node involvement, which is the most clinically important predictor of patient mortality.49 Although women presenting with lymph node-negative breast cancer have a good prognosis, those with high HIF-1α expression in the primary tumor biopsy have a greatly increased risk of mortality.12 In addition, HIF-1α levels are correlated with VEGF-C expression and peritumoral lymphatic vessel density in breast cancer.50 Taken together, these clinical studies suggest that HIF-1 may promote lymphatic metastasis of breast cancer cells.
An important factor that guides treatment decisions about adjuvant chemotherapy in stage I (lymph node-negative) breast cancer is the presence or absence of high-level expression of the estrogen (ER), progesterone (PR), and HER2neu receptors: women with ER/PR-expressing cancers (~70% of all breast cancers) are treated with tamoxifen or aromatase inhibitors, whereas women with HER2neu-overexpressing tumors (~20%) are treated with the monoclonal antibody trastuzumab.51 Women with triple-negative (i.e. ER−/PR−/HER2−) breast cancer are treated with cytotoxic chemotherapy, but have early recurrence and a poor prognosis,52 indicating a need for novel therapeutic approaches.
To identify lymphangiogenic factors that are induced by hypoxia in breast cancer cells, the triple-negative human breast cancer cell line MDA-MB-231 was incubated for 24 hours in 20% or 1% O2 and analyzed for the expression of mRNA encoding VEGF, PDGF, IGF, and FGF family members.53 Despite the reported association between HIF-1α and VEGF-C levels,50 hypoxia did not induce VEGF-C expression in MDA-MB-231 cells. VEGF-D expression is decreased in breast cancer as compared to normal breast, VEGF-D levels are inversely correlated with lymphatic metastasis54 and, interestingly, VEGF-D mRNA levels decreased ~16-fold in hypoxic MDA-MB-231 cells.53 In contrast, PDGF-B mRNA levels increased more than 4-fold in response to hypoxia in MDA-MB-231 and also in MDA-MB-435, another triple-negative breast cancer cell line,53 which was consistent with a report associating HIF-1α and PDGF-B expression in breast cancer55 and another clinical study linking expression of PDGF-B and PDGF receptor β to lymphatic metastasis in gastric cancer.56 HIF-1 (but not HIF-2) was shown to bind directly to a hypoxia response element located in intron 3 of the human PDGFB gene and activate its transcription.53
Overexpression of PDGF-B in a mouse fibrosarcoma cell line was previously reported to stimulate lymphangiogenesis and lymphatic metastasis through effects on LEC migration and proliferation.57 Conditioned medium from hypoxic breast cancer cells stimulated LEC migration and proliferation, an effect that was lost when HIF or PDGF activity was inhibited by genetic or pharmacologic means and these manipulations also significantly reduced lymphangiogenesis and lymph node metastasis following mammary fat pad implantation of MDA-MB-231 or MDA-MB-435 cells in immunodeficient mice.53 Immunohistochemical analysis of human biopsy sections revealed a highly significant co-localization of HIF-1α and PDGF-B expression in grades 2 and 3, but not in grade 1, breast cancer. Podoplanin+ lymphatic vessel area in the biopsies also correlated with HIF-1α and PDGF-B expression, and scoring for HIF-1α, PDGF-B, and podoplanin was sufficient to predict tumor grade. Taken together, the molecular, cellular, animal, and clinical studies indicate that HIF-1-dependent PDGF-B expression promotes lymphangiogenesis and lymphatic metastasis of hypoxic breast cancer cells. Treatment of tumor-bearing mice with digoxin to inhibit HIF-1α expression dramatically impaired primary tumor growth and lymph node metastasis,53 suggesting that women with HIF-1α+/PDGF-B+ grade 2-3 breast cancers may benefit from addition of a HIF inhibitor to their therapeutic regimen. Clinical trials are needed to test this hypothesis.
HIF-mediated functional interactions between breast cancer cells and BECs promote blood vessel metastasis
The process of blood vessel metastasis can be deconvoluted into several discrete steps (Figure 2): (i) intravasation, in which cancer cells invade through the endothelium of a tumor blood vessel; (ii) circulation, during which cancer cells must survive shear stress and the lack of cell-cell and cell-matrix attachments; (iii) margination, in which a circulating tumor cell arrests at a distant site by adhering to the luminal surface of a BEC; (iv) extravasation, in which the marginated cancer cell invades through the endothelial wall of a capillary at a distant site of metastasis; and (v) colonization, during which a single extravasated cancer cell proliferates to form a metastatic focus. The molecular mechanisms by which intratumoral hypoxia modulates each of these steps, specifically within the context of breast cancer dissemination to the lungs, are considered below. HIF-1 also promotes breast cancer metastasis to bone,58,59 but the underlying mechanisms have not been investigated.
Figure 2.

Deconvolution of steps involved in hematogenous metastasis of cancer cells. The process includes the invasion of a cancer cell into a blood vessel within the primary tumor (intravasation); survival of the cancer cell in the circulation; adherence of the cancer cell to the endothelium of a blood vessel in a distant tissue (margination); migration through the endothelium (extravasation); and cancer cell survival and proliferation at the distant site (colonization), which requires prior metastatic niche formation.
Intravasation
In many cancers, VEGF produced by hypoxic cancer cells play critical roles in promoting increased permeability of tumor blood vessels, thereby facilitating the intravasation of large numbers of cancer cells into the blood circulation.60 Hypoxia-induced ANGPT2 expression may also promote intravasation by reducing the coverage of BECs by pericytes in tumor vessels.40,61 Interactions between tumor-associated macrophages, cancer cells, and BECs appear to be critical for intravasation,62 but it is not known whether hypoxia modulates these interactions.
Circulation (and survival)
Epithelial cells require survival signals generated by interaction of integrins with the extracellular matrix (ECM) and, in the absence of such signals, cells undergo a form of apoptosis known as anoikis.63 Circulating cancer cells acquire resistance to anoikis by a variety of molecular mechanisms.64 Inhibition of ANGPTL4 expression in a skin cancer cell line resulted in increased susceptibility to anoikis and ANGPTL4 was shown to bind and activate integrins, which function as ECM sensors.65 As described below, ANGPTL4 expression is induced by hypoxia in human breast cancer cells and is required for hematogenous metastasis to the lungs following implantation into the mammary fat pad of severe combined immunodeficiency (SCID) mice.17 However, additional studies are required to determine whether ANGPTL4 protects hypoxic breast cancer cells from anoikis. CD24 is a HIF-1 target gene that was recently shown to promote the survival and metastasis of bladder and prostate cancer cells to the lungs after orthotopic, subcutaneous, or intravenous injection into immunodeficient mice,66 but it is not known whether CD24 specifically increases the survival of circulating tumor cells, nor whether it mediates cancer survival and metastasis in breast cancer. Expression of the hyaluronic acid receptor CD44, which is required for the survival of breast cancer cells that have invaded into the lung parenchyma,67 is also induced by hypoxia in a HIF-1-dependent manner.68
One potential source of confusion in understanding the impact of intratumoral hypoxia on vascular metastasis is that once a cancer cell enters the circulation, it is no longer in a hypoxic environment. However, although HIF activity is lost within minutes after reoxygenation,69 the proteins encoded by HIF target genes are much more stable. As a result, the hypoxic phenotype will persist for hours to days after the cancer cell has left the hypoxic microenvironment of the primary tumor and therefore a window exists during which the circulating cancer cell has an increased propensity for metastasis based on its prior exposure. HIF-1α expression can also be driven via O2-independent pathways that are activated by tumor suppressor loss-of-function or oncoprotein gain-of-function,10 which may explain why circulating tumor cells from over half of all breast cancer patients analyzed had detectable HIF-1α protein expression.70
Margination
In order to invade distant tissues, circulating cancer cells must first adhere to the luminal surface of a capillary by interacting with a BEC. This is analogous to margination, the process by which inflammatory cells arrest in the circulation. Exposure of human MDA-MB-231 triple-negative breast cancer cells to hypoxic culture conditions increased their ability to adhere to BECs, due to the HIF-1-dependent expression of the L1 cell adhesion molecule (L1CAM).17 When L1CAM expression was knocked down in these cells, adherence to BECs was inhibited ex vivo and in vivo, and spontaneous metastasis to the lungs following mammary fat pad implantation was dramatically reduced.
Extravasation
Cancer cells must migrate between BECs in order to exit from the capillary in which they have marginated. Exposure of breast cancer cells to hypoxic culture conditions leads to the HIF-mediated expression of ANGPTL4, which inhibits interactions between BECs as determined by measurement of transendothelial electrical resistance.17 Knockdown of ANGPTL4 expression inhibits transendothelial migration of cancer cells ex vivo and extravasation in vivo, and completely blocks spontaneous metastasis to the lungs following mammary fat pad implantation, while having no effect on primary tumor growth or intravasation.17,71 A HIF-1 binding site was identified in the 5′-flanking region of the ANGPTL4 gene and a 56-bp sequence spanning the site functioned as a hypoxia response element in transcriptional reporter assays.17 ANGPTL4 gene expression is significantly increased in the primary breast cancers of women with lung metastases as compared to those who are metastasis-free.71,72 Hypoxia-induced ANGPTL4 also contributes to the ex vivo transendothelial migration and pulmonary metastasis of hepatocellular carcinoma cells.73 It should be noted that the ANGPTL4 polypeptide has a remarkable range of metabolic, vascular, and cytoprotective (as noted above) functions depending upon whether it undergoes oligomerization or cleavage and depending upon the particular proteins with which it interacts.74 Because these processes are likely to be regulated in a cell-specific manner, the functional consequences of ANGPTL4 expression must be determined in a case-by-case manner.
HIF activity in BECs also plays an important role in extravasation. The formation of tumor foci in the lungs following tail vein injection of cancer cells is decreased in mice lacking HIF-1α expression in BECs, whereas loss of HIF-2α expression has the opposite consequence.75 The molecular basis for these effects appears to involve differential regulation of nitric oxide production by BECs: HIF-1 activates transcription of the NOS2 gene encoding inducible nitric oxide synthase, whereas HIF-2 activates transcription of the ARG1 gene encoding arginase, an enzyme that metabolizes arginine, which is one of the substrates required for production of nitric oxide by NOS2. Production of NO by BECs promotes transendothelial migration of cancer cells. Differential HIF-1→NOS2 and HIF-2→ARG1 signaling also occurs in macrophages,76 where it may promote intravasation (see above). These results suggest that even in cancers where HIF-2α overexpression in cancer cells is driving progression, proposed treatment with a HIF-2α-selective inhibitor77 may not be desirable, since it might stimulate pro-metastatic properties of BECs.
Colonization (and the role of metastatic niche formation)
Primary breast tumors secrete factors that remodel tissue at the site of subsequent metastases, a process known as metastatic niche formation because the remodeling is required to create a microenvironment that allows the survival and proliferation of metastatic cancer cells. These changes include alterations of the ECM (fibronectin deposition) and the recruitment of bone marrow-derived cells (BMDCs).78 Studies in xenograft models of melanoma and lung cancer indicated that production of VEGF-A, transforming growth factor-β, and tumor necrosis factor-α by the primary tumor induced the expression of the inflammatory proteins S100A8 and S100A9 in the lung parenchyma, which served as a signal for recruitment of Mac1+ (CD11b+) BMDCs.79 The effect of S100A8 and S100A9 was later shown to be indirect: they induced the pulmonary expression of serum amyloid A3, which bound directly to toll-like receptor 4 on BMDCs.80 Metastatic cancer cells homed specifically to areas of the lung that were populated by BMDCs.78
In addition to providing homing signals for the cancer cells, BMDCs in the metastatic niche may also serve as a source of growth/survival factors, angiogenic factors, and immunosuppressive factors, which are all required for clonal expansion of the cancer cell into a secondary tumor. It is likely that a functionally heterogeneous population of BMDCs is recruited to the metastatic niche, either in parallel or in series.
Subsequent studies demonstrated that the crosslinking of collagen fibers in the lungs by lysyl oxidase (LOX), which was secreted by primary breast tumors, was required for the recruitment of BMDCs.81 Expression of LOX was induced by hypoxia in breast cancer cells in a HIF-1-dependent manner,82 providing another mechanism by which intratumoral hypoxia promotes metastasis. LOX was found to colocalize with fibronectin in the lungs of tumor-bearing mice, prior to the arrival of CD11b+ BMDCs.81 Metastatic niche formation in the lungs of SCID mice showed a strikingly ordered and rapid progression following implantation of MDA-MB-435 breast cancer cells in the mammary fat pad: collagen crosslinking was observed by day 8, BMDC recruitment by day 16, and metastatic cancer cells were detected by day 24.18 This process is not an artifact of implanting human cancer cells into immunodeficient mice, as it was also observed after implantation of 4T1 mouse breast cancer cells into syngeneic immunocompetent hosts.81
In addition to LOX a family of LOX-like (LOXL) proteins also participate in metastatic niche formation. In different metastatic breast cancer cell lines, the expression of LOX, LOXL2, or LOXL4 was induced by hypoxia: in MDA-MB-231 cells HIF-1 and HIF-2 activate transcription of the LOX and LOXL4 genes, whereas in MDA-MB-435 cells HIF-1 alone activates LOXL2 expression.18 Analysis of human breast cancer biopsies also revealed heterogeneous expression of LOX/LOXL family members. Knockdown of LOX or LOXL4 in MDA-MB-231 cells and knockdown of LOXL2, but not LOX, in MDA-MB-435 cells blocked metastatic niche formation.18,81 Treatment of tumor-bearing mice with digoxin or acriflavine, which are drugs that inhibit HIF activity, blocked metastatic niche formation.21 Although studies in mouse models described above implicated HIF-dependent expression of LOX/LOXL family members in breast cancer cells, analysis of the Oncomine database revealed increased expression of LOXL2 and LOXL4 within stromal cells of invasive breast cancer as compared to normal breast tissue.21 Finally, it should be noted that in addition to the effects of LOX family members on ECM in the lung, these same proteins also remodel the ECM within the primary tumor and thereby facilitate the local invasion of cancer cells.21,82,83
Clinical implications
In the case of breast cancer, intratumoral hypoxia exerts a profound effect on cancer cells through the HIF-mediated production of secreted factors that mediate functional interactions with BMDACs, BECs, LECs, and BMDCs, which drive key aspects of cancer progression including angiogenesis, lymphangiogenesis, lymphatic metastasis, metastatic niche formation, and blood vessel metastasis (Figure 1). These recent findings provide direct molecular mechanisms underlying the clinical observation that intratumoral hypoxia promotes metastasis and patient mortality. The demonstration that HIFs play a master regulatory role in activating the transcription of the genes encoding all of the secreted factors involved in these processes provides a scientific foundation for the inclusion of HIF inhibitors in cancer therapy regimens, particularly in women with breast cancer who do not (yet) have lymph node involvement but have high HIF-1α levels in their primary tumor biopsy, which places them at greatly increased risk of cancer death.12 In addition, it should be noted that many of the studies described above were performed with the triple-negative breast cancer cell lines MDA-MB-231 and MDA-MB-435. Triple-negative breast cancer is currently treated with cytotoxic chemotherapy with rapid development of drug resistance and poor long-term survival.52 Combined treatment with digoxin and anthracycline chemotherapy increases primary tumor control in mouse xenograft models,17 suggesting that patients with triple-negative breast cancer may also benefit from the addition of digoxin or another HIF inhibitor to their therapeutic regimen.
Perspective and commentary
There are many other stromal cell types that interact with cancer cells to promote metastasis, including fibroblasts, immune cells (lymphocytes, macrophages, mast cells, neutrophils), and mesenchymal stem cells.3 Additional studies are required to investigate whether hypoxia induces HIF-mediated functional interactions between these stromal cells and cancer cells that also promote metastasis. It is striking that > 90% of cancer deaths are attributable to metastasis, yet < 3% of the National Cancer Institute (NCI) budget is devoted to investigating the cellular and molecular mechanisms of cancer metastasis.84 The profusion of clinical and experimental data linking intratumoral hypoxia to metastasis and patient mortality also stands in striking contrast to the paucity of NCI-funded initiatives that specifically target hypoxic cancer cells for therapy.
Acknowledgments
Cancer research in the author’s laboratory is supported by grants from the American Cancer Society, National Cancer Institute (U54-CA143868; Johns Hopkins Physical Sciences-Oncology Center), Susan G. Komen Breast Cancer Foundation, and the Johns Hopkins Institute for Cell Engineering. G.L.S. is the C. Michael Armstrong Professor at the Johns Hopkins University School of Medicine and an American Cancer Society Research Professor.
Footnotes
Conflict of interest
The author declares no conflict of interest.
References
- 1.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polyak K, Kalluri R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb Perspect Biol. 2010;2:a003244. doi: 10.1101/cshperspect.a003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 4.Höckel M, Schlenger K, Aral B, Mitze M, Schäfer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56:4509–4515. [PubMed] [Google Scholar]
- 5.Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR, et al. Tumor oxygenation predicts the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996;56:941–943. [PubMed] [Google Scholar]
- 6.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 7.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 9.Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaupel P, Mayer A, Höckel M. Tumor hypoxia and malignant progression. Meth Enzymol. 2004;381:335–354. doi: 10.1016/S0076-6879(04)81023-1. [DOI] [PubMed] [Google Scholar]
- 12.Bos R, van der Groep P, Greijer AE, Shvarts A, Meijer S, Pinedo HM, et al. Levels of hypoxia-inducible factor-1α independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer. 2003;97:1573–1581. doi: 10.1002/cncr.11246. [DOI] [PubMed] [Google Scholar]
- 13.Dales JP, Garcia S, Meunier-Carpentier S, Andrac-Meyer L, Haddad O, Lavaut MN, et al. Overexpression of hypoxia-inducible factor HIF-1α predicts early relapse in breast cancer: retrospective study in a series of 745 patients. Int J Cancer. 2005;116:734–739. doi: 10.1002/ijc.20984. [DOI] [PubMed] [Google Scholar]
- 14.Vergis R, Corbishley CM, Norman AR, Bartlett J, Jhavar S, Borre M, et al. Intrinsic markers of tumor hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: a retrospective analysis of two randomised radiotherapy trials and one surgical cohort study. Lancet Oncol. 2008;9:342–351. doi: 10.1016/S1470-2045(08)70076-7. [DOI] [PubMed] [Google Scholar]
- 15.Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- 16.Löfstedt T, Fredlund E, Holmquist-Mengelbier L, Pietras A, Ovenberger M, Poellinger L, et al. Hypoxia-inducible factor 2α in cancer. Cell Cycle. 2007;6:919–926. doi: 10.4161/cc.6.8.4133. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Wong CC, Wei H, Gilkes DM, Korangath P, Chaturvedi P, et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31:1757–1770. doi: 10.1038/onc.2011.365. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Wong CC, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci USA. 2011;108:16369–16374. doi: 10.1073/pnas.1113483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imamura T, Kikuchi H, Herraiz MT, Park DY, Mizukami Y, Mino-Kenduson M, et al. HIF-1α and HIF-2α have divergent roles in colon cancer. Int J Cancer. 2009 doi: 10.1002/ijc.24032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci USA. 2009;106:17910–17915. doi: 10.1073/pnas.0909353106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Wong CC, Zhang H, Gilkes DM, Chen J, Wei H, Chaturvedi P, et al. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J Mol Med (Berl) 2012;90:803–815. doi: 10.1007/s00109-011-0855-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luwor RB, Lu Y, Li X, Mendelsohn J, Fan Z. The antiepidermal growth factor receptor monoclonal antibody cetuximab/C225 reduces hypoxia-inducible factor-1α, leading to transcriptional inhibition of vascular endothelial growth factor expression. Oncogene. 2005;24:4433–4441. doi: 10.1038/sj.onc.1208625. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc Natl Acad Sci USA. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee K, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL. Anthracycline chemotherapy inhibits HIF-1 transcriptional activity and tumor-induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci U S A. 2009;106:2353–2358. doi: 10.1073/pnas.0812801106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1α-degradative pathway. J Biol Chem. 2002;277:29936–29944. doi: 10.1074/jbc.M204733200. [DOI] [PubMed] [Google Scholar]
- 26.Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF-1α and is required for O2-independent and HSP90 inhibitor-induced degradation of HIF-1α. Mol Cell. 2007;25:207–217. doi: 10.1016/j.molcel.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian DZ, Kachhap SK, Collis SJ, Verheul HM, Carducci MA, Atadja P, et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1α. Cancer Res. 2006;66:8814–8821. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 28.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu M, Howes A, Lesperance J, Stallcup WB, Hauser CA, Kadoya K, et al. Antitumor activity of rapamycin in a transgenic mouse model of ErbB2-dependent human breast cancer. Cancer Res. 2005;65:5325–5336. doi: 10.1158/0008-5472.CAN-04-4589. [DOI] [PubMed] [Google Scholar]
- 30.Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–127. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 31.Chintala S, Najrana T, Toth K, Cao S, Durrani FA, Pili R, et al. Prolyl hydroxylase 2 dependent and von Hippel-Lindau independent degradation of hypoxia-inducible factor 1 and 2 α by selenium in clear cell renal cell carcinoma leads to tumor growth inhibition. BMC Cancer. 2012;12:293. doi: 10.1186/1471-2407-12-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62:4316–4324. [PubMed] [Google Scholar]
- 33.Takano S, Kamiyama H, Mashiko R, Osuka S, Ishikawa E, Matsumura A. Metronomic treatment of malignant glioma xenografts with irinotecan (CPT-11) inhibits angiogenesis and tumor growth. J Neurooncol. 2010;99:177–185. doi: 10.1007/s11060-010-0118-8. [DOI] [PubMed] [Google Scholar]
- 34.Kummar S, Raffeld M, Juwara L, Horneffer Y, Strassberger A, Allen D, et al. Multihistology, target-driven pilot trial of oral topotecan as an inhibitor of hypoxia-inducible factor-1α in advanced solid tumors. Clin Cancer Res. 2011;17:5123–5131. doi: 10.1158/1078-0432.CCR-11-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- 38.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 39.Bosch-Marcé M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, et al. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res. 2007;101:1310–1318. doi: 10.1161/CIRCRESAHA.107.153346. [DOI] [PubMed] [Google Scholar]
- 40.Simon MP, Tournaire R, Pouyssegur J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J Cell Physiol. 2008;217:809–818. doi: 10.1002/jcp.21558. [DOI] [PubMed] [Google Scholar]
- 41.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 42.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegué E, et al. HIF-1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010;21:21–26. doi: 10.1016/j.cytogfr.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8:210–221. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fidler IJ. Critical determinants of cancer metastasis: rationale for therapy. Cancer Chemother Pharmacol. 1999;43:S3–S10. doi: 10.1007/s002800051091. [DOI] [PubMed] [Google Scholar]
- 47.Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 48.Cao Y. Emerging mechanisms of tumor lymphangiogenesis and lymphatic metastasis. Nat Rev Cancer. 2005;5:735–743. doi: 10.1038/nrc1693. [DOI] [PubMed] [Google Scholar]
- 49.Ran S, Volk L, Flister MJ. Lymphangiogenesis and lymphatic metastasis in breast cancer. Pathophysiology. 2010;17:229–251. doi: 10.1016/j.pathophys.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoppmann SF, Fenzl A, Schindl M, Bachleitner-Hofmann T, Nagy K, Gnant M, et al. Hypoxia inducible factor-1α correlates with VEGF-C expression and lymphangiogenesis in breast cancer. Breast Cancer Res Treat. 2006;99:135–141. doi: 10.1007/s10549-006-9190-3. [DOI] [PubMed] [Google Scholar]
- 51.Schwartz GF, Reis-Fihlo J, Pusztai L, Fentiman IS, Holland R, Bartelink H, et al. Adjuvant therapy in stage I carcinoma of the breast: the influence of multigene analyses and molecular phenotyping. Cancer. 2012;118:2031–2038. doi: 10.1002/cncr.27431. [DOI] [PubMed] [Google Scholar]
- 52.Pal SK, Childs BH, Pegram M. Triple negative breast cancer: unmet medical needs. Breast Cancer Res Treat. 2011;125:627–636. doi: 10.1007/s10549-010-1293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schito L, Rey S, Tafani M, Zhang H, Wong CC, Russo A, et al. Hypoxia-inducible factor 1-dependent expression of platelet-derived growth factor B promotes lymphatic metastasis of hypoxic breast cancer cells. Proc Natl Acad Sci USA. 2012;109:E2707–E2716. doi: 10.1073/pnas.1214019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koyama Y, Kaneko K, Akazawa K, Kanbayashi C, Kanda T, Hatakeyama K. Vascular endothelial growth factor-C and vascular endothelial growth factor-D messenger RNA expression in breast cancer: association with lymph node metastasis. Clin Breast Cancer. 2003;4:354–360. doi: 10.3816/cbc.2003.n.041. [DOI] [PubMed] [Google Scholar]
- 55.Bos R, van Diest PJ, de Jong JS, van der Groep P, van der Valk P, van der Wall E. Hypoxia-inducible factor-1α is associated with angiogenesis, and expression of bFGF, PDGF-BB, and EGFR in invasive breast cancer. Histopathology. 2005;46:31–36. doi: 10.1111/j.1365-2559.2005.02045.x. [DOI] [PubMed] [Google Scholar]
- 56.Kodama M, Kitadai Y, Sumida T, Ohnishi M, Ohara E, Tanaka M, et al. Expression of platelet-derived growth factor (PDGF)-B and PDGF-receptor β is associated with lymphatic metastasis in human gastric carcinoma. Cancer Sci. 2010;101:1984–1989. doi: 10.1111/j.1349-7006.2010.01639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao R, Björndahl MA, Religa P, Clasper S, Garvin S, Galter D, et al. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell. 2004;6:333–345. doi: 10.1016/j.ccr.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 58.Hiraga T, Kizaka-Kondoh S, Hirota K, Hiraoka M, Yoneda T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 2007;67:4157–4163. doi: 10.1158/0008-5472.CAN-06-2355. [DOI] [PubMed] [Google Scholar]
- 59.Dunn LK, Mohammad KS, Fournier PG, McKenna CR, Davis HW, Niewolna M, et al. Hypoxia and TGF-β drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS One. 2009;4:e6896. doi: 10.1371/journal.pone.0006896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol. 1999;237:97–132. doi: 10.1007/978-3-642-59953-8_6. [DOI] [PubMed] [Google Scholar]
- 61.Falcón BL, Hashizume H, Koumoutsakos P, Chou J, Bready JV, Coxon A, et al. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am J Pathol. 2009;175:2159–2170. doi: 10.2353/ajpath.2009.090391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 63.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim YN, Koo KH, Sung JY, Yun UJ, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. Int J Cell Biol. 2012;2012:306879. doi: 10.1155/2012/306879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu P, Tan MJ, Huang RL, Tan CK, Chong HC, Pal M, et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2−:H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell. 2011;19:401–415. doi: 10.1016/j.ccr.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 66.Thomas S, Harding M, Smith SC, Overdevest JB, Nitz MD, Frierson HF, Jr, et al. CD24 is an effector of HIF-1 driven primary tumor growth and metastasis. Cancer Res. 2012;72:5600–5612. doi: 10.1158/0008-5472.CAN-11-3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu Q, Toole BP, Stamenkovic I. Induction of apoptosis of metastatic mammary carcinoma cells in vivo by disruption of tumor cell surface CD44 function. J Exp Med. 1997;186:1985–1996. doi: 10.1084/jem.186.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krishnamachary B, Penet MF, Nimmagadda S, Mironchik Y, Raman V, Solaiyappan M, et al. Hypoxia regulates CD44 and its variant isoforms through HIF-1α in triple negative breast cancer. PLoS One. 2012;7:e44078. doi: 10.1371/journal.pone.0044078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268:21513–21518. [PubMed] [Google Scholar]
- 70.Kallergi G, Markomanolaki H, Giannoukaraki V, Papadaki MA, Strati A, Lianidou ES, et al. Hypoxia-inducible factor-1α and vascular endothelial growth factor expression in circulating tumor cells of breast cancer patients. Breast Cancer Res. 2009;11:R84. doi: 10.1186/bcr2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H, Ge C, Zhao F, Yan M, Hu C, Jia D, et al. Hypoxia-inducible factor 1α-activated angiopoietin-like protein 4 contributes to tumor metastasis via vascular cell adhesion molecule-1/integrin β1 signaling in human hepatocellular carcinoma. Hepatology. 2011;54:910–919. doi: 10.1002/hep.24479. [DOI] [PubMed] [Google Scholar]
- 74.Tan MJ, Teo Z, Sng MK, Zhu P, Tan NS. Emerging roles of angiopoietin-like 4 in human cancer. Mol Cancer Res. 2012;10:677–688. doi: 10.1158/1541-7786.MCR-11-0519. [DOI] [PubMed] [Google Scholar]
- 75.Branco-Price C, Zhang N, Schnelle M, Evans C, Katschinski DM, Liao D, et al. Endothelial cell HIF-1α and HIF-2α differentially regulate metastatic success. Cancer Cell. 2012;21:52–65. doi: 10.1016/j.ccr.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, et al. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Keith B, Johnson RS, Simon MC. HIF-1α and HIF-2α: sibling rivalry in hypoxic tumor growth and progression. Nat Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive hematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumor-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8:1369–1375. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 80.Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 81.Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15:35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Erler JT, Bennewith KL, Nicolau M, Dornhöfer N, Kong C, Le QT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 83.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.National Foundation for Cancer Research 2012 http://www.nfcr.org/?option=com_content&view=article&id=1041