Fetal-derived adrenomedullin mediates the innate immune milieu of the placenta (original) (raw)
Abstract
The remodeling of maternal uterine spiral arteries (SAs) is an essential process for ensuring low-resistance, high-capacitance blood flow to the growing fetus. Failure of SAs to remodel is causally associated with preeclampsia, a common and life-threatening complication of pregnancy that is harmful to both mother and fetus. Here, using both loss-of-function and gain-of-function genetic mouse models, we show that expression of the pregnancy-related peptide adrenomedullin (AM) by fetal trophoblast cells is necessary and sufficient to promote appropriate recruitment and activation of maternal uterine NK (uNK) cells to the placenta and ultimately facilitate remodeling of maternal SAs. Placentas that lacked either AM or its receptor exhibited reduced fetal vessel branching in the labyrinth, failed SA remodeling and reendothelialization, and markedly reduced numbers of maternal uNK cells. In contrast, overexpression of AM caused a reversal of these phenotypes with a concomitant increase in uNK cell content in vivo. Moreover, AM dose-dependently stimulated the secretion of numerous chemokines, cytokines, and MMPs from uNK cells, which in turn induced VSMC apoptosis. These data identify an essential function for fetal-derived factors in the maternal vascular adaptation to pregnancy and underscore the importance of exploring AM as a biomarker and therapeutic agent for preeclampsia.
Introduction
It is widely accepted that the placenta provides the venue for communication between the mother and fetus and that this communication is essential for their continued health throughout pregnancy. During midgestation, the maternal uterine spiral arteries (SAs) must provide increasing amounts of oxygen and nutrients to the growing fetus, and so they undergo active remodeling in order to transform into low-resistance, high-capacitance blood vessels. SA remodeling is a complex event that involves several distinct processes: (a) endothelial cell vacuolization and relaxation of VSMCs that lead to arterial dilation and growth, (b) induction of local placental factors, including MMPs, chemokines, and cytokines, which in turn cause (c) dissolution of the ECM, (d) dedifferentiation and apoptosis of VSMCs, and (e) replacement of VSMCs by invasive fetal trophoblast cells (1–5). Considering the complexity of this process, it is not surprising that preeclampsia — a life-threatening pregnancy complication caused by poor SA remodeling — is so prevalent and detrimental to both mother and fetus (6, 7). Since the fetus is the primary beneficiary of increased blood flow, it stands to reason that fetal signals may play an active role in enabling SA remodeling. However, very few fetal-derived trophoblast factors involved in SA remodeling have been identified (8–11).
On the other hand, maternal uterine NK (uNK) cells, which do not exhibit the cytotoxic behavior of peripheral NK cells, are known to be required for SA remodeling (4, 11, 12). uNK cells are the most abundant of all decidual leukocytes, accounting for approximately 70% of CD45+ cells, and are histologically identified by large cytoplasmic granules containing perforin and granzymes (1). uNK cells, which are transiently present in the decidua, are frequently aggregated around SAs and play a functional role in remodeling (12–14). Pioneering studies by Croy and colleagues have demonstrated that the SAs of transgenic mice that lack uNK cells show a persistence of VSMCs surrounding un-dilated SAs (1, 15, 16). Only very recently have sophisticated techniques for the isolation and enrichment of uNK cells permitted more in-depth evaluation of these cells (17, 18). uNK cells secrete a wide variety of growth factors, chemokines, cytokines, and MMPs (4) and thus represent a major constituent of the innate immune milieu of the placenta. Several recent studies have begun to reveal important pathophysiological roles for uNK cells in human pregnancy (19–22). However, much remains unknown about the precise molecular interactions between fetal trophoblast cells and uNK cells in the placenta. It is also unclear whether perturbations in this fetal-to-maternal communication can account for, or be diagnostic of, failed SA remodeling and preeclampsia.
Adrenomedullin (Adm; encoding AM) is a hypoxia-induced, angiogenic peptide vasodilator that is elevated approximately 5-fold in the maternal plasma of normal human pregnancies, but often blunted in pregnancies complicated by preeclampsia (23). Moreover, the direct transcriptional induction of Adm gene expression (and that of its receptor components) by estrogen confers high levels of this peptide signaling pathway in female reproductive organs, including the ovaries, uterus, uterine arteries, and placenta (24–27). Yallampalli and colleagues showed that systemic administration of an AM antagonist during late rat gestation caused a myriad of pregnancy abnormalities, including fetal growth restriction, placental and fetal membrane necrosis, and fetal edema (28). While the AM antagonist did not change maternal systemic blood pressure, it is likely that many of these fetoplacental sequelae developed as a consequence of acutely reduced uterine perfusion (29).
Using gene-targeted mice, we previously showed that full expression of maternal_Adm_ is required for the early establishment of pregnancy and placentation: modest haploinsufficiency for maternal Adm caused poor uterine receptivity, reduced pinopode formation, and subfertility, even when WT embryos were transferred to the uterus (30). Consequently, pregnant Adm+/– females exhibit abnormal implantation, ectopic placentation, and fetal growth restriction that results in a wave of embryonic lethality around E9.5 that is largely independent of fetal genotype (31). These findings highlight the importance of maternal Adm genetic dosage during the early stages of pregnancy, yet the function of fetal Adm expression during pregnancy remains unclear.
Here, using both loss-of-function and gain-of-function genetic mouse models, we showed that expression of Adm and its G protein–coupled receptor by fetal trophoblast cells was necessary and sufficient to promote appropriate recruitment and activation of maternal uNK cells to the placenta and ultimately facilitate the remodeling of maternal SAs.
Results
Adm-null placentas exhibit parietal trophoblast giant cell death and reduced labyrinth vessel branching.
We previously showed that Adm is expressed in trophectoderm cells of the preimplantation blastocyst and that its expression is increased when trophoblast stem cells are differentiated into the giant cell lineage (31). Using in situ hybridization, we found that this robust expression was maintained in vivo within fetal parietal trophoblast giant cells (TGCs; polyploidy derivatives of the primary mural trophectoderm cells that line the implantation site; refs. 32–34) at E9.5 and E13.5 (Figure 1, A–C), consistent with previous studies (35). There was also strong expression of Adm surrounding the invading ectoplacental cone at E9.5 and at the innermost border of the maternal-fetal interface (Figure 1, A and B). Adm expression persisted in TGCs at E13.5, and stromal cells of the maternal decidua expressed moderate levels of_Adm_ (Figure 1C). The chorionic plate, embryo, fetal membranes, labyrinth, and spongiotrophoblast-containing junctional zone expressed little to no_Adm_.
Figure 1. Fetal loss of Adm causes trophoblast apoptosis at the maternal-fetal interface.
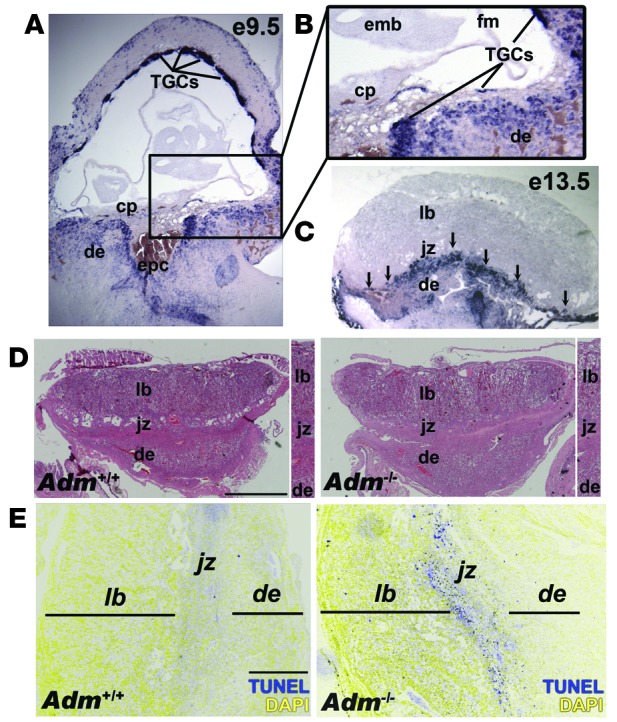
(A) In situ hybridization of Adm gene expression in WT E9.5 mouse placentas, revealing robust expression in parietal TGCs. (B) Digital zoom of boxed region in A (enlarged ×2-fold), showing punctuate Adm staining in TGCs lining the ectoplacental cone (epc) at the innermost border of the maternal-fetal interface and little to no expression in the chorionic plate (cp), embryo (emb), or fetal membranes (fm). (C) Adm expression persisted in TGCs at E13.5 (arrows) and was diffusely expressed in stromal cells throughout the maternal decidua (de). The labyrinth (lb) and spongiotrophoblast-containing junctional zone (jz) expressed little to no_Adm_. (D) H&E staining of placentas from Adm+/+ and_Adm–/–_ littermates revealed largely normal structures, with no difference in the thickness of the labyrinth layer (Supplemental Figure 1). Digital zoom of central part of placentas is shown at the right of each image (enlarged ×1.5-fold). (E) TUNEL staining of placentas from_Adm+/+_ and_Adm–/–_ littermates showed a prominent band of apoptosing cells in_Adm–/–_ placentas that colocalized to TGC location and correlated with the high level of_Adm_ expression in these cells. For better clarity and data presentation, the original colors from the captured images in E were inverted using Adobe Photoshop. Original magnification, ×4 (A–C). Scale bars: 1 mm (D); 500 μM (E).
Although Adm–/– mice die at midgestation with lymphatic vascular defects (36), we wondered whether genetic dosage of fetal _Adm_could also contribute to pregnancy outcomes. Histological and morphometric analyses showed that Adm–/– placentas appeared overtly normal, with well-formed layers and labyrinth thickness that were indistinguishable from those of Adm+/+ littermates (Figure 1D and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI67039DS1). Because a hallmark function of AM peptide is vasodilation, some have speculated that its expression in placenta may be associated with maintaining low-resistance blood flow through the placental vasculature and umbilical cord (37). Contrary to this hypothesis, we found that loss of Adm had no effect on placental impedance, as calculated by umbilical cord blood flow using in vivo Doppler ultrasound (Supplemental Figure 2).
Despite their overtly normal appearance and function, E13.5_Adm–/–_ placentas had a prominent band of apoptosing cells that spatially localized to the innermost border of the maternal-fetal interface (Figure 1E), where Adm_-expressing parietal TGCs are located, which suggests that AM is an essential survival factor for these cells. Consistent with this finding, qRT-PCR analysis of trophoblast marker genes revealed that_Adm–/– placentas had significantly reduced expression of placental lactogen 1 — a specific marker of parietal TGCs — while the expression of other TGC lineage markers and spongiotrophoblast markers did not differ significantly from that of WT placentas (Supplemental Figure 3). Even though Adm is not abundantly expressed in the labyrinth, we also found a modest, yet significant, reduction in the labyrinth-expressing genes Gcm1, JunB, and_Gab1_ (Supplemental Figure 3).
Further characterization of the labyrinth layer showed marked defects in the vascular patterning of fetal vessels in_Adm–/–_ placentas. Isolectin B4 staining of the ECM surrounding fetal labyrinth vessels revealed a highly branched network in Adm+/+ placentas, but abnormally large and underbranched fetal vessels in_Adm–/–_ placentas (Figure 2A). Using nucleated and non-nucleated blood cells as markers for fetal and maternal blood sinuses, respectively, we performed quantitative morphometric analysis of the labyrinth vasculature (Figure 2B). Consistent with our finding of normal labyrinth thickness and normal placental impedance, the total area of vascular space in Adm–/– labyrinth did not differ from that in Adm+/+ placentas (Figure 2C). However, we found that individual fetal sinuses of Adm–/– placentas were significantly larger than those of Adm+/+ placentas (P < 0.01; Figure 2D). This increase in the size of fetal sinuses was accompanied by a concomitant reduction in the size of maternal sinuses in_Adm–/–_ versus_Adm+/+_ placentas (P < 0.02; Figure 2D).
Figure 2. Reduced branching of fetal labyrinth vessels in_Adm–/–_ placentas.

(A) Isolectin B4 staining revealed a highly branched network in_Adm+/+_ placentas, in contrast to abnormally large and underbranched fetal vessels in_Adm–/–_ placentas. (B) Fetal (blue) and maternal (red) blood sinuses demarcated within the labyrinth layer. (C) Quantitative morphometric analysis of total sinus space per field. (D) Quantitation of individual sinus area revealed that fetal sinuses of_Adm–/–_ placentas were significantly larger than those of Adm+/+ placentas, with a concomitant reduction in size of maternal sinuses. n = 12 placentas per genotype. *P < 0.02;#P < 0.01. (E andF) Scanning electron microscopy of vascular corrosion casts of maternal placental vasculature, at low power (E) and higher magnification (F). The large holes (white arrows) were indicative of large fetal vessels. Images are representative of n = 4 placentas per genotype. (G) Alkaline phosphatase staining of labyrinth revealed no structural or quantitative differences in chorionic villus cells. Data are mean ± SEM. Scale bars: 50 μM (A, B, and G); 1 mm (E); 100 μM (F).
To better visualize the architecture and structure of the labyrinth vasculature, we made vascular corrosion casts of the placentas by perfusing pregnant female mice with a liquid polymer. Fetal placental tissue was enzymatically digested away from the hardened maternal casts in order to reveal the interdigitation of maternal and fetal vasculature within the labyrinth placentas of Adm+/+ and_Adm–/–_ littermates. Therefore, in the scanning electron micrographs of Figure 2, E and F, the open spaces represent the footprint of the fetal vasculature as it was interdigitated between the maternal blood sinuses within the central labyrinth layer. Adm–/–_placentas had large holes (Figure 2E, arrowheads) and higher-magnification views demonstrated vast spacing between the maternal sinuses (Figure 2F), reflective of the large and under-branched phenotype of_Adm–/– fetal vessels. Taken together, these data demonstrated a failure of fetal vessels to branch appropriately in Adm–/– placentas.
Because shallow invasion of chorionic villi can lead to blunted fetal vascular branching, we used alkaline phosphatase staining to evaluate the extent of chorionic villus formation and found no significant difference between WT and_Adm–/–_ placentas (integrated density, Adm+/+, 11.8 ± 2.0 AU;Adm–/–, 11.3 ± 2.0 AU; Figure 2G). This finding demonstrated that the fetal vascular branching defects of_Adm–/–_ placentas were inherent to the fetal vasculature, rather than secondary to defects in chorionic villus invasion or formation. Collectively, the data presented in Figures 1 and 2 showed that compared with those of their WT littermates,Adm–/– placentas exhibited increased apoptosis of parietal trophoblast cells accompanied by reduced fetal vessel branching.
Failed SA remodeling and reduced uNK cells in Adm-null placentas.
Because several clinical studies have shown that maternal levels of AM are reduced in preeclampsia (23), we evaluated another characteristic pathology of preeclampsia in_Adm–/–_ placentas: remodeling of maternal SAs. As expected, SAs of WT placentas progressively lost staining for SMA as they approached the maternal-fetal interface (Figure 3A). In contrast, SAs from_Adm–/–_ placentas retained a thick smooth muscle layer throughout the decidua that was nearly twice the thickness measured in WT placentas (P < 0.01; Figure 3, A and B). The shedding of VSMCs surrounding SAs is associated with localized degradation of the ECM, a process in which the cathepsin family of proteases has been shown to play a predominant role (10, 38). As expected, SAs from Adm+/+ placentas were tightly surrounded by a prominent band of cathepsin staining, whereas cathepsin staining was weak and diffuse throughout the deciduas of Adm–/–_placentas and did not surround the maternal SAs (Figure 3C), consistent with a failure of these SAs to shed their smooth muscle coverage. The enlargement of maternal SAs at midgestation is also associated with proliferation of vascular endothelial cells, a process termed reendothelialization. Using BrdU incorporation assays, we readily observed this robust endothelial cell proliferation in all SAs of Adm+/+ placentas, but this was rarely evident in the SAs of_Adm–/– placentas (Figure 3D).
Figure 3. Fetal Adm dosage influences maternal SA remodeling and placental uNK cell content.

(A) Anti–α-SMA staining showed that decidual SAs of E13.5 Adm–/– placentas retained thick coverage of VSMCs (arrowheads) compared with those of_Adm+/+_ littermates. (B) Morphometric analysis showed a statistically significant increase in the thickness of SMA staining surrounding the SAs of_Adm–/–_ versus littermate_Adm+/+_ placentas. *P < 0.01. (C) Cathepsin staining of decidual SAs. (D) BrdU incorporation assays showed marked proliferation of vascular endothelial cells (arrowheads) in all SAs of Adm+/+ placentas, but this was rarely evident in SAs of_Adm–/–_ placentas. (E) Perforin staining of uNK cells within deciduas at E13.5. (F) Quantitation of perforin+ uNK cells showed a significant reduction in_Adm–/–_ versus_Adm+/+_ placentas. *P < 0.05. For all analyses, n = 10 placentas per genotype. Scale bars: 50 μM.
Numerous studies have pointed to maternal uNK cells as essential mediators of SA remodeling, and the precise factors contributing to their recruitment, activation, and function remains an area of intense study. Therefore, we stained and counted the number of uNK cells using 2 different uNK markers that identify mature (perforin-containing) and/or activated (Ly49G2+) uNK cells. Consistent with the phenotype of failed SA remodeling, we found that deciduas of_Adm–/–_ placentas had half the number of perforin-containing uNK cells observed in_Adm+/+_ littermates (P < 0.05; Figure 3, E and F). Similarly, levels of Ly49G2+ uNK cells were also reduced by approximately half in_Adm–/–_ placentas, although the difference did not reach statistical significance.
Taken together, our findings demonstrated that lack of fetal AM caused numerous placental pathologies, including reduced fetal vessel branching, failed SA remodeling, and reduced number of uNK cells. We also noted that the incidence and degree of the Adm–/– placental phenotype was strictly and exclusively correlated with the genotype of the individual_Adm–/–_ conceptus, not influenced by the genotype of neighboring conceptuses.
Adm–/– placental phenotype is independent of maternal genotype.
A confounding factor to the present findings is that the maternal genotype used in the above studies was haploinsufficient for AM; we have previously shown that during early gestation, Adm+/– females exhibit reduced fertility due to abnormal implantation (30, 31). To eliminate this confounding factor, we performed ovary transplantations in which_Adm+/+_ ovaries or_Adm+/–_ ovaries (containing gene-targeted _Adm_-null gametes) were transplanted into WT recipient females (referred to herein as _Adm+/+_ovary→WT or Adm+/–_ovary→WT recipients, respectively), which were then bred to_Adm+/– males (Figure 4A). This approach allowed us to evaluate the consequences of loss of fetal AM within the context of a WT maternal uterus and immune system and to circumvent the inherently low offspring resulting from embryo transfer approaches that we previously experienced with the 129S6/SvEv strain (30). The average success rate of ovarian transplants (the percentage of pregnant mice relative to the total undergoing transplantation) was 57.5% and did not differ based on recipient or donor genotype. The transplanted ovaries exhibited overtly normal histology, with numerous ovarian follicles and corpora lutea (Supplemental Figure 4), suggestive of normal estrogenic function. In addition, compared with Adm+/+ donor ovaries, Adm+/– donor ovaries did not adversely affect the recipient females’ breeding fecundity, since both exhibited similar recovery times to first parturition (Adm+/+ ovary→WT recipients, 56.3 days;Adm+/– ovary→WT recipients, 50.6 days; Figure 4C) and similar litter sizes (Figure 4D).
Figure 4. Ovary transplantation reveals independence of maternal genotype on_Adm–/–_ placental phenotype.

(A) Ovary transplantation protocol. Donor ovaries from_Adm+/–_ or_Adm+/+_ female mice were surgically sutured in the ovarian bursa of WT 129S6/SvEv recipient mice after removal of their own ovaries. The recipient females were then bred to_Adm+/–_ male mice to generate_Adm+/+,Adm+/–, or_Adm–/– offspring. (B) Mendelian ratios of offspring were as expected, and_Adm–/–_ mice exhibited characteristic embryonic edema (arrowhead) at E13.5. (C andD) Recovery time to first parturition (C) and average litter size (D). Numbers within bars denote total number of litters analyzed per breeding. (E–I)Adm–/– placentas from_Adm+/–_ ovary→WT recipient females displayed the same pathological phenotypes of_Adm–/–_ placentas from_Adm+/–_ females — including (E) reduced fetal vessel branching in the labyrinth, (F) retention of SMCs (arrowheads) around maternal SAs, (G) reduction in endothelial cell proliferation (arrowheads) in maternal SAs, and (H and I) significantly reduced DBA+ uNK cell numbers — compared with_Adm+/+_ littermates. *P < 0.05. For ovary transplant studies, n = 6–10 placentas per genotype. Scale bars: 50 μM.
The cross of Adm+/– males to_Adm+/–_ ovary→WT recipient females resulted in the expected Mendelian ratio of fetal genotypes and the previously characterized phenotype of extreme embryonic edema and lethality for_Adm–/–_ fetuses (Figure4B and refs. 36, 39). Consistently, the placental phenotypes of underbranched fetal labyrinth vessels, poorly remodeled SAs, and significantly reduced uNK cell numbers were completely recapitulated in_Adm–/–_ placentas born to_Adm+/–_ ovary→WT recipient females compared with Adm+/+ littermate placentas and_Adm+/+_ placentas derived from_Adm+/+_ ovary→WT recipient females (Figure 4, E–I). Thus, loss of_Adm_ from fetal tissues directly contributed to the observed placental pathologies, even in the context of genotypically normal maternal_Adm_.
Recapitulation of phenotypes in placentas genetically lacking the AM receptor.
AM peptide can bind to and signal through the G protein–coupled receptor calcitonin receptor–like receptor (Calcrl; encoding CLR). We have previously shown that mice with genetic deletion of_Calcrl_ phenotypically recapitulate the embryonic lethality and lymphatic vascular defects observed in_Adm–/–_ mice (36, 40), thereby providing genetic and in vivo evidence to substantiate CLR as a bona fide AM receptor.
Using in situ hybridization, we consistently found high levels of_Calcrl_ expression in WT placentas that spatially and temporally colocalized with that of the peptide ligand Adm. Specifically,Calcrl expression was robustly present in parietal TGCs at both E9.5 and E13.5 (Figure 5, A–C). In contrast to the diffuse expression of Adm surrounding the ectoplacental cone and stromal decidual cells, expression of _Calcrl_was concentrated to endothelial-like cords of the E9.5 decidua (41) and to maternal endothelial cells lining the maternal SAs of E13.5 placentas (Figure 5C, inset).
Figure 5. Fetal loss of Calcrl recapitulates_Adm–/–_ placental phenotypes.

(A) In situ hybridization of Calcrl gene expression in WT E9.5 mouse placentas revealed expression in parietal TGCs, similar to the pattern of Adm expression. Unlike Adm,Calcrl was expressed at moderate levels in the embryo and fetal membranes. There was also robust expression of Calcrl in cord-like structures throughout the early decidua. (B) Digital zoom of boxed region in A (enlarged ×2-fold), showing punctuate Calcrl staining in TGCs lining the ectoplacental cone at the innermost border of the maternal-fetal interface and little to no expression in the chorionic plate. (C) Calcrl_expression was diffuse throughout the labyrinth at E13.5 and robustly expressed in the maternal endothelial cells lining the decidual SAs (arrows, inset; enlarged ×2-fold). (D) H&E staining of placentas from_Calcrl+/+ and_Calcrl–/–_ littermates revealed largely normal structures, with no appreciable differences in layers. Digital zoom of central part of placentas is shown at the right of each image (enlarged ×1.5-fold). (E–H)Calcrl–/– placentas at E13.5 displayed the same pathological phenotypes of_Adm–/–_ placentas — including (E) reduced fetal vessel branching in the labyrinth, as visualized by isolectin staining, (F) retention of SMCs around maternal SAs, and (G and H) significantly reduced number of DBA+ uNK cells — compared with_Calcrl+/+_ littermate placentas. *P < 0.001. n ≥ 6 placentas analyzed per genotype. Data are mean ± SEM. Original magnification, ×4 (A–C). Scale bars: 1 mm (D); 50 μM (E andG); 10 μM (F).
Like Adm–/– placentas, the_Calcrl–/–_ placentas appeared overtly normal and contained well-formed placental layers and normal histomorphometric features (Figure 5D). Nevertheless, the Calcrl–/–_placentas exhibited the same abnormal phenotypes of_Adm–/– placentas, including reduced fetal vessel branching, retention of SMCs surrounding maternal SAs, and significantly reduced uNK cell numbers compared with_Calcrl+/+_ littermate placentas (Figure 5, E–H). Thus, the recapitulation of the Adm–/– placental phenotype in_Calcrl–/–_ placentas further supports the role of CLR as an AM receptor within this tissue and indicates that lack of AM signaling within the fetal compartment is causally associated with these phenotypes.
Overexpression of fetal Adm reverses the placental phenotype and drives maternal uNK cell recruitment to the decidua.
The absence of apoptosis within decidual regions that contain uNK cells (Figure 1E) suggests that the reduced number of uNK cells in Adm–/– placentas may be caused by abnormalities in their recruitment to the tissue, rather than their loss through cell death. To determine whether fetal AM can directly influence the recruitment of uNK cells to the decidua, we generated and characterized a novel gene-targeted mouse model in which AM expression was increased approximately 3-fold (Figure 6, A–C). The gene targeting was designed to increase AM expression approximately 3-fold via stabilization of mRNA levels through genetic modification of the 3′ untranslated region (3′UTR) of the endogenous Adm gene.
Figure 6. Genetic overexpression of fetal Adm reverses the placental preeclampsia phenotypes and drives uNK recruitment to the decidua.

(A) Targeting vector for generation of_Admhi/hi_ mice consisted of (a) a 6-kb genomic fragment of the Adm gene isolated from a 129S6/SvEv genomic phage library and containing all 4 exons and 5′UTR and 3′UTR of the Adm gene, (b) the bovine growth hormone polyA sequence (bGH 3′UTR), (c) 2 tandem copies of the 1.2-kb 5′ insulator sequence from chicken β-globin gene (2XIns), (d) 1.3 kb of pMC1 promoter–driven neomycin (Neo), (e) 80 bp of AU/U-rich element of the mouse c-fos gene (ARE), and (f) 2 loxP recombination sites. The latter 5 elements were cloned as a cassette, 23 bp downstream of the endogenous_Adm_ stop codon. (B) Southern blot analysis on genomic DNA confirmed correct targeting of the Admhi_allele. (C) Adm gene expression in placentas from_Adm+/+ and Admhi/hi_mice, analyzed by quantitative RT-PCR, showed a significant 3-fold increase in gene expression level. *P < 0.05. (D–G)Admhi/hi placentas (D) appeared histologically comparable to Adm+/+ placentas and showed (E) highly branched fetal labyrinth vessels, (F) appropriate SA remodeling, and (G) reendothelialization. (H and I) DBA staining revealed that_Admhi/hi placentas had a significant 30% increase in uNK cell numbers compared with _Adm+/+_littermate placentas. *P < 0.05. Data are mean ± SEM. Scale bars: 1 mm (D); 50 μM (E–H).
Adm+/hi mice were viable and fertile and were intercrossed in order to compare placental phenotypes between_Adm+/+_ and Admhi/hi_littermates. The average litter size of Admhi/hi females was 6.04 live births, which was not significantly different than the 6.60 pups per litter observed for WT C57BL/6J mice of the comparable genetic background.Admhi/hi placentas born to_Adm+/hi intercrossed mice appeared overtly normal and had appropriately branched labyrinth fetal vessels that were indistinguishable from Adm+/+ littermates (Figure 6, D and E). Moreover, the SAs of_Admhi/hi_ placentas were appropriately remodeled and did not differ from those of Adm+/+ littermates (Figure6, F and G). Interestingly, Dolichos biflorus agglutinin (DBA) staining and counting of uNK cells revealed that Admhi/hi placentas had a significant 30% increase in the number of uNK cells within the decidua compared with that of_Adm+/+_ littermate placentas (Figure 6, H and I). These data indicate that 3-fold overexpression of Adm is compatible with normal placental development and, moreover, that fetal-derived AM can actively promote the recruitment of uNK cells to the placenta.
AM dose-dependently alters the chemokine, cytokine, and MMP profiles of uNK cells in vivo and in vitro.
uNK cells constitute the largest proportion of immune cells in the decidua, and so we expected and found that the dynamic fluctuations in uNK cell recruitment between_Adm–/–_ and_Admhi/hi_ placentas were reflected by concomitant changes in the expression of numerous chemokines and cytokines. Specifically, the expression of Ccl7, Ccl17, Cxcl9, Cxcl10, Xcl1, and TNF were downregulated in_Adm–/–_ placentas and concomitantly upregulated in Admhi/hi placentas compared with their respective WT controls (Figure 7A). To determine whether AM peptide directly causes secretion of chemokines and cytokines from uNK cells, we isolated uNK cells and first determined that these cells expressed high levels of Calcrl and survived up to 48 hours in culture without evidence of apoptosis, as evaluated by TUNEL and trypan blue exclusion (Supplemental Figure 5 and data not shown). Then, using a Luminex-based detection system, we found that treatment of uNK cells with 10 nM AM stimulated marked secretion of a variety of chemokines and cytokines (Figure 7B), several of which have established functions in reproductive immunology (i.e., CXCL10, GM-CSF, and IL-23) (42). Furthermore, we found that AM treatment dose-dependently increased the gene expression and activity of MMP9, but not MMP2, in uNK cells (Figure 7, C–E), consistent with the previously described functions of uNK-derived MMP9 in SA remodeling (43). Collectively, these data suggest that fetal-derived AM potently influences the immune milieu of the placenta in 2 ways: first by recruiting, then by activating, uNK cells to secrete chemokines, cytokines, and MMPs, which are important contributors to SA remodeling.
Figure 7. AM is a direct activator of uNK cells in vivo and in vitro.

(A) Using a mouse Chemokines and Receptors array platform (see Methods), the expression of 85 chemokines, cytokines, and related proteins was evaluated in placental RNA extracts from_Adm–/–_,Admhi/hi, and respective WT littermates.Admhi/hi placentas showed significant elevations in numerous cytokines and chemokines that were concomitantly downregulated in_Adm–/–_ placentas. *P < 0.05. (B) uNK cells isolated from E10.5–E12.5 WT placentas were cultured in vitro and treated with 10 nM AM. Screening for more than 25 chemokines and cytokines from media samples revealed significant 2- to 4-fold increases in secretion of numerous chemokines and cytokines and a prominent reduction in the secretion of CCL5, IL-16, and IL-23 in response to AM treatment. All changes were statistically significant compared with untreated control (P < 0.05). (C) Isolated WT uNK cells exhibited dose-dependent increases in _Mmp9_gene expression, which (D and E) correlated with increased MMP9 zymography activity. Mmp2 gene expression levels and zymography activity were not affected by AM treatment. *P< 0.05.
AM-treated uNK cells stimulate VSMC apoptosis.
Finally, we sought to determine whether the effects of AM on isolated uNK cells directly relate to the phenotype of reduced SA remodeling in_Adm–/–_ placentas. To test this, we developed an in vitro cell culture system in which primary mouse VSMCs were treated with control media or uNK-conditioned media that had been supplemented or not with 10 nM AM peptide. As predicted, treatment of VSMCs with control media and AM-supplemented control media had no obvious effects on cell growth, morphology, or apoptosis (Figure 8, A and B). In contrast, treatment of VSMCs with uNK-conditioned media resulted in dramatic changes in cellular morphology and apoptosis rate. Importantly, these effects were markedly and significantly exacerbated when the uNK-conditioned media was supplemented with AM, with a nearly 4-fold increase in the ratio of proapoptotic Bax to antiapoptotic Bcl2 gene expression (Figure 8C). Thus, we conclude that AM can promote the secretion of a cocktail of chemokines and cytokines from uNK cells that can in turn induce apoptosis of VSMCs.
Figure 8. Conditioned media from AM-treated uNK cells causes VSMC apoptosis.

(A and B) Confluent monolayers of cultured mouse VSMCs were placed for 12 hours in control media or uNK-conditioned media treated or not with 10 nM AM peptide, then imaged (A) and stained with TUNEL (B). (C) After treatment with uNK-conditioned media, expression levels of the proapoptotic Bax and antiapoptotic_Bcl2_ genes were analyzed from VSMC RNA lysates by quantitative RT-PCR; data are expressed as Bax/Bcl2 ratio. *P < 0.05, 1-way ANOVA. Data are mean ± SEM. Original magnification, ×20 (A).
Discussion
Using both loss-of-function and gain-of-function genetic mouse models, we found that fetal AM, acting through CLR, serves as a trophoblast-derived factor critical for fetal placental vascularization and for the maternal vascular adaptation to pregnancy (Figure9). There were several pleiotropic consequences of absent AM/CLR signaling within the trophoblast compartment, which could be difficult to causally distinguish from one another. However, the precise replication of the_Adm–/–_ phenotype in_Calcrl–/–_ placentas demonstrated that the predominant factor in initiating the complex phenotype is the absence of ligand signaling and secretion in the trophoblast cell. Ultimately, the mechanisms of AM action appear to be largely mediated through its local effects on the decidual content and activation of maternal uNK cells, which in our experiments expressed either WT or hemizygous levels of CLR. Therefore, the phenotypes associated with Adm–/– and_Calcrl–/–_ placentas, and their reversal by Adm overexpression, demonstrated that the dosage of AM provided by the fetus is a critical communication factor that has a profound effect on the innate immune milieu of the placenta.
Figure 9. Model of fetal-derived AM action at the maternal-fetal interface.

AM and its G protein–coupled receptor, CLR, are highly expressed in parietal TGCs at the maternal-fetal interface at midgestation. The genetic dosage of fetal AM is directly proportional to the content of maternal uNK cells and their secreted chemokines, cytokines, and MMPs within the placenta. As a consequence, AM expression in fetal trophoblast cells is necessary for appropriate branching of the fetal vasculature and remodeling of maternal SAs. Overexpression of AM within the fetal compartment is sufficient to promote uNK recruitment and activation in the decidua, a process that likely contributes to AM-induced activation of MMP9 and maternal SA remodeling.
It is worth noting that nearly all of the characteristic features of_Adm–/–_ and_Calcrl–/–_ placentas were similar to the pathological features that have been characterized in human preeclampsia. Although it is imperative to consider the anatomical and functional differences between placentas of humans and mice (44–46), the reduction in fetal vascular branching, failed SA remodeling, and lack of SA reendothelialization we describe here have long been associated with preeclampsia in humans (47). More recently, additional studies demonstrating deregulation of uNK cells and the levels of placental chemokines and cytokines have also been appreciated in human preeclampsia (20, 22). Finally, some of the most clinically relevant breakthroughs in recent years have been discoveries of numerous angiogenic factors and their soluble receptors as predictive biomarkers of preeclampsia, including soluble Fms-like tyrosine kinase 1 (sFlt1) and soluble endoglin (sEng) (48). In this regard, it is important to appreciate that AM signaling — by virtue of its regulation by hypoxia, high levels of receptor expression in endothelium, and requirement for vascular development — serves as an additional and powerful angiogenic factor within the placenta, with potential importance in the pathological presentation of preeclampsia.
An obvious question that extends from these findings is whether_Adm+/–_ dams exhibit the maternal systemic symptoms of preeclampsia, including elevated blood pressure and proteinuria, as a result of renal dysfunction. In fact, our data demonstrated that_Adm+/–_ dams did not exhibit overt preeclampsia. Using telemetric recordings of maternal blood pressure, we found no significant differences in basal blood pressure between pregnant_Adm+/–_ and WT control dams throughout gestation (49), and there were also no differences in urine protein levels during late gestation (data not shown). Although an explanation for a lack of maternal preeclampsia in_Adm+/–_ dams could be built around the differences between mouse and human gestation, a much more simplified explanation is likely. Namely, Adm–/– fetuses die between E13.5 and E14.5, and so their defective placentas are resorbed at midgestation, thus precluding systemic toxicity to the mother. In addition, since only one-quarter of the litter was of the Adm–/–_genotype, it is unlikely that a minority of placentas could induce the presentation of overt maternal preeclampsia early in gestation. Future studies of genetic deletion of_Adm and/or Calcrl exclusively in TGCs may help address this shortcoming (50).
Another interesting avenue for future studies will be to determine the mechanism by which the genetic dosage of fetal-derived AM is proportionally related to the number of uNK cells recruited to the decidua. A remarkable aspect of these findings is that the local dosage of AM peptide within an individual conceptus profoundly influenced decidual uNK cell content compared with a neighboring conceptus within the same litter, independent of maternal genotype. Therefore, this implies that localized fetal-derived mechanisms for trafficking and recruitment of uNK cells to the decidua must exist. It is worth noting that although AM peptide can act as a vasodilator on uterine blood vessels (29), and thereby possibly increase localized maternal blood flow and uNK cell delivery to the decidua, the more prevalent role of AM appears to be on lymphatic vessels. Indeed, global deletion of the AM receptor,Calcrl, in adult animals results in a multitude of systemic defects associated with lymphatic dysfunction (51), and we and others have consistently shown a preferential and prevalent role for AM in regulating lymphatic vessel permeability and proliferation (36, 52, 53). Considering that we are still only beginning to understand the complex processes that regulate uNK cell trafficking to the placenta (11, 54), it stands to reason that additional studies incorporating the function of uterine and decidual lymphatic vessels as conduits for the delivery, maturation, and clearance of immune cells should be explored (55).
Based on the mouse phenotypes presented herein, it will be of interest to determine whether perturbations in AM signaling during pregnancy in humans might be associated with, or even causative of, poor pregnancy outcomes. Numerous studies over the past decade have used an assortment of “homemade” and commercial kits for the measurement of plasma AM levels during human pregnancy. While there has been general concordance among studies that AM is elevated during a normal pregnancy, but blunted in preeclampsia, there has also been significant variation among studies, due to the inherently poor stability of AM peptide and differences in assays used (reviewed in ref. 23). However, measurement of the highly stable peptide hormone precursor of AM, midregional proadrenomedullin (MR-proAM), can now be performed with such a high level of accuracy that it is showing great clinical value in the context of predicting poor outcomes after myocardial infarction, sepsis, and community-acquired pneumonia (56). Therefore, reevaluation of plasma AM levels during pregnancy and in complications of pregnancy using these new technological advances is warranted. In addition, these data can be linked to recent findings revealing several SNPs in the human Adm gene (some of which correlate directly with plasma AM levels) that are associated with hypertension and proteinuria (57, 58).
An inherent caveat to these types of clinical studies, which is somewhat incongruent with our current model, is that they are often focused on maternal plasma levels and genotype (albeit by necessity). Our data clearly demonstrated that the fetal levels and genotype of Adm were the critical determinants of its function at the maternal-fetal interface. Therefore, it becomes of importance to identify genetic, physiological, and/or environmental factors that regulate _Adm_expression within the fetal trophoblast cell. It has long been appreciated that estrogen and hypoxia can potently upregulate Adm expression in a variety of cell types, including those of reproductive relevance (24, 59). Most interestingly, it has recently become clear that cigarette smoke, likely acting through 2 functional xenobiotic response elements proximal to the Adm start codon (60), can also potently induce Adm_expression in trophoblast cells (61). Moreover, exposure of humans and mice to cigarette smoke during pregnancy has recently been linked to a statistically significant increase in uNK cell numbers (62). Thus, although cigarette smoking during pregnancy has a wide range of deleterious effects, the cigarette smoke–induced upregulation of_Adm expression in fetal trophoblast cells, and the potential subsequent increase in uNK cells, may provide the mechanistic link for explaining the decades-old clinical observation that moderate smoking during pregnancy is actually protective against preeclampsia (63, 64).
In summary, our present findings revealed a critical function for a fetal-derived trophoblast factor in the maternal adaptation to pregnancy. The increased clinical usage of MR-proAM plasma levels as a biomarker for a variety of cardiovascular conditions underscores the importance of exploring AM as a biomarker for preeclampsia. Because CLR is already being exploited as a pharmacological target for the treatment of migraine (65, 66), it is conceivable that direct targeting of the AM/CLR axis may hold tremendous clinical benefit for the amelioration of pregnancy outcomes.
Methods
Mice.
Generation of mice with a targeted, homozygous deletion of the _Adm_and Calcrl genes has been previously described (39, 40). These mice were maintained on an isogenic 129S6/SvEv background.
Admhi/hi mice were generated by standard gene targeting protocols. The endogenous 3′UTR of the Adm gene was replaced with a cassette (parts of which were provided by M. Kakoki, The University of North Carolina at Chapel Hill) containing the bovine growth hormone UTR (bGH 3′UTR) in E14 ES cells. Southern blot analysis was performed by digesting genomic DNA with BamHI, which revealed an 11.4-kb fragment representing the WT allele and a 3.9-kb fragment corresponding to the Admhi allele. Several chimeric animals were generated, and germline transmission of the targeted allele was established on a mixed genetic background. F1 heterozygotes from male chimera–to–female C57BL/6J breedings were backcrossed to C57BL/6J for more than 10 generations. Routine genotyping of the_Admhi_ allele was performed by a 3-primer, PCR-based strategy (primer 1, 5′-AACCTTACACCTTGCTGAGACATTC-3′; primer 2, 5′-TTTATTAGGAAAGGACAGTGGGAGTG-3′; primer 3, 5′-CCCACATTCGTGTCAAACGCTAC-3′). Primers 1 and 3 amplify a 760-bp WT allele, while primers 2 and 3 amplify a 600-bp targeted allele.
For timed pregnancies, heterozygous male and female_Adm+/–_ or_Adm+/hi_ intercrosses were established, and the day when the vaginal plug was detected was considered E0.5. Fetal genotypes were determined by established allele-specific, PCR-based assays (39, 40) using genomic DNA isolated from fetal membranes or tail biopsies.
Ovary transplantation.
Ovary transplantation experiments were performed as previously described (67) using donor and recipient animals 8–16 weeks old on the 129S6/SvEv genetic background. Hemiovaries from_Adm+/–_ or_Adm+/+_ mice were surgically sutured into the ovarian bursa of recipient WT mice after bilateral removal of the recipient’s own ovaries. 2 weeks after surgery, _Adm+/–_ovary→WT and Adm+/+ ovary→WT recipients were used in experiments.
Doppler ultrasound.
Intravital Doppler ultrasound was performed on anesthetized female mice at E12.5 using the VisualSonics Vevo 770 system with a 40-MHz probe. Placental impedance was calculated from umbilical cord waveforms by dividing the arterial venous delay by the cycle length.
Placental vascular corrosion casting and scanning electron microscopy.
Vascular corrosion casting of E13.5 Adm+/+ and_Adm–/–_ placentas from littermates of Adm+/– male and female intercrosses were performed as previously described (68). The castings were processed for scanning electron microscopy as previously described (30) and were viewed and photographed with a Cambridge Stereoscan S200 scanning electron microscope (LEO Electron Microscopy Inc.) using an accelerating voltage of 20 kV.
In situ hybridization.
In situ hybridization was performed as previously described (30) using nonradioactive in situ hybridization reagents (1745816, 1585762, 1093274; Roche Diagnostics).
Cytokine expression arrays and cytokine antibody array.
RNA was extracted from whole placentas using TRIzol reagent (15596-026; Invitrogen) and purified with Qiagen RNeasy Mini Kit (74104; Qiagen). cDNA was made using the RT First Strand Kit (C-03; SABiosciences), and cytokine expression was determined using RT Profiler PCR Array System for Mouse Chemokines and Receptors (PAMM-022A-2; SABiosciences). Data were analyzed using the ΔΔCt method. Analysis for the simultaneous detection of cytokines and chemokines was performed with the Cytokine Antibody Array I (ARY006; R&D) following the manufacturer’s instructions.
TUNEL and BrdU staining.
TUNEL in situ cell death detection kit (1 684 795; Roche Applied Science) was used to identify apoptotic cells. Cell proliferation was marked with a single intraperitoneal injection of 100 mg BrdU per kilogram body weight (B9285; Sigma-Aldrich). BrdU Staining kit (93-3943; Zymed) was used to detect BrdU+ cells, which were quantified using Image J software.
uNK cell isolation and culture of primary aortic VSMCs.
uNK cells were isolated as previously described using the Dynal CELLection Biotin Binder Kit (115.33D; Dynal) in conjunction with biotinylated DBA lectin (L6533; Sigma-Aldrich) (17). Isolated uNK cells were cultured in complete RPMI media with 10% FCS for 24 hours and then supplemented with 10 nM AM or PBS vehicle. Cells and media were collected for analysis 24–48 hours after isolation. Aortic VSMCs were isolated from C57BL/6J mice and cultured as described previously (69).
Immunohistochemical antibodies.
Antibodies were as follows: anti–α-SMA (A2547; Sigma-Aldrich), anti-perforin (804-057-C100; Alexis Biochemicals), Ly49G2+(55-5314; BD Biosciences — Pharmingen), anti–placental alkaline phosphatase (CBL207; Millipore), cathepsin S M-19 (6505; Santa Cruz), FITC-conjugated isolectin B4 (L2895; Sigma-Aldrich), FITC-conjugated DBA lectin (L9142; Sigma-Aldrich).
Statistics.
In all figures, data are presented as mean ± SEM. Statistical analyses were performed with JMP software (SAS). 2-tailed Student’s_t_ test was used to compare 2 groups; 1-way ANOVA was used to compare more than 2 groups. A P value less than 0.05 was considered significant. Exact P values and n for each experiment are provided in the figure legends.
Study approval.
All experiments using animals were approved by the Institutional Animal Care and Use Committee of The University of North Carolina at Chapel Hill.
Supplementary Material
Supplemental data
Acknowledgments
We are grateful to members of the Caron lab, C. Grotegut (Duke University Medical Center), and members of the NIH-sponsored Collaborative Team on Implantation Biology for critically evaluating our manuscript and providing helpful discussions. We thank Xiu Xu for technical assistance with animal genotyping. The following School of Medicine Core Facilities of The University of North Carolina at Chapel Hill provided technical assistance and access to specialized equipment: Histology Research (K. McNaughton); Animal Models (R. Thresher); In Situ Hybridization (M. Aita); Mouse Cardiovascular Models Core (M. Rojas); Microscopy Services Laboratory (C.R. Bagnell and V.J. Madden). This work was supported by NIH grants HD46970, HD060860, and ARRA-HD060860-01S1, by the March of Dimes Birth Defects Foundation, and by the Burroughs Wellcome Fund (to K.M. Caron).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Citation for this article: J Clin Invest. 2013;123(6):2408–2420. doi:10.1172/JCI67039.
References
- 1.Bulmer JN, Williams PJ, Lash GE. Immune cells in the placental bed. Int J Dev Biol. 2009;54(2–3):281–294. doi: 10.1387/ijdb.082763jb. [DOI] [PubMed] [Google Scholar]
- 2.Smith SD, Dunk CE, Aplin JD, Harris LK, Jones RL. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am J Pathol. 2009;174(5):1959–1971. doi: 10.2353/ajpath.2009.080995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitley GS, Cartwright JE. Trophoblast-mediated spiral artery remodelling: a role for apoptosis. J Anat. 2009;215(1):21–26. doi: 10.1111/j.1469-7580.2008.01039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lash GE, Robson SC, Bulmer JN. Review: Functional role of uterine natural killer (uNK) cells in human early pregnancy decidua. Placenta. 2010;31(suppl):S87–S92. doi: 10.1016/j.placenta.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 5.Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. 2006;27(9–10):939–958. doi: 10.1016/j.placenta.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308(5728):1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 7.Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376(9741):631–644. doi: 10.1016/S0140-6736(10)60279-6. [DOI] [PubMed] [Google Scholar]
- 8.Hu D, Cross JC. Ablation of Tpbpa-positive trophoblast precursors leads to defects in maternal spiral artery remodeling in the mouse placenta. Dev Biol. 2011;358(1):231–239. doi: 10.1016/j.ydbio.2011.07.036. [DOI] [PubMed] [Google Scholar]
- 9.Madeja Z, et al. Paternal MHC expression on mouse trophoblast affects uterine vascularization and fetal growth. Proc Natl Acad Sci U S A. 2011;108(10):4012–4017. doi: 10.1073/pnas.1005342108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Screen M, Dean W, Cross JC, Hemberger M. Cathepsin proteases have distinct roles in trophoblast function and vascular remodelling. Development. 2008;135(19):3311–3320. doi: 10.1242/dev.025627. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Chen Z, Smith GN, Croy BA. Natural killer cell-triggered vascular transformation: maternal care before birth? Cell Mol Immunol. 2011;8(1):1–11. doi: 10.1038/cmi.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanna J, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. 2006;12(9):1065–1074. doi: 10.1038/nm1452. [DOI] [PubMed] [Google Scholar]
- 13.Burke SD, et al. Uterine NK cells, spiral artery modification and the regulation of blood pressure during mouse pregnancy. Am J Reprod Immunol. 2008;63(6):472–481. doi: 10.1111/j.1600-0897.2010.00818.x. [DOI] [PubMed] [Google Scholar]
- 14.Croy BA HH. Uterine natural killer cells: insights into their cellular and molecular biology from mouse modelling. Reproduction. 2003;126(2):149–160. doi: 10.1530/rep.0.1260149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Croy BA, van den Heuvel MJ, Borzychowski AM, Tayade C. Uterine natural killer cells: a specialized differentiation regulated by ovarian hormones. Immunol Rev. 2006;214:161–185. doi: 10.1111/j.1600-065X.2006.00447.x. [DOI] [PubMed] [Google Scholar]
- 16.Greenwood JD, et al. Ultrastructural studies of implantation sites from mice deficient in uterine natural killer cells. Placenta. 2000;21(7):693–702. doi: 10.1053/plac.2000.0556. [DOI] [PubMed] [Google Scholar]
- 17.Croy BA, Zhang J, Tayade C, Colucci F, Yadi H, Yamada AT. Analysis of uterine natural killer cells in mice. Methods Mol Biol. 2010;612:465–503. doi: 10.1007/978-1-60761-362-6_31. [DOI] [PubMed] [Google Scholar]
- 18.Yadi H, Burke S, Madeja Z, Hemberger M, Moffett A, Colucci F. Unique receptor repertoire in mouse uterine NK cells. J Immunol. 2008;181(9):6140–6147. doi: 10.4049/jimmunol.181.9.6140. [DOI] [PubMed] [Google Scholar]
- 19.Bao SH, Shuai W, Tong J, Wang L, Chen P, Sun J. Increased expression of Toll-like receptor 3 in decidual natural killer cells of patients with unexplained recurrent spontaneous miscarriage. Eur J Obstet Gynecol Reprod Biol. 2012;165(2):326–330. doi: 10.1016/j.ejogrb.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Fraser R, et al. Impaired decidual natural killer cell regulation of vascular remodelling in early human pregnancies with high uterine artery resistance. J Pathol. 2012;228(3):322–332. doi: 10.1002/path.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundara YT, Jordanova ES, Hernowo BS, Gandamihardja S, Fleuren GJ. Decidual infiltration of FoxP3(+) regulatory T cells, CD3(+) T cells, CD56(+) decidual natural killer cells and Ki-67 trophoblast cells in hydatidiform mole compared to normal and ectopic pregnancies. Mol Med Report. 2012;5(1):275–281. doi: 10.3892/mmr.2011.633. [DOI] [PubMed] [Google Scholar]
- 22.Fukui A, et al. Uterine and circulating natural killer cells and their roles in women with recurrent pregnancy loss, implantation failure and preeclampsia. J Reprod Immunol. 2011;90(1):105–110. doi: 10.1016/j.jri.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 23.Lenhart PM, Caron KM. Adrenomedullin and pregnancy: perspectives from animal models to humans. Trends Endocrinol Metab. 2012;23(10):524–532. doi: 10.1016/j.tem.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hewitt SC, Collins J, Grissom S, Deroo B, Korach KS. Global uterine genomics in vivo: microarray evaluation of the estrogen receptor alpha-growth factor cross-talk mechanism. Mol Endocrinol. 2005;19(3):657–668. doi: 10.1210/me.2004-0142. [DOI] [PubMed] [Google Scholar]
- 25.Ross GR, Chauhan M, Gangula PR, Reed L, Thota C, Yallampalli C. Female sex steroids increase adrenomedullin-induced vasodilation by increasing the expression of adrenomedullin2 receptor components in rat mesenteric artery. Endocrinology. 2006;147(1):389–396. doi: 10.1210/en.2005-0664. [DOI] [PubMed] [Google Scholar]
- 26.Thota C, Gangula PR, Dong YL, Yallampalli C. Changes in the expression of calcitonin receptor-like receptor, receptor activity-modifying protein (RAMP) 1, RAMP2, and RAMP3 in rat uterus during pregnancy, labor, and by steroid hormone treatments. Biol Reprod. 2003;69(4):1432–1437. doi: 10.1095/biolreprod.103.015628. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe H, et al. The estrogen-responsive adrenomedullin and receptor-modifying protein 3 gene identified by DNA microarray analysis are directly regulated by estrogen receptor. J Mol Endocrinol. 2006;36(1):81–89. doi: 10.1677/jme.1.01825. [DOI] [PubMed] [Google Scholar]
- 28.Witlin AG, Li ZY, Wimalawansa SJ, Grady JJ, Grafe MR, Yallampalli C. Placental and fetal growth and development in late rat gestation is dependent on adrenomedullin. Biol Reprod. 2002;67(3):1025–1031. doi: 10.1095/biolreprod.101.002196. [DOI] [PubMed] [Google Scholar]
- 29.Ross GR, Yallampalli C. Vascular hyperresponsiveness to adrenomedullin during pregnancy is associated with increased generation of cyclic nucleotides in rat mesenteric artery. Biol Reprod. 2007;76(1):118–123. doi: 10.1095/biolreprod.106.053777. [DOI] [PubMed] [Google Scholar]
- 30.Li M, Wu Y, Caron KM. Haploinsufficiency for adrenomedullin reduces pinopodes and diminishes uterine receptivity in mice. Biol Reprod. 2008;79(6):1169–1175. doi: 10.1095/biolreprod.108.069336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li M, Yee D, Magnuson TR, Smithies O, Caron KM. Reduced maternal expression of adrenomedullin disrupts fertility, placentation, and fetal growth in mice. J Clin Invest. 2006;116(10):2653–2662. doi: 10.1172/JCI28462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu D, Cross JC. Development and function of trophoblast giant cells in the rodent placenta. Int J Dev Biol. 2010;54(2–3):341–354. doi: 10.1387/ijdb.082768dh. [DOI] [PubMed] [Google Scholar]
- 33.Simmons DG, Cross JC. Determinants of trophoblast lineage and cell subtype specification in the mouse placenta. Dev Biol. 2005;284(1):12–24. doi: 10.1016/j.ydbio.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 34.Simmons DG, Fortier AL, Cross JC. Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev Biol. 2007;304(2):567–578. doi: 10.1016/j.ydbio.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Yotsumoto S, Shimada T, Cui CY, Nakashima H, Fujiwara H, Ko MS. Expression of adrenomedullin, a hypotensive peptide, in the trophoblast giant cells at the embryo implantation site in mouse. Dev Biol. 1998;203(2):264–275. doi: 10.1006/dbio.1998.9073. [DOI] [PubMed] [Google Scholar]
- 36.Fritz-Six KL, Dunworth WP, Li M, Caron KM. Adrenomedullin signaling is necessary for murine lymphatic vascular development. J Clin Invest. 2008;118(1):40–50. doi: 10.1172/JCI33302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shindo T, et al. Vascular abnormalities and elevated blood pressure in mice lacking adrenomedullin gene. Circulation. 2001;104(16):1964–1971. doi: 10.1161/hc4101.097111. [DOI] [PubMed] [Google Scholar]
- 38.Mason RW. Emerging functions of placental cathepsins. Placenta. 2008;29(5):385–390. doi: 10.1016/j.placenta.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Caron KM, Smithies O. Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc Natl Acad Sci U S A. 2001;98(2):615–619. doi: 10.1073/pnas.98.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dackor RT, Fritz-Six K, Dunworth WP, Gibbons CL, Smithies O, Caron KM. Hydrops fetalis, cardiovascular defects, and embryonic lethality in mice lacking the calcitonin receptor-like receptor gene. Mol Cell Biol. 2006;26(7):2511–2518. doi: 10.1128/MCB.26.7.2511-2518.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Croy BA, Chen Z, Hofmann AP, Lord EM, Sedlacek AL, Gerber SA. Imaging of vascular development in early mouse decidua and its association with leukocytes and trophoblasts. Biol Reprod. 2012;87(5):125. doi: 10.1095/biolreprod.112.102830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makrigiannakis A, Petsas G, Toth B, Relakis K, Jeschke U. Recent advances in understanding immunology of reproductive failure. J Reprod Immunol. 2011;90(1):96–104. doi: 10.1016/j.jri.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 43.Naruse K, et al. Localization of matrix metalloproteinase (MMP)-2, MMP-9 and tissue inhibitors for MMPs (TIMPs) in uterine natural killer cells in early human pregnancy. Hum Reprod. 2009;24(3):553–561. doi: 10.1093/humrep/den408. [DOI] [PubMed] [Google Scholar]
- 44.Maltepe E, Bakardjiev AI, Fisher SJ. The placenta: transcriptional, epigenetic, and physiological integration during development. J Clin Invest. 2010;120(4):1016–1025. doi: 10.1172/JCI41211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pijnenborg R, Robertson WB, Brosens I, Dixon G. Review article: trophoblast invasion and the establishment of haemochorial placentation in man and laboratory animals. Placenta. 1981;2(1):71–91. doi: 10.1016/S0143-4004(81)80042-2. [DOI] [PubMed] [Google Scholar]
- 46.Watson ED, Cross JC. Development of structures and transport functions in the mouse placenta. Physiology (Bethesda). 2005;20:180–193. doi: 10.1152/physiol.00001.2005. [DOI] [PubMed] [Google Scholar]
- 47.Moffett A, Loke C. Immunology of placentation in eutherian mammals. Nat Rev Immunol. 2006;6(8):584–594. doi: 10.1038/nri1897. [DOI] [PubMed] [Google Scholar]
- 48.Steinberg G, Khankin EV, Karumanchi SA. Angiogenic factors and preeclampsia. Thromb Res. 2009;123(suppl 2):S93–99. doi: 10.1016/S0049-3848(09)70020-9. [DOI] [PubMed] [Google Scholar]
- 49.Caron K, Hagaman J, Nishikimi T, Kim HS, Smithies O. Adrenomedullin gene expression differences in mice do not affect blood pressure but modulate hypertension-induced pathology in males. Proc Natl Acad Sci U S A. 2007;104(9):3420–3425. doi: 10.1073/pnas.0611365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ouseph MM, et al. Atypical E2F repressors and activators coordinate placental development. Dev Cell. 2012;22(4):849–862. doi: 10.1016/j.devcel.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoopes SL, Willcockson HH, Caron KM. Characteristics of multi-organ lymphangiectasia resulting from temporal deletion of calcitonin receptor-like receptor in adult mice. PLoS One. 2012;7(9):e45261. doi: 10.1371/journal.pone.0045261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dunworth WP, Fritz-Six KL, Caron KM. Adrenomedullin stabilizes the lymphatic endothelial barrier in vitro and in vivo. Peptides. 2008;29(12):2243–2249. doi: 10.1016/j.peptides.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin D, Harada K, Ohnishi S, Yamahara K, Kangawa K, Nagaya N. Adrenomedullin induces lymphangiogenesis and ameliorates secondary lymphoedema. Cardiovasc Res. 2008;80(3):339–345. doi: 10.1093/cvr/cvn228. [DOI] [PubMed] [Google Scholar]
- 54.Manaster I, Mandelboim O. The unique properties of uterine NK cells. Am J Reprod Immunol. 2010;63(6):434–444. doi: 10.1111/j.1600-0897.2009.00794.x. [DOI] [PubMed] [Google Scholar]
- 55.Red-Horse K, et al. Cytotrophoblast induction of arterial apoptosis and lymphangiogenesis in an in vivo model of human placentation. J Clin Invest. 2006;116(10):2643–2652. doi: 10.1172/JCI27306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Melander O, et al. Novel and conventional biomarkers for prediction of incident cardiovascular events in the community. JAMA. 2009;302(1):49–57. doi: 10.1001/jama.2009.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishimitsu T, et al. Microsatellite DNA polymorphism of human adrenomedullin gene in normotensive subjects and patients with essential hypertension. Hypertension. 2001;38(1):9–12. doi: 10.1161/01.HYP.38.1.9. [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi Y, Nakayama T, Sato N, Izumi Y, Kokubun S, Soma M. Haplotype-based case-control study revealing an association between the adrenomedullin gene and proteinuria in subjects with essential hypertension. Hypertens Res. 2005;28(3):229–236. doi: 10.1291/hypres.28.229. [DOI] [PubMed] [Google Scholar]
- 59.Marinoni E, et al. Regulation by hypoxia of adrenomedullin output and expression in human trophoblast cells. Eur J Obstet Gynecol Reprod Biol. 2011;154(2):146–150. doi: 10.1016/j.ejogrb.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 60.Portal-Nunez S, et al. Aryl hydrocarbon receptor-induced adrenomedullin mediates cigarette smoke carcinogenicity in humans and mice. Cancer Res. 2012;72(22):5790–5800. doi: 10.1158/0008-5472.CAN-12-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beiswenger TR, Feng L, Brown HL, Heine RP, Murtha AP, Grotegut CA. The effect of cigarette smoke extract on trophoblast cell viability and migration: the role of adrenomedullin. Reprod Sci. 2012;19(5):526–533. doi: 10.1177/1933719111426600. [DOI] [PubMed] [Google Scholar]
- 62.Prins JR, et al. Smoking during pregnancy influences the maternal immune response in mice and humans. Am J Obstet Gynecol. 2012;207(1):76.e1–76.e14. doi: 10.1016/j.ajog.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 63.Conde-Agudelo A, Althabe F, Belizan JM, Kafury-Goeta AC. Cigarette smoking during pregnancy and risk of preeclampsia: a systematic review. Am J Obstet Gynecol. 1999;181(4):1026–1035. doi: 10.1016/S0002-9378(99)70341-8. [DOI] [PubMed] [Google Scholar]
- 64.Lindqvist PG, Marsal K. Moderate smoking during pregnancy is associated with a reduced risk of preeclampsia. Acta Obstet Gynecol Scand. 1999;78(8):693–697. doi: 10.1080/j.1600-0412.1999.780806.x. [DOI] [PubMed] [Google Scholar]
- 65.Doods H, Arndt K, Rudolf K, Just S. CGRP antagonists: unravelling the role of CGRP in migraine. Trends Pharmacol Sci. 2007;28(11):580–587. doi: 10.1016/j.tips.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 66.Salvatore CA, et al. Pharmacological characterization of MK-0974 [N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxamide], a potent and orally active calcitonin gene-related peptide receptor antagonist for the treatment of migraine. J Pharmacol Exp Ther. 2008;324(2):416–421. doi: 10.1124/jpet.107.130344. [DOI] [PubMed] [Google Scholar]
- 67.Russell WL, Hurst JG. Pure strain mice born to hybrid mothers following ovarian transplantation. Proc Natl Acad Sci U S A. 1945;31(9):267–273. doi: 10.1073/pnas.31.9.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adamson SL, et al. Interactions between trophoblast cells and the maternal and fetal circulation in the mouse placenta. Dev Biol. 2002;250(2):358–373. doi: 10.1006/dbio.2002.0773. [DOI] [PubMed] [Google Scholar]
- 69.Sayers RL, et al. FRNK expression promotes smooth muscle cell maturation during vascular development and after vascular injury. Arterioscler Thromb Vasc Biol. 2008;28(12):2115–2122. doi: 10.1161/ATVBAHA.108.175455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental data