Microfluidic single-cell whole-transcriptome sequencing (original) (raw)
Significance
RNA sequencing of single cells enables measurement of biological variation in heterogeneous cellular populations and dissection of transcriptome complexity that is masked in ensemble measurements of gene expression. The low quantity of RNA in a single cell, however, hinders efficient and consistent reverse transcription and amplification of cDNA, limiting accuracy and obscuring biological variation with high technical noise. We developed a microfluidic approach to prepare cDNA from single cells for high-throughput transcriptome sequencing. The microfluidic platform facilitates single-cell manipulation, minimizes contamination, and furthermore, provides improved detection sensitivity and measurement precision, which is necessary for differentiating biological variability from technical noise.
Keywords: genomics, lab on chip, embryonic stem cell
Abstract
Single-cell whole-transcriptome analysis is a powerful tool for quantifying gene expression heterogeneity in populations of cells. Many techniques have, thus, been recently developed to perform transcriptome sequencing (RNA-Seq) on individual cells. To probe subtle biological variation between samples with limiting amounts of RNA, more precise and sensitive methods are still required. We adapted a previously developed strategy for single-cell RNA-Seq that has shown promise for superior sensitivity and implemented the chemistry in a microfluidic platform for single-cell whole-transcriptome analysis. In this approach, single cells are captured and lysed in a microfluidic device, where mRNAs with poly(A) tails are reverse-transcribed into cDNA. Double-stranded cDNA is then collected and sequenced using a next generation sequencing platform. We prepared 94 libraries consisting of single mouse embryonic cells and technical replicates of extracted RNA and thoroughly characterized the performance of this technology. Microfluidic implementation increased mRNA detection sensitivity as well as improved measurement precision compared with tube-based protocols. With 0.2 M reads per cell, we were able to reconstruct a majority of the bulk transcriptome with 10 single cells. We also quantified variation between and within different types of mouse embryonic cells and found that enhanced measurement precision, detection sensitivity, and experimental throughput aided the distinction between biological variability and technical noise. With this work, we validated the advantages of an early approach to single-cell RNA-Seq and showed that the benefits of combining microfluidic technology with high-throughput sequencing will be valuable for large-scale efforts in single-cell transcriptome analysis.
Although cells from the same organism are genetically similar, no two cells are identical (1, 2). Variation in gene expression can be found in cells from the same tissue as well as cells of the same type. This heterogeneity in cellular populations plays an important role in many biological processes, including cell fate determination (3, 4), cancer development and relapse (5, 6), embryonic development (7, 8), immune response (9), and neuron networking (10). Transcriptome analysis at the single-cell level is critical for uncovering this heterogeneity, which is obscured in conventional ensemble measurements, and identifying rare subpopulations defined by unique gene expression profiles (5, 11). To this end, differential gene expression in single cells has been studied using various methods, including fluorescent in situ hybridization (12, 13), microarray technology (14), and quantitative multiplex RT-PCR (15). Amplification of cDNA followed by high-throughput transcriptome sequencing (RNA-Seq) has recently become popular, because this approach provides the most comprehensive analysis of the transcriptome as well as the potential to discover novel genes, transcripts, or long noncoding RNAs (16).
Tang et al. (16) previously developed a single-cell RNA-Seq technology (Tang2009 protocol) that used oligo(dT) primers to reverse transcribe mRNA with poly(A) tails into cDNA. Recently, there has been a number of new approaches for low-quantity RNA-Seq (17–21), all with unique advantages and limitations. A notable approach, Smart-Seq, was developed to provide better coverage of full-length cDNAs for long mRNA molecules (19), and has undergone successive improvements since its inception (22, 23), including a recent demonstration of absolute mRNA counting (24). One limitation that remains among most current single-cell RNA-Seq methods, however, is sensitivity. Efficient and reproducible reverse transcription and cDNA amplification are difficult with the extremely low quantity of total RNA in a single cell (around 10 pg in a typical mammalian cell) (11), and insufficient reverse transcription efficiency and bias to highly expressed genes during amplification impede accurate quantification of low-abundance transcripts (25). Similarly with recent reports of quantifying variation in gene expression within homogeneous populations of cells using single-cell RNA-Seq, it is apparent that technical noise still poses significant limitations to the technology (26–28). Additional challenges to single-cell RNA-Seq include the precise sample manipulation necessary to isolate a single cell from a suspended population or tissue sample and effects of contamination, which are amplified with such few RNA transcripts in a single cell.
Here, we present a microfluidic-based system to prepare cDNA from single cells for RNA sequencing with improved precision and sensitivity. We used the Tang2009 protocol for reverse transcription and cDNA amplification outlined in ref. 29. This approach was recently shown to detect roughly 37% more genes than the Smart-Seq method when used with human ES cells (30). Our goal was to improve this method with microfluidic technology, which often offers both quantitative and qualitative advantages over traditional bench-top techniques (31). Implementing single-cell RNA-Seq in a microfluidic platform is promising for a number of reasons. (1) Performing reactions in parallel nanoliter volumes predefined by photolithography ensures high reproducibility by removing stochastic variation caused by pipetting error and variability in handling associated with bench-top experimentation. (2) Executing cell trapping, sorting, and lysis within a closed microfluidic device minimizes the chance for exogenous RNA and RNase contamination during this otherwise labor-intensive and hands-on procedure in the bench-top format. (3) It has been shown that performing amplification in nanoliter volumes improves reaction efficiency (32). Wu et al. (27) recently evaluated the performance of a commercial microfluidic single-cell RNA-Seq platform (C1; Fluidigm) and showed that implementation of a cDNA preparation protocol in microfluidic chambers offers advantages over tube-based approaches, including improved detection sensitivity.
In this report, we investigated gene expression in mouse embryonic cells using microfluidic-facilitated RNA-Seq to analyze 56 single mouse ES cell (mESC) transcriptomes and 6 single mouse embryonic fibroblast (MEF) transcriptomes. To quantitatively evaluate the sensitivity and precision of our technique, we also sequenced 23 libraries from extracted mESC RNA, representing three sets of technical replicates with varying starting amounts of material. Our technique enabled the identification of coding and noncoding genes that provided a clear distinction between pluripotent and differentiated mouse embryonic cells from a heterogeneous population. The high detection sensitivity and precision also allowed for quantification of variation within cells of the same type. By characterizing the technical variation of microfluidic-based RNA-Seq, we were able to measure true biological variation in a population of mESCs at the single-cell level.
Results and Discussion
Microfluidic cDNA Preparation.
Multilayer microfluidic devices with integrated valves provide an ideal platform for single-cell manipulation and analysis (33). Previously, microfluidic technology was used to perform whole-genome amplification with single cells (34), including single bacterial cells (35) as well as single human metaphase cells (36) and sperm cells (37). The basic procedure involved taking advantage of a microfluidic peristaltic pump to direct a single cell in suspension to an isolated sorting chamber. The cell was then pushed into successive chambers, where cell lysis and subsequent multistep amplification reactions could be performed in sequence. We adapted this technology to prepare double-stranded cDNA from mRNA of eight single cells in parallel using the protocol described in ref. 29, which was modified for compatibility with a microfluidic environment (Fig. 1 and SI Materials and Methods). A single-cell suspension was prepared from cultured mouse embryonic cells and injected into the microfluidic inlet channel. Single cells were trapped between two valves and then injected into the sorting chamber with a PBS solution (Fig. 1_B_). Each cell was then stored in its respective sorting chamber while the following cells were trapped and sorted. Single-cell trapping was performed manually under a stereomicroscope (Fig. S1). After all eight lanes were loaded, the chip was placed on a temperature-controlled platform (Fig. S1_A_), where the cDNA preparation reactions were completed for each cell in parallel. Before each reaction step, the appropriate reagent mix was manually loaded onto the device, and the reagent input line was purged and filled. A semiautomated protocol provided defined and consistent loading and mixing times, which minimized technical variation between each single-cell reaction (Fig. S2) and removed the need for highly trained technicians to carry out experimental protocols. The total reaction volume of all preparation steps was 140 nL, which is an over 600-fold decrease from the bench-top protocol (90 µL). After second-strand cDNA synthesis, the lanes were independently flushed with 5 µL nuclease-free water, which was recovered along with the cDNA using gel-loading pipette tips. Additional amplification, followed by purification and library preparation, was performed in a tube using conventional bench-top techniques (SI Materials and Methods). cDNA libraries representing whole single-cell transcriptomes were then sequenced on a next generation sequencing platform (Fig. 1_C_).
Fig. 1.

Device schematic and experimental pipeline. (A) Micrograph of the microfluidic device filled with colored dye. Blue lines are the control channels, and purple lines are the flow channels. The single-cell suspension was injected into cell input, and reagents were injected into reagent input. Double-stranded cDNA was recovered from the output ports. (B) Detailed diagram of a single-reaction pipeline. After a single cell was trapped in the trapping chamber (0.86 nL), it was pushed into the sorting chamber (S; 1.35 nL) and then, consecutive reactions for cell lysis (1; 3.82 nL), reverse transcription (2; 3.82 nL), polyA tailing (3; 2.70 nL), primer digestion (4; 10.1 nL), and second-strand cDNA synthesis (5; 128 nL). (C) Complete experimental pipeline. Off-chip amplification and library preparation are explained in SI Materials and Methods.
Transcriptome Analysis of Single mESCs, MEFs, and Bulk Extracted RNA.
We used the Illumina HiSeq 2500 platform to sequence 94 cDNA libraries generated on-chip from single cells and bulk RNA extracted from mESCs. In all, we analyzed 56 mESCs, 6 MEFs, 3 technical replicates of 40-pg bulk RNA, 16 technical replicates of 8-pg bulk RNA, 4 technical replicates of 2-pg bulk RNA, and 9 negative controls (Fig. 2 and Table S1). Each library was sequenced, on average, to 10 million paired-end reads (2 × 100 bp), which were trimmed, filtered, and mapped to annotated genes in the mouse reference sequence (Refseq) downloaded from the University of California, Santa Cruz genome browser (38) using the Burrows–Wheeler Aligner (39). Relative gene expression was estimated by calculating reads per kilobase transcript per million mapped reads (RPKM). Throughout this study, reliably detected genes were defined by RPKM > 1 unless stated otherwise. Because we did not initially discriminate cell state or survival during sorting, some of the libraries may have come from dead or unhealthy cells with nonrepresentative mRNA distributions (Fig. S3 A and B). For most of the following analysis, we discarded libraries in which less than 40% of the reads were mapped to the mouse reference transcriptome (Fig. S3_C_).
Fig. 2.
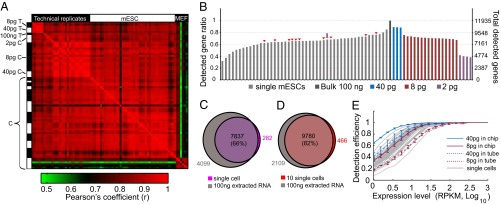
Single-cell transcriptome sequencing sensitivity. (A) Pairwise Pearson correlation coefficient between expression levels of all genes with RPKM > 1 for each library (80 in total). Discarded cells and negative control libraries were excluded (Table S1). The left vertical and top horizontal axes labels identify library type. T represents tube experiments, and C represents chip experiments. Black and white in the left vertical axis denote cDNA prepared in separate microfluidic devices. (B) Gene detection in 48 single-cell and 23 technical replicate libraries ranked by their total number of detected genes and compared with 100-ng bulk extracted RNA. The single-cell data are plotted with sample name labels as in Fig. S4_C_. (C) Comparison of genes detected with RPKM > 1 in a typical cell (indicated with a purple arrow in B) with the genes detected in the 100-ng bulk sample. All genes within the purple circle were detected in at least 2 of 48 single cells. The percent is the ratio of the overlapping region to the entire bulk circle. (D) Genes detected in 10 randomly selected cells (indicated with red arrows in B) after randomly sampling 200,000 reads from each library and mapping them to the reference sequence (red circle) compared with the genes detected in 2 million reads from the 100-ng bulk sample (gray circle). (E) The ratio of genes detected in the single-cell libraries (gray lines) and technical replicate libraries to the genes detected in the bulk library binned by expression level. Error bars indicate SD in the following technical replicates: solid blue line, 40 pg in chip (n = 3); dashed blue line, 40 pg in tube (n = 3); solid red line, 8 pg in chip (n = 16); dashed red line, 8 pg in tube (n = 3).
For technical replicate experiments, purified RNA extracted from 500,000 mESCs was diluted and injected into the cell loading channel (SI Materials and Methods). The eight sets of trapping valves were then simultaneously actuated, and the content of each trapping chamber was pushed into their respective sorting chambers in parallel. The total RNA mass was determined by multiplying the concentration of diluted bulk RNA by the volume of the trapping chamber. After performing a microfluidic cDNA preparation experiment, results were validated by quantitative real-time PCR measurement of reference genes and pluripotency-related genes (for the mESCs) before purification and library preparation (Fig. S3_A_). We also sequenced libraries from extracted mESC RNA prepared in a tube for comparison, including three technical replicates of 40 pg, three technical replicates of 8 pg, and one library from 100 ng, which was used as an estimate of the complete mESC transcriptome. The heat map in Fig. 2_A_ displays the correlation coefficient between all single-cell and technical replicate libraries. As expected, the technical replicates were generally more correlated than individual ES cells. MEF cells correlated weakly (r < 0.8) with single mESCs and extracted mESC RNA (Fig. S4_A_).
Gene Detection Sensitivity.
Sensitivity of RNA-Seq is defined by the detection efficiency of a single transcript. Single-molecule detection efficiency, however, is not necessarily consistent across transcript homology and length; therefore, in whole-transcriptome analysis, sensitivity can be practically understood as the global detection efficiency or the total number of genes detected. Fig. 2_B_ displays the total number of reliably detected genes in each of the single mESC libraries and technical replicates. The ratio of genes detected in each library to the total genes detected in the 100-ng bulk sample is also represented. On average, 8,000 genes were detected in single mESCs, which were 65% of the genes detected in the bulk sample. The Venn diagram in Fig. 2_C_ shows the overlap between reliably detected genes in an average single cell and the bulk sample. On average, there were over 200 genes that were detected in single cells but not the bulk sample. These are likely transcripts that were expressed in a small fraction of cells or low-abundance transcripts that were below the detection limit in ensemble measurements.
Because of heterogeneity in gene expression between single cells, it is possible to partially reconstruct the bulk mRNA distribution by sequencing multiple single cells. We pooled transcriptomes of 10 randomly selected single mESCs sequenced to a depth of 0.2 million reads each and compared genes reproducibly detected in single cells to genes mapped from 2 million bulk sample reads. With the same total number of reads, 10 single-cell transcriptomes accounted for over 80% of the bulk mRNA population, with good correlation between expression levels (Fig. 2_D_ and Fig. S4_B_). This showed that, with our microfluidic approach, it is possible to construct a representative sample of the bulk transcriptome and collect 10 single-cell transcriptomes for the same cost as sequencing a single bulk library.
It is challenging, however, to accurately assess sensitivity with single-cell libraries, because the number of genes detected in a cell depends on the total amount of mRNA present in that cell, which is variable. We, therefore, further evaluated sensitivity by comparing the number of detected genes in technical replicate libraries with the bulk sample across the full range of expression levels (Fig. 2_E_). Our microfluidic RNA-Seq technology consistently detected more genes than conventional cDNA preparations performed in tube. For low-abundance transcripts with RPKM = 1, we were able to detect 35% of genes in the 8-pg sample and over 60% of genes in the 40-pg sample. The 8-pg replicates contained a total number of genes that was comparable to the single-cell libraries (Fig. 2 B and E). These results show that the microfluidic approach provides a twofold increase in sensitivity for detection of low-abundance genes within single cells.
Single-Cell RNA-Seq Measurement Precision.
Variation between single-cell cDNA library preparations is caused by random experimental error, stochastic variability in the RNA-Seq protocol, and biological variation between cells. Sources of random error typically include variation in pipetting volumes, timing, mixing, and reaction temperature. These noise sources can potentially limit precision in any RNA-Seq technique, and a reduction of the noise floor would improve the sensitivity of measurements to biological variation. With microfluidics, it is possible to minimize the technical noise associated with human handling by carrying out reactions semiautomatically in parallel reaction chambers with lithographically defined nanoliter volumes. We characterized the stochastic variation of our method with the technical replicate samples. Because these replicates were prepared from the same bulk extracted RNA sample, variation in relative gene counts between replicates represented a combination of technical noise and stochastic sampling effects. We first compared variation between pairs of replicates at decreasing starting amounts of RNA (Fig. 3 A–C); 40-pg replicates show strong correlation in their gene count distribution (Pearson r > 0.99). Two-picogram replicates showed comparable gene counts among the more abundant genes; however, such low RNA quantities presented a practical limit on precision, because 76% of genes had a coefficient of variation (CV) greater than one (Fig. S5_A_). In general, the microfluidic approach is slightly more reproducible than the tube-based protocol, showing stronger correlation between replicates of both 40- and 8-pg samples (Fig. 3_D_).
Fig. 3.

Assessment of microfluidic single-cell RNA-Seq reproducibility. Scatter plots show the correlation between technical replicates of extracted mESC RNA diluted to (A) 40 pg (B40-2 and B40-3), (B) 8 pg (B8-12 and B8-15), and (C) 2 pg (B2-1 and B2-2) with the Pearson correlation coefficient (r). (D) For each extracted RNA dilution, the correlation was measured for all pairs of replicates. The mean of these correlation coefficients is displayed for 40 pg (blue) and 8 pg (red) in both chip (solid bars) and tube (striped bars). The number of replicates for each dataset is the same as in Fig. 2_E_. (E) The CV (SD normalized by the mean) is plotted against the log10-transformed geometric mean of expression for all genes detected in 44 single mESCs (gray dots), 16 8-pg technical replicates (red dots), and 3 40-pg technical replicates (blue dots). The spread in variation for the extracted RNA samples represents technical noise, whereas the variation in single-cell expression is a combination of technical and biological variation.
Precision in quantifying the abundance of any gene depends on the absolute number of mRNA molecules present in the sample. RPKM, however, is a measure of relative abundance, which is why genes with similar RPKM values show less variation between 40-pg replicates than they do in 8-pg replicates (Fig. 3_E_ and Fig. S5_A_). This observation is a reminder that the limiting parameter in RNA-Seq experiments is the total number of RNA molecules and not the number of cells involved. An average mESC has about 10–20 pg mRNA (SI Materials and Methods), and therefore, we used 8-pg technical replicates to make a conservative estimate of the technical noise for single-cell quantities of starting material. Genes that had a CV between single cells that was 3 SDs higher than the mean CV of genes with the same RPKM in the 8-pg sample were concluded to show biological variability above the technical noise (Fig. S5_B_ and Dataset S1). Among these 475 genes was Dppa3, which is known to exhibit high cell-to-cell variability in stem cells (40). These genes showed a large range of variation between cells (Fig. S5_C_). Such heterogeneity can be a feature of cell size or phase, can be caused by intrinsically stochastic processes during transcription, or may be related to complex regulatory networks. It is only in the absence of comparable technical variation that gene expression distribution characteristics can be identified.
Accuracy of mRNA Abundance Measurements.
To assess accuracy, it is important to have an estimate of the true value of a measured quantity. Single-cell gene expression measurements with RNA-Seq have been validated with quantitative real-time PCR (27) and compared with known quantities of an exogenous spike-in (17, 28). We added the External RNA Controls Consortium (ERCC) mRNA spike-ins (Ambion; Life Technologies), a set of 92 synthetic mRNA molecules covering a range of concentrations, to 35 of the single-cell reactions (Table S1). We then compared the measured mean abundance with the number of starting molecules in three of these experiments that had comparable ratios of reads mapped to the ERCC reference (Fig. 4_A_ and Fig. S6 A and B). The results confirmed a strong correlation (Pearson r > 0.98) between measured and predicted abundance of spike-in molecules. Low-quantity ERCC transcripts, however, showed increased noise levels (possibly caused by degradation as a result of dilution). To evaluate sensitivity and accuracy at low-molecule levels, we used a set of three exogenous genes encoding red fluorescent protein (RFP), green fluorescent protein (GFP), and cre recombinase (Cre) that we purified and quantified for spike-in applications (SI Materials and Methods). We added a known amount of these purified transcripts to the lysis buffer with ERCC before initiating the reaction. This small subset of spike-in genes was diluted and added to 27 samples, such that there were, on average, two Cre molecules in each single-cell reaction. Assuming a Poisson distribution, the predicted fraction of experiments containing at least one Cre molecule was 0.86 or 23 of 27 samples that contained spike-in. After sequencing, Cre was detected (RPKM > 0) in 21 experiments (Fig. 4_B_). This result indicates that, with an average of two molecules, we were able to successfully detect the presence of one or more Cre molecules over 90% of the time. Low-copy number detection in single-cell experiments is useful for finding rare gene expression events, which may be masked in bulk measurements. Additionally, high single-molecule detection efficiency is a critical measure of accuracy that ultimately determines how much useful information can be gained with single-cell RNA-Seq (28).
Fig. 4.

Assessment of accuracy with RNA spike-in. (A) Mean expression of ERCC spike across three samples: mESC42 (negative control), mESC43 (negative control), and mESC44 (discarded cell). Samples were chosen because of their large number and high ratio of reads mapped to the ERCC reference (Fig. S6 A and B). (B) Detection of Cre spike-in abundance in 27 samples that each contained two Cre molecules on average.
Biological Variation Between Cell Types.
Cell type can be distinguished many ways, including morphology, response to functional assays, cell surface markers, and gene expression profile. Many of these approaches require sorting and isolating cells as well as a priori knowledge for which indicators to look. Single-cell whole-transcriptome profiling, however, can be applied in heterogeneous populations, and sophisticated statistical methods can be used to identify differences between cells based on relative gene expression patterns (5). We explored the ability of our microfluidic approach to reveal subtle differences between mESCs and MEFs. To make this comparison, we sequenced six MEF cell transcriptomes and compared them with the mESC population in an attempt to characterize the relationship between a cell’s gene expression profile and identity.
We grouped 6 MEF cells with 6 typical mESCs and calculated the CV for detected genes across all 12 cells as a function of mean expression (Fig. 5_A_). In this heterogeneous group, large variation in transcript abundance is expected in genes that have strong differential expression between MEFs and mESCs. Additionally, genes with high cell-to-cell variation within mESCs and MEFs will also display large CVs. To account for variability between mESCs, we compared variation in the heterogeneous population with variation in a homogeneous population of 12 single mESCs. We found over 600 genes that had a significantly higher CV (P value < 0.003) in the heterogeneous cell mixture. Unsupervised hierarchical clustering of 12 cells in the heterogeneous group was performed using the expression levels of these 689 variable genes (Fig. 5_B_, SI Materials and Methods, and Dataset S1). Twelve cells were sorted into two distinct groups that accurately reflected their known type. As expected, there were three general subsets of genes: genes that showed high differential expression in mESCs, genes that showed high differential expression in MEFs, and genes that seemed to show large variation among all of the cells.
Fig. 5.

Variation in gene expression within and between mESCs and MEFs. (A) The CV in genes expressed in a group of 12 single mESCs (gray dots) and a group of 6 mESCs and 6 MEFs (red dots) plotted against mean expression. Genes in the heterogeneous population exhibiting a CV greater than 3 SDs above the mean CV of 12 mESCs were considered to show high variability and are colored turquoise (Dataset S1). (B) Unsupervised hierarchical clustering of six mESCs and six MEFs based on the expression levels of 689 genes found to show high variation between the two cell types in A. (C) Unsupervised hierarchical clustering of six mESCs and six MEFs based on expression levels of 38 long noncoding RNAs that showed high variability between the two cell types (Fig. S6_C_ and Dataset S1). (D) Histogram of expression levels of Dppa3 in 44 single mESC libraries. (E) Correlation of CV between genes expressed with RPKM > 50 in six MEF cells and six typical mESCs (labeled purple in Fig. S4_C_). The percentages represent the fraction of genes with higher CV among MEF cells than mESCs (blue) and lower CV among MEF cells than mESCs (red). Similar plots with various sets of six mESCs are shown in Fig. S7.
We used the annotated gene ontology database DAVID (41) to classify groups of genes identified by clustering (Table S2). Genes that were enriched in the mESCs included stem cell maintenance-related genes (for example, Klf4 and Sox2) and transcription factors associated with undifferentiated stem cells (like Pou5f1 and Utf1). Similarly, among genes that were enriched in MEFs were genes important in differentiation and collagen production. There was also a large subset of genes that was highly expressed in both cell types (RPKM > 300) but consistently enriched in mESCs. This group of genes included many subsets related to ribosomal processes and components. We took the same approach to find differences in the expression of long noncoding RNA between the two cell types and found 38 transcripts that showed more variability within a heterogeneous population of mESCs and MEFs than within a homogeneous population of mESCs (Fig. S6_C_ and Dataset S1). Unsupervised clustering of 12 cells in the heterogeneous population and their expression levels of these long noncoding RNAs accurately resolved the two cell types as well (Fig. 5_C_).
Biological Variation Within Cell Type.
The mESCs and MEF cells showed substantial differential expression in large sets of genes. Characterizing variation within cell type, however, can be more challenging because it often requires detection of much smaller changes in transcript abundance. Fig. 3_E_ reveals hundreds of genes that showed variability in mESCs that was above the measurement’s technical noise. Some of these genes, like Dppa3, have broad or bimodal expression distributions at the single-cell level with over 1,000-fold change in expression levels (Fig. 5_D_ and Fig. S5_C_).
The clustering diagram in Fig. 5_B_ reveals noticeable heterogeneity between the six MEF cells as well. This observation is corroborated by a weaker correlation among MEF cells compared with mESCs (Fig. 2_A_ and Fig. S4_A_). We investigated this heterogeneity further to understand the nature of cell-to-cell variation in these two cell types. Genes expressed at low levels in one of two cell types were expected to showed more cell-to-cell variation (Fig. 3_E_). For that reason, we compared the CV of genes, which were highly expressed (RPKM > 50) in both mESCs and MEF cells. Among these roughly 1,000 genes, 64% exhibited higher variation in six MEF cells, on average, compared with sets of six mESCs (Fig. 5_E_ and Fig. S7). A possible explanation for the increased variation in MEF cells is the diversity of tissue origin within the fibroblasts, which were isolated from the whole mouse embryo.
Conclusion
Currently, sensitivity and precision present some of the major obstacles for the development of single-cell RNA-Seq technologies. In this report, we addressed these limitations with a microfluidic platform coupled with high-throughput sequencing for single-cell whole-transcriptome analysis. The microfluidic approach facilitates single-cell manipulation, minimizes contamination, eliminates operational errors, and increases experimental efficiency and throughput with parallel reaction pipelines. We sequenced technical replicates of extracted total RNA and 62 single mouse embryonic cells to benchmark the performance of this technology and showed improvements in precision and sensitivity. There, inevitably, is bias associated with capture and nonlinear amplification of miniscule amounts of mRNA (25). Using oligo(dT) primers for reverse transcription, for example, limits the ability to capture full-length transcripts. However, bias to the 3′ end of the transcript is, in fact, reduced in the microfluidic format (Fig. S8). Additionally, many applications of single-cell RNA-Seq benefit from a thoroughly annotated reference genome, like the mouse or the human reference genome, and do not require complete transcript coverage. Here, we sacrificed transcript coverage for the increased mRNA detection sensitivity of oligo(dT) primers and showed that, by sequencing 10 single cells to an average of 200,000 reads each, we were able to effectively reconstruct a large portion of the bulk transcriptome. We also showed the ability to identify differentially expressed genes in a heterogeneous population of cells, which were, in turn, used to distinguish cell type. In addition to improved sensitivity and precision, there are many practical advantages of performing cDNA preparation in a microfluidic device. With the throughput reported here, reagent costs can be cut by up to 10 times, because we can use less reagent to process eight cells than is required to process one cell with the bench-top approach. Additionally, this approach is scalable, and the throughput could be doubled without much added complexity in chip design, fabrication, and operation. In this aspect, throughput is limited by the time required to trap and sort individual cells. Some microfluidic devices (42), including Fluidigm’s C1, take advantage of passive cell trapping to achieve an order of magnitude higher throughput. An advantage of active trapping, however, is the ability to actively select cells of interest or discard unwanted cells. Furthermore, with refined cell suspension preprocessing, the capture rate in our configuration can, in theory, approach 100%. This feat would require careful device engineering to ensure that no cells are lost between injection and trapping. A high capture rate is particularly valuable for applications that require transcriptome analysis of rare cells. Coupled with fluorescent labeling and microscopy, our microfluidic platform presents the possibility of actively selecting rare cells of interest and performing whole-transcriptome sequencing with higher throughput and reproducibility than is possible in a bench-top format.
Materials and Methods
Microfluidic devices were fabricated using standard multilayer soft lithography. cDNA preparation was based on the protocol outlined in ref. 29. A detailed description of microfluidic device fabrication and operation along with a reagent list and protocols for cDNA preparation and library preparation can be found in SI Materials and Methods.
Supplementary Material
Supporting Information
Acknowledgments
The authors thank A. R. Wu and B. X. Feng for helpful discussions. This work was supported by Ministry of Science and Technology of China Grants 2011CB809106, 2012CB966704, and 2012AA02A101; National Natural Science Foundation of China Grants 21222501, 91313302, 31271543, and 21305007; China Postdoctoral Science Foundation Grant 20100470165; and the Fok Ying Tung Educational Foundation. A.M.S. was supported by the Whitaker International Biomedical Engineering Scholarship.
Footnotes
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE47835).
References
- 1.Kalisky T, Blainey P, Quake SR. Genomic analysis at the single-cell level. Annu Rev Genet. 2011;45:431–445. doi: 10.1146/annurev-genet-102209-163607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li GW, Xie XS. Central dogma at the single-molecule level in living cells. Nature. 2011;475(7356):308–315. doi: 10.1038/nature10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo G, et al. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell. 2010;18(4):675–685. doi: 10.1016/j.devcel.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 4.Losick R, Desplan C. Stochasticity and cell fate. Science. 2008;320(5872):65–68. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalerba P, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol. 2011;29(12):1120–1127. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marks H, Veenstra GJ, Stunnenberg HG. Insightful tales from single embryonic cells. Cell Stem Cell. 2010;6(5):397–398. doi: 10.1016/j.stem.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 8.Tang F, et al. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell. 2010;6(5):468–478. doi: 10.1016/j.stem.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. 2010;20(1):4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ståhlberg A, et al. Defining cell populations with single-cell gene expression profiling: Correlations and identification of astrocyte subpopulations. Nucleic Acids Res. 2011;39(4):e24. doi: 10.1093/nar/gkq1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang F, Lao K, Surani MA. Development and applications of single-cell transcriptome analysis. Nat Methods. 2011;8(4) Suppl:S6–S11. doi: 10.1038/nmeth.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Femino AM, Fay FS, Fogarty K, Singer RH. Visualization of single RNA transcripts in situ. Science. 1998;280(5363):585–590. doi: 10.1126/science.280.5363.585. [DOI] [PubMed] [Google Scholar]
- 13.Taniguchi Y, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329(5991):533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurimoto K, et al. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res. 2006;34(5):e42. doi: 10.1093/nar/gkl050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taniguchi K, Kajiyama T, Kambara H. Quantitative analysis of gene expression in a single cell by qPCR. Nat Methods. 2009;6(7):503–506. doi: 10.1038/nmeth.1338. [DOI] [PubMed] [Google Scholar]
- 16.Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 17.Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012;2(3):666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Islam S, et al. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011;21(7):1160–1167. doi: 10.1101/gr.110882.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramsköld D, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30(8):777–782. doi: 10.1038/nbt.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasagawa Y, et al. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013;14(4):R31. doi: 10.1186/gb-2013-14-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan X, et al. Two methods for full-length RNA sequencing for low quantities of cells and single cells. Proc Natl Acad Sci USA. 2013;110(2):594–599. doi: 10.1073/pnas.1217322109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Picelli S, et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10(11):1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 23.Picelli S, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9(1):171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 24.Islam S, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014;11(2):163–166. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- 25.Ozsolak F, Milos PM. RNA sequencing: Advances, challenges and opportunities. Nat Rev Genet. 2011;12(2):87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brennecke P, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods. 2013;10(11):1093–1095. doi: 10.1038/nmeth.2645. [DOI] [PubMed] [Google Scholar]
- 27.Wu AR, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11(1):41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marinov GK, et al. From single-cell to cell-pool transcriptomes: Stochasticity in gene expression and RNA splicing. Genome Res. 2014;24(3):496–510. doi: 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang F, et al. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc. 2010;5(3):516–535. doi: 10.1038/nprot.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan L, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20(9):1131–1139. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- 31.Streets AM, Huang Y. Chip in a lab: Microfluidics for next generation life science research. Biomicrofluidics. 2013;7(1):11302. doi: 10.1063/1.4789751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marcy Y, et al. Nanoliter reactors improve multiple displacement amplification of genomes from single cells. PLoS Genet. 2007;3(9):1702–1708. doi: 10.1371/journal.pgen.0030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lecault V, White AK, Singhal A, Hansen CL. Microfluidic single cell analysis: From promise to practice. Curr Opin Chem Biol. 2012;16(3-4):381–390. doi: 10.1016/j.cbpa.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 34.Marcy Y, et al. Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci USA. 2007;104(29):11889–11894. doi: 10.1073/pnas.0704662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pamp SJ, Harrington ED, Quake SR, Relman DA, Blainey PC. Single-cell sequencing provides clues about the host interactions of segmented filamentous bacteria (SFB) Genome Res. 2012;22(6):1107–1119. doi: 10.1101/gr.131482.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan HC, Wang J, Potanina A, Quake SR. Whole-genome molecular haplotyping of single cells. Nat Biotechnol. 2011;29(1):51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Fan HC, Behr B, Quake SR. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell. 2012;150(2):402–412. doi: 10.1016/j.cell.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karolchik D, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayashi K, Lopes SM, Tang F, Surani MA. Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell. 2008;3(4):391–401. doi: 10.1016/j.stem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 42.White AK, et al. High-throughput microfluidic single-cell RT-qPCR. Proc Natl Acad Sci USA. 2011;108(34):13999–14004. doi: 10.1073/pnas.1019446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information