Ezrin Enhances EGFR Signaling and Modulates Erlotinib Sensitivity in Non–Small Cell Lung Cancer Cells (original) (raw)
Abstract
Ezrin is a scaffolding protein that is involved in oncogenesis by linking cytoskeletal and membrane proteins. Ezrin interacts with epidermal growth factor receptor (EGFR) in the cell membrane, but little is known about the effects of this interaction on EGFR signaling pathway. In this study, we established the biological and functional significance of ezrin-EGFR interaction in non–small cell lung cancer (NSCLC) cells. Endogenous ezrin and EGRF interaction was confirmed by co-immunoprecipitation and immunofluorescent staining. When expression of ezrin was inhibited, EGFR activity and phosphorylation levels of downstream signaling pathway proteins ERK and STAT3 were decreased. Cell fractionation experiments revealed that nuclear EGFR was significantly diminished in ezrin-knockdown cells. Consequently, mRNA levels of EGFR target genes AURKA, COX-2, cyclin D1, and iNOS were decreased in ezrin-depleted cells. A small molecule inhibitor of ezrin, NSC305787, reduced EGF-induced phosphorylation of EGFR and downstream target proteins, EGFR nuclear translocation, and mRNA levels of nuclear EGFR target genes similar to ezrin suppression. NSC305787 showed synergism with erlotinib in wild-type EGFR-expressing NSCLC cells, whereas no synergy was observed in EGFR-null cells. Phosphorylation of ezrin on Y146 was found as an enhancer of ezrin-EGFR interaction and required for increased proliferation, colony formation, and drug resistance to erlotinib. These findings suggest that ezrin-EGFR interaction augments oncogenic functions of EGFR and that targeting ezrin may provide a potential novel approach to overcome erlotinib resistance in NSCLC cells.
Introduction
Epidermal growth factor receptor (EGFR) activation positively regulates growth of epithelial cells. The overexpression of EGFR has been observed in both premalignant and malignant tumors of the lung and occurs in 40% to 80% of patients with non–small cell lung cancer (NSCLC) [1], [2]. Ligand binding to EGFR results in receptor dimerization at the plasma membrane leading to activation of the tyrosine kinase domain and autophosphorylation of the cytoplasmic tail. Phosphorylation of the cytoplasmic domain of EGFR generates docking sites for several oncogenic proteins that induce Ras and PI3K. In addition, Src family tyrosine kinases, phospholipase C-gamma, protein kinase C, and signal transducers and activators of transcription (STAT) proteins have been shown to interact with EGFR either directly or indirectly [3], [4]. Activation of EGFR triggers receptor internalization and results in degradation or recycling of the receptor back to the cell surface [5], [6].
Nuclear localization of EGFR has been detected in many cancers including NSCLC cells [7], and research over the last decade characterized the steps for nuclear EGFR transport. After internalization, full-length EGFR interacts with importin β1 in the early endosomes via its nuclear localization sequence. This complex then moves through the Golgi apparatus and endoplasmic reticulum before it shuttles to the nucleus [8], [9].
Nuclear EGFR (nEGFR) interacts with STAT proteins in the nucleus and acts as a transcriptional coactivator that regulates expression of several tumor-promoting genes including cyclin D1, inducible nitric oxide synthase (iNOS), Aurora kinase A (AURKA), B-Myb, COX-2, c-Myc, breast cancer related protein, and GRP78 [10], [11], [12]. nEGFR phosphorylates proliferating cell nuclear antigen to promote cell proliferation and DNA repair [13]. Interaction of nEGFR with DNA-dependent protein kinase leads to repair of radiation-induced double-strand breaks in DNA of bronchial carcinoma cells [14].
High levels of nEGFR correlate with poor clinical outcome in breast, ovarian, oropharyngeal, esophageal, gallbladder, and NSCLC patients [15]. nEGFR is also related to resistance to chemotherapy [7] and radiotherapy [16]. Therefore, it is important to understand the mechanisms of nuclear transport of EGFR to target its nuclear functions.
Ezrin is a member of the membrane-cytoskeleton linker Ezrin-Radixin-Moesin (ERM) family of proteins, which are involved in the assembly of specialized domains of the membrane. ERM proteins were originally characterized as structural components of the cell cortex but were later shown to participate in signaling pathways [17]. Through their dynamic and reversible interactions with membrane proteins and actin filaments, ERM proteins coordinate multiple signal transduction pathways. In particular, ezrin is involved in trafficking events to and from the plasma membrane and recycling of transmembrane proteins such as α1-β adrenergic receptor [18], NHE3 [19], and H-K-ATPase [20]. Ezrin also facilitates nuclear import of β-dystroglycan [21].
Ezrin overexpression enhances malignant behavior of various cancer cells. In NSCLC, a high level of ezrin expression is associated with higher metastatic potential and lower patient survival [22]. Expression levels of ezrin were found to be significantly higher in primary cancer tissues than matched normal lung tissues [23]. Suppression of ezrin expression in NSCLC cell lines leads to reduced migration and invasion, anchorage-independent growth ability, and increased drug sensitivity [24].
Tyrosine phosphorylation of ezrin at Y146 and Y353 after EGF treatment [25], [26], [27], and the interaction between ezrin and EGFR have been demonstrated previously [28]. Recent findings showed that C terminal truncated forms of ezrin prohibited EGF-induced Ras pathway activation [29], and ezrin homolog ERM-1 in Caenorhabditis elegans regulated EGFR localization and signaling in vulvar cells [30]. Ezrin is also required for the delivery of EGFR to the lysosomes [31].
In this study, we show that ezrin enhances signaling and nuclear transport of EGFR in NSCLC cell lines. We further demonstrate synergism between a small molecule inhibitor of ezrin, NSC305787, and an EGFR inhibitor, erlotinib, in these cell lines, which may influence future therapy directions in NSCLC.
Methods
Cell Culture, Oligo Transfection, and Drug Treatment
A549, H292, H520, and H1944 cells were obtained from Georgetown University Tissue Culture Core Facility, and H2073 cells were kindly provided by Dr. Michael Peyton (UT Southwestern Medical Center); all cell lines were maintained in RPMI-1640 (Life Technologies #11875) supplemented with 10% fetal bovine serum (Sigma-Aldrich, #F2442). Ezrin siRNA (Ambion Life, #s14796) and ON-TARGETplus nontargeting control siRNA (Dharmacon, #D001818-02) were transfected using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. Briefly, Lipofectamine 2000 and siRNA were diluted in reduced-serum media, OptiMEM, and incubated for 10 minutes at room temperature. Diluted siRNA and Lipofectamine 2000 reagent were then combined in one tube and incubated for another 15 minutes before being added to the cell plate. For 96-well plate proliferation, cytotoxicity, and synergy assays, cells were transfected in 10-cm dishes, split after 48 hours, serum starved, and treated with EGF or HGF (R&D Systems Inc.) after an additional 24 hours. For the demonstration of ezrin knockdown via Western blot, cells were lysed 72 hours after transfection.
Erlotinib and etoposide were purchased from Selleckchem, and NSC305787 was obtained from Developmental Therapeutic Program of the National Institutes of Health (Bethesda, MD, USA). For growth factor–induced experiments, cells were treated with 5 μM NSC305787 30 minutes before growth factors. For immunoflourescence and nuclear fractionation experiments, NSC305787 was given at 1-μM concentration for 24 hours.
Antibodies
The following antibodies were used for Western blot, immunoprecipitation, and immunofluorescence: Ezrin (E8897) from Sigma-Aldrich; EGFR (#4267), Lamin A/C (#2032), Tubulin (#2144), phospho-STAT3 (#9131), phospho-ERK (#4370), ERK (#9107), phospho-EGFR Y1068 (#2236), phospho-EGFR Y845 (#2231), Myc-tag (#2276), and phospho-ezrin Tyr353 (#3144) from Cell Signaling; actin (C-11) HRP (sc1615) and mouse IgG (sc2025) from Santa Cruz Biotechnology; STAT3 (ab50761) and phospho-ezrin Y146 (ab92507) from Abcam; and Alexa-Fluor mouse (A21235) and rabbit (A11034) secondary antibodies from Thermo Fisher.
Immunoprecipitation, Cell Fractionation, and Western Blot
Subconfluent cells were lysed with RIPA buffer (50 mM Tris HCl pH 7.4, 1% Nonidet P-40, 0.5% Na-deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA, 50 mM NaF) supplemented with protease and phosphatase inhibitors and DNase. After protein quantification using bicinchoninic acid assay, 1 mg of protein was incubated for 2 hours with the indicated antibodies and then for 1 hour at 4°C with A/G Plus-agarose beads (#sc2001, Santa Cruz Biotechnology). After each incubation period, samples were washed three times with IP wash buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 0.5% Nonidet P-40, 0.2 mM sodium vanadate, and 0.2 mM PMSF). To elute the protein bound to the beads, samples were boiled in SDS-PAGE sample buffer for 5 minutes. Qproteome Cell Compartment Kit (Qiagen #37502) was used for cell fractionation according to kit instructions and the previously described method [32]. Western blot signals were quantitated by ImageJ64 1.47v software (NIH, USA).
Immunofluorescence and Microscopy
Coverslips were placed in 12-well plates and precoated with collagen. Cells were grown on the coverslips, treated if necessary, and washed twice with 37°C PBS before fixation with 3.7% paraformaldehyde in PBS for 15 minutes. The coverslips were then washed with PBS three times each for 5 minutes, and cells were permeablized and blocked with 1% BSA, 10% goat serum, and 0.5% Triton X-100 in PBS for 30 minutes. Next, coverslips were incubated with primary antibodies diluted in 1% BSA in PBS for 1 hour at room temperature and washed three times with PBS. Lastly, coverslips were incubated for 30 minutes in the dark at room temperature with secondary antibodies and DAPI diluted in PBS. Before mounting with Flourogel with Tris Buffer (#17985, Electron Microscopy Sciences), the coverslips were washed three times with PBS and two times with ddH2O. Images were acquired using a Nikon Eclipse Ti-S microscope with Nikon Intensilight C-HGFI fiber illuminator (Nikon Instruments Inc.).
Quantitative Polymerase Chain Reaction (PCR) and Primers
Total RNA was extracted from cell lines using an RNAeasy mini kit (Qiagen) and reverse transcribed using Transcriptor First Strand cDNA Synthesis Kit (Roche) according to manufacturer’s protocols. Quantitative PCR was done on an Eppendorf Mastercycler using SYBR Green mix (Sigma Aldrich). Reactions were performed in triplicate in a 96-multiwell plate (Eppendorf, #30132700). Gene expressions were normalized to 18S rRNA, and fold differences were calculated using the comparative CT method: 2−(ΔΔCT), where ΔΔCT refers to (normalized control sample) − (normalized treated sample). Primer pairs were as follows—18S rRNA: sense: 5′-cttagagggacaagtggcg-3′, antisense: 5′-acgctgagccagtcagtgta-3′; aurora kinase A: sense: 5′-tctagtcctccttaaccacttatct-3′, antisense: 5′-gacacatggcctcttctgtatc-3′; COX2: sense: 5′-tgtacccggacaggattcta-3′, antisense: 5′-cccttgaagtgggtaagtatgt-3′; iNOS: sense: 5′-aaggtctacgttcaagacatcc-3′, antisense: 5′-gcacatcgccacaaacatag-3′; cyclin D1: sense: 5′-gggttgtgctacagatgatgag-3′, antisense: 5′-agacgcctcctttgtgttaat-3′.
Cell Viability, Proliferation, and Synergy Experiments
NSCLC cells (3.5 × 103 cells/well) were seeded to 96-well plates. After overnight growth, 0 to 50 mM erlotinib or 0 to 100 μM NCS305787 (in DMSO) was added; the DMSO level in each sample was adjusted to 2% for erlotinib and 1% for NSC305787. After 24 hours of incubation, 0.5 mg/ml thiazolyl blue tetrazolium bromide (MTT; Sigma-Aldrich, #M5655) was added to the media and incubated for an additional 4 hours. Then, the media was aspirated, and 100 μl of isopropanol was added to each well. The plates were shaken before light absorbance was measured at 570 and 650 nm using a plate reader, and IC50 values were calculated using GraphPad Prism 6 software (GraphPad Software, Inc.).
For synergy experiments, 50 μM erlotinib, 20 μM NSC305787, and 40 μM etoposide were used for the maximum concentrations, and cells were treated with drugs in serial dilutions in seven concentrations either alone or in combined form. DMSO concentrations in synergy plates were adjusted to the highest DMSO concentration (2.5%) in the plate for each synergy experiment. After absorbance measurements were taken, affected fractions (Fa) were calculated separately for each triplicate treatment according to the Chou-Talalay method [33] using the formula 1 − (X - blank)/(DMSO − blank): X represents the average of each triplicated treatment, and blank represents the background measurement at 650 nm. Combination Index (CI) calculations were made using CompuSyn software (ComboSign, Inc.).
Plasmid Constructs and Site-Directed Mutagenesis
The pcDNA3.1 plasmids encoding myc- and _his_-tagged ezrin Y146E, Y146F, Y353E, and Y353F were created by site-directed mutagenesis using WT ezrin as a template. The following primers were designed using the QuikChange Primer Design web tool (Agilent Technologies): Y146E: sense: 5′-ccgctcagagctgagttccccagacttgtgcac-3′, antisense: 5′-gtgcacaagtctggggaactcagctctgagcgg-3′; Y146F: sense: 5′-ccgctcagagctgagttccccagacttgtgcac-3′, antisense: 5′-gtgcacaagtctggggaactcagctctgagcgg-3′; Y354E: sense: 5′-ttctttgtcttctcctcttcgtcctgcagccgcagc-3′, antisense: 5′-gctgcggctgcaggacgaagaggagaagacaaagaa-3′; Y354F: sense: 5′-cttctcctcaaagtcctgcagccgcagca-3′, antisense: 5′-tgctgcggctgcaggactttgaggagaag-3′. QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) was used according to the manufacturers’ instructions. Cells were transfected using FuGENE 6 transfection reagent (Promega Corporation) and selected with G418 (Geneticin; Life Technologies) for 7 to 10 days before splitting for experiments.
Soft Agar Colony Formation Assay
The bottom layer was prepared with 0.6% agar-containing media in 12 well plates, and 1 × 103 cells were mixed with 0.4% agar-containing media and added to the plates in the top layer. Colonies were detected after 3 weeks of incubation at 37°C in a humidified atmosphere. Pictures of soft agar colonies were analyzed using ImageJ64 1.47v software (NIH, USA).
Statistical Analysis
Statistical analyses were performed using GraphPad Prisim 6 (GraphPad Software, Inc, CA, USA). Unpaired t test with Welch’s correction was used for comparison of two different conditions.
Results
Ezrin-EGFR Interaction in NSCLC Cells
We first validated the interaction between endogenous ezrin and EGFR in two wild-type (wt) EGFR-expressing NSCLC cell lines, A549 and H292, in the presence and absence of the ligand EGF (Figure 1A). We were able to detect ezrin and EGFR in the same protein complex that was immunoprecipitated by an anti-ezrin antibody (Figure 1A, lane 2 compared with lane 3), and this interaction was increased in EGF-stimulated cells (Figure 1A, lane 3 compared with lane 9). This enhancement may be due to the increase in the total level of EGFR in EGF-stimulated cells (Figure 1A, lane 1 compared with lane 7).
Figure 1.

Ezrin and EGFR interacts in NSCLC cell lines. (A) H292 cells were serum starved overnight and pretreated with 5 μM ezrin inhibitor NSC305787 or DMSO for 2 hours followed by 100-ng/ml EGF treatment for 5 minutes. Cell lysates were immunoprecipitated using an antiezrin antibody or control mouse total IgG. Precipitates were evaluated by Western blotting using anti-EGFR (upper panel) and antiezrin (lower panel) antibodies. EGFR interaction with ezrin was enhanced with EGF stimulation, and NSC305787 inhibited this interaction in both the presence and absence of the ligand. (B) Immunofluorescence staining of EGFR (red), ezrin (green), and DAPI (blue) in DMSO-treated (upper images) and NSC305787-treated (bottom images) A549 cells was analyzed by fluorescence microscopy. Ezrin and EGFR colocalization (yellow) in the membrane (arrow) and perinuclear area (arrow head) is indicated. NSC305787 treatment inhibited the colocalization of ezrin and EGFR.
Next, we evaluated the effect of NSC305787, a small molecule inhibitor of ezrin, which functions through preventing specific protein-protein interactions of ezrin [5], [32]. NSC305787 inhibited EGFR binding to ezrin in both the presence and absence of EGF (Figure 1A, lane 3 compared with lane 6 and lane 9 compared with lane 12). To further examine the effect of NSC305787 on the interaction between ezrin and EGFR, we analyzed localization patterns of ezrin and EGFR by immunofluorescence staining. Ezrin-EGFR colocalization was visible on both the plasma membrane and the perinuclear area, which was inhibited by NSC305787 treatment. We also observed morphological changes in NSC305787-treated cells, which possibly result from reduced ezrin levels around the plasma membrane (Figure 1B, lower panel).
Effects of Ezrin on EGFR Signaling
To evaluate the effect of ezrin on EGFR phosphorylation, we silenced ezrin expression in A549 and H292 by small interfering RNAs. Cells were first serum starved for 24 hours and then treated with EGF for 5 minutes. Immunoblot analysis revealed that suppression of ezrin expression resulted in 50% and 40% decrease in phosphorylation of EGFR (Tyr 1068 and Tyr 845 residues), respectively (Figure 2A). We also observed an increase in the total EGFR levels in some of the siEZRIN-treated cells with an average of 40% (± 10%). Inhibition of ezrin expression also resulted in reduced phosphorylation of two EGFR downstream pathway proteins: ERK and STAT3 (Figure 2B). Reduced levels of phosphorylation of ERK and STAT3 upon NSC305787 treatment further validated dependency of the EGFR signaling on ezrin. NSC305787 inhibited EGF-mediated phosphorylation of both STAT3 and ERK in NSCLC cell lines (Figure 2Bx) but did not affect ERK phosphorylation levels in two EGFR-null cell lines (Supplementary Figure S1). NSC305787 also did not affect basal ERK phosphorylation levels in H292 cells (Supplementary Figure S4).
Figure 2.

Reduced ezrin expression inhibits EGFR and phosphorylation of downstream target proteins in NSCLC cells. (A) Expression of ezrin was inhibited using siRNA oligos in A549 and H292 cells. Serum-starved cells were stimulated with 100 ng/ml of EGF for 5 minutes. EGFR activity was determined by immunoblot using phosphospecific antibodies for Y1068 and Y845. (B) Phosphorylation of EGFR downstream pathway proteins ERK and STAT3 was determined in cell lysates from panel A. Similar to EGFR, phospho-STAT3 and phospho-ERK levels were decreased in ezrin-depleted cells. (C) Cells were serum starved for 24 hours and treated with 5 μM NSC305787 or DMSO for 30 minutes before 5 minutes of EGF treatment. Phospho-STAT3 and phospho-ERK protein levels were decreased in NSC305787-treated cells.
Supplementary Figure S1.

Ezrin inhibitor NSC305787 shows no effect on ERK phosphorylation in two EGFR-null cell lines. (A) Western blot analysis of EGFR levels in H1944 and H520 cells. A549 cell line was used as a positive control. (B) Cells were serum starved for 24 hours and treated with 5 μM NSC305787 or DMSO for 30 minutes before EGF treatment for 5 minutes. (C) Cells were serum starved for 24 hours and treated with 5 μM NSC305787 or DMSO for 30 minutes before HGF treatment for 5 minutes.
Supplementary Figure S4.
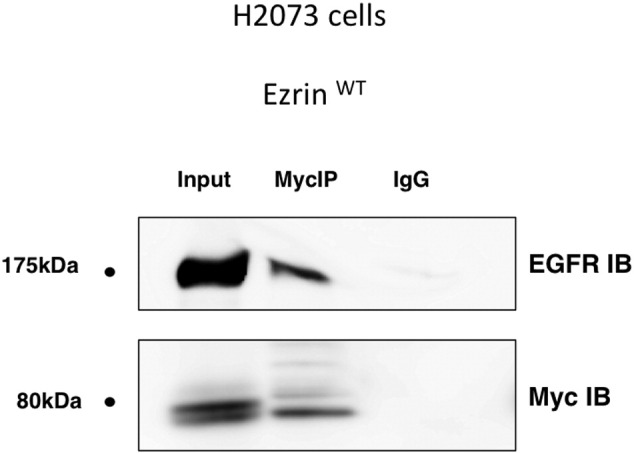
Ezrin wt did interact with EGFR. Ezrin-EGFR interaction in H2073 cells transfected with cDNAs coding for ezrin wt was analyzed by immunoprecipitation using anti-myc antibody followed by Western blotting.
Nuclear EGFR Levels in Ezrin Knockdown Cells
Earlier work suggested that ezrin might regulate endosome transport and nuclear import of several of its protein binding partners [21], [31]. We hypothesized that ezrin may also have an effect on the nuclear shuttling of EGFR. To this end, we determined nEGFR levels in A549 and H292 cells following siRNA-mediated knockdown of ezrin expression (Figure 3A). Using Image J64 software, we observed 55% and 30% reductions in the pixels of nEGFR protein levels in response to ezrin siRNA in A549 and H292 cell lines, respectively. We also observed an increase in the cytoplasmic and total EGFR levels, which might be induced by a positive feedback mechanism. The decrease in nEGFR levels in response to ezrin depletion was also validated by immunofluorescence staining in A549 cells (Figure 3B). Antiezrin compound NSC305787 inhibited the nuclear translocation of EGFR as well (Figure 3C).
Figure 3.

Inhibition of ezrin expression causes decreased levels of EGFR in nucleus in NSCLC cells. (A) Ezrin expression was inhibited using siRNA oligos in A549 and H292 cells, and whole cell lysates were subjected to cell fractionation and immunoblotting with antiezrin and anti-EGFR antibodies. Lamin A/C and tubulin antibodies were used as control for nuclear and non-nuclear fractions, respectively. (B) Immunofluorescence staining of EGFR, ezrin, and DAPI in control siRNA–treated (left panel) and ezrin siRNA–treated (right panel) A549 cells. Nuclear and perinuclear EGFR localization was decreased in ezrin-depleted cells. (C) Cells were serum starved for 24 hours and treated with 5 μM NSC305787 or DMSO for 2 hours before EGF stimulation for 30 minutes. NSC305787 decreased EGF-induced nuclear translocation of EGFR in H292 cells.
Lack of Ezrin Effects nEGFR Target Gene Expression
Because reduced ezrin expression caused a decrease in nEGFR levels in NSCLC cell lines, we evaluated the mRNA levels of nEGFR target genes AURKA, COX-2, cyclin D1 (CCND1), and iNOS in the same cell lines (Figure 4A). RNAi-mediated knockdown of ezrin in A549 cells decreased AURKA, COX-2, and iNOS expression more than 50%. A moderate but consistent 20% reduction in CCND1 (cyclin D1) mRNA levels was also observed. Pharmacological inhibition of ezrin by NSC305787 also decreased AURKA and COX-2 expression in H292 cells (Figure 4B). There was no significant decrease in nEGFR target gene expression in NSC305787-treated H520 cells, which do not express any EGFR, or in H292 cells without EGF induction (Supplementary Figure S5). Because those genes are associated with cell proliferation, we checked the proliferation of all three NSCCLC cell lines after transfection of cells with siRNA for ezrin (Figure 4C). Inhibition of ezrin expression significantly decreased proliferation of wt EGFR-expressing A549 and H292 cells but had no effect on EGFR-null H520 cells (Figure 4D).
Figure 4.

Suppression of ezrin expression and its pharmacological inhibition by NSC305787 causes decreased mRNA levels of nEGFR target genes in EGFR wt NSCLC cells_._ (A) mRNA expression levels of indicated genes were analyzed by quantitative PCR in ezrin-depleted A549 cells. CT values were normalized to 18S rRNA, and fold differences were calculated using the comparative CT method. (B) NSC305787 treatment did not decrease mRNA levels of nEGFR target genes in EGFR-null H520 cells, whereas AURKA and COX-2 levels were decreased in wt EGFR H292 cells. (C) Ezrin and EGFR protein expression levels were determined by immunoblotting after transfection of A549, H292, and H520 cells with siRNA oligos targeting ezrin. (D) Proliferation of A549, H292, and H520 cells after transfection with siRNA oligos targeting ezrin was determined by MTT assay. Ezrin knockdown significantly decreased proliferation of wt EGFR-expressing cells, A549 and H292, and did not affect EGFR-null cells, H520. (*P < .01, unpaired t test with Welch’s correction).
Supplementary Figure S5.
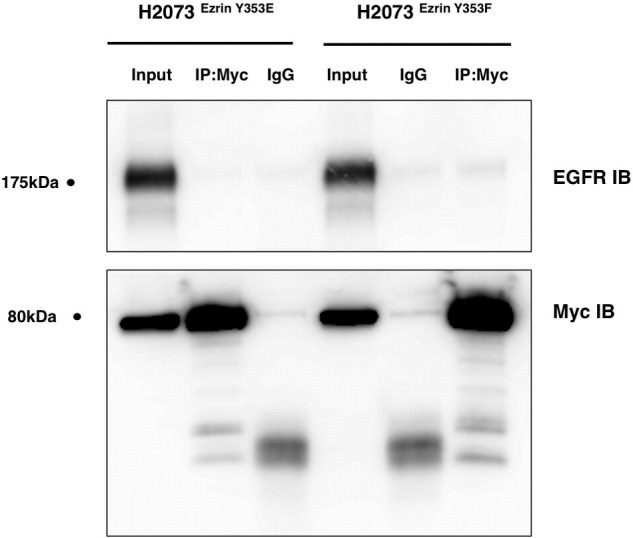
Ezrin Y353E or Y353F mutants do not interact with EGFR. Ezrin-EGFR interaction in H2073 cells transfected with cDNAs coding for ezrin Y353E and Y353F mutants was analyzed by immunoprecipitation using anti-myc antibody followed by Western blotting.
Ezrin and Erlotinib Sensitivity
Erlotinib functions by inhibiting EGFR tyrosine kinase activity in both EGFR wt and mutant cancer cells. There are several mechanisms independent of kinase activity that may result in erlotinib resistance in NSCLC cells including nuclear accumulation of EGFR. Thus, we hypothesized that suppression of ezrin expression might sensitize cells to erlotinib treatment. To test this hypothesis, two NSCLC cells were transfected with siRNA oligos targeting ezrin and incubated with increasing concentrations of erlotinib for 24 hours. Ezrin knockdown significantly decreased the viability of cells and caused a reduction in IC50 values for erlotinib in both EGFR-expressing cell lines but not the EGFR-null cell line (Figure 5, A and B). Having demonstrated that silencing of ezrin expression sensitizes cells to erlotinib treatment, we then tested the potential synergy between NSC305787 and erlotinib. We treated the cells with erlotinib alone and in combination with NSC305787 and determined the cell viability. Calculation of CIs was performed using Compu-Syn software based on the Chou-Talalay method [33], where CI < 1 indicates synergy. We observed synergy between NSC305787 and erlotinib in A549 (Figure 5C) but not in H520 cell line (Figure 5D). Etoposide was used as a non-Tyrosine kinase inhibitor control drug, and it did not show any synergy with NSC305787 (Figure 5E).
Figure 5.

Ezrin knockdown sensitizes NSCLC cells to erlotinib treatment. MTT test was used to evaluate the cell viability. (A) Nonlinear curve fit and (B) IC50 values of control siRNA (siCTRL)– and siEZR-treated A549, H292, and H520 cells are given. Reduced ezrin expression caused a decreased IC50 value for erlotinib in A549 and H292 cells compared with H520 cells. (C) Ezrin inhibitor NSC305787 shows synergism with erlotinib in A549 cells. No synergy was observed in H520 cells treated with erlotinib-NSC305787 combination (D) and in A549 cells treated with etoposide-NSC305787 combination (E).
Ezrin Y146 Phosphorylation and EGFR Binding
Previous data and our findings in this study show that EGF treatment might increase the interaction between ezrin and EGFR. It has been demonstrated that EGF induces tyrosine phosphorylation of ezrin at Y146 and Y353. We validated increased ezrin phosphorylation on Y146 and Y353 residues in A549 cells following EGF treatment (Figure 6A). We then investigated whether these phosphorylation events can affect ezrin-EGFR interaction. We transfected erlotinib-sensitive H2073 cells with mammalian expression vectors containing cDNA for wt ezrin (Ezrinwt) and mutant forms of ezrin (Y146E, Y146F, Y353E, and Y353F) with a myc-tag (Figure 6B). Immunoprecipitation experiments of ectopic ezrin with anti-myc antibody revealed that wt (Supplementary Figure S6) and phospho-mimicking ezrin Y146E mutant strongly interacts with EGFR compared with phospho-null mutant Y146F (Figure 6C). Ezrin Y353E and Y353F mutants did not interact with EGFR in co-IP experiments (Supplementary Figure S2). We then compared the IC50 values of erlotinib for mock-transfected, Ezrinwt-, Y146E-, and Y146F-expressing H2073 cells. Ezrinwt- and Y146E-expressing cells showed resistance to erlotinib treatment unlike ezrin Y146F-expressing cells (Figure 6D). Overexpression of ectopic ezrin Y146E mutant in wt EGFR-expressing H2073 cells caused an increased number of colonies in a soft agar colony formation assay (Figure 6E) and cell proliferation (Figure 6F). EGFR-null H520 cells, on the other hand, did not show any significant difference with any of the ezrin expression constructs (Supplementary Figure S3).
Figure 6.

Y146 phosphorylation of ezrin affects its interaction with EGFR. (A) The 100-ng/ml EGF treatment induces tyrosine phosphorylation of ezrin on Y146 and Y353 residues in A549 cells. (B) Protein expression levels of myc-tagged Ezrinwt, Y146E, and Y146F were determined by immunoblotting following transfection of H2073 cells with corresponding expression constructs. (C) Ezrin-EGFR interaction in H2073 cells transfected with cDNAs coding for ezrin Y146E and Y146F mutants was determined by immunoprecipitation using anti-myc antibody followed by Western blotting. (D) Ectopic expression of ezrinwt and ezrin Y146E mutant sensitizes cells to erlotinib in H2073 cells. IC50 values are given in the table, and cell viability curves are shown in the graph. (E) The number of colonies in H2073 cells transfected with cDNAs coding for ezrin Y145E and Y145F mutants was determined by soft agar colony formation assay (*P < .01, unpaired t test with Welch’s correction). (F) Ectopic overexpression of ezrinwt and ezrin Y146E mutant caused an increased proliferation of H2073 cells compared with ezrin Y146F mutant.
Supplementary Figure S6.
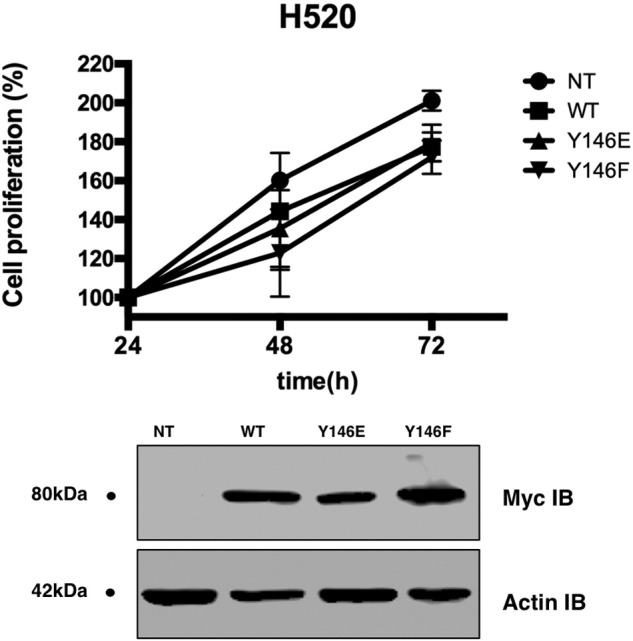
Ezrin Y146E mutant does not affect proliferation of H520 cells. EGFR-null H520 cells were transfected with cDNAs coding for Ezrin wt and Y146E and Y146F mutant forms, and cell proliferation was assessed by MTT assay after 72 hours. Western blot analysis 25 shows levels of ezrin expression after transfection of H520 cells with corresponding expression constructs.
Supplementary Figure S2.
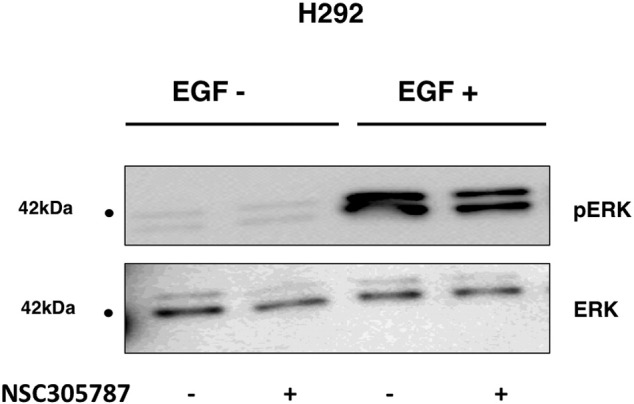
_N_SC305787 did not affect pERK levels in EGF − H292 cells. H292 cells starved overnight and treated with NSC305787 or DMSO in EGF − and EGF + conditions. There was no effect on pERK levels in EGF − H292 cells.
Supplementary Figure S3.
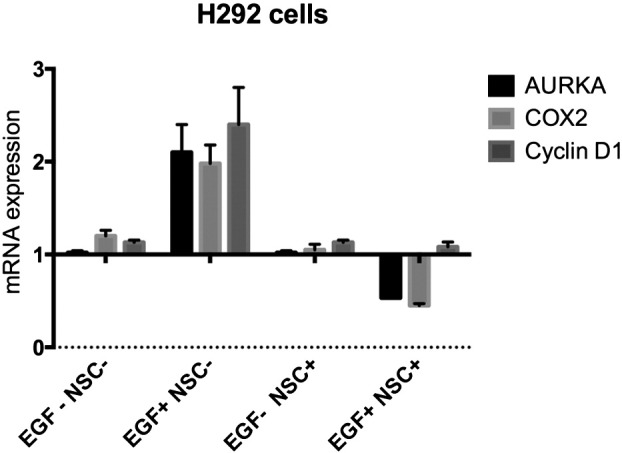
NSC305787 did not affect mRNA levels of nEGFR target genes in EGF − H292 cells. H292 cells starved overnight and treated with NSC305787 or DMSO in EGF − and EGF + conditions. There was no effect on mRNA levels in EGF − H292 cells.
Discussion
Promising characteristics of EGFR as a molecular target for cancer therapy have inspired the development of targeted inhibitors against EGFR and its signaling components. It has been reported that combining distinct classes of EGFR inhibitors may not only potentiate cellular toxicity but also assist to overcome inherent or acquired resistance to a single class of EGFR inhibitors [34]. In the current study, we showed for the first time that targeting ezrin function with small molecule inhibitor might provide synergism in combination with EGFR inhibition.
Ezrin is linked to different oncogenic phenotypes in different tumor models [23]. Specifically, its role as a key regulator of metastasis in osteosarcoma is well established [35]. Ezrin is involved in a variety of cellular functions such as cell cytoskeleton organization, cell motility, proliferation, cell signaling, and mRNA translation [32], [36], [37]. But ezrin functioning on the plasma membrane may be an independent process from its activities in the cytoplasm [29], [38]. It is likely that ezrin may regulate particular signaling pathways such as Ras, Akt, and VEGFR in certain tumor types that may result in diverse outcomes There are also accumulating data on the correlation between ezrin expression and clinical outcomes in epithelial tumors; however, the exact molecular mechanisms that result in these oncologic phenotypes still remain unclear. In this study, we used two wt EGFR-expressing NSCLC cell lines and showed that ezrin interacts with EGFR and a small molecule inhibitor of ezrin, NSC305787, inhibits this interaction. Downregulation of ezrin and its pharmacological inhibition by NSC305787 caused a decreased Y1068 and Y845 phosphorylation of EGFR and of common EGFR downstream target proteins, ERK and STAT3, following EGF treatment. Our findings support the growing list of unique ezrin functions that contribute to oncogenesis.
Ezrin together with the homotypic fusion and protein sorting complex regulates the maturation of early endosomes. Initiation of the nuclear translocation of EGFR starts in early endosomes after receptor internalization via its interaction with importin β1, and depletion of ezrin causes a delay in EGFR trafficking in the endosomes [31]. We observed decreased levels of nEGFR in ezrin-knockdown NSCLC cells, although there were increases in the total and cytoplasmic EGFR levels which might be a result of positive feedback mechanisms or the act of Ezrin as a translation regulator [32], [36]. We also showed that expression of nEGFR target genes AURKA, COX-2, and iNOS was significantly decreased in response to ezrin knockdown. These observations established that ezrin interaction with EGFR has functional significance. Experiments with an EGFR-null cell line further validated that the observed biological phenotypes in response to ezrin modulation were through EGFR interaction.
The ezrin inhibitor NSC305787 has been shown to reduce metastasis in experimental osteosarcoma model [35] and inhibit cell survival in colorectal cancer cells [39]. Our data revealed that NSC305787 diminished ezrin-EGFR interaction and caused decreased phosphorylation levels of downstream target genes ERK and STAT3. NSC305787 also inhibited membranous localization of ezrin and caused morphological changes in the cells.
Phosphorylation of ezrin is required both for conformational activation and for signaling to downstream events [26]. C-terminal threonine phosphorylation at T567 is the best known activating phosphorylation site as a downstream target of Rho pathway [40]. Ezrin has been initially identified as a substrate for tyrosine phosphorylation by EGFR [26], [28], and recent studies have demonstrated that growth factors EGF, PDGF, and HGF and Src kinases are able to induce tyrosine phosphorylation of ezrin [41], [42]. Phosphorylation of ezrin at Y353 is related to signal survival of PI3K/Akt pathway [43] and has been shown to relate with late-stage and poor differentiation in NSCLC [44]. Ezrin phosphorylation at Y146 is associated with adhesion-mediated signaling in A431 cells [42], but the effect of this phosphorylation on EGFR interaction was unknown. In this study, we showed that ezrin Y146E mutant more strongly interacts with EGFR compared with phospho-null Y146F mutant. Ectopic overexpression of ezrin Y146E mutant in wt EGFR-expressing H2073 cells produced increased cell proliferation and colony formation and caused erlotinib resistance.
Even though the discovery of the targeted receptor tyrosine kinase inhibitors led to promising improvement in cancer treatment, they failed to bring benefit as monotherapeutic drugs. This could be the result of drug resistance or coactivation of other tyrosine kinase receptors [45]. Particularly in NSCLC, different tyrosine kinase receptors are active in cell lines and human tumor samples, and these receptors mostly share common signaling pathways [46]. Therefore, targeting intersecting pathways such as EGFR–c-MET or EGFR-PDGFR pathways was beneficial in NSCLC and ovarian cancer animal models [47], [48], [49]. Our findings were in support of this hypothesis, as targeting ezrin function in NSCLC cells treated with EGFR inhibitor shows synergism.
Overall, our findings show that ezrin regulates EGFR signaling and trafficking to the nucleus; therefore, inhibiting ezrin function with small molecule inhibitors may provide an effective combination with EGFR inhibitors in EGFR-positive NSCLC. Further evaluation of this synergism in in vivo models may accelerate clinical translation of our findings for potential clinical applications.
The following are the supplementary data related to this article.
Acknowledgements
We thank the Microscopy and Imaging Shared Resource at the Lombardi Comprehensive Cancer Center (Georgetown University), which is supported by a grant P30 CA51008 (PI Louis Weiner) from the National Cancer Institute. We also like to thank Dr. Michael Payton (UT Southwestern Medical Center) for kindly providing us the H2073 cell line.
Footnotes
1
Conflict of interest: Georgetown University has filed a patent application for using NSC305787 and related compounds in cancer therapy, where J. A. T. and A. U. are listed as inventors.
References
- 1.Grandis JR, Sok JC. Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol Ther. 2004;102(1):37–46. doi: 10.1016/j.pharmthera.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Merrick DT, Kittelson J, Winterhalder R, Kotantoulas G, Ingeberg S, Keith RL, Kennedy TC, Miller YE, Franklin WA, Hirsch FR. Analysis of c-ErbB1/epidermal growth factor receptor and c-ErbB2/HER-2 expression in bronchial dysplasia: evaluation of potential targets for chemoprevention of lung cancer. Clin Cancer Res. 2006;12(7 Pt 1):2281–2288. doi: 10.1158/1078-0432.CCR-05-2291. [DOI] [PubMed] [Google Scholar]
- 3.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12(18):5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 4.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366(1):2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 5.Wiley HS. Trafficking of the ErbB receptors and its influence on signaling. Exp Cell Res. 2003;284(1):78–88. doi: 10.1016/s0014-4827(03)00002-8. [DOI] [PubMed] [Google Scholar]
- 6.Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2008;314(17):3093–3106. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28(43):3801–3813. doi: 10.1038/onc.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brand TM, Iida M, Luthar N, Starr MM, Huppert EJ, Wheeler DL. Nuclear EGFR as a molecular target in cancer. Radiother Oncol. 2013;108(3):370–377. doi: 10.1016/j.radonc.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 9.Wang YN, Yamaguchi H, Huo L, Du Y, Lee HJ, Lee HH, Wang H, Hsu JM, Hung MC. The translocon Sec61beta localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus. J Biol Chem. 2010;285(49):38720–38729. doi: 10.1074/jbc.M110.158659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin beta1 and CRM1. J Cell Biochem. 2006;98(6):1570–1583. doi: 10.1002/jcb.20876. [DOI] [PubMed] [Google Scholar]
- 11.Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog. 2006;45(1):10–17. doi: 10.1002/mc.20147. [DOI] [PubMed] [Google Scholar]
- 12.Hung LY, Tseng JT, Lee YC, Xia W, Wang YN, Wu ML, Chuang YH, Lai CH, Chang WC. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008;36(13):4337–4351. doi: 10.1093/nar/gkn417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol. 2006;8(12):1359–1368. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- 14.Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Raju U, Milas L, Chen DJ, Kehlbach R, Rodemann HP. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem. 2005;280(35):31182–31189. doi: 10.1074/jbc.M506591200. [DOI] [PubMed] [Google Scholar]
- 15.Traynor AM, Weigel TL, Oettel KR, Yang DT, Zhang C, Kim K, Salgia R, Iida M, Brand TM, Hoang T. Nuclear EGFR protein expression predicts poor survival in early stage non–small cell lung cancer. Lung Cancer. 2013;81(1):138–141. doi: 10.1016/j.lungcan.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liccardi G, Hartley JA, Hochhauser D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res. 2011;71(3):1103–1114. doi: 10.1158/0008-5472.CAN-10-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002;3(8):586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- 18.Stanasila L, Abuin L, Diviani D, Cotecchia S. Ezrin directly interacts with the alpha1b-adrenergic receptor and plays a role in receptor recycling. J Biol Chem. 2006;281(7):4354–4363. doi: 10.1074/jbc.M511989200. [DOI] [PubMed] [Google Scholar]
- 19.Zhao H, Shiue H, Palkon S, Wang Y, Cullinan P, Burkhardt JK, Musch MW, Chang EB, Turner JR. Ezrin regulates NHE3 translocation and activation after Na+-glucose cotransport. Proc Natl Acad Sci U S A. 2004;101(25):9485–9490. doi: 10.1073/pnas.0308400101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao X, Ding X, Guo Z, Zhou R, Wang F, Long F, Wu F, Bi F, Wang Q, Fan D. PALS1 specifies the localization of ezrin to the apical membrane of gastric parietal cells. J Biol Chem. 2005;280(14):13584–13592. doi: 10.1074/jbc.M411941200. [DOI] [PubMed] [Google Scholar]
- 21.Vasquez-Limeta A, Wagstaff KM, Ortega A, Crouch DH, Jans DA, Cisneros B. Nuclear import of beta-dystroglycan is facilitated by ezrin-mediated cytoskeleton reorganization. PLoS One. 2014;9(3):e90629. doi: 10.1371/journal.pone.0090629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen QY, Yan J, Hu HZ, Chen FY, Song J, Jiang ZY, Jiao DM, Wu YQ. Expression of ezrin in human non–small cell lung cancer and its relationship with metastasis and prognosis. Zhonghua Zhong Liu Za Zhi. 2012;34(6):436–440. [PubMed] [Google Scholar]
- 23.Li Q, Gao H, Xu H, Wang X, Pan Y, Hao F, Qiu X, Stoecker M, Wang E. Expression of ezrin correlates with malignant phenotype of lung cancer, and in vitro knockdown of ezrin reverses the aggressive biological behavior of lung cancer cells. Tumour Biol. 2012;33(5):1493–1504. doi: 10.1007/s13277-012-0400-9. [DOI] [PubMed] [Google Scholar]
- 24.Chen QY, Xu W, Jiao DM, Wu LJ, Song J, Yan J, Shi JG. Silence of ezrin modifies migration and actin cytoskeleton rearrangements and enhances chemosensitivity of lung cancer cells in vitro. Mol Cell Biochem. 2013;377(1-2):207–218. doi: 10.1007/s11010-013-1586-x. [DOI] [PubMed] [Google Scholar]
- 25.Gould KL, Cooper JA, Bretscher A, Hunter T. The protein-tyrosine kinase substrate, p81, is homologous to a chicken microvillar core protein. J Cell Biol. 1986;102(2):660–669. doi: 10.1083/jcb.102.2.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bretscher A. Rapid phosphorylation and reorganization of ezrin and spectrin accompany morphological changes induced in A-431 cells by epidermal growth factor. J Cell Biol. 1989;108(3):921–930. doi: 10.1083/jcb.108.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper JA, Bowen-Pope DF, Raines E, Ross R, Hunter T. Similar effects of platelet-derived growth factor and epidermal growth factor on the phosphorylation of tyrosine in cellular proteins. Cell. 1982;31(1):263–273. doi: 10.1016/0092-8674(82)90426-3. [DOI] [PubMed] [Google Scholar]
- 28.Krieg J, Hunter T. Identification of the two major epidermal growth factor–induced tyrosine phosphorylation sites in the microvillar core protein ezrin. J Biol Chem. 1992;267(27):19258–19265. [PubMed] [Google Scholar]
- 29.Sperka T, Geissler KJ, Merkel U, Scholl I, Rubio I, Herrlich P, Morrison HL. Activation of Ras requires the ERM-dependent link of actin to the plasma membrane. PLoS One. 2011;6(11):e27511. doi: 10.1371/journal.pone.0027511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haag A, Gutierrez P, Buhler A, Walser M, Yang Q, Langouet M, Kradolfer D, Frohli E, Herrmann CJ, Hajnal A. An in vivo EGF receptor localization screen in C. elegans identifies the Ezrin homolog ERM-1 as a temporal regulator of signaling. PLoS Genet. 2014;10(5):e1004341. doi: 10.1371/journal.pgen.1004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chirivino D, Del Maestro L, Formstecher E, Hupe P, Raposo G, Louvard D, Arpin M. The ERM proteins interact with the HOPS complex to regulate the maturation of endosomes. Mol Biol Cell. 2011;22(3):375–385. doi: 10.1091/mbc.E10-09-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Celik H, Sajwan KP, Selvanathan SP, Marsh BJ, Pai AV, Saygideger Kont Y, Han J, Minas TZ, Rahim S, Erkizan HV. Ezrin binds to DEAD-box RNA helicase DDX3 and regulates its function and protein level. Mol Cell Biol. 2015;35(18):3145–3162. doi: 10.1128/MCB.00332-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 34.Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res. 2004;64(15):5355–5362. doi: 10.1158/0008-5472.CAN-04-0562. [DOI] [PubMed] [Google Scholar]
- 35.Bulut G, Hong SH, Chen K, Beauchamp EM, Rahim S, Kosturko GW, Glasgow E, Dakshanamurthy S, Lee HS, Daar I. Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene. 2012;31(3):269–281. doi: 10.1038/onc.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Briggs JW, Ren L, Nguyen R, Chakrabarti K, Cassavaugh J, Rahim S, Bulut G, Zhou M, Veenstra TD, Chen Q. The ezrin metastatic phenotype is associated with the initiation of protein translation. Neoplasia. 2012;14(4):297–310. doi: 10.1593/neo.11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu KL, Khan S, Lakhe-Reddy S, Jarad G, Mukherjee A, Obejero-Paz CA, Konieczkowski M, Sedor JR, Schelling JR. The NHE1 Na +/H + exchanger recruits ezrin/radixin/moesin proteins to regulate Akt-dependent cell survival. J Biol Chem. 2004;279(25):26280–26286. doi: 10.1074/jbc.M400814200. [DOI] [PubMed] [Google Scholar]
- 38.Agarwal E, Chaudhuri A, Leiphrakpam PD, Haferbier KL, Brattain MG, Chowdhury S. Akt inhibitor MK-2206 promotes anti-tumor activity and cell death by modulation of AIF and Ezrin in colorectal cancer. BMC Cancer. 2014;14:145–157. doi: 10.1186/1471-2407-14-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leiphrakpam PD, Rajput A, Mathiesen M, Agarwal E, Lazenby AJ, Are C, Brattain MG, Chowdhury S. Ezrin expression and cell survival regulation in colorectal cancer. Cell Signal. 2014;26(5):868–879. doi: 10.1016/j.cellsig.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol. 1998;140(3):647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crepaldi T, Gautreau A, Comoglio PM, Louvard D, Arpin M. Ezrin is an effector of hepatocyte growth factor-mediated migration and morphogenesis in epithelial cells. J Cell Biol. 1997;138(2):423–434. doi: 10.1083/jcb.138.2.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srivastava J, Elliott BE, Louvard D, Arpin M. Src-dependent ezrin phosphorylation in adhesion-mediated signaling. Mol Biol Cell. 2005;16(3):1481–1490. doi: 10.1091/mbc.E04-08-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gautreau A, Poullet P, Louvard D, Arpin M. Ezrin, a plasma membrane-microfilament linker, signals cell survival through the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96(13):7300–7305. doi: 10.1073/pnas.96.13.7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin T, Jin J, Li X, Zhang S, Choi YH, Piao Y, Shen X, Lin Z. Prognostic implications of ezrin and phosphorylated ezrin expression in non–small cell lung cancer. BMC Cancer. 2014;14:191–199. doi: 10.1186/1471-2407-14-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu AM, Huang PH. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010;70(10):3857–3860. doi: 10.1158/0008-5472.CAN-10-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 47.Stabile LP, Rothstein ME, Keohavong P, Lenzner D, Land SR, Gaither-Davis AL, Kim KJ, Kaminski N, Siegfried JM. Targeting of both the c-Met and EGFR pathways results in additive inhibition of lung tumorigenesis in transgenic mice. Cancers (Basel) 2010;2(4):2153–2170. doi: 10.3390/cancers2042153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu H, Stabile LP, Gubish CT, Gooding WE, Grandis JR, Siegfried JM. Dual blockade of EGFR and c-Met abrogates redundant signaling and proliferation in head and neck carcinoma cells. Clin Cancer Res. 2011;17(13):4425–4438. doi: 10.1158/1078-0432.CCR-10-3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y, Zhang M, Zhang X, Cai Q, Hong S, Jiang W, Xu C. Synergistic effects of combined platelet-activating factor receptor and epidermal growth factor receptor targeting in ovarian cancer cells. J Hematol Oncol. 2014;7:39–50. doi: 10.1186/1756-8722-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]