Pseudomonas aeruginosa High-Level Resistance to Polymyxins and Other Antimicrobial Peptides Requires cprA, a Gene That Is Disrupted in the PAO1 Strain (original) (raw)
Abstract
The arn locus, found in many Gram-negative bacterial pathogens, mediates resistance to polymyxins and other cationic antimicrobial peptides through 4-amino-l-arabinose modification of the lipid A moiety of lipopolysaccharide. In Pseudomonas aeruginosa, several two-component regulatory systems (TCSs) control the arn locus, which is necessary but not sufficient for these resistance phenotypes. A previous transposon mutagenesis screen to identify additional polymyxin resistance genes that these systems regulate implicated an open reading frame designated PA1559 in the genome of the P. aeruginosa PAO1 strain. Resequencing of this chromosomal region and bioinformatics analysis for a variety of P. aeruginosa strains revealed that in the sequenced PAO1 strain, a guanine deletion at the end of PA1559 results in a frameshift and truncation of a full-length open reading frame that also encompasses PA1560 in non-PAO1 strains, such as P. aeruginosa PAK. Deletion analysis in the PAK strain showed that this full-length open reading frame, designated cprA, is necessary for polymyxin resistance conferred by activating mutations in the PhoPQ, PmrAB, and CprRS TCSs. The cprA gene was also required for PmrAB-mediated resistance to other cationic antimicrobial peptides in the PAK strain. Repair of the mutated cprA allele in the PAO1 strain restored polymyxin resistance conferred by an activating TCS mutation. The deletion of cprA did not affect the _arn_-mediated lipid A modification, indicating that the CprA protein is necessary for a different aspect of polymyxin resistance. This protein has a domain structure with a strong similarity to the extended short-chain dehydrogenase/reductase family that comprises isomerases, lyases, and oxidoreductases. These results suggest a new avenue through which to pursue targeted inhibition of polymyxin resistance.
INTRODUCTION
Polymyxins (Pm) are acylated cationic peptides active against Pseudomonas aeruginosa and other Gram-negative pathogens. As pharmaceuticals, Pm B sulfate (PMB) and colistimethate, the prodrug form of colistin (also known as Pm E), are increasingly used to treat multidrug-resistant strains of P. aeruginosa that cause serious infections in critically ill patients and in those with cystic fibrosis (CF) (1, 2). Multidrug-resistant strains emerge when first-line agents, such as antipseudomonal β-lactams, aminoglycosides, and fluoroquinolones, are used repeatedly, imposing major selective pressure (3–5). Unfortunately, the prevalence of clinical isolates of P. aeruginosa and other Gram-negative pathogens resistant to PMB and colistin is increasing (6–13).
Pm binds to lipopolysaccharide (LPS), the major constituent of the Gram-negative outer membrane, promoting its own uptake and diffusion through the periplasm to the inner membrane, where it disrupts cellular respiration and leads to cell lysis (14). The resistance of Gram-negative pathogens to Pm and other cationic antimicrobial peptides (CAPs) is associated with covalent modification of LPS, namely, the addition of 4-amino-l-arabinose (l-Ara4N) to phosphate groups within its lipid A and core oligosaccharide components (13, 15–17). This amino-sugar modification likely hinders charge interactions between phosphate groups within LPS and amino groups within CAPs. Genes of the arnBCADTEFpmrE operon encode enzymes responsible for synthesis and transfer of l-Ara4N to LPS (18, 19).
Several two-component systems (TCSs) of P. aeruginosa, including PhoPQ, PmrAB, ParRS, CprRS, and ColRS, convergently regulate Pm resistance. These systems generally induce arn operon transcription in response to CAP exposure or divalent cation depletion (18–21), or as a consequence of mutation (12, 17, 22–29). However, disruption of the cprRS locus (deletion or transposon insertion) results in a partial loss of PhoPQ-mediated Pm resistance without a loss of l-Ara4N from lipid A (29). Moreover, some clinical strains of P. aeruginosa lose Pm resistance after drug-free passage without the concomitant loss of l-Ara4N from lipid A (12). These observations indicate that in P. aeruginosa, Pm resistance requires not only l-Ara4N modification of lipid A, but also the expression of one or more CprRS-regulated gene products.
Prior studies in the P. aeruginosa PAO1 reference strain have shown that the annotated PA1559 and PA1560 open reading frames (ORFs) (30) are positively regulated through the CprRS (31), ParRS (27), and PmrAB TCSs (32) and are induced by CAPs, such as indolicidin (27) and LL-37 (33). We previously observed, in a Pm-resistant (Pmr) phoQ mutant of the P. aeruginosa PAK reference strain, that transposon insertion within the PA1559 ORF is associated with a loss of Pm resistance (29). We hypothesized that PhoPQ-, PmrAB-, and CprRS-mediated Pm resistance in P. aeruginosa is dependent on the PA1559/PA1560 ORFs, in addition to the known dependence of this resistance phenotype on the arn locus. The objectives of this study were to assess the degree of interdependence among the PhoPQ, PmrAB, and CprRS systems in mediating Pm resistance and to define the role of the PA1559/PA1560 ORFs in TCS-induced resistance to Pm and other CAPs.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The laboratory-adapted wild-type (WT) strains and clinical isolates of P. aeruginosa used in this study are listed in Table 1. The institutional review board of Massachusetts General Hospital reviewed and approved the use of CF clinical isolates of P. aeruginosa for this study. Escherichia coli strain DH5α was used as host for plasmid manipulation, and E. coli strain SM10 was used for conjugative transfer of plasmids into P. aeruginosa. The strains were stored at −80°C in lysogeny broth (LB) supplemented with 16% glycerol and grown at 30°C or 37°C on lysogeny agar (LA) or in LB with aeration. Antibiotics were used at the following concentrations for plasmid selection and maintenance: 50 mg/liter kanamycin or 10 mg/liter gentamicin (GEN) for E. coli and 50 to 100 mg/liter GEN for P. aeruginosa. For conjugative transfer of plasmids, Pseudomonas isolation agar (PIA) containing appropriate antibiotics and low-salt LA (10 g/liter tryptone, 5 g/liter yeast extract, and 15 g/liter agar) containing 5% sucrose were used. For induction, 0.1% l-arabinose (l-Ara) was included in LA or LB.
TABLE 1.
Strains of P. aeruginosa used in this work
| Strain | Strain description (source/resistance profile) | Reference |
|---|---|---|
| 1026 | PAK laboratory-adapted WT (S. Lory) | 65 |
| 1027 | PAO1 laboratory-adapted WT (S. Lory) | 46 |
| 1555 | PAO1 laboratory-adapted WT (M. Franklin) | 66 |
| 2114 | PAO1 laboratory-adapted WT (C. Manoil) | 67 |
| 2178 | PAO1 laboratory-adapted WT (M. Vasil) | 68 |
| 1016 | Clinical isolate (Aarhus CF patient 2, 1996) | 12 |
| 1020 | Clinical isolate (Copenhagen CF patient 6, 1998) | 12 |
| 1603 | Clinical isolate (Copenhagen CF patient 14, 2003) | 12 |
| 1611 | Clinical isolate (Copenhagen CF patient 15, 2001) | 12 |
| 4240 | PAK Δ_cprRS_ pJN105D::cprR+S21 (GENr) | 29 |
| 4537 | PAK Δ_phoPQ_ Δ_cprRS_ pJN105D::cprR+S21(GENr) | This study |
| 4541 | PAK Δ_phoPQ_ Δ_cprRS_ pJN105 (GENr) | This study |
| 4472 | PAK Δ_pmrAB_ Δ_cprRS_ pJN105D::cprR+S21 (GENr) | This study |
| 4470 | PAK Δ_pmrAB_ Δ_cprRS_ pJN105 (GENr) | This study |
| 4543 | PAK Δ_phoPQ_ Δ_pmrAB_ Δ_cprRS_ pJN105D::cprR+S21 (GENr) | This study |
| 4547 | PAK Δ_phoPQ_ Δ_pmrAB_ Δ_cprRS_ pJN105 (GENr) | This study |
| 4232 | PAK Δ_pmrAB_ Δ_cprRS_ pJN105D::pmrAB12 (GENr) | This study |
| 4234 | PAK Δ_pmrAB_ Δ_cprRS_ pJN105 (GENr) | This study |
| 4343 | PAK Δ_cprA2_ | This study |
| 4458 | PAK Δ_cprA2_ pJN105 (GENr) | This study |
| 4460 | PAK Δ_cprA2_ pJN105D::cprA2 (GENr) | This study |
| 2326 | PAK Δ_phoQ_ | 24 |
| 4374 | PAK Δ_cprA2_ Δ_phoQ_ | This study |
| 4614 | PAK Δ_cprA2_ Δ_phoQ_ pJN105D::cprA2 (GENr) | This study |
| 4497 | PAK Δ_cprA2_ Δ_phoQ_ pJN105 (GENr) | This study |
| 3420 | PAK Δ_pmrAB_ pJN105 (GENr) | This study |
| 2735 | PAK Δ_pmrAB_ pJN105D::pmrAB12 (GENr) | 12 |
| 4519 | PAK Δ_cprA2_ Δ_pmrAB_ pJN105D::pmrAB12 (GENr) | This study |
| 4521 | PAK Δ_cprA2_ Δ_pmrAB_ pJN105 (GENr) | This study |
| 4425 | PAK Δ_cprA2_ Δ_cprRS_ pJN105D::cprR+S21 (GENr) | This study |
| 4447 | PAK Δ_cprA2_ Δ_cprRS_ pJN105 (GENr) | This study |
| 4200 | PAK Δ_cprRS_ Δ_phoQ_ pJN105D::cprR+S21 (GENr) | 12 |
| 4608 | PAK Δ_cprA2_ Δ_cprRS_ Δ_phoQ_ pJN105D::cprR+S21 (GENr) | This study |
| 4612 | PAK Δ_cprA2_ Δ_cprRS_ Δ_phoQ_ pJN105 (GENr) | This study |
| 4581 | PAK Δ_arnC_ Δ_cprRS_ pJN105D::cprR+S21(GENr) | This study |
| 4586 | PAK Δ_arnC_ Δ_cprRS_ pJN105 (GENr) | This study |
| 4734 | PAO1 Δ_pmrAB_ pJN105D::pmrAB12 (GENr) | This study |
| 4763 | PAO1 Δ_pmrAB_ Δ_cprA1_::cprA+ pJN105D::pmrAB12 (GENr) | This study |
| 4765 | PAO1 Δ_pmrAB_ Δ_cprA1_::cprA+ pJN105 (GENr) | This study |
Bacterial DNA sequencing and bioinformatics.
For Sanger sequencing of the cprA gene, the primers used are listed in Table S1 in the supplemental material. For whole-genome sequencing, a random-fragment library was constructed from each genome using a custom protocol, as described previously (34). Briefly, genomic DNA was purified using the MCD 85201 kit (Epicentre Biotechnologies, Madison, WI), and 2 μg was sheared using a Covaris S2 sonicator (5% intensity; 10% duty cycle; 200 cycles/burst, processing time, 4 min). The ends were repaired using T4 DNA polymerase, T4 polynucleotide kinase, DNA polymerase large Klenow fragment, and deoxynucleoside triphosphate (dNTP) mix (New England BioLabs, Beverly, MA). The reaction was purified on MinElute columns (Qiagen, Valencia, CA). The repaired fragments were subjected to A-tailing using Klenow fragment 3′-5′ exonuclease and dATP (New England BioLabs), followed by treatment with RNase (Roche Diagnostics, Indianapolis, IN) and purification using SPRI beads (Beckman Coulter, Indianapolis, IN). Custom “Y” adaptors (Illumina, San Diego, CA) were ligated to A-tailed fragments using T4 DNA ligase, indexed using a universal primer combined with one of 24 index primers (Illumina) and the Phusion high-fidelity PCR master mix (New England BioLabs), followed by purification using SPRI beads. The libraries were analyzed and quantified using the HiSeq 2100 bioanalyzer (Illumina) and high-sensitivity DNA kit (Agilent Technologies, Santa Clara, CA). After pooling and sequencing of the libraries, alignment of demultiplexed short reads was performed using the Burrows-Wheeler Aligner (BWA) (35), SAMtools (36), and Picard (http://broadinstitute.github.io/picard). Calling of single-nucleotide polymorphisms and short insertions/deletions was performed using the GATK UnifiedGenotyper (37) and SnpEff (38).
The nucleotide sequences of PAO1 strains from various laboratories and the single PAK strain were aligned with cprA reference sequences from the PAO1 genome (PA1559 and PA1560 ORFs, in the genomic region between positions 1697188 and 1698344) and from the PA14 genome (PA14_44311 ORF) via Clustal W2 (39). The cprA reference nucleotide sequences were obtained from the Pseudomonas Genome database. BioPHP (www.biophp.org) was used to translate the nucleotide sequences to corresponding protein sequences in six different reading frames. Protein BLAST and Nucleotide BLAST were performed using the NCBI website (40).
Bacterial strain construction.
Expression plasmids and chromosomal deletions were constructed as described previously (24, 29) using the Gateway cloning system (Invitrogen, Carlsbad, CA), with the specific oligonucleotide primers listed in Table S1 in the supplemental material. A unique HindIII restriction endonuclease site was used to mark and confirm the deletions. For strains in which the phoQ gene was deleted, this was performed as the final step of strain construction to minimize the occurrence of secondary suppressor mutations (24).
To perform allelic replacement of cprA on the bacterial chromosome, the native cprA allele was first deleted as described above. The allelic replacement construct was produced through PCR amplification of the replacement cprA allele and flanking chromosomal sequences (∼1 kb on each side) from genomic DNA using specific oligonucleotide primers (see Table S1 in the supplemental material). This DNA fragment was inserted into the Gateway entry vector pDONR221 (Invitrogen) via a BP recombinase reaction and then transferred into the Gateway destination vector pEXGmGW (41) via an LR recombinase reaction. Allelic replacement plasmids introduced into P. aeruginosa were selected for chromosomal insertion on LA containing GEN and then counterselected for loss of the plasmid backbone on LA containing 5% sucrose (42). To confirm allelic replacement, the cprA coding sequence was PCR amplified from chromosomal DNA and subjected to Sanger sequencing.
For the PAO1 cprA1 allele, the QuikChange site-directed mutagenesis method (Agilent Technologies) was used, according to the manufacturer's instructions, to reinsert a deleted guanine nucleotide at codon 224, thereby restoring the WT translational reading frame. The PCR amplification step was performed using specific oligonucleotide primers (see Table S1 in the supplemental material), with plasmid pDONR221::cprA1 as the template. To eliminate the parental DNA template, the amplification product was incubated with DpnI (New England BioLabs) at 37°C for 1.5 h. To confirm the site-directed mutagenesis product, Sanger sequencing was performed using specific oligonucleotide primers (see Table S1).
Antibiotic susceptibility testing.
For all susceptibility assays, LA and LB were supplemented with 1 mM MgCl2 to avoid inducing resistance through Mg2+ depletion. When preparing inocula of P. aeruginosa strains containing expression plasmids, the media were also supplemented with 0.1% l-Ara and 50 mg/liter GEN.
Pm agar dilution testing and the PMB plate assay were performed and interpreted as described previously (24, 29). For the PMB plate assay of strains containing expression plasmids, prolonged (24-h) strong induction with 0.1% l-Ara was performed as described previously (29).
For the quantitative bactericidal assay (17), strains containing expression plasmids were streaked onto LA containing 0.1% l-Ara and GEN and then incubated at 30°C for 20 h. Colonies of each strain were suspended in LB containing 0.1% l-Ara and GEN to achieve an optical density at 600 nm (OD600) of 0.5, back-diluted 1:10 (strains with empty vectors) or 1:5 (strains with episomal TCS mutant alleles), and incubated at 30°C for 3 h to achieve an OD600 of 0.8 to 1.2. Each inoculum was then diluted in Mueller-Hinton broth to a final density of 1 × 104 to 2 × 104 CFU per ml and incubated at 37°C for 30 min in 2-fold serial dilutions of PMB (0.5 to 500 mg/liter) and a Pm-free control, or for 120 min in 2-fold serial dilutions of C18G (0.5 to 500 mg/liter) or protegrin-1 (0.0625 to 64 mg/liter) and a CAP-free control. To enumerate the surviving CFU, the cells were then diluted 1:10, spread on LA, and incubated at 37°C for 16 to 20 h.
Lipid A isolation and analysis.
LPS was isolated from cells grown in LB, supplemented with 1 mM MgCl2, 0.1% l-Ara, and GEN when required (43). Lipid A was prepared as described previously (29) from hydrolyzed LPS (44). The lipid A structure was analyzed using matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) in negative-ion linear mode (45). All MALDI-TOF MS analyses were performed on an autoflex speed LRF mass spectrometer (Bruker Daltonics, Billerica, MA).
Reverse transcription-quantitative PCR.
Cultures of WT and mutant strains were grown in LA and LB media supplemented with 1 mM MgCl2 and 0.1% l-Ara. The media for the expression strains also included 50 mg/liter GEN; such strains were induced for 24 h, as described previously (29). The strains were grown in 25 ml of medium with aeration at 30°C to an OD600 of 0.2 to 0.5. Total RNA was extracted using TRI reagent (Life Technologies, Grand Island, NY), bromo-chloropropane (Fisher), and the RNeasy kit (Qiagen), with DNase I (Qiagen) treatment performed as part of the RNeasy column purification step. A reverse transcription kit (Qiagen) was used to synthesize cDNA from 100 ng of total RNA; to analyze 50 ng of cDNA, Phusion master mix (New England BioLabs), 5× SYBR green (Invitrogen), ROX passive reference dye (U.S. Biochemical Corporation, Cleveland, OH), and 400 nM oligonucleotide primers (see Table S1 in the supplemental material) designed with IDT PrimerQuest were used in an Applied Biosystems 7500 Fast quantitative PCR (qPCR) system (Life Technologies), as recommended by the manufacturer. cDNA derived from the PA4268 ORF, which encodes the 30S ribosomal protein S12, was used as an internal control to normalize the results (32), and the comparative cycle threshold was used for quantification. For each qPCR, technical duplicates were performed; for each strain analyzed, biological triplicates were performed. Two-sided P values were calculated using the one-sample t test.
Nucleotide sequence accession numbers.
New genomic DNA sequences for the cprA region of P. aeruginosa strain 1026 (PAK) and four clinical strains (1020, 1603, 1016, and 1611) have been deposited in GenBank under accession numbers KR131719 to KR131723, respectively.
RESULTS
Pm resistance mediated through the P. aeruginosa CprRS two-component system is partially dependent on the PhoPQ and PmrAB systems.
Previous work demonstrated that the Pm resistance conferred by phoQ mutation in the P. aeruginosa PAK strain is partially dependent on an intact CprRS TCS (29). That work also defined a mutant allele, cprRS21, which confers Pm resistance when overexpressed in a PAK Δ_cprRS_ strain. To assess the extent to which Pm resistance conferred by cprRS21 is dependent on the PhoPQ TCS, this mutant allele was overexpressed in a PAK strain in which the WT phoPQ and cprRS genes had been deleted from the chromosome. The resulting strain exhibited a partial loss of Pm resistance in comparison to the positive control (Fig. 1A).
FIG 1.
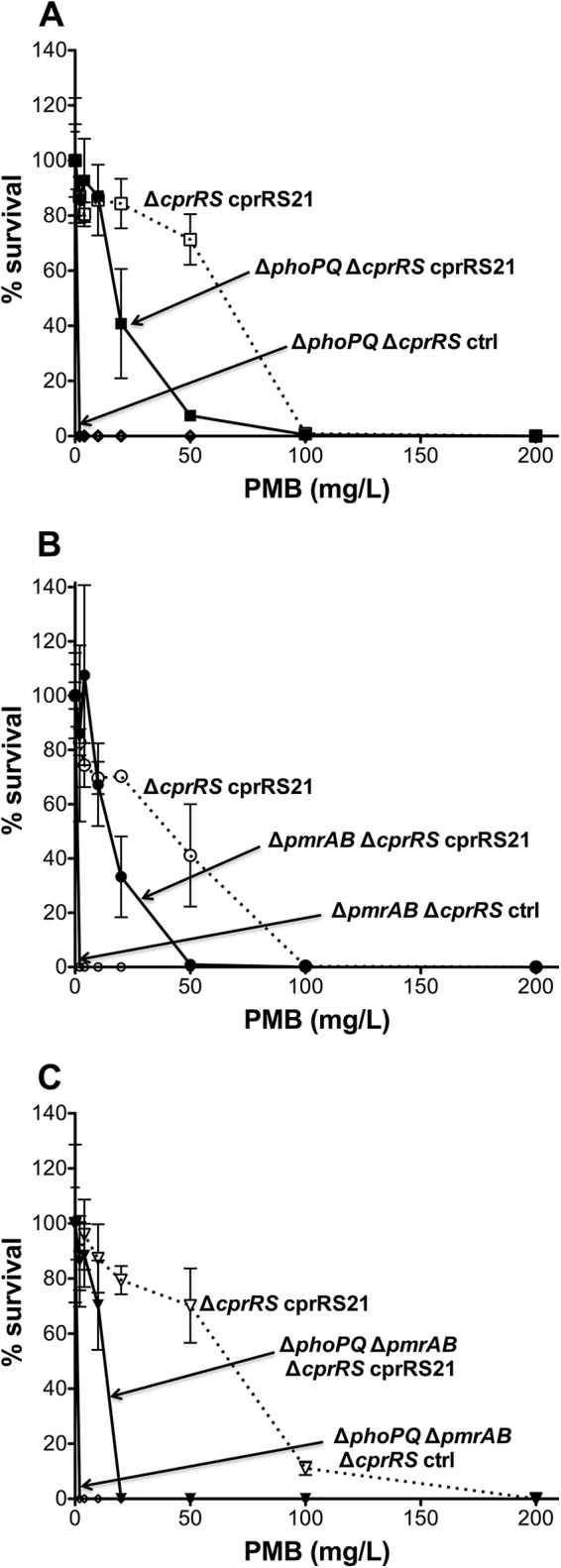
Effect of phoPQ, pmrAB, or phoPQ and pmrAB deletion on Pm resistance in a PAK Δ_cprRS_ cprRS21 background. For this and subsequent figures, Pm resistance experiments were performed twice; if discrepancies were seen, the experiment was performed a third time. Each panel shows a representative experiment, with the results expressed as means from three technical replicates. The error bars represent the standard deviations (SD). The nonitalicized allele name indicates the presence of an episomal version, i.e., an inducible expression strain (ctrl, empty vector control). Results from a PMB plate assay of strains induced with 0.1% l-Ara for 24 h are shown for strain 4537 (Δ_phoPQ_ Δ_cprRS_ cprRS21) with strain 4240 (Δ_cprRS_ cprRS21) as a positive control and strain 4541 (Δ_phoPQ_ Δ_cprRS_ ctrl) as a negative control (A), strain 4472 (Δ_pmrAB_ Δ_cprRS_ cprRS21) with strain 4240 (Δ_cprRS_ cprRS21) as a positive control and strain 4470 (Δ_pmrAB_ Δ_cprRS_ ctrl) as a negative control (B), and strain 4543 (Δ_phoPQ_ Δ_pmrAB_ Δ_cprRS_ cprRS21) with strain 4240 (Δ_cprRS_ cprRS21) as a positive control and strain 4547 (Δ_phoPQ_ Δ_pmrAB_ Δ_cprRS_ ctrl) as a negative control (C).
To broaden these observations to the PmrAB TCS, which also regulates Pm resistance, the extent to which resistance conferred by the mutant allele pmrAB12 (17) is dependent on the CprRS TCS was assessed. This allele was overexpressed in a PAK strain in which the WT pmrAB and cprRS genes had been deleted. The resulting strain exhibited a partial loss of Pm resistance (see Fig. S1 in the supplemental material), the extent of which was dependent on the conditions used to induce expression of the pmrAB12 allele; prolonged induction partially overcame the effect of cprRS deletion. To determine the reciprocal relationship, i.e., the degree to which resistance conferred by the cprRS21 mutant allele is dependent on the PmrAB system, the cprRS21 allele was overexpressed in the same pmrAB cprRS deletion strain. The resulting strain exhibited a partial loss of Pm resistance in comparison to the positive control (Fig. 1B).
To assess the extent to which Pm resistance conferred by cprRS21 is dependent on having at least one of the other TCSs intact, this mutant allele was overexpressed in a PAK strain in which the phoPQ, pmrAB, and cprRS genes had all been deleted. The resulting triple-deletion overexpression strain exhibited substantial but incomplete loss of Pm resistance in comparison to the positive-control and the double-deletion overexpression strains (Fig. 1C). Thus, even though the deletion of both the PhoPQ and PmrAB TCSs has a combined effect on CprRS-mediated Pm resistance, in their absence, the cprRS21 mutant allele still conferred some residual Pm resistance. These findings are consistent with the possibility of cross talk among these TCSs but also indicate that their regulatory activities are not completely interdependent.
CprRS, PhoPQ, and PmrAB systems jointly regulate PA1559 and PA1560, distinct ORFs in the PAO1 strain that together constitute a single ORF in PAK and other P. aeruginosa strains.
Having established the partial interdependence of the CprRS, PhoPQ, and PmrAB TCSs with respect to Pm resistance, we sought to define loci, in addition to the arn operon, jointly regulated by these TCSs; the availability of transcriptional array data for each TCS facilitated this task (31, 32). One such jointly regulated locus in those analyses is the PA1559 ORF, which we previously defined as being required for PhoPQ-mediated Pm resistance in the PAK strain (29). Bioinformatics analysis of completely assembled P. aeruginosa genomes available in the Pseudomonas Genome Database (30) as of April 2015 revealed that in all non-PAO1 genomes (n = 12), PA1559 corresponds to the 5′ end of a conserved ORF. However, only PAO1 genomes were found to have a distinct adjacent ORF corresponding to the annotated PA1560. In the non-PAO1 genomes, PA1560 instead corresponds to the 3′ end of the conserved ORF that contains PA1559-encoded residues at its 5′ end (see Fig. S2 in the supplemental material).
To confirm the unitary relationship of the PA1559 and PA1560 ORFs in a non-PAO1 strain, Sanger sequencing of the genomic region corresponding to these ORFs was performed for the PAK strain of P. aeruginosa (1026). This analysis revealed a single ORF of 1,158 bp encoding a hypothetical protein comprised of 385 amino acids; the completely assembled genome of the PA14 strain and those of 11 other P. aeruginosa strains in the Pseudomonas Genome database (30) also contain this single ORF. Considering the role that the CprRS TCS plays in regulating PA1559 and PA1560 of the PAO1 strain (31) and the role that this locus plays in cationic peptide resistance (this work), we designated it the cprA gene.
Allelic forms of the cprA gene.
Analysis of assembled whole-genome sequences from the cprA genomic region for four Pmr CF clinical isolates of P. aeruginosa (1020, 1603, 1016, and 1611) with colistin agar dilution MICs of >512 mg/liter (12) revealed amino acid sequences identical to those of 13 completely assembled genomes of non-PAO1 strains in publicly available databases (see Table S2 in the supplemental material). We designated the cprA allele of these clinical strains and the other non-PAO1 strains cprA + (WT).
Sanger sequencing of cprA for four Pm-susceptible (Pms) PAO1 substrains (1027, 1555, 2114, and 2178) revealed a single guanine deletion in codon 224 that results in a translational frameshift and premature termination at codon 244 (see Fig. S2 and Table S2 in the supplemental material). This mutation, which is responsible for the annotation of cprA as adjacent ORFs (PA1559 and PA1560) within the published PAO1 genome (30, 46), is also found in three completely assembled PAO1 genomes in the public NCBI database (see Table S2). We have designated the cprA allele of the PAO1 strain cprA1.
Sanger sequencing of cprA for the Pms PAK parental strain revealed nonsynonymous changes at codons 192 (Glu→Gln) and 196 (Gly→Asp) relative to cprA +; one non-PAO1 genome in the NCBI database has these same amino acid changes (see Table S2 in the supplemental material). We designated the cprA allele of the PAK strain cprA2.
Predicted structure of the P. aeruginosa cprA gene product.
The predicted CprA structure is typical of an extended short-chain dehydrogenase/reductase (SDR) family member that (i) is >250 amino acids, (ii) contains a Rossmann fold having an N-terminal dinucleotide cofactor binding motif with consensus [S/T]GXXGXXG (CprA, TGATGFLG, codons 22 to 29), and (iii) possesses a catalytic residue motif with consensus YXXXK (CprA, YTASK, codons 179 to 183) (47). In addition to these conserved motifs, the SDR enzymes typically have a low pairwise sequence identity of 20 to 30% (48) and are functionally diverse (47). Protein BLAST searches performed versus other Gram-negative bacteria indicate that CprA has ∼50% sequence identity with a Klebsiella pneumoniae protein, hemolysin F (HlyF). An alphanumeric search of the Pseudomonas Genome database revealed 69 proteins annotated as SDRs in the PAO1 genome (30). The enzymatic function is known for 15 of these proteins; eight are involved in LPS biosynthesis.
Deletion of cprA abrogates Pm resistance conferred by phoQ, pmrB, and cprS mutant alleles.
To define the role of cprA in Pm resistance, clean deletions of this gene were constructed in the Pms PAK parental strain and in various Pmr derivatives. The cprA gene does not appear to be part of an operon; thus, deleting or otherwise disrupting it has minimal potential for polar effects on adjacent loci.
In the PAK parental background, neither deletion of the cprA2 allele nor in trans expression of this allele under prolonged strong induction (0.1% l-Ara) had a detectible effect on Pm susceptibility (data not shown). In contrast, deletion of cprA2 abrogated the Pm resistance of a PAK phoQ deletion mutant, as shown in a PMB plate assay (Fig. 2A). The Pm resistance of the Δ_cprA2_ Δ_phoQ_ strain was similar to that of the PAK parental strain, indicating that the loss of Pm resistance was more complete than that previously seen for strains in which the CprRS TCS or the ColRS TCS had been deleted in a phoQ mutant background (29). Moreover, in trans expression of cprA2 under prolonged strong induction in the Δ_cprA2_ Δ_phoQ_ strain partially restored Pm resistance (Fig. 2A). Decreasing the l-Ara concentration used for induction to 0.025%, which enhanced the resistance phenotype that an episomal cprRS21 allele conferred in a phoQ mutant (29), did not enhance this partial complementation (data not shown).
FIG 2.
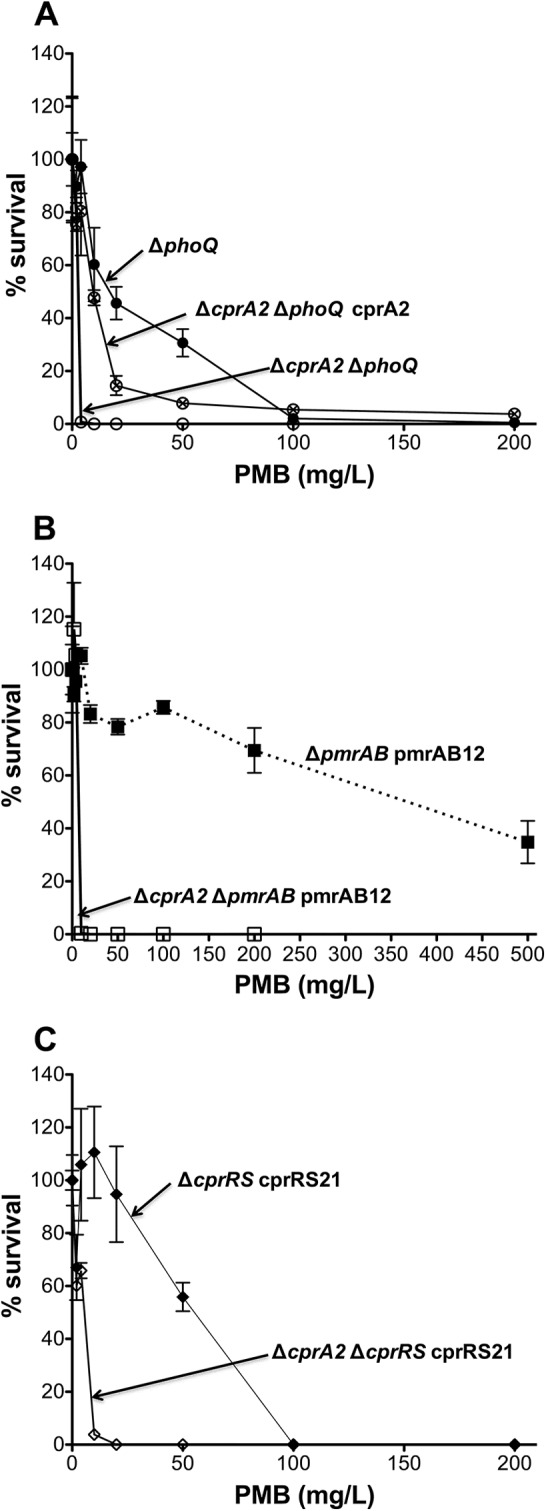
Effect of cprA2 deletion on Pm resistance in various PAK strain backgrounds. Shown are the results of a PMB plate assay of strain 4374 (Δ_cprA2_ Δ_phoQ_) and strain 4614 (Δ_cprA2_ Δ_phoQ_ cprA2) induced with 0.1% l-Ara for 24 h, with strain 2326 (Δ_phoQ_) as a positive control (A), strain 4519 (Δ_cprA2_ Δ_pmrAB_ pmrAB12) induced with 0.1% l-Ara for 24 h, with strain 2735 (Δ_pmrAB_ pmrAB12) as a positive control (B), and strain 4425 (Δ_cprA2_ Δ_cprRS_ cprRS21) induced with 0.1% l-Ara for 24 h, with strain 4240 (Δ_cprRS_ cprRS21) as a positive control (C). Empty vector control strains 4497 (Δ_cprA2_ Δ_phoQ_ pJN105), 4521 (Δ_cprA2_ Δ_pmrAB_ pJN105), and 4447 (Δ_cprA2_ Δ_cprRS_ pJN105) had Pm susceptibility values similar to those of the corresponding parental strains (not shown).
The deletion of cprA2 led to a loss of Pm resistance in a PAK Δ_pmrAB_ strain that carries a resistance-conferring pmrAB12 allele in trans (12) (Fig. 2B). The deletion of cprA2 also caused a substantial loss of Pm resistance (∼10-fold) in a PAK Δ_cprRS_ strain that carries a resistance-conferring cprRS21 allele in trans (29) (Fig. 2C). The deletion of cprA2 caused a similar loss of resistance in a PAK Δ_phoQ_ Δ_cprRS_ strain expressing cprRS21 in trans (see Fig. S3 in the supplemental material).
Deletion of cprA abrogates CAP resistance conferred by a pmrB mutant allele.
Pm resistance-conferring mutations in the P. aeruginosa pmrB gene, such as that found in the pmrAB12 allele, also confer cross-resistance to CAPs, such as protegrin-1 and C18G (12), as measured through a standard CAP killing assay (17). To determine whether resistance to CAPs other than Pm is dependent on cprA, we used the killing assay to test resistance to protegrin-1 and C18G of PAK Δ_pmrAB_ and PAK Δ_cprA2_ Δ_pmrAB_ mutants harboring an episomal l-Ara-inducible version of the pmrAB12 allele. The expression of pmrAB12 in the Δ_pmrAB_ strain (positive control) resulted in a >256-fold increase in resistance (FIR) to Pm, relative to that of the negative-control strains, and FIRs of 96 and 128 to protegrin-1 and C18G, respectively (Table 2). Expression of this allele in the Δ_cprA2_ Δ_pmrAB_ strain resulted in a 16-fold decrease in Pm resistance relative to the positive control, similar to the effect seen in the PMB plate assay (Fig. 2B), and in 6- and 4-fold decreases in resistance to protegrin-1 and C18G, respectively (Table 2). These results indicate that the CAP resistance of P. aeruginosa pmrB mutants is partially dependent on the functionality of the cprA gene.
TABLE 2.
Effect of cprA2 deletion in the PAK strain on _pmrAB12_-induced cationic antimicrobial peptide resistance
| Strain/genotype | PMBa | Protegrin-1 | C18G | |||
|---|---|---|---|---|---|---|
| EC50 (mg/liter) | FIR | EC50 (mg/liter) | FIR | EC50 (mg/liter) | FIR | |
| PAK WT | <0.125 | 1 | 0.125 | 1 | 2 | 1 |
| PAK Δ_pmrAB_ pJN105 | <0.125 | 1 | 0.125 | 1 | 2 | 1 |
| PAK Δ_pmrAB_ pJN105D::pmrAB12 | 32 | >256 | 12 | 96 | 256 | 128 |
| PAK Δ_cprA2_ Δ_pmrAB_ pJN105D::pmrAB12 | 2 | >16 | 2 | 16 | 64 | 32 |
Repair of cprA1 mutant allele in the PAO1 strain markedly increases PmrAB-mediated Pm resistance.
In the PAO1 strain, increased Pm resistance is conferred by mutational activation of the PhoPQ system or is induced by specific environmental conditions (e.g., Mg2+ depletion or subinhibitory CAP exposure) that act through the PmrAB, ParRS, and CprRS systems (20, 21, 23, 27, 31, 32). However, the degree of Pm resistance seen with the PAO1 strain is much more modest than that with the PAK strain or clinical strains (12, 17, 24, 29, 45). To test the hypothesis that the difference in Pm resistance levels seen with these strains is attributable to the frameshift mutation in the cprA1 allele, its guanine deletion was repaired in a PAO1 Δ_pmrAB_ strain through plasmid-based site-directed mutagenesis and chromosomal allelic replacement. The pJN105D::pmrAB12 plasmid was introduced into the cprA_-repaired PAO1 Δ_cprA1::cprA+ Δ_pmrAB_ strain, and as a control, into a PAO1 Δ_pmrAB_ strain in which the cprA1 allele had not been repaired. A PAO1 Δ_cprA1_::cprA+ Δ_pmrAB_ strain carrying an empty plasmid served as an additional control. Restoration of the cprA + allele in the pmrAB12_-expressing PAO1 was associated with a marked increase in Pm resistance: an effective concentration yielding 50% lethality (EC50) of ≥500 mg/liter PMB for the repaired strain versus ∼6 mg/liter PMB for the control strain (Fig. 3). The level of Pm resistance that episomal pmrAB12 expression conferred on the cprA_-repaired PAO1 Δ_pmrAB strain was comparable to that conferred on the PAK Δ_pmrAB strain (Fig. 2B). Thus, the difference in Pm resistance between the PAO1 and PAK strains is largely attributable to the frameshift mutation in the cprA1 allele of the PAO1 strain.
FIG 3.
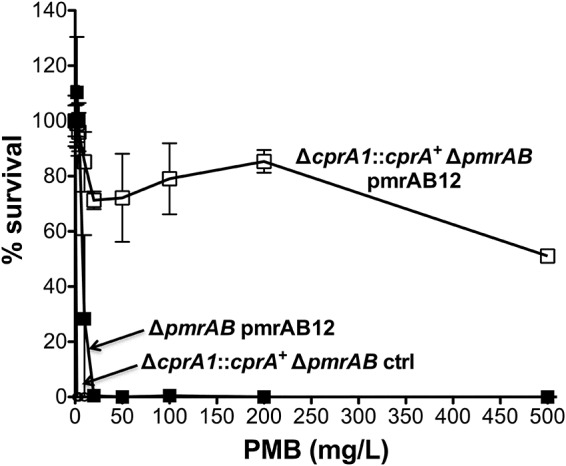
Repair of the cprA1 mutant allele promotes high-level Pm resistance in the PAO1 Δ_pmrAB_ strain expressing pmrAB12 in trans. Shown are the results of a PMB plate assay for strain 4763 (Δ_cprA1_::cprA + Δ_pmrAB_ pmrAB12) induced with 0.1% l-Ara for 24 h, with strains 4734 (Δ_pmrAB_ pmrAB12) and 4765 (Δ_cprA1_::cprA + Δ_pmrAB_ ctrl) as negative controls.
Deletion of cprA does not abolish lipid A modifications conferred by phoQ, pmrB, and cprS mutant alleles.
Covalent modification of lipid A is a biochemical hallmark of resistance to Pm and other CAPs in Gram-negative bacilli (13, 45, 49, 50) and is commonly analyzed using mass spectral methods, such as MALDI-TOF MS (18). Analysis of lipid A from the Pms PAK parental strain revealed unmodified penta- and hexa-acylated species (Fig. 4A, top) corresponding to major mass spectral peaks at mass/charge ratio (m/z) 1,447 and 1,617 (Fig. 4B). In contrast, analysis of lipid A from the Pmr PAK Δ_phoQ_ strain had modified species with the addition of one or two l-Ara4N residues (Δ_m/z_, +131 or +262) to the 1- and 4′-phosphates of the penta- and hexa-acylated species (Fig. 4A, middle and bottom), corresponding to peaks at m/z 1,578, 1,748, and 1,879 (Fig. 4C). The deletion of cprA2 in the PAK Δ_phoQ_ strain did not qualitatively alter the complex pattern of modified lipid A species resulting from the addition of l-Ara4N in this strain background (Fig. 4D), indicating that the cprA dependence of Pm resistance in the Δ_phoQ_ background is not attributable to a loss of this lipid A modification. This is not surprising in light of our previous observation that deletion of the CprRS TCS in a Δ_phoQ_ background did not abolish the robust addition of l-Ara4N to lipid A, despite a substantial loss of Pm resistance associated with the cprRS deletion (29).
FIG 4.

Deletion of cprA2 does not affect l-Ara4N modification of lipid A from the PAK strain. (A) Structure of lipid A with baseline hexa-acylation (m/z, 1,617), single l-Ara4N addition (m/z, 1,748), and double l-Ara4N addition (m/z, 1,879). Also shown are the MALDI-TOF spectra of lipid A isolated from strain 1026 (WT) (B), strain 2326 (Δ_phoQ_) (C), and strain 4374 (Δ_cprA2_ Δ_phoQ_) (D).
Analysis of lipid A from the Pmr PAK Δ_pmrAB_ pJN105D::pmrAB12 expression strain revealed unmodified penta-acylated species with one or two 2-hydroxylauroyl residues, corresponding to minor peaks at m/z 1,447 and 1,463, and unmodified hexa-acylated species with one or two 2-hydroxylauroyl residues, corresponding to major peaks at m/z 1,617 and 1,633 (see Fig. S4A in the supplemental material). This lipid A also had modified species with one l-Ara4N residue added, corresponding to minor peaks at m/z 1,578 and 1,594 and major peaks at m/z 1,748 and 1,764, with adjacent Na+ adduct peaks at m/z 1,771 and 1,787 (Δ_m/z_, +23). Peaks corresponding to Na+ adducts at m/z 1,902 and 1,918 dwarfed the species from which they derived that had two l-Ara4N residues, corresponding to minor peaks at m/z 1,879 and 1,895. The deletion of cprA2 in the Δ_pmrAB_ pJN105D::pmrAB12 expression background did not alter the pattern of modified lipid A species, except that the peaks corresponding to the Na+ adducts were minimal (see Fig. S4B in the supplemental material).
Similarly, analysis of lipid A from the Pmr PAK Δ_cprRS_ pJN105D::cprRS21 expression strain revealed unmodified penta-acylated species with one or two 2-hydroxylauroyl residues, corresponding to minor peaks at m/z 1,447 and 1,463, and unmodified hexa-acylated species with one or two 2-hydroxylauroyl residues, corresponding to major peaks at m/z 1,617 and 1,633 (see Fig. S4C in the supplemental material). This lipid A also had modified species with one or two l-Ara4N residues, corresponding to minor peaks at m/z 1,578 and 1,594, major peaks at m/z 1,748 and 1,764, and minor peaks at m/z 1,879 and 1,895. The deletion of cprA2 in the Δ_cprRS_ pJN105D::cprRS21 expression background did not alter this modification pattern (see Fig. S4D in the supplemental material).
Taken together, these results indicate that in pmrB and cprS mutant backgrounds, as in the phoQ mutant background, the dependence of Pm resistance on cprA is not attributable to a loss of l-Ara4N addition or other modifications detectible through MALDI-TOF MS analysis of lipid A derived from hydrolyzed LPS.
Both cprA and arn are required for Pm resistance.
We previously showed that the deletion of the arnC gene, which encodes an enzyme that transfers l-Ara4N from UDP to undecaprenyl phosphate at the inner membrane (51), causes a loss of lipid A l-Ara4N modification and a loss of Pm resistance in a P. aeruginosa clinical strain with a resistance-conferring pmrB mutation (12). Thus, PmrAB-mediated Pm resistance requires both cprA (Fig. 2B) and arnC. Pm resistance conferred through phoQ deletion requires cprA (Fig. 2A) and presumably also arnC, although to our knowledge, the dependence of PhoPQ-mediated resistance on arnC has not been directly documented in the published literature. Given that CprRS-mediated Pm resistance requires cprA (Fig. 2C), we examined the extent to which Pm resistance also depends on the arnC locus in the cprRS21 expression background. The deletion of arnC resulted in a marked decrease in Pm resistance (Fig. 5), indicating that similarly to PmrAB-mediated Pm resistance, CprRS-mediated resistance requires both cprA and arnC. However, the cprRS21 expression strain still had a minor degree of residual Pm resistance (∼10 to 15% survival at 2 mg/liter Pm) compared to that of the negative control (0% survival at 2 mg/liter Pm), indicating that arnC deletion is not epistatic to the resistance-conferring effect of the cprRS21 allele.
FIG 5.
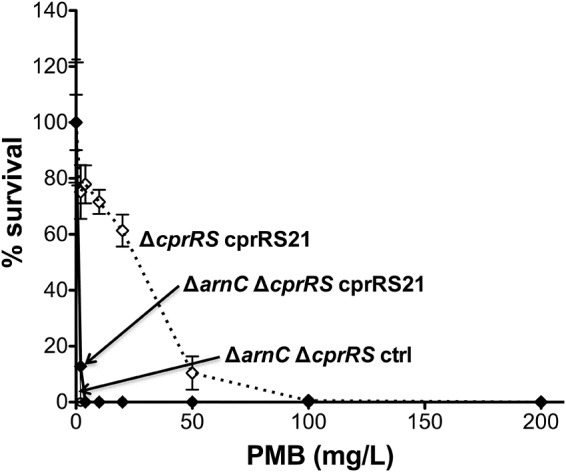
Effect of arnC deletion in the PAK Δ_cprRS_ cprRS21 strain. Shown are the results of a PMB plate assay of strain 4581 (Δ_arnC_ Δ_cprRS_ cprRS21) induced with 0.1% l-Ara for 24 h, with strain 4240 (Δ_cprRS_ cprRS21) as a positive control and strain 4586 (Δ_arnC_ Δ_cprRS_ ctrl) as a negative control.
To quantify the correlation of cprA and arnC expression, reverse transcription-quantitative PCR (RT-qPCR) was used to measure the abundance of their mRNA transcripts in a phoQ deletion strain, a pmrAB12 expression strain, a cprRS21 expression strain, a combined phoQ deletion/cprRS21 expression strain, and cprRS21 expression strains in which either cprA2 or arnC had been deleted. The cprA and arnC transcripts were both increased in the pmrAB12 and cprRS21 expression strains (Table 3). In contrast, only the arnC transcript was increased in the phoQ deletion strain. Considering that CprA is required for Pm resistance conferred through phoQ mutation (Fig. 2A), this indicates that a basal level of cprA expression is permissive for resistance in the Δ_phoQ_ strain. The deletion of either arnC or cprA in the cpRS21 expression background did not significantly alter the mRNA expression level of the other locus (Table 3). Both transcripts were increased when cprRS21 was expressed in the phoQ cprRS double-deletion background, indicating that phoQ deletion does not have a suppressive effect on CprRS-mediated cprA expression.
TABLE 3.
Relative abundance of cprA and arnC mRNA transcripts in phoQ deletion and pmrAB12 or cprRS21 expression backgrounds, determined by RT-qPCR analysis (n = 3 biological replicates)
| Genotype | Relative mRNA abundance ± SDa | |
|---|---|---|
| cprA | arnC | |
| PAK Δ_phoQ_ | 1 ± 0.4 | 34 ± 17 |
| PAK Δ_pmrAB_ pJN105D::pmrAB12 | 94 ± 26 | 21 ± 7 |
| PAK Δ_cprRS_ pJN105D::cpRS21 | 165 ± 37 | 34 ± 18 |
| PAK Δ_cprA2_ Δ_cprRS_ pJN105D::cpRS21 | NA | 30 ± 9 |
| PAK Δ_arnC_ Δ_cprRS_ pJN105D::cpRS21 | 172 ± 70 | NA |
| PAK Δ_phoQ_ Δ_cprRS_ pJN105D::cprRS21 | 236 ± 162 | 51 ± 40 |
DISCUSSION
Previous studies have established that the Pm resistance of P. aeruginosa is dependent on arn_-mediated addition of l-Ara4N to lipid A (12). However, recent work has shown that to achieve substantial Pm resistance, additional loci are required. For example, loss of the CprRS TCS was found to diminish PhoPQ-mediated resistance without a loss of l-Ara4N addition (29). That study used transposon mutagenesis of a Pm-resistant PAK mutant (phoQ6 Δ_pmrAB) to define such additional loci, including PA1559, an ORF in the PAO1 genome which transcriptional analysis had previously defined as being CprRS regulated (31). Prior PAO1 transcriptional analyses indicated that the ParRS and PmrAB systems also regulate the expression of PA1559 (27, 32). Nonetheless, experiments performed in the PAO1 strain background failed to demonstrate a role for PA1559 in inducible Pm resistance (27), even though this prior work with the PAO1 strain clearly established that specific TCSs mediate differential CAP sensing and thus trigger transcription from the arn promoter (27, 31). Bioinformatics analysis and resequencing of the chromosomal region containing the PA1559 and PA1560 ORFs has revealed that in P. aeruginosa strains other than PAO1, this region contains only a single ORF (annotated PA14_44311 in the PA14 genome) that we named cprA. A comparison of cprA alleles in silico reveals that this discrepancy is attributable to a frameshift mutation and associated premature termination codon in the cprA1 allele of the PAO1 strain, resulting in annotation as adjacent ORFs.
The relatively low levels of mutational and adaptive resistance to Pm and other CAPs that have been reported for the PAO1 strain (20, 21, 23, 27, 31) are likely attributable to its nonfunctional cprA1 allele and may thus need to be revisited. This work has shown that in P. aeruginosa, cprA is required not only for robust Pm resistance mediated through phoQ mutation but also for resistance mediated through activating mutations in cprS and pmrB. Moreover, cprA is likely critical for the induction of adaptive Pm resistance via TCS sensing of subinhibitory CAP concentrations and other physiological conditions, responses that appear to be impaired in the PAO1 strain. Because PAO1 has undergone genotypic and phenotypic microevolution over decades under laboratory conditions (52), it may not accurately represent the adaptive capacity of typical pathogenic strains in vivo, in which P. aeruginosa encounters CAPs as a front-line component of host innate immunity.
Several TCSs mediate resistance to Pm and other CAPs (Fig. 6), controlling P. aeruginosa loci that encode LPS-modifying enzymes, such as those necessary for the synthesis and transfer of l-Ara4N to lipid A. Key insights into this regulatory complexity have gradually emerged over the past 15 years. Initial work focused on the role of the P. aeruginosa PhoPQ TCS in Pm resistance (21, 45). Subsequent work defined a stronger effect on this resistance phenotype by the PmrAB TCS, which appears to be independent of PhoPQ in P. aeruginosa (17, 20). More recently, the ParRS, ColRS, and CprRS TCSs have been recognized as making additional contributions to Pm resistance, although the ParRS effect is modest, and the ColRS effect appears to be dependent on the PhoPQ TCS (27–29, 31). The ColRS TCS was also recently shown to promote phosphoethanolamine addition at the 4′ position of lipid A under specific environmental conditions, through positive regulation of the newly defined eptA gene (corresponding to the PA1972 ORF) and negative regulation of arn, but without a detected effect on Pm resistance (53). These prior studies indicate that most of these TCSs regulate both the arn operon and the cprA gene, although some of the regulatory effects may be indirect. However, the extent to which Pm resistance induced via a given TCS is dependent on the functionality of the other TCSs and their regulons has not been comprehensively defined.
FIG 6.

Two-component regulatory systems mediating resistance to Pm and other CAPs modulate expression of arn and eptA, loci that enable specific lipid A modifications, and cprA, a locus potentially implicated in other LPS modification(s) and vesiculation from the outer membrane.
The present work increases our understanding of these regulatory relationships, showing that the ability of an activating mutation in cprS to confer Pm resistance is partially dependent on the presence of functional PmrAB and PhoPQ TCSs. Conversely, the ability of an activating mutation in pmrB to confer Pm resistance is partially dependent on the presence of a functional CprRS TCS. However, the induction of l-Ara4N addition by such pmrB mutations is not strictly CprRS dependent. Collectively, these findings corroborate the observation that in addition to the arn locus, other key components of the CprRS regulon, such as cprA, are required for maximal Pm resistance. The deletion or disruption of the cprA coding sequence causes diminished Pm resistance in several different Pmr strain backgrounds and results in diminished resistance to other CAPs.
Activating the PmrAB or CprRS TCS through a resistance-conferring mutation of pmrB or cprS increases the transcription of both arn and cprA in a PAK strain background (Table 3). In contrast, activating the PhoPQ TCS through the deletion of phoQ increases arn transcription and Pm resistance but does not increase cprA transcription relative to the WT background (Table 3). Nonetheless, experiments with the Pmr phoQ deletion strain demonstrate that its Pm resistance is dependent on cprA (Fig. 2A). This suggests that a basal level of cprA transcription prevails in the Δ_phoQ_ strain background, enough to permit an l-Ara4N-mediated increase in Pm resistance, albeit at a lower level than that seen with the pmrAB12 or cprRS21 alleles that actively promote cprA transcription.
Understanding the biochemical role of the CprA protein in Pm resistance is of paramount importance, given the genetic and transcriptional evidence implicating cprA in this phenotype. Bioinformatics analysis of P. aeruginosa CprA indicates strong similarity to the extended SDR enzyme family, which comprises isomerases (e.g., glycosyl epimerases), lyases (e.g., glycosyl dehydratases), and some oxidoreductases not classified as classical SDRs (e.g., certain multifunctional enzymes with both dehydrogenase and isomerase activities) (54). Several functionally defined SDRs participate in LPS biosynthesis in P. aeruginosa, such as WbpM and WbpK (55); CprA may similarly function as an LPS-modifying enzyme. However, the deletion of cprA in Pmr strain backgrounds had no effect on arn transcription or on mass spectrometric detection of the l-Ara4N lipid A modification, indicating that the CprA protein does not affect this specific LPS modification.
A comparison of CprA to the genomes of other sequenced bacterial species also indicates strong similarity to hemolysin F (hlyF), a K. pneumoniae gene sometimes carried on large plasmids found in pathogenic clinical isolates of E. coli (56, 57). Hemolysin F was recently shown to contribute to E. coli virulence in an avian model of colibacillosis (58). Moreover, hemolysin F appears to enhance the production of outer membrane vesicles (OMVs) and release of the cytolysin ClyA, potentially explaining its hemolytic phenotype. Among Gram-negative pathogens, outer membrane turnover and OMV production provide a general mechanism of protection against environmental sources of envelope stress, such as antimicrobials and agents of host innate immunity (59–61). Of potential relevance to the work reported here, exposure of the P. aeruginosa PA14 strain to subinhibitory concentrations of PMB was found to increase OMV production via unknown mechanisms (62). Thus, CprA may participate directly in P. aeruginosa OMV production. Alternatively, if CprA has enzyme activity typical of extended SDRs, it may function as a nucleoside diphosphate-sugar epimerase and thus play a role in LPS synthesis (as suggested above). This might have an indirect effect on P. aeruginosa OMV production, which was shown to be dependent on the synthesis of B-band LPS (62).
Our discovery that P. aeruginosa resistance to Pm and other CAPs requires both the arn locus and an active CprA protein moves the field beyond a narrow focus on enzymes involved in l-Ara4N synthesis (63, 64), opening the way to novel resistance inhibition strategies. The observation that laboratory-adapted strains with mutated cprA alleles, such as PAO1, have diminished resistance suggests the need to examine how Pm resistance correlates with CprA expression and function in clinical isolates of P. aeruginosa. Moreover, we found that several of the TCSs regulating cprA transcription influence Pm resistance in a convergent and partially interdependent fashion, suggesting that an analysis of clinical isolates may reveal additional correlations between patterns of TCS mutations and levels of CprA protein expression. CprA is predicted to be an extended SDR that appears not to affect lipid A modification but might participate in the modification of other LPS moieties (O antigen or core oligosaccharide), thus influencing OMV production or another aspect of outer membrane remodeling. We are currently pursuing functional characterization of CprA to delineate its enzymatic activity and assess its viability as a potential drug target.
Supplementary Material
Supplemental material
ACKNOWLEDGMENTS
We thank Virendar Kaushik (Broad Institute, Cambridge, MA) for assistance with MALDI-TOF MS and Jane Burns, Joanna Goldberg, Talia Ramsdell, and Matthew Wargo for valuable feedback on the manuscript.
This work was supported by grant MOSKOW13P0 to S.M.M. from the CF Foundation.
Footnotes
REFERENCES
- 1.Michalopoulos A, Falagas ME. 2008. Colistin and polymyxin B in critical care. Crit Care Clin 24:377–391. doi: 10.1016/j.ccc.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Moskowitz SM, Silva SJ, Mayer-Hamblett N, Pasta DJ, Mink DR, Mabie JA, Konstan MW, Wagener JS, Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis (ESCF). 2008. Shifting patterns of inhaled antibiotic use in cystic fibrosis. Pediatr Pulmonol 43:874–881. doi: 10.1002/ppul.20873. [DOI] [PubMed] [Google Scholar]
- 3.Alonso A, Campanario E, Martinez JL. 1999. Emergence of multidrug-resistant mutants is increased under antibiotic selective pressure in Pseudomonas aeruginosa. Microbiology 145(Pt 10):2857–2862. [DOI] [PubMed] [Google Scholar]
- 4.Henrichfreise B, Wiegand I, Pfister W, Wiedemann B. 2007. Resistance mechanisms of multiresistant Pseudomonas aeruginosa strains from Germany and correlation with hypermutation. Antimicrob Agents Chemother 51:4062–4070. doi: 10.1128/AAC.00148-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foweraker JE, Laughton CR, Brown DF, Bilton D. 2009. Comparison of methods to test antibiotic combinations against heterogeneous populations of multiresistant Pseudomonas aeruginosa from patients with acute infective exacerbations in cystic fibrosis. Antimicrob Agents Chemother 53:4809–4815. doi: 10.1128/AAC.00269-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams MD, Nickel GC, Bajaksouzian S, Lavender H, Murthy AR, Jacobs MR, Bonomo RA. 2009. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob Agents Chemother 53:3628–3634. doi: 10.1128/AAC.00284-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denton M, Kerr K, Mooney L, Keer V, Rajgopal A, Brownlee K, Arundel P, Conway S. 2002. Transmission of colistin-resistant Pseudomonas aeruginosa between patients attending a pediatric cystic fibrosis center. Pediatr Pulmonol 34:257–261. doi: 10.1002/ppul.10166. [DOI] [PubMed] [Google Scholar]
- 8.Falagas ME, Bliziotis IA. 2007. Pandrug-resistant Gram-negative bacteria: the dawn of the post-antibiotic era? Int J Antimicrob Agents 29:630–636. doi: 10.1016/j.ijantimicag.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Hawley JS, Murray CK, Jorgensen JH. 2008. Colistin heteroresistance in Acinetobacter and its association with previous colistin therapy. Antimicrob Agents Chemother 52:351–352. doi: 10.1128/AAC.00766-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansen HK, Moskowitz SM, Ciofu O, Pressler T, Høiby N. 2008. Spread of colistin resistant non-mucoid Pseudomonas aeruginosa among chronically infected Danish cystic fibrosis patients. J Cyst Fibros 7:391–397. doi: 10.1016/j.jcf.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE, Liolios L. 2006. Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 50:2946–2950. doi: 10.1128/AAC.00103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moskowitz SM, Brannon MK, Dasgupta N, Pier M, Sgambati N, Miller AK, Selgrade SE, Miller SI, Denton M, Conway SP, Johansen HK, Høiby N. 2012. PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob Agents Chemother 56:1019–1030. doi: 10.1128/AAC.05829-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olaitan AO, Morand S, Rolain JM. 2014. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol 5:643. doi: 10.3389/fmicb.2014.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Storm DR, Rosenthal KS, Swanson PE. 1977. Polymyxin and related peptide antibiotics. Annu Rev Biochem 46:723–763. doi: 10.1146/annurev.bi.46.070177.003451. [DOI] [PubMed] [Google Scholar]
- 15.Boll M, Radziejewska-Lebrecht J, Warth C, Krajewska-Pietrasik D, Mayer H. 1994. 4-Amino-4-deoxy-l-arabinose in LPS of enterobacterial R-mutants and its possible role for their polymyxin reactivity. FEMS Immunol Med Microbiol 8:329–341. doi: 10.1111/j.1574-695X.1994.tb00460.x. [DOI] [PubMed] [Google Scholar]
- 16.Nummila K, Kilpeläinen I, Zähringer U, Vaara M, Helander IM. 1995. Lipopolysaccharides of polymyxin B-resistant mutants of Escherichia coli are extensively substituted by 2-aminoethyl pyrophosphate and contain aminoarabinose in lipid A. Mol Microbiol 16:271–278. [DOI] [PubMed] [Google Scholar]
- 17.Moskowitz SM, Ernst RK, Miller SI. 2004. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J Bacteriol 186:575–579. doi: 10.1128/JB.186.2.575-579.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunn JS, Lim KB, Krueger J, Kim K, Guo L, Hackett M, Miller SI. 1998. PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol Microbiol 27:1171–1182. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 19.Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. 2000. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar Typhimurium. Infect Immun 68:6139–6146. doi: 10.1128/IAI.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McPhee JB, Lewenza S, Hancock RE. 2003. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol Microbiol 50:205–217. doi: 10.1046/j.1365-2958.2003.03673.x. [DOI] [PubMed] [Google Scholar]
- 21.Macfarlane EL, Kwasnicka A, Ochs MM, Hancock RE. 1999. PhoP-PhoQ homologues in Pseudomonas aeruginosa regulate expression of the outer-membrane protein OprH and polymyxin B resistance. Mol Microbiol 34:305–316. doi: 10.1046/j.1365-2958.1999.01600.x. [DOI] [PubMed] [Google Scholar]
- 22.Roland KL, Martin LE, Esther CR, Spitznagel JK. 1993. Spontaneous pmrA mutants of Salmonella Typhimurium LT2 define a new two-component regulatory system with a possible role in virulence. J Bacteriol 175:4154–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macfarlane EL, Kwasnicka A, Hancock RE. 2000. Role of Pseudomonas aeruginosa PhoP-PhoQ in resistance to antimicrobial cationic peptides and aminoglycosides. Microbiology 146:2543–2554. [DOI] [PubMed] [Google Scholar]
- 24.Miller AK, Brannon MK, Stevens L, Johansen HK, Selgrade SE, Miller SI, Høiby N, Moskowitz SM. 2011. PhoQ mutations promote lipid A modification and polymyxin resistance of Pseudomonas aeruginosa found in colistin-treated cystic fibrosis patients. Antimicrob Agents Chemother 55:5761–5769. doi: 10.1128/AAC.05391-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abraham N, Kwon DH. 2009. A single amino acid substitution in PmrB is associated with polymyxin B resistance in clinical isolate of Pseudomonas aeruginosa. FEMS Microbiol Lett 298:249–254. doi: 10.1111/j.1574-6968.2009.01720.x. [DOI] [PubMed] [Google Scholar]
- 26.Barrow K, Kwon DH. 2009. Alterations in two-component regulatory systems of phoPQ and pmrAB are associated with polymyxin B resistance in clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother 53:5150–5154. doi: 10.1128/AAC.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fernández L, Gooderham WJ, Bains M, McPhee JB, Wiegand I, Hancock RE. 2010. Adaptive resistance to the “last hope” antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob Agents Chemother 54:3372–3382. doi: 10.1128/AAC.00242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller C, Plesiat P, Jeannot K. 2011. A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and β-lactams in Pseudomonas aeruginosa. Antimicrob Agents Chemother 55:1211–1221. doi: 10.1128/AAC.01252-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gutu AD, Sgambati N, Strasbourger P, Brannon MK, Jacobs MA, Haugen E, Kaul RK, Johansen HK, Høiby N, Moskowitz SM. 2013. Polymyxin resistance of Pseudomonas aeruginosa phoQ mutants is dependent on additional two-component regulatory systems. Antimicrob Agents Chemother 57:2204–2215. doi: 10.1128/AAC.02353-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernández L, Jenssen H, Bains M, Wiegand I, Gooderham WJ, Hancock RE. 2012. The two-component system CprRS senses cationic peptides and triggers adaptive resistance in Pseudomonas aeruginosa independently of ParRS. Antimicrob Agents Chemother 56:6212–6222. doi: 10.1128/AAC.01530-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McPhee JB, Bains M, Winsor G, Lewenza S, Kwasnicka A, Brazas MD, Brinkman FS, Hancock RE. 2006. Contribution of the PhoP-PhoQ and PmrA-PmrB two-component regulatory systems to Mg2+-induced gene regulation in Pseudomonas aeruginosa. J Bacteriol 188:3995–4006. doi: 10.1128/JB.00053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strempel N, Neidig A, Nusser M, Geffers R, Vieillard J, Lesouhaitier O, Brenner-Weiss G, Overhage J. 2013. Human host defense peptide LL-37 stimulates virulence factor production and adaptive resistance in Pseudomonas aeruginosa. PLoS One 8:e82240. doi: 10.1371/journal.pone.0082240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowman SK, Simon MD, Deaton AM, Tolstorukov M, Borowsky ML, Kingston RE. 2013. Multiplexed Illumina sequencing libraries from picogram quantities of DNA. BMC Genomics 14:466. doi: 10.1186/1471-2164-14-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 40.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 41.Wolfgang MC, Lee VT, Gilmore ME, Lory S. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell 4:253–263. doi: 10.1016/S1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 42.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 43.Yi EC, Hackett M. 2000. Rapid isolation method for lipopolysaccharide and lipid A from Gram-negative bacteria. Analyst 125:651–656. doi: 10.1039/b000368i. [DOI] [PubMed] [Google Scholar]
- 44.Caroff M, Tacken A, Szabó L. 1988. Detergent-accelerated hydrolysis of bacterial endotoxins and determination of the anomeric configuration of the glycosyl phosphate present in the “isolated lipid A” fragment of the Bordetella pertussis endotoxin. Carbohydr Res 175:273–282. doi: 10.1016/0008-6215(88)84149-1. [DOI] [PubMed] [Google Scholar]
- 45.Ernst RK, Yi EC, Guo L, Lim KB, Burns JL, Hackett M, Miller SI. 1999. Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science 286:1561–1565. doi: 10.1126/science.286.5444.1561. [DOI] [PubMed] [Google Scholar]
- 46.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 47.Kavanagh KL, Jörnvall H, Persson B, Oppermann U. 2008. Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol Life Sci 65:3895–3906. doi: 10.1007/s00018-008-8588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kallberg Y, Oppermann U, Persson B. 2010. Classification of the short-chain dehydrogenase/reductase superfamily using hidden Markov models. FEBS J 277:2375–2386. doi: 10.1111/j.1742-4658.2010.07656.x. [DOI] [PubMed] [Google Scholar]
- 49.Helander IM, Nummila K, Kilpeläinen I, Vaara M. 1995. Increased substitution of phosphate groups in lipopolysaccharides and lipid A of polymyxin-resistant mutants of Salmonella Typhimurium and Escherichia coli. Prog Clin Biol Res 392:15–23. [PubMed] [Google Scholar]
- 50.Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, Miller SI. 1998. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 95:189–198. doi: 10.1016/S0092-8674(00)81750-X. [DOI] [PubMed] [Google Scholar]
- 51.Raetz CR, Reynolds CM, Trent MS, Bishop RE. 2007. Lipid A modification systems in Gram-negative bacteria. Annu Rev Biochem 76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klockgether J, Munder A, Neugebauer J, Davenport CF, Stanke F, Larbig KD, Heeb S, Schöck U, Pohl TM, Wiehlmann L, Tümmler B. 2010. Genome diversity of Pseudomonas aeruginosa PAO1 laboratory strains. J Bacteriol 192:1113–1121. doi: 10.1128/JB.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nowicki EM, O'Brien JP, Brodbelt JS, Trent MS. 2015. Extracellular zinc induces phosphoethanolamine addition to Pseudomonas aeruginosa lipid A via the ColRS two-component system. Mol Microbiol 97:166–178. doi: 10.1111/mmi.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kallberg Y, Oppermann U, Jörnvall H, Persson B. 2002. Short-chain dehydrogenases/reductases (SDRs). Eur J Biochem 269:4409–4417. doi: 10.1046/j.1432-1033.2002.03130.x. [DOI] [PubMed] [Google Scholar]
- 55.Burrows LL, Charter DF, Lam JS. 1996. Molecular characterization of the Pseudomonas aeruginosa serotype O5 (PAO1) B-band lipopolysaccharide gene cluster. Mol Microbiol 22:481–495. doi: 10.1046/j.1365-2958.1996.1351503.x. [DOI] [PubMed] [Google Scholar]
- 56.Zienkiewicz M, Kern-Zdanowicz I, Gołebiewski M, Zyliñska J, Mieczkowski P, Gniadkowski M, Bardowski J, Cegłowski P. 2007. Mosaic structure of p1658/97, a 125-kilobase plasmid harboring an active amplicon with the extended-spectrum beta-lactamase gene _bla_SHV-5. Antimicrob Agents Chemother 51:1164–1171. doi: 10.1128/AAC.00772-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peigne C, Bidet P, Mahjoub-Messai F, Plainvert C, Barbe V, Medigue C, Frapy E, Nassif X, Denamur E, Bingen E, Bonacorsi S. 2009. The plasmid of Escherichia coli strain S88 (O45:K1:H7) that causes neonatal meningitis is closely related to avian pathogenic E. coli plasmids and is associated with high-level bacteremia in a neonatal rat meningitis model. Infect Immun 77:2272–2284. doi: 10.1128/IAI.01333-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin P, Murase K, Porcheron G, Houle S, Helloin E, Nougayrède J-P, Dozois C, Hayashi T, Oswald E. 2014. Role for hemolysin F in the virulence of Escherichia coli, abstr B-1539 114th Gen Meet Am Soc Microbiol, 17 to 20 May 2014, Boston, MA. [Google Scholar]
- 59.Tan TT, Morgelin M, Forsgren A, Riesbeck K. 2007. Haemophilus influenzae survival during complement-mediated attacks is promoted by Moraxella catarrhalis outer membrane vesicles. J Infect Dis 195:1661–1670. doi: 10.1086/517611. [DOI] [PubMed] [Google Scholar]
- 60.Kulp A, Kuehn MJ. 2010. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol 64:163–184. doi: 10.1146/annurev.micro.091208.073413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manning AJ, Kuehn MJ. 2011. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol 11:258. doi: 10.1186/1471-2180-11-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Macdonald IA, Kuehn MJ. 2013. Stress-induced outer membrane vesicle production by Pseudomonas aeruginosa. J Bacteriol 195:2971–2981. doi: 10.1128/JB.02267-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gatzeva-Topalova PZ, May AP, Sousa MC. 2005. Crystal structure and mechanism of the Escherichia coli ArnA (PmrI) transformylase domain. An enzyme for lipid A modification with 4-amino-4-deoxy-l-arabinose and polymyxin resistance. Biochemistry 44:5328–5338. doi: 10.1021/bi047384g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Williams GJ, Breazeale SD, Raetz CR, Naismith JH. 2005. Structure and function of both domains of ArnA, a dual function decarboxylase and a formyltransferase, involved in 4-amino-4-deoxy-l-arabinose biosynthesis. J Biol Chem 280:23000–23008. doi: 10.1074/jbc.M501534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strom MS, Lory S. 1986. Cloning and expression of the pilin gene of Pseudomonas aeruginosa PAK in Escherichia coli. J Bacteriol 165:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sarkisova S, Patrauchan MA, Berglund D, Nivens DE, Franklin MJ. 2005. Calcium-induced virulence factors associated with the extracellular matrix of mucoid Pseudomonas aeruginosa biofilms. J Bacteriol 187:4327–4337. doi: 10.1128/JB.187.13.4327-4337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gallagher LA, Manoil C. 2001. Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol 183:6207–6214. doi: 10.1128/JB.183.21.6207-6214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ochsner UA, Vasil AI, Johnson Z, Vasil ML. 1999. Pseudomonas aeruginosa_fur_ overlaps with a gene encoding a novel outer membrane lipoprotein, OmlA. J Bacteriol 181:1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material