Phase II Study of the MEK1/MEK2 Inhibitor Trametinib in Patients With Metastatic BRAF-Mutant Cutaneous Melanoma Previously Treated With or Without a BRAF Inhibitor (original) (raw)
Abstract
Purpose
BRAF mutations promote melanoma cell proliferation and survival primarily through activation of MEK. The purpose of this study was to determine the response rate (RR) for the selective, allosteric MEK1/MEK2 inhibitor trametinib (GSK1120212), in patients with metastatic _BRAF_-mutant melanoma.
Patients and Methods
This was an open-label, two-stage, phase II study with two cohorts. Patients with metastatic _BRAF_-mutant melanoma previously treated with a BRAF inhibitor (cohort A) or treated with chemotherapy and/or immunotherapy (BRAF-inhibitor naive; cohort B) were enrolled. Patients received 2 mg of trametinib orally once daily.
Results
In cohort A (n = 40), there were no confirmed objective responses and 11 patients (28%) with stable disease (SD); the median progression-free survival (PFS) was 1.8 months. In cohort B (n = 57), there was one (2%) complete response, 13 (23%) partial responses (PRs), and 29 patients (51%) with SD (confirmed RR, 25%); the median PFS was 4.0 months. One patient each with BRAF K601E and BRAF V600R had prolonged PR. The most frequent treatment-related adverse events for all patients were skin-related toxicity, nausea, peripheral edema, diarrhea, pruritis, and fatigue. No cutaneous squamous cell carcinoma was observed.
Conclusion
Trametinib was well tolerated. Significant clinical activity was observed in BRAF-inhibitor–naive patients previously treated with chemotherapy and/or immunotherapy. Minimal clinical activity was observed as sequential therapy in patients previously treated with a BRAF inhibitor. Together, these data suggest that BRAF-inhibitor resistance mechanisms likely confer resistance to MEK-inhibitor monotherapy. These data support further evaluation of trametinib in BRAF-inhibitor–naive _BRAF_-mutant melanoma, including rarer forms of _BRAF_-mutant melanoma.
INTRODUCTION
Mutations in the serine/threonine protein kinase B-Raf (BRAF) are common in cutaneous melanoma, occurring in 40% to 60% of patients.1–3 Approximately 70% to 90% of these mutations cause a substitution of glutamic acid for valine (V600E), and an additional 10% to 30% cause a substitution of lysine for valine (V600K); rare BRAF mutations occur in approximately 6% to 7% of _BRAF_-mutated melanoma.4–7 BRAF mutations constitutively activate MEK and ERK1/ERK2 proteins, which are downstream of BRAF in the mitogen-activated protein kinase (MAPK) pathway.2
Recently, two BRAF inhibitors, vemurafenib (PLX4032/RG7204) and dabrafenib (GSK2118436), have demonstrated response rates (RRs) of approximately 50% to 60% and progression-free survival (PFS) benefit over dacarbazine in _BRAF V600E_–mutant melanoma3,8,9; vemurafenib has also shown a superior overall survival (OS).3,8,10 These data confirm that _BRAF V600E_–mutated tumors are responsive to BRAF-inhibitor therapy; however, resistance to BRAF-inhibitor therapy is common and rapidly acquired in the majority of patients.8,9 To the best of our knowledge, no MEK inhibitor has demonstrated significant clinical activity in _BRAF_-mutated melanoma in the phase II setting; selumetinib (AZD6244) had an RR of only 10% in _BRAF_-mutant melanoma,11 and PD0325901 was poorly tolerated.12,13 In vitro studies and analyses of predose and postprogression tumor biopsies in clinical trials have shown both MEK-dependent and MEK-independent resistance following exposure to a BRAF inhibitor.14–20 Because MEK is the downstream effector of BRAF, MEK inhibition is an attractive mechanism for blocking activation of the MAPK pathway and could also potentially block reactivation of the MAPK pathway in BRAF-inhibitor–resistant disease.19
Trametinib is a reversible, selective, allosteric inhibitor of MEK1/MEK2 activation and kinase activity, with a half-maximal inhibitory concentration of 0.7 to 14.9 nmol/L for MEK1/MEK2.21 In vitro studies have demonstrated that trametinib decreases cell proliferation, causes G1 cell cycle arrest, and induces apoptosis.21 In the first-in-human (FIH) study of trametinib (Open-label Study to Investigate the Safety, PK, and Pharmacodynamics of GSK1120212 in Subjects With Solid Tumors or Lymphoma [NCT00687622]), 2 mg administered once daily was selected as the recommended phase II dose on the basis of safety, pharmacokinetic, pharmacodynamic, and efficacy data.22 In this study, trametinib also demonstrated a 33% confirmed RR among 30 patients with _BRAF_-mutant melanoma.23 Moreover, in six other patients with _BRAF_-mutant melanoma who were previously treated with BRAF-inhibitor therapy, one patient with an unconfirmed partial response (PR) and four patients with stable disease (SD) were observed. These data supported further analysis of trametinib in two patient populations: patients who previously received a BRAF inhibitor and those who did not.
The results of a phase III study demonstrating improved rates of both PFS and OS for trametinib compared with chemotherapy has recently been reported.24 Here we report the results of a multicenter phase II study designed to evaluate the clinical efficacy of trametinib monotherapy in patients with metastatic _BRAF_-mutant melanoma previously treated either with or without a BRAF inhibitor. Long-term follow-up data are also included.
PATIENTS AND METHODS
Patient Selection
Key eligibility criteria included metastatic cutaneous _BRAF_-mutant melanoma (amended later to allow BRAF V600E, V600K, or V600D mutations only), 18 years of age or older, Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2 (amended to 0 to 1), adequate organ function, and one or more prior systemic therapies. Patients with brain metastasis previously treated with surgery or stereotactic radiosurgery and with confirmed SD for ≥ 8 weeks were allowed. Prior MEK-inhibitor therapy, a history of predisposing factors to retinal vein occlusion (RVO) or central serous retinopathy (CSR), and a cardiac symptom or event within 24 weeks were exclusion criteria. All patients provided written informed consent, and the protocol was approved by local ethics committees.
Study Design
This was an open-label, two-cohort, multicenter, phase II study to evaluate the clinical activity of trametinib in patients with _BRAF_-mutant metastatic melanoma (Fig 1). The protocol was approved by the institutional review board at each participating institution. Patients were enrolled onto one of two cohorts: cohort A, previously treated with a BRAF inhibitor (either vemurafenib or dabrafenib) or cohort B, previously treated with chemotherapy and/or immunotherapy but not with a BRAF inhibitor.
Fig 1.

Trial design.
Patients received 2 mg of oral trametinib once daily. Safety assessments and laboratory tests were performed predose on day 15, day 28, and every 4 weeks thereafter; laboratory tests were also performed on day 8. ECGs were obtained predose and every 4 weeks after the first dose. An echocardiogram or multiple-gated acquisition scan was performed predose, at week 4, and every 12 weeks thereafter (or more frequently, if clinically indicated). Blood samples for pharmacokinetic analysis were collected on day 15 (predose, 0.5 to 2, 2 to 4, and 4 to 8 hours postdose) and predose at weeks 4, 8, and 12 (Appendix Fig A1, online only).
Disease assessment was performed at baseline and every 8 weeks according to Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST).25 A planned interim analysis for futility was performed approximately 12 weeks after enrollment of the thirtieth patient in each cohort, which was 4 weeks after the first tumor assessment.
The severity of toxicity was graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0.26 Patients were allowed to enroll on the basis of BRAF testing results obtained from local laboratories. However, tumor sample submission to a central laboratory (Response Genetics, Inc [RGI], Los Angeles, CA) was required before enrollment. Tissue submitted to RGI was evaluated by using an allele-specific polymerase chain reaction assay that identifies BRAF V600E and V600K mutations. Genotyping reports of BRAF V600D from local laboratories and discrepant results between local laboratories and RGI were to be further evaluated by DNA sequence analysis at RGI; no reports of BRAF V600D were received.
Statistical Analysis
The primary end point for both cohorts was objective RR as determined by the investigator; each cohort was assessed separately. The null hypothesis was an RR of ≤ 10%; the alternative hypothesis was an RR of ≥ 25%. To allow early termination for futility, a two-stage Green-Dahlberg design was used.27 If fewer than three responses were observed in the first 30 patients at the interim analysis, enrollment in that cohort would be terminated. If three or more responses were observed, enrollment would continue to 55 patients to achieve the desired type I (< 5%) and type II (> 90%) error rates. PFS was defined as the interval between the date of first dose and the date of disease progression or death, whichever occurred first. PFS and OS were summarized descriptively by using Kaplan-Meier medians and quartiles. Summary statistics were reported for plasma trametinib concentrations by cohort and the time of assessments.
RESULTS
Patient Characteristics
Between December 2009 and December 2010, a total of 97 patients were enrolled (cohort A, n = 40; cohort B, n = 57). Patient baseline characteristics (Table 1) were similar for both cohorts except for prior therapy due to trial design. Seventy percent were male and all but one had an ECOG performance status of 0 or 1. All patients except one had stage IV melanoma, of which 75% had American Joint Committee on Cancer (AJCC) stage M1c disease. Pre-existent treated brain metastases were reported for 13% of cohort A and 21% of cohort B. The frequency of V600E (81%) and V600K (12%) mutations was within the expected range. A K601E mutation was identified in two patients; four patients had discrepant results from the same tumor tissue between local laboratories and RGI (Table 1). Three patients in each cohort had received prior ipilimumab treatment; these six patients also had received prior chemotherapy. In cohort A, all patients had received either dabrafenib or vemurafenib before study enrollment. Among these patients, approximately 50% had received BRAF-inhibitor therapy for less than 24 weeks. For cohort A, 50% of the patients received three or more prior therapies, and in cohort B, 38% received chemotherapy and 19% received immunotherapy in the advanced/metastatic setting.
Table 1.
Baseline Patient Characteristics
| Characteristic | Cohort A (n = 40) | Cohort B (n = 57) | All Patients (N = 97) | |||
|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | |
| Age, years, | ||||||
| Mean | 55.6 | 54.0 | 54.7 | |||
| Median | 58.0 | 54.0 | 55.0 | |||
| Range | 23–76 | 26–79 | 23–79 | |||
| Male sex | 25 | 63 | 43 | 75 | 68 | 70 |
| ECOG performance status* | ||||||
| 0 | 19 | 48 | 42 | 74 | 61 | 63 |
| 1 | 20 | 50 | 15 | 26 | 35 | 36 |
| Stage | ||||||
| IIIc | 0 | 1 | 2 | 1 | 1 | |
| M1a | 5 | 13 | 7 | 12 | 12 | 12 |
| M1b | 6 | 15 | 6 | 11 | 12 | 12 |
| M1c | 29 | 73 | 43 | 75 | 72 | 74 |
| Serum LDH at baseline | ||||||
| ≤ ULN | 15 | 38 | 32 | 56 | 47 | 48 |
| > ULN | 22 | 55 | 24 | 42 | 46 | 47 |
| Unknown | 3 | 8 | 1 | 2 | 4 | 4 |
| No. of prior systemic therapies | ||||||
| 1-2 | 20 | 50 | 50 | 88 | 70 | 72 |
| ≥ 3 | 20 | 50 | 7 | 12 | 27 | 28 |
| Chemotherapy | 25 | 63 | 49 | 86 | 74 | 76 |
| Immunotherapy | 17 | 43 | 31 | 54 | 48 | 49 |
| Both chemotherapy and immunotherapy† | 13 | 33 | 23 | 40 | 36 | 37 |
| BRAF mutation status | ||||||
| V600E | 33 | 83 | 46 | 81 | 79 | 81 |
| V600K | 4 | 10 | 8 | 14 | 12 | 12 |
| K601E | 0 | 1 | 2 | 1 | 1 | |
| K601E/V600E‡ | 1 | 3 | 0 | 1 | 1 | |
| V600E/V600K§ | 1 | 3 | 1 | 2 | 2 | 2 |
| V600K/V600R¶ | 0 | 1 | 2 | 1 | 1 | |
| Unknown∥ | 1 | 3 | 0 | 1 | 1 | |
| Prior BRAF inhibitors | ||||||
| Vemurafenib | 23 | 58 | N/A | |||
| Dabrafenib | 17 | 43 | N/A | |||
| Duration of prior BRAF inhibitor, weeks | ||||||
| < 24 | 19 | 48 | N/A | |||
| ≥ 24 | 19 | 48 | N/A | |||
| Unknown | 2 | 5 | N/A | |||
| Best response during prior BRAF inhibitor therapy | ||||||
| CR | 2 | 5 | N/A | |||
| PR | 16 | 40 | N/A | |||
| SD | 10 | 25 | N/A | |||
| PD | 6 | 15 | N/A | |||
| Unknown | 6 | 15 | N/A |
Treatment
The median time receiving study drug was 56 days for cohort A and 120 days for cohort B. Across both arms, 80% of patients discontinued study treatment because of disease progression, and four patients (4%) discontinued study treatment because of adverse events (AEs; two ejection fraction decreases, one intestinal perforation, and one pulmonary embolism).
Efficacy at Interim Analysis
There were no responses at the interim analysis among the first 30 patients enrolled in cohort A; enrollment was therefore terminated due to futility. (Ten additional patients were enrolled because they had given consent before the thirtieth patient was dosed.) Among the 30 patients of cohort B in the first stage, six PRs were observed at the interim analysis. Consequently, enrollment continued toward a target of 55 patients. (Two additional patients were enrolled because they had given consent before the fifty-fifth patient was dosed.)
Overall Efficacy
The median follow-up at the planned analysis was 12 months for cohort A and 10 months for cohort B. Table 2 summarizes the clinical efficacy in both cohorts. None of the 40 patients in cohort A had a confirmed clinical response at the time of data cutoff, although eight patients (20%) experienced tumor reduction (Fig 2A). The best unconfirmed response was one complete response (CR; 3%), one PR (3%), and 11 SD (28%); five patients (13%) received trametinib for ≥ 16 weeks. The patient with a CR at week 8 progressed at week 16, and the patient with a PR, which became a confirmed PR after the data cutoff, was ongoing at week 44; both of these patients had discontinued prior BRAF-inhibitor therapy after less than 24 weeks of treatment because of an AE rather than because of disease progression. In contrast, 36 patients had discontinued BRAF-inhibitor therapy because of disease progression. The median PFS for this cohort was 1.8 months (95% CI, 1.8 to 2.0 months; Fig 3). The median OS for cohort A was 5.8 months (95% CI, 4.1 to 9.0 months).
Table 2.
Clinical Efficacy
| Cohort | No. of Patients | Confirmed RR (CR + PR) | Median PFS | Median OS | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Outcome | RR (%) | 95% CI | ||||||||
| CR | PR | SD | RR (months) | 95% CI | RR (months) | 95% CI | ||||
| A | 40 | 0 | 0 | 11 | 0 | 1.8 | 1.8 to 2.0 | 5.8 | 4.1 to 9.0 | |
| B | 57 | 1 | 13 | 29 | 25 | 14.1 to 37.8 | 4.0 | 3.6 to 5.6 | 14.2 | 11.3 to N/R |
| BRAF V600E without prior brain metastases | 36 | 1 | 9 | 20 | 28 | 14.2 to 45.2 | 5.3 | 3.6 to 7.4 | N/R |
Fig 2.
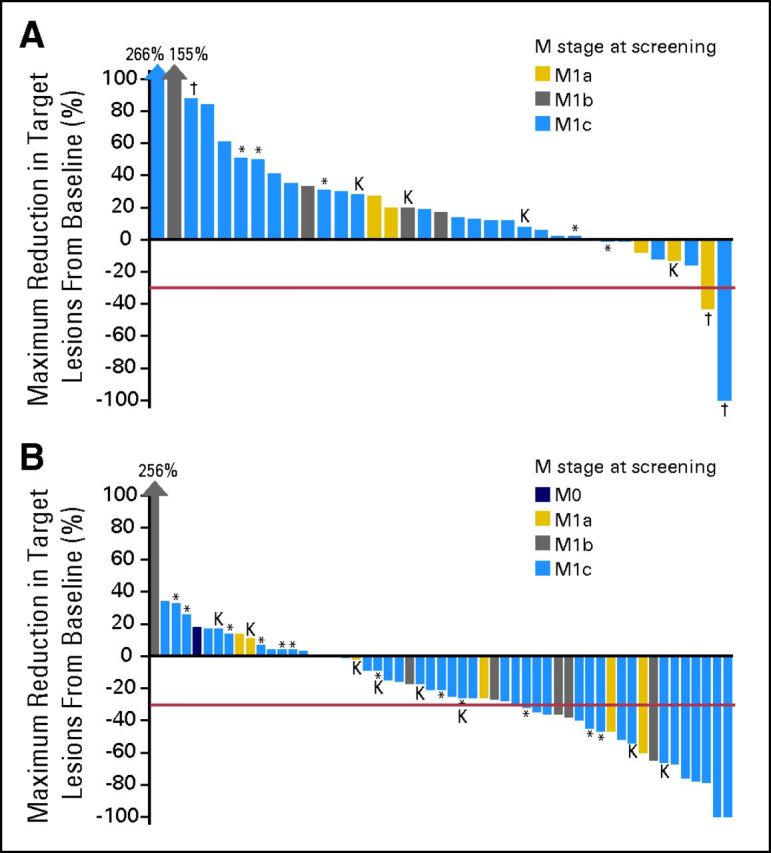
Best percentage change from baseline in target lesions for (A) cohort A (radiographic scan data was incomplete or unavailable for 5 patients) and (B) cohort B (radiographic scan data was incomplete or unavailable for two patients). Staging was according to the American Joint Committee on Cancer (AJCC) system. (*) Patients with prior brain metastases; (†) patients who discontinued prior BRAF inhibitor because of toxicity; (K) patients with the V600K mutation.
Fig 3.

Kaplan-Meier estimated survival for all patients by cohort: (A) progression-free survival (PFS) for cohort A, (B) PFS for cohort B, (C) overall survival (OS) for cohort A, and (D) OS for cohort B. The bars represent the date of the last adequate tumor assessment before the data cutoff among patients with censored data without disease progression or death.
Among the 57 patients in cohort B, the best confirmed responses were one CR (2%) and 13 PRs (23%), resulting in a best overall RR of 25% (95% CI, 14.1% to 37.8%). The disease control rate (CR + PR + SD/total number of patients) was 75%, and tumor reduction was observed in 37 patients (65%; Fig 2B). Eighteen patients (32%) were treated for ≥ 24 weeks without disease progression. On the basis of this RR, the null hypothesis was rejected in favor of the alternative hypothesis. The median PFS was 4.0 months (95% CI, 3.6 to 5.6 months; Fig 3), and the median duration for response was 5.7 months (95% CI, 3.7 to 9.2 months). Among the 12 patients (21%) with a history of brain metastases, five (42%) had disease progression in the brain or the CNS. Subgroup analysis of patients with a BRAF V600E mutation and without prior brain metastases showed a confirmed RR of 28% (95% CI, 14.2% to 45.2%) and median PFS of 5.3 months (95% CI, 3.6 to 7.4 months).
There were 10 patients with non-V600E mutations in cohort B. Among the eight patients with a BRAF V600K mutation, one patient had an unconfirmed PR, and four patients had SD as the best response. The only patient in this cohort with a BRAF K601E mutation had a confirmed PR with a PFS of 32 weeks. One patient with discrepant results for BRAF V600K (analyzed at a local laboratory) and BRAF V600R (sequence analysis at RGI) had a confirmed PR with a PFS of at least 57 weeks.
Overall survival data for cohort B were not mature at the time of data cutoff. However, on the basis of data collected after the original data cutoff, the 6- and 12-month survival rates for all patients enrolled in cohort B, including patients with a V600K mutation and patients with prior brain metastases, were 79% (95% CI, 66% to 87%) and 59% (95% CI, 45% to 71%), respectively. The median OS was 14.2 months (95% CI, 11.3 months to upper boundary not reached; Fig 3). The median follow-up time was 20.8 months.
Treatment-Related Toxicity
All 97 patients across both cohorts received at least one dose of trametinib. The most frequent treatment-related AEs across both cohorts are listed in Table 3. There were no treatment-related deaths and only one grade 4 treatment-related AE (pulmonary embolism). Skin-related toxicity (10%), diarrhea (4%), and peripheral edema (3%) were the only grade ≥ 3 AEs that occurred in more than 2% of the patients. CSR was reported in two patients (2%); both were reversible. No cases of RVO were observed. Three patients (3%) had asymptomatic and reversible grade 3 left ventricular ejection fraction reduction. Two patients (2%) experienced reversible asymptomatic grade 3 increase of serum aminotransferases. AEs leading to dose reduction occurred in 15 patients (15%), most frequently for skin-related toxicity. There were no reports of treatment-related cutaneous squamous cell carcinoma. Treatment-related hypertension was reported for six patients (6%); none were grade ≥ 3. However, blood pressure evaluations demonstrated an overall trend toward increased values.
Table 3.
Treatment-Related Adverse Events Occurring in ≥ 10% of Patients (n = 97)
| Adverse Event | All Grades | Grade 3 | Grade 4 | |||
|---|---|---|---|---|---|---|
| No. | % | No. | % | No. | % | |
| Overall | 89 | 92 | 25 | 26 | 1 | 1* |
| Rash/dermatitis acneiform | 73 | 75 | 9 | 9 | 0 | |
| Diarrhea | 50 | 52 | 4 | 4 | 0 | |
| Nausea | 29 | 30 | 0 | 0 | ||
| Peripheral edema | 28 | 29 | 3 | 3 | 0 | |
| Pruritis | 26 | 27 | 1 | 1 | 0 | |
| Fatigue | 25 | 26 | 2 | 2 | 0 | |
| Dry skin | 21 | 22 | 0 | 0 | ||
| Vomiting | 17 | 18 | 0 | 0 | ||
| Abdominal pain | 15 | 15 | 0 | 0 | ||
| Constipation | 14 | 14 | 0 | 0 | ||
| Decreased appetite | 11 | 11 | 1 | 1 | 0 | |
| Dry mouth | 11 | 11 | 0 | 0 |
Trametinib Plasma Concentrations
The mean predose concentrations of trametinib in all patients ranged from 11.6 to 12.6 ng/mL across study visits, with no large differences between cohorts and no trends over the 12-week pharmacokinetics study. The mean concentrations were highest in the 2- to 4-hour collection interval (21.7 ng/mL), consistent with a time-to-peak-concentration of approximately 2 hours. The peak:trough concentration ratio based on mean day 15 concentrations was approximately 1.8, which is consistent with the relatively long half-life of trametinib.22
DISCUSSION
Since the discovery of frequent BRAF mutations in melanoma,2 there has been considerable progress in the therapeutic approaches for this disease. Following favorable results in the trametinib FIH study,22 trametinib was further evaluated in this phase II study in patients with _BRAF_-mutant melanoma who had been previously treated either with or without a BRAF inhibitor. Vemurafenib had not been approved by the US Food and Drug Administration at the start of this study.
In this study, the patients previously treated with chemotherapy and/or immunotherapy but not with a BRAF inhibitor (cohort B) had significant pretreatment with standard therapies and had primarily AJCC stage M1c disease (75%); 21% of the patients in this cohort had a history of treated brain metastases. The confirmed RR (25%), including one CR in a patient with stage M1c disease, and the number of patients with measurable tumor reduction demonstrate substantial clinical activity in this patient population. The RR, median duration of response, and PFS were similar to those of both the FIH23 and phase III studies.24 Although objective RRs for trametinib are lower than those reported for BRAF inhibitors in similar patient populations,3,8,9 the estimated 6- and 12-month survival rates and median OS reported here are comparable. The basis for the different RR observed with trametinib and BRAF inhibitors is not clear, but it may be because of different modes of action, degree of pathway inhibition, or acquired drug-resistance mechanisms. In this cohort, 54% of the patients received a BRAF inhibitor or ipilimumab postprogression, some of whom are still alive more than 1 year after disease progression on trametinib. Objective and durable responses were not limited in this study to BRAF V600E or V600K mutations, suggesting that trametinib may be broadly active in _BRAF_-mutant melanoma.
The mean, steady-state, predose trametinib concentrations in this study were generally comparable to those previously observed22 and above the target concentration of 10.4 ng/mL, based on preclinical findings ( GlaxoSmithKline, unpublished data). These data suggest that target inhibition is maintained throughout the entire dosing interval and, combined with its unique exposure profile among clinically evaluated MEK inhibitors,22 may explain why trametinib demonstrated a higher RR than those reported for other MEK inhibitors in similar patient populations.28,29
This study demonstrated that trametinib had minimal clinical activity as sequential therapy in patients that had progressed on BRAF-inhibitor therapy (cohort A). Only five patients (13%) received trametinib for ≥ 16 weeks. The two patients in this cohort who responded to trametinib were not BRAF-inhibitor refractory but were BRAF-inhibitor intolerant. The minimal activity in this cohort may be due to several factors, including heavy pretreatment (50% had received three or more prior therapies). Alternatively, acquired BRAF-inhibitor resistance mechanisms might also confer resistance to trametinib monotherapy, corroborating data from preclinical modeling.30
Trametinib treatment was well tolerated, with patients experiencing mostly grade 1 or 2 AEs. The majority of patients tolerated a daily dose of 2 mg of trametinib without dose reduction, and only 3% of the patients discontinued trametinib because of treatment-related AEs. The spectrum of AEs was consistent with the class effect of MEK inhibitors. CSR was rarely observed, and there were no RVO events. Skin-related toxicity and diarrhea, the most common AEs, were adequately managed with supportive care only; importantly, there were no reports of treatment-related cutaneous squamous cell carcinoma, commonly seen with BRAF-inhibitor treatment. Treatment-related hypertension was observed in six patients (6%) and may be a class effect, since hypertension was also reported in 8.1% of patients administered selumetinib.11 Additional studies will be required to understand trametinib-related hypertension. Importantly, cardiac, ophthalmologic, and hepatic events were uncommon and reversible on interruption of trametinib treatment.
The data presented here demonstrate that trametinib is well tolerated and clinically active in patients with previously treated metastatic _BRAF_-mutant melanoma who have not received or have not progressed on BRAF-inhibitor therapy. Activity was broad, with objective responses observed in patients with BRAF V600E, V600K, and rare BRAF mutations. Additional clinical studies are needed to understand the level of activity in patients with rare BRAF mutations and the individual role of trametinib in patients who have melanoma with BRAF V600E or V600K mutations. The efficacy and safety results reported here are consistent with those of the phase III study (GSK1120212 vs Chemotherapy in Advanced or Metastatic BRAF V600E/K Mutation-positive Melanoma [NCT01245062]), which demonstrated significant improvement in both PFS and OS in patients with BRAF V600E- or _V600K_-mutant metastatic melanoma compared with chemotherapy.24 In addition, the results from this study demonstrate that trametinib monotherapy has minimal clinical activity as sequential therapy in patients with _BRAF-_mutant melanoma who have progressed on BRAF-inhibitor therapy. Taken together, these data suggest that trametinib monotherapy could be useful for patients with BRAF V600E- or _V600K-_mutant melanoma, patients who cannot tolerate a BRAF inhibitor, and for patients who have melanoma with rare BRAF mutations. Sequential therapy with trametinib monotherapy followed by BRAF inhibitors or ipilimumab have not been formally evaluated.
Trametinib may also be useful in reducing toxicity and increasing efficacy when combined with a BRAF inhibitor in patients who have BRAF V600E- or V600K-mutant melanoma.31 Phase II (Investigate Safety, Pharmacokinetics and Pharmacodynamics of GSK2118436 & GSK1120212 [NCT01072175]) and phase III (A Study Comparing Trametinib and Dabrafenib Combination Therapy to Dabrafenib Monotherapy in Subjects With BRAF-mutant Melanoma [NCT01584648] and Dabrafenib Plus Trametinib vs Vemurafenib Alone in Unresectable or Metastatic BRAF V600E/K Cutaneous Melanoma (COMBI-v) [NCT01597908]) studies comparing trametinib and dabrafenib combination therapy with either dabrafenib or vemurafenib monotherapy in _BRAF_-mutant melanoma are ongoing.
Acknowledgment
Presented at Perspectives in Melanoma XV, New York, NY, September 16–17, 2011, and at the 8th International Congress of the Society of Melanoma Research, Tampa, FL, November 9–13, 2011.
We thank all the patients and their families who participated in this study; all the investigators for their efforts on behalf of their patients; Arthur Clements, Alex Menzies, Natalie Byrne, Deirdre d'Souza, and Vicky Wegener (Westmead Hospital, Australia) for their intellectual input in this study; Peng Sun ( GlaxoSmithKline) for statistical assistance; and the entire MEK113583 study team.
Appendix
Fig A1.
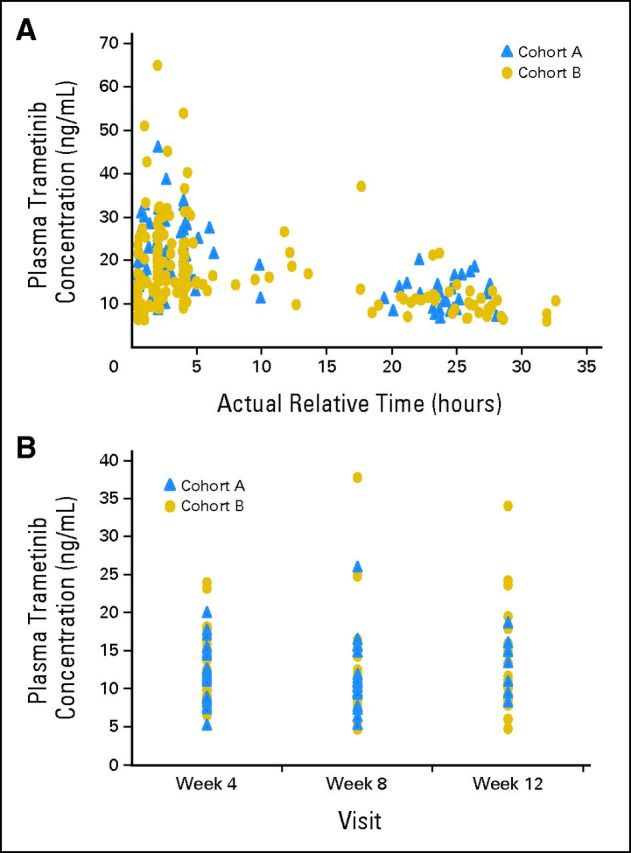
Pharmacokinetic analysis. (A) Individual plasma trametinib concentration versus actual time postdose observed on day 15. (B) Individual plasma predose trametinib concentration observed at weeks 4, 8, and 12.
Footnotes
See accompanying article on page 499; listen to the podcast by Dr Flaherty at www.jco.org/podcasts
Supported by GlaxoSmithKline, Philadelphia, PA.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: Olivia S. Gardner, GlaxoSmithKline (C); Daniele Ouellet, GlaxoSmithKline (C); Yanmei Xu, GlaxoSmithKline (C); Douglas J. DeMarini, GlaxoSmithKline (C); Ngocdiep T. Le, GlaxoSmithKline (C); Kiran Patel, GlaxoSmithKline (C) Consultant or Advisory Role: Richard Kefford, GlaxoSmithKline (C); Jeffrey R. Infante, GlaxoSmithKline (U); Antoni Ribas, GlaxoSmithKline (U); Jeffrey A. Sosman, Roche/Genentech (C), Millennium Pharmaceuticals (C); Leslie A. Fecher, Bristol-Myers Squibb (U); Michael Millward, GlaxoSmithKline (C); Grant A. McArthur, Plexxikon/Roche/Genentech (U), GlaxoSmithKline/Novartis (U), Millennium Pharmaceuticals (U); Rene Gonzalez, GlaxoSmithKline (C); Georgina V. Long, GlaxoSmithKline (C), Roche (C), Bristol-Myers Squibb (C) Stock Ownership: Olivia S. Gardner, GlaxoSmithKline; Daniele Ouellet, GlaxoSmithKline; Yanmei Xu, GlaxoSmithKline; Douglas J. DeMarini, GlaxoSmithKline; Ngocdiep T. Le, GlaxoSmithKline; Kiran Patel, GlaxoSmithKline Honoraria: Jeffrey A. Sosman, Roche, Millennium Pharmaceuticals; Georgina V. Long, Roche Research Funding: Kevin B. Kim, GlaxoSmithKline, AstraZeneca, Roche; Jeffrey A. Sosman, Roche, GlaxoSmithKline, Millennium Pharmaceuticals; Leslie A. Fecher, GlaxoSmithKline, Novartis, Bristol-Myers Squibb, Roche-Genentech; Grant A. McArthur, Pfizer; Rene Gonzalez, GlaxoSmithKline; Georgina V. Long, Roche; Karl D. Lewis, GlaxoSmithKline Expert Testimony: None Other Remuneration: Leslie A. Fecher, GlaxoSmithKline
AUTHOR CONTRIBUTIONS
Conception and design: Kevin B. Kim, Olivia S. Gardner, Daniele Ouellet, Yanmei Xu, Douglas J. DeMarini, Ngocdiep T. Le, Kiran Patel
Provision of study materials or patients: Richard Kefford, Antoni Ribas, Jeffrey A. Sosman, Michael Millward, Rene Gonzalez, Georgina V. Long
Collection and assembly of data: Kevin B. Kim, Richard Kefford, Anna C. Pavlick, Jeffrey R. Infante, Antoni Ribas, Jeffrey A. Sosman, Leslie A. Fecher, Michael Millward, Grant A. McArthur, Patrick Hwu, Rene Gonzalez, Georgina V. Long, Olivia S. Gardner, Yanmei Xu, Douglas J. DeMarini, Karl D. Lewis
Data analysis and interpretation: Kevin B. Kim, Richard Kefford, Anna C. Pavlick, Jeffrey R. Infante, Antoni Ribas, Leslie A. Fecher, Michael Millward, Grant A. McArthur, Patrick Hwu, Patrick A. Ott, Georgina V. Long, Olivia S. Gardner, Daniele Ouellet, Yanmei Xu, Douglas J. DeMarini, Ngocdiep T. Le, Kiran Patel, Karl D. Lewis
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 2.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 3.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239–1246. doi: 10.1200/JCO.2010.32.4327. [DOI] [PubMed] [Google Scholar]
- 5.Thomas NE, Edmiston SN, Alexander A, et al. Number of nevi and early-life ambient UV exposure are associated with BRAF-mutant melanoma. Cancer Epidemiol Biomarkers Prev. 2007;16:991–997. doi: 10.1158/1055-9965.EPI-06-1038. [DOI] [PubMed] [Google Scholar]
- 6.Cheng S, Chu P, Hinshaw M, et al. Frequency of mutations associated with targeted therapy in malignant melanoma patients. J Clin Oncol. 2011;29(suppl):549s. abstr 8597. [Google Scholar]
- 7.Jakob JA, Bassett RL, Jr, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014–4023. doi: 10.1002/cncr.26724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 10.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirkwood JM, Bastholt L, Robert C, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–567. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LoRusso PM, Krishnamurthi SS, Rinehart JJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16:1924–1937. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 13.Haura EB, Ricart AD, Larson TG, et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010;16:2450–2457. doi: 10.1158/1078-0432.CCR-09-1920. [DOI] [PubMed] [Google Scholar]
- 14.Fedorenko IV, Paraiso KH, Smalley KS. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem Pharmacol. 2011;82:201–209. doi: 10.1016/j.bcp.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long GV, Wilmott JS, Howle JR, et al. Morphologic and immunohistochemical (IHC) changes in metastatic melanoma (MM) tissue and associations with clinical outcome in patients (pts) on BRAF inhibitors (BRAFi) J Clin Oncol. 2011;29(suppl):536s. abstr 8542. [Google Scholar]
- 17.McArthur GA, Ribas A, Chapman PB, et al. Molecular analyses from a phase I trial of vemurafenib to study mechanism of action (MOA) and resistance in repeated biopsies from BRAF mutation-positive metastatic melanoma patients (pts) J Clin Oncol. 2011;29(suppl):526s. abstr 8502. [Google Scholar]
- 18.Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 22.Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–781. doi: 10.1016/S1470-2045(12)70270-X. [DOI] [PubMed] [Google Scholar]
- 23.Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–789. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 25.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 26.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v3.0. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf.
- 27.Green SJ, Dahlberg S. Planned versus attained design in phase II clinical trials. Stat Med. 1992;11:853–862. doi: 10.1002/sim.4780110703. [DOI] [PubMed] [Google Scholar]
- 28.Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–2146. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol. 2005;23:5281–5293. doi: 10.1200/JCO.2005.14.415. [DOI] [PubMed] [Google Scholar]
- 30.Atefi M, von Euw E, Attar N, et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One. 2011;6:e28973. doi: 10.1371/journal.pone.0028973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weber JS, Flaherty KT, Infante JR, et al. Updated safety and efficacy results from a phase I/II study of the oral BRAF inhibitor dabrafenib (GSK2118436) combined with the oral MEK 1/2 inhibitor trametinib (GSK1120212) in patients with BRAFi-naive metastatic melanoma. J Clin Oncol. 2012;30(suppl):542s. abstr 8510. [Google Scholar]