A focused library synthesis and cytotoxicity of quinones derived from the natural product bolinaquinone (original) (raw)
Abstract
Bolinaquinone is a natural product that is a structurally complex, cytotoxic sesquiterpene quinone. A scaffold simplification and focused library approach using a microwave-assisted Suzuki coupling gave 32 bolinaquinone analogues with good-to-excellent cytotoxicity profiles. Mono-arylbenzoquinones, Library A, were preferentially toxic towards BE2-C (neuroblastoma) cells with growth inhibition (GI50) values of 4–12 µM; only the 3,4-dimethoxyphenyl 23 and 3-biphenyl 28 variants were broad-spectrum active—HT29 (colon carcinoma), U87 and SJ-G2 (glioblastoma), MCF-7 (breast carcinoma), A2780 (ovarian carcinoma), H460 (lung carcinoma), A431 (skin carcinoma), Du145 (prostate carcinoma), BE2-C (neuroblastoma), MIA (pancreatic carcinoma) and SMA (spontaneous murine astrocytoma). Library B with a second aryl moiety exhibited broad-spectrum cytotoxicity with MCF-7 cells’ GI50 values of 5.6 ± 0.7 and 5.1 ± 0.5 µM for 2,5-dimethoxy-3-(naphthalene-1-yl)-6-(naphthalene-3-yl) 33 and 2,5-dimethoxy-3-(biaryl-2-yl)-6-(naphthalene-3-yl) 36, respectively. Similar potencies were also noted with 2,5-dimethoxy-3,6-diphenyl 30 against A2780 (GI50 = 5.9 ± 0.0 µM) and with 2,5-dimethoxy-3-(biaryl-3-yl)-6-(naphthalene-3-yl) 37 against HT29 (GI50 = 5.4 ± 0.4 µM), while the 3,4-dimethoxy mono-aryl analogue 23 exhibited good levels of activity against A2780 (GI50 = 3.8 ± 0.75 µM), the neuroblastoma cell line BE2-C (GI50 = 3 ± 0.35 µM) and SMA (GI50 = 3.9 ± 0.54 µM). Introduction of the amino-substituted Library C gave 2-(naphthalen-1-yl)-5-(naphthalen-3-yl)-3,6-bis(propylamino) 43, with excellent activity against HT29 (0.08 ± 0.0 µM), MCF-7 (0.17 ± 0.1 µM), A2780 (0.14 ± 0.1 µM), A431 (0.11 ± 0.0 µM), Du145 (0.16 ± 0.1 µM), BE2-C (0.08 ± 0.0 µM) and MIA (0.1 ± 0.0 µM).
Keywords: bolinaquinone, Suzuki coupling, microwave-assisted synthesis, cytotoxicity
1. Introduction
Cancer can be defined as a collection of diseases in which cells undergo aberrant division and at times invasive proliferation. Broadly speaking, there are over 100 cancer types spanning carcinoma, sarcoma, leukaemia, lymphoma and myeloma, and central nervous system cancers http://www.cancer.gov/cancertopics/cancerlibrary/what-is-cancer (accessed 25 May 2017). In Australia, cancer is the second most common cause of death after cardiovascular diseases with lifetime mortality risks of one in four for males and one in six for females http://www.cancer.org.au/about-cancer/what-is-cancer/facts-and-figures.html (accessed 25 May 2017). In 2014, there was an estimated 128 000 new Australian cancer diagnoses, a number predicted to reach 150 000 annually by 2020 [1].
Over the past decade, our group has explored two general approaches to the development of potential anti-cancer agents: (i) target-based approach and (ii) phenotypic screening that relies on cytotoxicity assays across a broad panel of cancer cell lines. Our target-derived explorations have led to the development of novel cytotoxic clathrin and dynamin GTPase [2–4], and protein phosphate 1 and 2A inhibitors [5–8]. Our phenotypic approach has identified the cytotoxic acrylonitriles [9,10], steroids [11], natural product-derived leads [12,13] and more recently inhibitors of the hedgehog signalling pathway [14,15].
Given that the phenotypic screening has seen resurgence in use over the past few years [16], we sought to expand our efforts in this area. Here our interest focused on the cytotoxic natural product bolinaquinone (1, figure 1). Bolinaquinone has been shown to inhibit the growth of HCT-116 colon carcinoma cells with a GI50 = 5.3 µM (50% growth inhibition) [17]. Cytotoxicity has also been reported for related marine sesquiterpene quinones such as avarone (2), mamanuthaquinone (3), epi-ilimaquinone (4), nakijiquinone A (5), dysidaminone E-5 (6) and 18-phenethylaminoavarone (7) (figure 1). These quinones also reveal a wide range of cytotoxicity against a diverse panel of cancer cell lines: 2: HeLa (GI50 = 26.8 ± 3.0 µM), L5178Y mouse lymphoma cells (ED50 = 0.62 ± 0.11 µM) [18,19]; 3: HCT-116 (GI50 = 5.58 µM) [20]; 4: P-388 leukaemia cells (GI50 = 6.14 µM) and different solid tumours: A-549 lung (GI50 = 2.51 µM), HT-29 colorectal (GI50 = 9.48 µM) and melanoma B16/F10 (GI50 = 3.1 µM) [21]; 5: leukaemia L-1210 and epidermoid KB cell lines with (GI50 = 9.46 and 18.92 µM, respectively) [22]; 6: HepG2 hepatoma cancer cell line (GI50 = 0.94 µM) and 7: NCI-H929 myeloma (GI50 = 1.76 µM) [23]. Quinones have been previously explored in clinical development [24,25] and are believed to elicit their cytotoxic action via a redox cycling mechanism. Alongside this mode of action, quinones are reported to be promiscuous, especially those analogues capable of both redox cycling (electron scavenging) and forming Michael adducts [26,27]. Despite the potential PAIN nature of quinones, it has been proposed that they be retained in phenotypic screening libraries, with a key argument being that the core motif is present in numerous clinically used drugs, e.g. the anthracyclines, mitomycin and mitoxantrone. Despite their mechanism of action remaining unknown, these agents are successfully used in the treatment of multiple cancers, pneumonia and multiple sclerosis. Quinone potentially provide leads, but these require careful examination for promiscuity, which arguably is the role of medicinal chemistry—to remove toxicity and enhance selectivity [28].
Figure 1.

The chemical structures of selected sesquiterpene quinones: bolinaquinone (1), avarone (2), mamanuthaquinone (3), epi-ilimaquinone (4), nakijiquinone A (5), dysidaminone E-5 (6) and 18-phenethylaminoavarone (7), and their reported cytotoxicity towards different cancer cell lines.
Synthetic routes to sesquiterpenes (1–7) are either complex and low yielding, or yet to be reported [29–32]. Thus, herein we explore a scaffold simplification approach in our efforts to develop the cytotoxicity structure–activity relationship of bolinaquinone analogues.
2. Procedures
2.1. Synthetic
We viewed our lead, bolinaquinone (1) as comprising three readily modifiable regions: the decalin or hydrophobic moiety, the linker and quinone moieties (figure 2). Based on this, three focused libraries were designed to explore the effect of the modification of each region. In our initial design considerations, given that there was no prior structure activity data in this area, we were keen to simplify access to bolinaquinone analogues. To this end, we envisaged analogues that retained the central quinone core with at least one methoxy moiety and a range of pendant aromatic moieties fulfilling the role of the hydrophobic decalin moiety of 1. The lack of a polar moiety associated with the pendant parent decalin of 1 and analogues 2–7 supported the introduction of simple aromatic moieties only. We were cognisant of the potentially detrimental effect on compound physico-chemical properties on the introduction of multiple lipophilic groups, but viewed that this may also improve cellular uptake. Thus Library A sought to explore the effect of decalin core replacement with less complex hydrophobic moieties such as simple aromatic rings (Type A analogues, figure 2). This route allowed the use of well-established Suzuki coupling approaches [33,34]. The introduction of a methoxy moiety offered the possibility of synthesis simplification and direct access to Library B through further scaffold arylation (Type B analogues, figure 2); in doing so, the hydroxyl moiety of 1 was modified to a methoxy moiety (Type A analogues, figure 2). Finally, in the development of dynamin inhibitors we had shown that the introduction of amino moieties increased inhibition and analogue solubility, and viewed that this approach would potentially enhance cytotoxicity and ameliorate the poor physico-chemical characteristics of the hydrophobic analogues planned in Libraries A and B. Thus amine displacement of the methoxy moieties afforded Library C (Type C analogues, figure 2).
Figure 2.

The proposed structural simplification of the lead, bolinaquinone 1, to afford three bolinaquinone libraries: A, B and C.
Library A synthesis commenced with the treatment of 2,5-dihydroxy-1,4-benzoquinone (8) in methanol in the presence of catalytic H2SO4 at room temperature. This afforded, after work-up, the desired 2,5-dimethoxy-1,4-benzoquinone (9) in an 86% yield [35]. Bromination of 9 through warming the reaction mixture in DMF at 60°C (then 25°C) with portion-wise addition of excess of _N_-bromosuccinimide (NBS) gave a readily separable mixture of 3-bromo-2,5-dimethoxy-1,4-benzoquinone (10) and 3,6-dibromo-2,5-dimethoxy-1,4-benzoquinone (11) in a 42% and 40% yield, respectively (scheme 1) [36,37].
Scheme 1.

Reagents and conditions: (i) MeOH, H2SO4, 25°C, 6 h; (ii) NBS, DMF, 60°C (then 25°C), 5–8 h; (iii) arylboronic acid (Ar-B(OH)2), Pd(dppf)Cl2, K2CO3, dioxane, µW, 120°C, 20 min, or arylboronic acid (R-B(OH)2), Pd(dppf)Cl2, K2CO3, toluene, reflux, 72 h.
By varying the bromination conditions, preferential access to the mono- or dibrominated products was possible. This simplified product isolation. For 3-bromo-2,5-dimethoxy-1,4-benzoquinone (10), the quinone was dissolved in DMF and heated to 100°C followed by the addition of 1 equivalent of NBS in a single portion. The dibrominated product, 3,6-dibromo-2,5-dimethoxy-1,4-benzoquinone (11), was accessed through the addition of 2 equivalents of NBS in DMF at 60°C. In both approaches, the desired product was isolated by flash chromatography. Subsequent application of a microwave-assisted Suzuki coupling approach with 10 gave facile access to Library A analogues 12–29 [33]. With the exception of sterically encumbered and aliphatic boronic acids, e.g. anthracene and cyclohexyl, microwave irradiation at 120°C for 20 min gave these analogues in 60–75% yield (scheme 1 and table 1). Within this library those reactions that displayed poor conversion to the desired analogue (14, 18, 19, 22 and 23) were also accessed by reflux approaches as extended microwave irradiation led to increasing levels of the undesired des-methoxy analogues. In general, these batch reactions were lower yielding when compared with the microwave conditions. Moreover, we noted that (in our hands) the Suzuki coupling failed to provide useful quantities of the target aryl-coupled quinones with the 2-hydroxy-, 3-hydroxy- and 4-methoxy- phenyl boronic acids. The 2-tolyl boronic acid analogue failed to react to completion and gave rise to a complex, inseparable mixture; as such, this analogue was not pursued further.
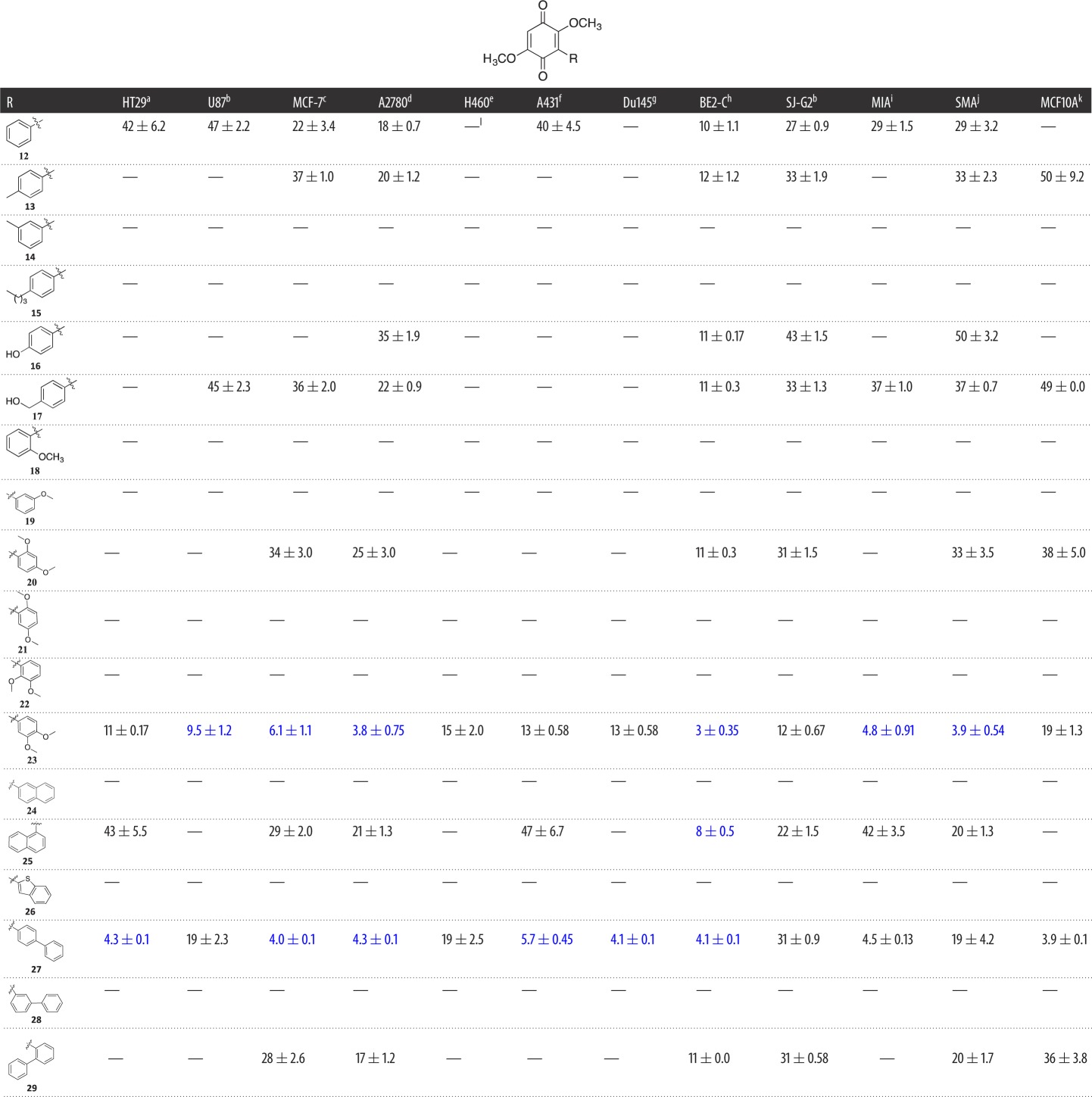
Table 1.
Evaluation of the cytotoxicity, GI50 values (µM) of 3-aryldimethoxybenzoquinone derivatives 12–29 (Library A), against a panel of 11 cancer cell lines and one normal cell line. GI50 is the concentration of drug that reduces cell growth by 50% relative to an untreated control.
 |
|---|
Library B leveraged our ability to simultaneously conduct a Suzuki coupling with either two equivalents of an arylboronic acid or sequentially with one equivalent of two different arylboronic acids (scheme 2). Symmetrical 3,6-diaryl-dimethoxybenzoquinones were synthesized on treatment of 11 with two equivalents of an arylboronic acid and microwave irradiation at 120°C for 20 min as previously described, which afforded 30 and 31. Asymmetric analogues 32–37 were accessed by treatment of 11 with one equivalent of arylboronic acid under microwave conditions, and once TLC analysis had confirmed consumption of the starting materials, with ensuing addition of an equivalent of a second arylboronic acid and microwave irradiation at 120°C for 20 min as previously described. Analogues 30–37 were obtained in 65–70% yields after flash chromatography (scheme 2 and table 2). In some instances these Suzuki couplings failed, e.g. with the 1-naphthyl, 2-naphthyl and 4-biphenyl boronic acids as one of the substituents, which was most probably a consequence of steric hindrance. The asymmetric analogues were purified only in the last step to omit time-consuming purification of intermediates.
Scheme 2.

Reagents and conditions: (i) 2 equivalents of arylboronic acid (R-B(OH)2), Pd(dppf)Cl2, K2CO3, dioxane, μW, 120°C, 20 min; (ii) 1 equivalent of arylboronic acid (R-B(OH)2), Pd(dppf)Cl2, K2CO3, dioxane, μW, 120°C, 20 min, then 1 equivalent of arylboronic acid (R'-B(OH)2), 120°C, 20 min.
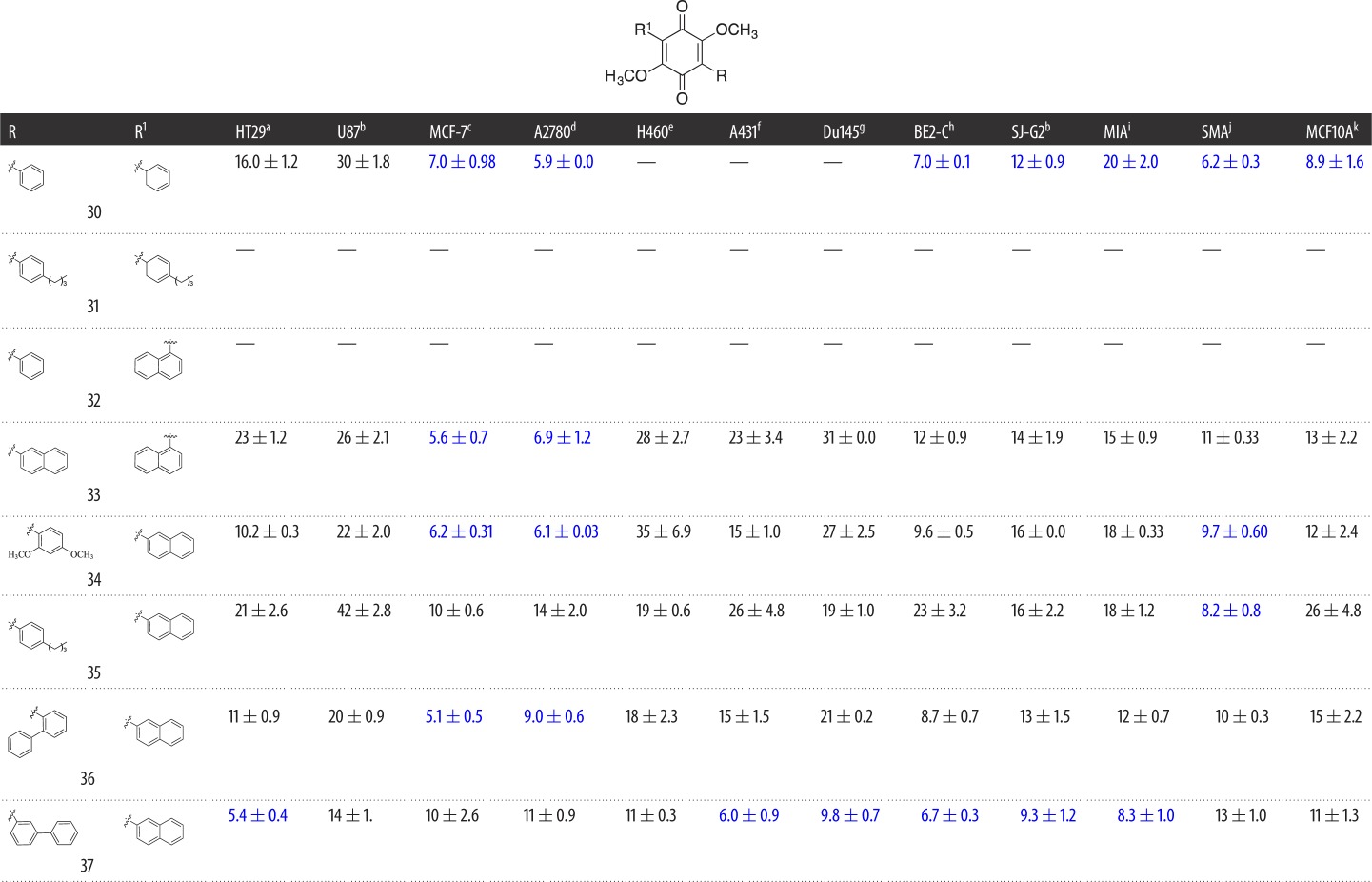
Table 2.
Evaluation of the cytotoxicity, GI50 values (μM) of 3-aryldimethoxybenzoquinone derivatives 30–37 (Library B), against a panel of 11 cancer cell lines and one normal cell line. GI50 is the concentration of drug that reduces cell growth by 50% relative to an untreated control.
 |
|---|
In the construction of Library C, a total of six analogues from Library A and Library B were treated with either _n_-propyl amine or _N,N_-dimethylpropane-1,3-diamine in methanol at room temperature to afford excellent yields (85%–95%) of the diaminoquinones (38–43) (scheme 3 and table 3).
Scheme 3.

Reagent and conditions: (i) _n_-propyl amine or _N,N_-dimethylpropane-1,3-diamine, MeOH, 25°C, 1 h.
Table 3.
Evaluation of the cytotoxicity, GI50 values (μM) of 3-aryldimethoxybenzoquinone derivatives 38–43 (Library C), against a panel of 11 cancer cell lines and one normal cell line. GI50 is the concentration of drug that reduces cell growth by 50% relative to an untreated control.
2.2. Cytotoxicity
With the desired bolinaquinone analogues 12–43 in hand, we examined their cytotoxicity against a panel of 11 tumour cell lines: HT29 (colon carcinoma), U87 and SJ-G2 (glioblastoma), MCF-7 (breast carcinoma), A2780 (ovarian carcinoma), H460 (lung carcinoma), A431 (skin carcinoma), Du145 (prostate carcinoma), BE2-C (neuroblastoma), MIA (pancreatic carcinoma), SMA (spontaneous murine astrocytoma) and a normal breast cell line (MCF10A). The inclusion of the MCF-10A cell line allowed a determination of a crude measure of relative toxicity while noting that the ultimate arbiter of toxicity requires in vivo evaluation [38,39]. All analogues were initially screened at a 25 µM drug concentration [40,41]. Those analogues returning 80–100% growth inhibition across all cell lines, or those displaying cell line specificity were subjected to full dose response, GI50, evaluation. These data are given in tables 1–3.
3. Results and discussion
Our investigation of the bolinaquinone SAR commenced with an application of a scaffold simplification and focused library approach that saw the synthesis of mono-arylated dimethoxybenzoquinones (Library A). Of this initial library, analogues 14, 15, 18, 19, 21, 22, 24, 26 and 28 were deemed not sufficiently active at the initial screening concentration of 25 µM to proceed to GI50 determination (table 1; electronic supplementary material). Of the other Library A members, phenyl 12 and 4-toluoyl 13 were essentially equipotent across the cell line panel with GI50 values ranging from 10 ± 1.1 µM, (12, BE2-C) to greater than 50 µM (12, H460; and 13, HT29, U87, H460, A431 and MIA). However, 13 showed no activity (defined here as a GI50 value greater than 50 µM) against HT29, U87, A431 and MIA cell lines, and was 50 µM potent against the normal cell line, MCF10A. The 3-toluoyl 14 analogue was inactive as was 4-butyl 15, which suggested that the position and nature of the simple aliphatic substituent affected the potency of these analogues with a 3-alkyl moiety and a larger 4-alkyl moiety not tolerated. Polar substituents on the introduced phenyl moiety such as 4-OH 16, 4-CH2OH 17 and 2,4-di-OCH3 20 showed moderate activity against BE2-C with all GI50 values approximately 11 µM, and were equipotent with 12 and 13 with similar activity profiles across the cell lines examined. The mono-OCH3 analogues 18 and 19 were inactive. Of the di-OCH3 analogues 20–23 examined, only 3,4-di-OCH3 20 was active. Notably, of analogues 20–23, analogue 20 was the only active 2-substituted di-OCH3 analogue, albeit modestly (except against BE2-C cells), which suggests that a 2-substituent may be detrimental to activity. The most active of the compounds with single aromatic moieties on the quinone scaffold was found to be 3,4-di-OMe 23, which was also the first broad-spectrum cytotoxic analogue in this series displaying GI50 values of 3–15 µM across the cancer cell lines examined.
Within Library A the introduction of a larger aromatic moiety resulted in a modest increase in cytotoxicity with 1-naphthyl 25 showing preferential activity towards the BE2-C cell line, with a GI50 value of 8 ± 0.5 µM; however, 2-naphthyl 24 was inactive. In a similar manner, the benzothiophene 26 and 3-biphenyl 28 analogues did not proceed to GI50 determination. However, the introduction of a 2- and especially a 4-biphenyl moiety with 29 and 27, respectively, resulted in increased cytotoxicity with the latter returning broad-spectrum activity from 3.9 to 19 µM (table 1), but also a significant increase in toxicity towards the MCF10A cell line with 27 returning a GI50 of 3.9 µM. This in turn suggested that the orientation of the aromatic or hydrophobic moiety relative to the core quinone moiety is critical to the retention of cytotoxicity. The two most active compounds in this library, 4-biphenyl 27 and 3,4-di-OMe 23, both contain 4-disposed moieties. The introduction of a 2-substituent results in the aromatic moieties twisting out of plane relative to the quinone moiety, e.g. 18 and 20–22, which has an adverse impact on the observed cytotoxicity (not shown).
Library A demonstrated that the introduction of large aromatic moieties resulted in good levels of broad-spectrum cytotoxicity with 27; as such, with Library B we sought to explore the effect of increasing the aromatic content of these analogues on compound cytotoxicity. Interestingly, the introduction of a second aryl moiety with Library B gave rise to a different activity profile to that observed with Library A. The parent diphenyl (30) analogue showed moderate-to-good broad-spectrum cytotoxicity, e.g. A2780 GI50 = 5.9 ± 0.0 µM and SMA GI50 = 6.2 ± 0.3 µM, except with the H460, A431 and Du145 cell lines (table 2; GI50 > 50 µM). The introduction of an alkyl chain with 31 was detrimental to activity (table 2). However, this did not appear to be a consequence of a steric clash as the naphthyl substituted 32–37, even in the presence of the 2- and 3-biphenyl moieties, returned good-to-excellent cytotoxicity across the cell lines examined. In these instances, the GI50 values ranged from 5.1 ± 0.5 µM (36; MCF-7) to 35 ± 6.9 µM (34; H460). The bis-naphthyl (33) saw an overall reduction in cytotoxicity, except with the ovarian and breast cell lines MCF-7 and A2780, with GI50 values of 5.6 ± 0.7 and 6.9 ± 1.2 µM, respectively. The replacement of the decalin moiety of 1 with a phenyl moiety (12) resulted in a decrease in cLogP (5.10 to 1.85) and polar surface area (PSA, 48.97 to 41.65 Å2); a 4-biphenyl (27) moiety resulted in cLogP of 3.65 and a PSA of 41.90 Å2 (see the electronic supplementary material). The introduction of a second aromatic moiety was highly detrimental to cLogP with 37 displaying a cLogP of 6.71. However, as we suspected from our earlier dynamin studies, the introduction of amino moieties improved the cLogP values of the resultant analogues such as 41 (cLogP 5.21), but the presence of the amino moieties failed to overcome the effect of two naphthyl moieties with 43 (cLogP 7.91). Despite this significant deterioration in cLogP, we noted no effect of compound precipitation during our cytotoxicity studies, with all analogues soluble in DMSO at 40 mM and maintaining solubility in the MTT assay.
Having established the effects of aryl substitutions on the cytotoxicity of modified bolinaquinone analogues, we next examined the displacement of the –OCH3 moieties with simple amines to generate Library C. Of the six Library C analogues, only 41 and 43 returned sufficient activity at the initial 25 µM screening dose to proceed to GI50 determination (table 3). However, both 41 and 43 displayed considerable increase in cytotoxicity, returning the highest level of activity of any of the analogues developed thus far with average GI50 values of 2.5 µM and 0.69 µM, respectively. Comparison of 41 and 43 with the parent analogues 29 (table 1) and 33 (table 2), respectively, highlights a greater than or equal to 20-fold potency enhancement on the introduction of the amine substituents, potentially suggesting the presence of additional binding domains that these substituents are capable of accessing. Of 41 and 43, the tri-substituted quinone moiety of 41 is capable of both DNA adduction and redox cycling, whereas the tetra-substituted quinone moiety of 43 is not.
While the exact cytotoxic mechanism was not determined, these data support 43 mediating its effects through a redox-cycling mechanism and validate our phenotypic approach to the identification of cytotoxic compounds. Within the cell lines examined, 41 displayed no obvious cell line preference. The bis-naphthylamino-substituted 43 analogue returned nanomolar potency against HT29 (colon), MCF-7 (breast), A2780 (ovarian), H460 (lung), A431 (skin), Du145 (prostate) and MIA (pancreatic). However, while 43 returned a GI50 value of 0.07 ± 0 µM against the normal breast cell line MCF-10A, it is critical to note that an in vitro assay is a poor determinant of human toxicity, noting that compound toxicity can only really be measured in vivo [38]. It is accepted that animal models are required to predict potential clinical toxicity.
4. Conclusion
The natural product bolinaquinone was reported as moderately cytotoxic (HCT-116, GI50 = 5.3 µM). The lead was deemed to be structurally too complex to facilitate rapid focused library development while retaining the parent hydrophobic core. However, application of a scaffold simplification approach focusing on the hydrophobic core combined with a microwave-assisted Suzuki coupling (and, in some cases, reflux conditions) permitted the synthesis of 32 modified bolinaquinone analogues. These novel analogues spanned three focused libraries. Simple mono-arylbenzoquinones displayed preferential toxicity towards the BE2-C neuroblastoma cell line with GI50 values of 3–12 µM, apart from the 3,4-dimethoxy (23) and 4-biphenyl (27), which returned broad-spectrum cytotoxicity with an average GI50 of 10.2 µM. The introduction of a second aryl moiety with Library B failed to enhance BE2-C specificity, but resulted in enhanced broad-spectrum cytotoxic activity in general. Despite the transition towards broad-spectrum cytotoxicity, good levels of activity were apparent against the MCF-7 breast cancer cell lines with GI50 values of 5.6 ± 0.7 and 5.1 ± 0.5 for 33 and 36, respectively. Similar potencies were also noted with 30 against the ovarian cell line A2780 (GI50 = 5.9 ± 0.0 µM), with 37 against the colon cell line HT29 (GI50 = 5.4 ± 0.4 µM), and with 23 against the ovarian cell line A2780 (GI50 = 3.8 ± 0.75 µM), the neuroblastoma cell line BE2-C (GI50 = 3 ± 0.35 µM) and the glioblastoma cell line SMA (GI50 = 3.9 ± 0.54 µM).
Introduction of the amino substituents with Library C resulted in the development of the most potent broad-spectrum analogues with 41 and 43 yielding average GI50 values of 2.3 and 0.69 µM, respectively. As previously noted herein, enhanced broad-spectrum activity resulted from the introduction of a second aryl moiety, as is the case with 41 and 43. In addition, 43 displayed excellent activity against HT29 (0.08 ± 0.0 µM), MCF-7 (0.17 ± 0.1 µM), A2780 (0.14 ± 0.1 µM), A431 (0.11 ± 0.0 µM), Du145 (0.16 ± 0.1 µM), BE2-C (0.08 ± 0.0 µM) and MIA (0.1 ± 0.0 µM). Of the analogues reported herein, the highest levels of cytotoxicity were observed in those with bulky aromatic moieties such as a biphenyl and naphthyl, especially in the presence of additional propyl amine moieties. These modified bolinaquinone analogues are promising leads in the search for new cytotoxic agents and we will report on further developments in due course.
5. Experimental
5.1. Biology
5.1.1. Cell culture and stock solutions
Stock solutions were prepared as follows and stored at −20°C: drugs were stored as 40 mM solutions in DMSO. All cell lines were cultured in a humidified atmosphere 5% CO2 at 37°C. The cancer cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) (Trace Biosciences, Australia) supplemented with 10% fetal bovine serum, 10 mM sodium bicarbonate, penicillin (100 IU ml−1), streptomycin (100 µg ml−1) and glutamine (4 mM). The non-cancer MCF10A cell line was cultured in DMEM:F12 (1 : 1) cell culture media, 5% heat-inactivated horse serum, supplemented with penicillin (50 IU ml−1), streptomycin (50 µg ml−1), 20 mM Hepes, L-glutamine (2 mM), epidermal growth factor (20 ng ml−1), hydrocortisone (500 ng ml−1), cholera toxin (100 ng ml−1) and insulin (10 ug ml−1).
5.1.2. In vitro growth inhibition assay
Cells in logarithmic growth were transferred to 96-well plates. Cytotoxicity was determined by plating cells in duplicate in 100 µl medium at a density of 2500–4000 cells per well. On day 0 (24 h after plating), when the cells were in logarithmic growth, 100 μl of medium with or without the test agent was added to each well. After 72 h, drug exposure growth inhibitory effects were evaluated using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide) assay and the absorbance read at 540 nm. Percentage growth inhibition was determined at a fixed drug concentration of 25 µM. A value of 100% is indicative of complete cell growth inhibition. Those analogues showing appreciable percentage growth inhibition underwent further dose–response analysis allowing for the calculation of a GI50 value. This value is the drug concentration at which cell growth is 50% inhibited based on the difference between the optical density values on day 0 and those at the end of drug exposure [40,41].
5.2. Chemistry
5.2.1. General methods
All reactions were performed using standard laboratory equipment and glassware. Solvents and reagents were purchased from Sigma Aldrich, Lancaster International or TCI and used as received. Organic solvents were of bulk quality, and were distilled from glass prior to use. Organic solvent extracts were dried with magnesium sulfate (MgSO4), and dried under reduced pressure with either Büchi or Heidolph rotary evaporators. Melting points were recorded in open capillaries on a Stuart SMP11 melting point apparatus. Where available, literature values are provided and appropriately referenced. Electrospray mass spectra were recorded using 10% DMSO/H2O or HPLC-grade methanol or acetonitrile as carrier solvents on a Shimadzu LC-MS spectrometer, or an Agilent Technologies 1260 Infinity UPLC system with a 6120 Quadrupole LC/MS in electrospray ionization (ESI) positive and negative modes. TLC was performed on Merck silica gel 60 F254 pre-coated aluminium plates with a thickness of 0.2 mm. Column chromatography was performed under ‘flash’ conditions on Merck silica gel 60 (230–400 mesh).
Nuclear magnetic resonance (NMR) spectroscopy was performed on a Brüker Avance III 400 MHz spectrometer, where proton NMR (1H NMR) spectra and carbon NMR (13C NMR) spectra were acquired at 400 and 100 MHz, respectively. All spectra were recorded in deuterated dimethyl sulfoxide (DMSO-d6), deuterated acetone (acetone-d6) or deuterated chloroform (CDCl3) obtained from Sigma Aldrich or Cambridge Isotope Laboratories Inc. Chemical shifts (δ) were measured in parts per million (ppm) and referenced against the internal reference peaks. Coupling constants (J) were measured in hertz (Hz). NMR assignments were determined through the interpretation of one- and two-dimensional spectra. Multiplicities are denoted as singlet (s), broad singlet (s, br), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), triplet of doublets (td), doublet of triplets (dt) and multiplet (m). Peaks are listed in decreasing chemical shift in the following format: chemical shift (integration (1H), multiplicity (1H), coupling constant (1H). The Biotage® initiator+ was used to perform Suzuki coupling (Technical information available on: http://www.biotage.com).
Synthesis of 2,5-dimethoxycyclohexa-2,5-diene-1,4-dione (9).
To a solution of 2,5-dihydroxy-1,4-benzoquinone 8 (2.80 g, 20.0 mmol) in MeOH (150 ml), H2SO4 (98%, 4 ml) was added dropwise, and the resultant mixture allowed to stir at room temperature for 6 h. The precipitate was collected, washed with MeOH : water (9 : 1, 2 × 20 ml) and dried in vacuo. The title compound was obtained as a yellow solid with identical spectral data to the previous report [29]. Yield 2.89 g (86%); m.p. 245–247°C (Lit. 240–242°C) [31].
1H NMR (DMSO-d6) δ 6.03 (s, 2H), 3.78 (s, 6H); 13C NMR (DMSO-d6) δ 181.9 (2C), 159.7 (2C), 106.0 (2C), 57.5 (2C); IR _υ_max/cm−1: 1652 (C=O), 1579 (C=C), 1328 (C–O); LRMS (ESI+) m/z: 168 (M+).
Synthesis of 3-bromo-2,5-dimethoxycyclohexa-2,5-diene-1,4-dione (10) and 3,6-dibromo-2,5-dimethoxycyclohexa-2,5-diene-1,4-dione (11).
A suspension of 9 (0.34 g, 2.02 mmol) in DMF (15 ml) was stirred for 10 min at 60°C and then NBS (0.79 g, 4.50 mmol) was added quickly in one portion to the solution. The reaction mixture was cooled to room temperature (25°C) and stirred for 8 h. Next, water (50 ml) was added and the mixture was extracted with EtOAc (2 × 30 ml), dried over MgSO4 and the solvent evaporated in vacuo. The crude product was purified by flash chromatography (5% EtOAc—95% hexanes). The title compounds were obtained as orange and red crystalline solids, respectively, with identical spectral data to the previous report [31,32].
(10) Yield 0.21 g (42%); m.p. 135–137°C (Lit. 136–138°C) [31]. 1H NMR (DMSO-d6) δ 5.99 (s, 1H), 4.21 (s, 3H), 3.89 (s, 3H); 13C NMR (DMSO-d6) δ 180.8, 175.0, 158.8, 157.5, 114.5, 105.5, 61.3, 56.6; IR _υ_max/cm−1: 1647 (C=O), 1557 (C=C), 1331 (C–O); LRMS (ESI+) m/z: 247 (M+).
(11) Yield 0.26 g (40%); m.p. 166–168°C (Lit. 168–170°C) [31]. 1H NMR (DMSO-d6) δ 4.10 (s, 6H); 13C NMR (DMSO-d6) δ 174.9 (2C), 157.1 (2C), 115.4 (2C), 62.2 (2C); IR _υ_max/cm−1: 1651 (C=O), 1558 (C=C), 1330 (C–O); LRMS (ESI+) m/z: 325 (M+).
General procedure for synthesis of 2,5-dimethoxy-3-aryl-2,5-diene-1,4-diones by Suzuki coupling (1).
(A) A Biotage microwave vial was charged with 10 (0.25 g, 1.01 mmol), requisite boronic acid (1.20 mmol), K2CO3 (0.34 g, 2.50 mmol) and Pd(dppf)Cl2 (0.07 g, 0.01 mmol), and then dioxane (5 ml) was added to this mixture. The reaction vessel was sealed and microwave-irradiated at 120°C for 20 min (holding time) with magnetic bar stirrer. After cooling with compressed air to 40°C, the reaction mixture was diluted with dioxane (10 ml) and filtered through the Celite®. The reaction crude was dried in vacuo and subjected to column chromatography for purification (15% EtOAc–85% hexanes) to afford an orange-red solid.
(B) Where none of the desired product or large amounts of starting material was found after microwave irradiation, the reactions were investigated under reflux conditions. A mixture of 10, the requisite boronic acid (1.20 equiv), K2CO3 (2.5 equiv), Pd(dppf)Cl2 (0.2 equiv) and toluene (15 ml) was heated at reflux and monitored by TLC and LCMS until the starting material had been consumed. When the reaction was deemed complete, the reaction mixture was absorbed onto silica and purified by automated column chromatography (0–100% EtOAc in hexanes).
2,5-Dimethoxy-3-phenylcyclohexa-2,5-diene-1,4-dione (12).
Synthesized according to the general procedure 1A from phenyl boronic acid (0.14 g, 1.20 mmol). The title compound was obtained as a yellow solid with identical spectral data to the previous report [30]. Yield 0.18 g (73%); m.p. 122–124°C (Lit. 124–126°C) [31].
1H NMR (acetone-_d_6) δ 7.43–7.36 (m, 3), 7.31–7.28 (m, 2H), 6.01 (s, 1H), 3.88 (s, 3H), 3.86 (s, 3H); 13C NMR (acetone-_d_6) δ 183.2, 181.2, 158.9, 155.3, 131.0, 130.5, 128.5, 128.0, 127.5, 125.9, 124.6, 105.7, 60.8, 56.1; IR _υ_max/cm−1: 1648 (C=O), 1587 (C=C), 1332 (C–O); LRMS (ESI+) m/z: 245 (M+H), HRMS (ESI+) m/z calculated for C14H13O4 (M+H) 245.0813; found 245.0810.
2,5-Dimethoxy-3-p-tolylcyclohexa-2,5-diene-1,4-dione (13).
Synthesized according to the general procedure 1A from p-tolylboronic acid (0.16 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.19 g (74%); m.p. 121–123°C.
1H NMR (acetone-d6) δ 7.23 (d, J = 7.7 Hz, 2H), 7.18 (dd, J = 7.72, 2.2 Hz, 2H), 6.00 (s, 1H), 3.88 (s, 3H), 3.85 (s, 3H), 2.36 (s, 3H); 13C NMR (acetone-d6) δ 184.1, 182.2, 159.8, 156.1, 138.7, 131.3 (2C), 129.1 (2C), 128.8, 128.5, 106.5, 61.7, 57.0, 21.3; IR _υ_max/cm−1: 1643 (C=O), 1593 (C=C), 1329 (C–O); LRMS (ESI+) m/z: 259 (M+H), HRMS (ESI+) m/z calculated for C15H15O4 (M+H) 259.0970; found 259.0967.
2,5-Dimethoxy-3-m-tolylcyclohexa-2,5-diene-1,4-dione (14).
Synthesized according to the general procedure 1B from 10 (0.17 g, 0.67 mmol) and m-tolylboronic acid (0.11 g, 0.80 mmol) under reflux for 72 h. The title compound was obtained as a yellow/orange solid. Yield 0.052 g (30%); m.p. 74–77°C.
1H NMR (400 MHz, CDCl3) δ 7.30 (t, J = 7.9 Hz, 1H), 7.19 (d, J = 7.5 Hz, 1H), 7.06 (d, J = 7.2 Hz, 2H), 5.89 (s, 1H), 3.85 (s, 3H), 3.78 (s, 3H), 2.38 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 183.6, 181.9, 159.0, 155.4, 137.7, 131.3, 130.2, 129.6, 128.0, 127.7, 126.6, 105.7, 61.7, 56.7, 21.6; IR _υ_max/cm−1: 2852, 1664, 1634, 1581, 1443, 780; LRMS (ESI+): m/z: 259 (C15H15O4) [M + H].
3-(4-Butylphenyl)-2,5-dimethoxycyclohexa-2,5-diene-1,4-dione (15).
Synthesized according to the general procedure 1A from (4-butylphenyl)boronic acid (0.21 g, 1.20 mmol). The title compound was obtained as a yellow solid. Yield 0.26 g (71%); m.p. 130–132°C.
1H NMR (acetone-d6) δ 7.24 (dd, J = 7.72, 2.2 Hz, 2H), 7.20 (dd, J = 7.72, 2.2 Hz, 2H), 6.00 (s, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 2.65 (t, J = 7.6 Hz, 2H), 1.67–1.58 (m, 2H), 1.43–1.34 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H); 13C NMR (acetone-d6) δ 184.2, 182.2, 159.8, 156.1, 143.7, 131.4, 129.0, 128.52 (2C), 128.46 (2C), 106.5, 61.7, 57.0, 36.0, 34.4, 23.1, 14.2; IR _υ_max/cm−1: 1644 (C=O), 1591 (C=C), 1332 (C–O); LRMS (ESI+) m/z: 301 (M+H), HRMS (ESI+) m/z calculated for C18H21O4 (M+H) 300.1440; found 301.1428.
3-(4-Hydroxyphenyl)-2,5-dimethoxycyclohexa-2,5-diene-1,4-dione (16).
Synthesized according to the general procedure 1A from 4-hydroxyphenylboronic acid (0.16 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.17 g (68%); m.p. 165–167°C.
1H NMR (acetone-d6) 1H NMR (400 MHz, acetone-d6) δ 7.16 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.8 Hz, 12H), 5.97 (s, 1H), 3.87 (s, 3H), 3.83 (s, 3H); 13C NMR (acetone-d6) δ 184.2, 182.4, 159.8, 158.4, 155.8, 132.9 (2C), 128.6, 122.5 (2C), 115.4, 106.5, 61.5, 56.9; IR _υ_max/cm−1: 3330 (O–H), 1631 (C=O), 1593 (C=C), 1300 (C–O); LRMS (ESI+) m/z: 261 (M+H), HRMS (ESI+) m/z calculated for C14H13O5 (M+H) 261.0762; found 261.0758.
3-(4-(Hydroxymethyl)phenyl)-2,5-dimethoxycyclohexa-2,5-diene-1,4-diene (17).
Synthesized according to the general procedure 1A from 4-hydroxymethylphenylboronic acid (0.18 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.15 g (62%); m.p. 116–118°C.
1H NMR (acetone-d6) δ 7.39 (d, J = 8.2 Hz, 2H), 7.25 (d, J = 8.2 Hz, 2H), 6.00 (s, 1H), 4.66 (s, 2H), 4.25–4.19 (s, br, OH) 3.86 (s, 3H), 3.85 (s, 3H); 13C NMR (acetone-d6): δ 184.1, 182.2, 159.8, 156.2, 143.5, 131.2 (2C), 130.3, 128.5, 126.6 (2C), 106.6, 64.5, 61.7, 57.0; IR (KBr) _υ_max/cm−1: 3249 (OH), 1644 (C=O), 1590 (C=C), 1332 (C–O); LRMS (ESI+) m/z: 275 (M+H), HRMS (ESI+) m/z calculated for C15H15O5 (M+H) 275.0919; found 275.0911.
3-(2-Methoxyphenyl)-2,5-dimethoxycyclohexa-2,5-diene-1,4-diene (18).
Synthesized according to the general procedure 1B from 10 (0.15 g, 0.58 mmol) and 2-methoxyphenylboronic acid (0.11 g, 0.70 mmol) under reflux for 72 h. The title compound was obtained as a yellow/orange solid. Yield 0.053 g (33%); m.p. 162–164°C.
1H NMR (400 MHz, CDCl3) δ 7.41–7.35 (m, 1H), 7.10 (dd, J = 7.5, 1.7 Hz, 1H), 6.98 (ddd, J = 14.5, 10.5, 4.6 Hz, 2H), 5.89 (s, 1H), 3.84 (s, 3H), 3.78 (s, 3H), 3.70 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 183.4, 181.2, 159.3, 157.5, 155.8, 131.8, 130.5, 122.7, 120.4, 120.1, 111.1, 105.6, 60.9, 56.6, 55.8; IR _υ_max/cm−1: 3004, 2952, 2853, 1656, 1589, 1492, 1437, 1016; LRMS (ESI+) m/z: 275 (M+H), HRMS (ESI+) m/z calculated for C15H15O5 (M+H) 275.0919; found 275.0931.
3-(3-Methoxyphenyl)-2,5-dimethoxycyclohexa-2,5-diene-1,4-diene (19).
Synthesized according to the general procedure 1B from 10 (0.15 g, 0.62 mmol) and 3-methoxyphenylboronic acid (0.11 g, 0.75 mmol) under reflux for 72 h. The title compound was obtained as an orange crystalline solid. Yield 0.088 g (48%); m.p. 72–75°C.
1H NMR (400 MHz, CDCl3) δ 7.32 (t, J = 8.0 Hz, 1H), 6.95–6.91 (m, 1H), 6.85 (d, J = 7.6 Hz, 1H), 6.83–6.80 (m, 1H), 5.90 (s, 1H), 3.85 (s, 3H), 3.81 (s, 3H), 3.80 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 183.4, 181.7, 159.2, 159.0, 155.4, 131.5, 129.1, 126.1, 123.2, 116.3, 114.5, 105.7, 61.7, 56.7, 55.4; IR _υ_max/cm−1: 3061, 2943, 2847, 1645, 1591, 1483, 1433, 1213, 970; LRMS (ESI+) m/z: 275 (M+H), HRMS (ESI+) m/z calculated for C15H15O5 (M+H) 275.0919; found 275.0925.
2,5-Dimethoxy-3-(2,4-dimethoxyphenyl)cyclohexa-2,5-diene-1,4-dione (20).
Synthesized according to the general procedure 1A from 2,4-dimethoxyphenylboronic acid (0.22 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.22 g (75%); m.p 190–192°C.
1H NMR (acetone-d6) δ 7.01 (d, J = 8.4 Hz, 1H), 6.61 (d, J = 2.4 Hz, 1H), 6.56 (dd, J = 8.4, 2.4 Hz, 1H), 5.97(s, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 3.75 (s, 6H); 13C NMR (acetone-d6) δ 183.9, 181.7, 162.5, 160.1, 159.4, 156.5, 132.9, 125.3, 113.6, 106.4, 105.4, 99.1, 60.9, 56.9, 55.9, 55.7; IR _υ_max/cm−1: 1649 (C=O), 1575 (C=C), 1329 (C–O); LRMS (ESI+) m/z: 305 (M+H), HRMS (ESI+) m/z calculated for C16H17O6 (M+H) 305.1024; found 305.1020.
2,5-Dimethoxy-3-(2,5-dimethoxyphenyl)cyclohexa-2,5-diene-1,4-dione (21).
Synthesized according to the general procedure 1A from 2,5-dimethoxyphenylboronic acid (0.22 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.22 g (74%); m.p. 180–182°C.
1H NMR (acetone-d6) δ 6.98 (d, J = 8.6 Hz, 1H), 6.93 (dd, J = 8.6, 2.8 Hz, 1H), 6.73 (d, J = 2.8 Hz, 1H), 5.99 (s, 1H), 3.87 (s, 3H), 3.78 (s, 3H), 3.75 (s, 3H), 3.71 (s, 3H); 13C NMR (acetone-_d_6) δ 183.9, 181.7, 162.5, 160.1, 159.4, 156.5, 132.9, 125.3, 113.6, 106.4, 105.4, 99.1, 60.9, 56.9, 55.9, 55.7; IR _υ_max/cm−1: 1644 (C=O), 1587 (C=C), 1326 (C–O); LRMS (ESI+) m/z: 305 (M+H), HRMS (ESI+) m/z calculated for C16H17O6 (M+H) 305.1024; found 305.1020.
2,5-Dimethoxy-3-(2,3-dimethoxyphenyl)cyclohexa-2,5-diene-1,4-dione (22).
Synthesized according to the general procedure 1B from 10 (0.16 g, 0.64 mmol) and 2,3-dimethoxyphenylboronic acid (0.14 g, 0.76 mmol) under reflux for 72 h. The title compound was obtained as a yellow/orange solid. Yield 0.050 g (26%); m.p. 122–125°C.
1H NMR (400 MHz, CDCl3) δ 7.07 (t, J = 7.9 Hz, 1H), 6.97 (dd, J = 8.2, 1.4 Hz, 1H), 6.69 (dd, J = 7.6, 1.5 Hz, 1H), 5.90 (s, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.77 (s, 3H), 3.71 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 183.2, 181.6, 159.2, 155.8, 152.7, 147.3, 125.2, 123.7, 123.0, 122.7, 113.3, 105.7, 61.0, 60.9, 56.7, 55.8; IR _υ_max/cm−1: 2852, 1664, 1634, 1581, 1443, 780; LRMS (ESI+) m/z: 305 (M + H), HRMS (ESI+) m/z calculated for C16H17O6 (M+H) 305.1024; found 305.1030.
2,5-Dimethoxy-3-(3,4-dimethoxyphenyl)cyclohexa-2,5-diene-1,4-dione (23).
Synthesized according to the general procedure 1B from 10 (0.16 g, 0.65 mmol) and 3,4-dimethoxyphenylboronic acid (0.14 g, 0.78 mmol) under reflux for 72 h. The title compound was obtained as a yellow/orange solid. Yield 0.042 g (22%); m.p. 135–137°C.
1H NMR (400 MHz, CDCl3) δ 6.93–6.87 (m, 2H), 6.83 (d, J = 1.7 Hz, 1H), 5.89 (s, 1H), 3.91 (s, 3H), 3.86 (d, J = 4.9 Hz, 6H), 3.80 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 183.5, 182.1, 158.9, 155.3, 149.5, 148.5, 126.3, 123.7, 122.4, 114.0, 110.7, 105.7, 61.5, 56.7, 56.1, 56.0; IR _υ_max/cm−1: 2837, 1671, 1633, 1592, 1434, 796; LRMS (ESI+) m/z: 305 (M + H), HRMS (ESI+) m/z calculated for C16H17O6 (M + H) 305.1024; found 305.1011.
2,5-Dimethoxy-3-(naphthalene-3-yl)cycloheza-2,5-diene-1,4-dione (24).
Synthesized according to the general procedure 1A from 2-naphthylboronic acid (0.20 g, 1.20 mmol). The title compound was obtained as a yellow solid. Yield 0.20 g (70%). m.p. 137–139°C.
1H NMR (acetone-d6) δ 7.94–7.90 (m, 3H), 7.84 (s, 1H), 7.57–7.50 (m, 2H), 7.42 (dd, J = 8.2, 1.6 Hz, 1H), 6.05 (s, 1H), 3.90 (s, 3H), 3.88 (s, 3H); 13C NMR (acetone-d6) δ 184.1, 182.1, 159.8, 156.4, 133.9, 133.8, 130.9, 129.5, 129.2, 129.0, 128.5, 128.2, 127.8, 127.3, 127.0, 106.6, 61.8, 57.0; IR _υ_max/cm−1: 1644 (C=O), 1583 (C=C), 1293 (C–O); MS (ESI+) m/z: 295 (M+H), HRMS (ESI+) m/z calculated for C18H15O4 (M+H) 295.0970; found 295.0965.
2,5-dimethoxy-3-(naphthalene-1-yl)cyclohexa-2,5-diene-1,4-dione (25).
Synthesized according to the general procedure 1A from 1-naphthylboronic acid (0.20 g, 1.20 mmol). The title compound was obtained as a yellow solid. Yield 0.19 g (65%); m.p. 134–137°C.
1H NMR (acetone-d6) δ 7.95 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.0 Hz, 1H), 7.57–7.46 (m, 3H), 7.38 (dd, J = 8.0, 1.2 Hz, 1H), 6.10 (s, 1H), 3.90 (s, 3H), 3.67 (s, 3H); 13C NMR (acetone-d6) δ 183.2, 181.3, 158.9, 155.5, 133.0, 132.9, 129.9, 128.6, 128.3, 128.1, 127.6, 127.3, 126.9, 126.4, 126.1, 105.8, 60.9, 56.2; IR _υ_max/cm−1: 1650 (C=O), 1587 (C=C), 1331 (C–O); LRMS (ESI+) m/z: 295 (M+H), HRMS (ESI+) m/z calculated for C18H15O4 (M+H) 295.0970; found 295.0965.
3-(Benzo[b]thiophen-2-yl)-2,5-dimethoxycyclohexa-2,5-diene-1,4-dione (26).
Synthesized according to the general procedure 1A from 2-benzothienylboronic acid (0.21 g, 1.20 mmol). The title compound was obtained as a red solid. Yield 0.19 g (64%); m.p. 205–207°C.
1H NMR (acetone-d6) δ 8.27 (s, 1H), 7.96–7.90 (m, 2H), 7.41–7.37 (m, 2H), 6.04 (s, 1H), 4.24 (s, 3H), 3.92 (s, 3H); 13C NMR (acetone-d6) δ 183.3, 181.4, 159.9, 155.6, 141.8, 139.9, 132.4, 129.2, 126.2, 125.2, 125.1, 122.4, 121.5, 106.5, 62.0, 57.3; IR _υ_max/cm−1: 1667 (C=O), 1563 (C=C), 1208 (C–O); LRMS (ESI+) m/z: 301 (M + H), HRMS (ESI+) m/z calculated for C16H13O4S (M+H) 301.0534; found 301.0528.
3-Biphenyl-4-yl-2,5-dimethoxycyclohexa-2,5-diene-1,4-dione (27).
Synthesized according to the general procedure 1A from 4-biphenylboronic acid (0.21 g, 1.20 mmol). The title compound was obtained as a red solid with identical spectral data to those previously reported [30]. Yield 0.22 g (69%); m.p. 178–180°C (Lit. 182–184°C) [31].
1H NMR (acetone-d6) δ 7.73–7.69 (m, 4H), 7.50–7.46 (m, 2H), 7.42–7.36 (m, 3H), 6.03 (s, 1H), 3.92 (s, 3H), 3.89 (s, 3H); 13C NMR (acetone-_d_6) δ 184.1, 182.1, 159.8, 156.3, 141.5, 141.4, 132.0 (2C), 130.9, 129.8 (2C), 128.4, 128.1, 127.8 (2C), 126.9 (2C), 106.6, 61.8, 57.0; IR _υ_max/cm−1: 1669 (C=O), 1582 (C=C), 1325 (C–O); LRMS (ESI+) m/z: 321 (M + H), HRMS (ESI+) m/z calculated for C20H17O4 (M+H) 321.1127; found 321.1120.
3-Biphenyl-3-yl-2,5-dimethoxycyclohexa-2,5-diene-1,4-dione (28).
Synthesised according to the general procedure 1A from 3-biphenylboronic acid (0.21 g, 1.20 mmol). The title compound was obtained as a red solid. Yield 0.21 g (68%); m.p. 172–174°C.
1H NMR (acetone-_d_6) δ 7.69–7.64 (m, 3H), 7.59 (t, J = 1.6 Hz, 1H), 7.53–7.45 (m, 3H), 7.37 (t, J = 8.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 6.03 (s, 1H), 3.92 (s, 3H), 3.89 (s, 3H); 13C NMR (acetone-d6) δ 184.1, 182.1, 159.9, 156.3, 141.6, 141.3, 132.5, 130.4, 130.0 (2C), 129.8, 129.1, 128.3, 127.8 (2C), 127.5 (2C), 106.6, 61.8, 57.0; IR _υ_max/cm−1: 1670 (C=O), 1581 (C=C), 1336 (C–O); LRMS (ESI+) m/z: 321 (M+H), HRMS (ESI+) m/z calculated for C20H17O4 (M+H) 321.1127; found 321.1120.
3-Biphenyl-2-yl-2,5-dimethoxycyclohexa-2,5-diene-1,4-dione (29).
Synthesized according to the general procedure 1A from 2-biphenylboronic acid (0.21 g, 1.20 mmol). The title compound was obtained as a red solid. Yield 0.21 g (67%); m.p. 186–188°C.
1H NMR (acetone-_d_6) δ 7.47 (td, J = 8.0, 1.6 Hz, 1H), 7.41 (td, J = 8.0, 1.6 Hz, 2H), 7.35–7.31 (m, 2H), 7.29–7.22 (m, 4H), 5.89 (s, 1H), 3.81 (s, 3H), 3.68 (s, 3H); 13C NMR (acetone-d6) δ 183.9, 181.9, 159.7, 156.0, 143.2, 142.4, 131.9, 130.9, 130.3, 129.4, 129.3 (2C), 128.9 (2C), 128.6, 128.0, 127.6, 106.5, 61.2, 57.0; IR _υ_max/cm−1: 1646 (C=O), 1589 (C=C), 1325 (C–O); LRMS (ESI+) m/z: 321 (M+H), HRMS (ESI+) m/z calculated for C20H17O4 (M+H) 321.1127; found 321.1121.
General procedure 2 for synthesis of symmetrical 2,5-dimethoxy-3,6-diaryl-2,5-diene-1,4-diones by Suzuki coupling.
A Biotage microwave vial was filled with 11 (0.32 g, 1.00 mmol), relevant boronic acid (2.40 mmol), K2CO3 (0.34 g, 2.50 mmol) and Pd(dppf)Cl2 (0.07 g, 0.01 mmol), and then dioxane (5 ml) was added to this mixture. The reaction vessel was sealed and irradiated with microwaves at 120°C for 20 min (holding time) with a magnetic bar stirrer. After cooling with compressed air to 40°C, the reaction mixture was diluted with dioxane (10 ml) and filtered through the Celite®. The reaction crude was dried in vacuo and subjected to column chromatography for purification (10% EtOAc-90% hexane) to afford an orange-red solid.
2,5-Dimethoxy-3,6-diphenylcyclohexa-2,5-diene-1,4-dione (30).
Synthesized according to the general procedure 2 from phenylboronic acid (0.29 g, 2.40 mmol). The title compound was obtained as a yellow solid with identical spectral data to those of the previous report [31]. Yield 0.22 g (70%); m.p. 150–152°C (Lit. 148–150°C) [31].
1H NMR (CDCl3): δ 7.47–7.40 (m, 6H), 7.38–7.34 (m, 4H), 3.83 (s, 6H); 13C NMR (CDCl3): δ 183.6 (2C), 154.8 (2C), 130.7 (4C), 130.3 (2C), 128.7 (2C), 128.1 (4C), 126.7 (2C), 61.7 (2C); IR _υ_max/cm−1: 1648 (C=O), 1592(C=C), 1295 (C–O); LRMS (ESI+) m/z: 321 (M + H), HRMS (ESI+) m/z calculated for C20H17O4 (M + H) 321.1126; found 321.1118.
2,5-Bis(4-butylphenyl)-3,6-dimethoxycyclohexa-2,5-diene-1,4-dione (31).
Synthesized according to the general procedure 2 from 4-butylphenylboronic acid (0.39 g, 2.40 mmol). The title compound was obtained as an orange solid. Yield 0.27 g (65%); m.p. 177–179°C.
1H NMR (acetone-d6) δ 7.30–7.25 (m, 8H), 3.85 (s, 6H), 3.77 (t, J = 7.6 Hz, 4H), 1.65 (m, 4H), 1.40 (m, 4H), 0.95 (t, J = 7.2 Hz, 6H); 13C NMR (acetone-d6) δ 184.3 (2C), 155.7 (2C), 143.8 (2C), 131.5 (4C), 129.0 (2C), 128.8 (2C), 128.5 (4C), 61.6 (2C), 36.1 (2C), 34.4 (2C), 23.1 (2C), 14.2 (2C); IR _υ_max/cm−1: 1647 (C=O), 1583 (C=C), 1301 (C–O); LRMS (ESI+) m/z: 433 (M+H), HRMS (ESI+) m/z calculated for C28H33O4 (M+H) 433.2378; found 433.2370.
General procedure 3 for synthesis of asymmetrical 2,5-dimethoxy-3,6-diaryl-2,5-diene-1,4-diones by Suzuki coupling.
A 5 ml Biotage microwave vial was filled with 11 (0.32 g, 1.00 mmol), 1-naphthylboronic acid or 2-naphthylboronic acid (0.20 g, 1.20 mmol), K2CO3 (0.34 g, 2.50 mmol) and Pd(dppf)Cl2 (0.07 g, 0.01 mmol), and then dioxane (5 ml) was added to this mixture. The reaction vessel was sealed and irradiated with microwaves at 120°C for 20 min (holding time) with a magnetic bar stirrer. After cooling with compressed air to 40°C, the second relevant boronic acid (1.20 mmol) with a similar portion of catalyst was added to the reaction crude, and irradiation was continued for further 20 min at 120°C. The microwave vial was cooled with compressed air and then the mixture was diluted with dioxane (10 ml) and filtered through the Celite®. The reaction crude was dried in vacuo and subjected to column chromatography for purification (10% EtOAc-90% hexane) to afford an orange-red solid.
2,5-Dimethoxy-3-(naphthalen-1-yl)-6-phenylcyclohexa-2,5-diene-1,4-dione (32).
Synthesized according to the general procedure 3 from 1-naphthylboronic acid (0.20 g, 1.20 mmol) and phenylboronic acid (0.14 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.25 g (70%); m.p. 168–170°C.
1H NMR (acetone-d6) δ 7.99–7.97 (m, 2H), 7.58 (dd, J = 8.0, 1.2 Hz, 1H), 7.59 (t, J = 8.0, 1H), 7.56–7.48 (m, 2H), 7.46–7.40 (m, 5H), 3.85 (s, 3H), 3.68 (s, 3H); 13C NMR NMR (acetone-d6) δ 184.2, 183.8, 156.9, 156.0, 134.4, 133.3, 132.0, 131.6 (2C), 130.4, 129.5, 129.24, 129.20, 129.0, 128.9, 128.5 (2C), 127.1, 126.74, 126.69, 126.2, 126.0, 61.7, 61.1; IR _υ_max/cm−1: 1647 (C=O), 1587 (C=C), 1297 (C–O); LRMS (ESI+) m/z: 371 (M+H), HRMS (ESI+) m/z calculated for C24H19O4 (M+H) 371.1283; found 371.1278.
2,5-Dimethoxy-3-(naphthalene-1-yl)-6-(naphthalene-3-yl)cyclohexa-2,5-diene-2,5-diene-1,4-dione (33).
Synthesized according to the general procedure 3 from 1-naphthylboronic acid (0.20 g, 1.20 mmol) and 2-naphthyl boronic acid (0.20 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.27 g (66%); m.p. 163–165°C.
1H NMR (acetone-d6) δ 8.00–7.94 (m, 6H), 7.89 (dd, J = 8.0, 1.2 Hz, 1H), 7.61–7.51 (m, 6H), 7.47 (dd, J = 8.0, 1.2 Hz, 1H), 3.88 (s, 3H), 3.69 (s, 3H); 13C NMR (acetone-d6) δ 184.3, 183.9, 156.9, 156.3, 134.5, 134.0, 133.8, 133.3, 131.1, 130.5, 129.6, 129.5, 129.3, 129.28, 129.27, 129.1, 128.7, 128.5, 127.8, 127.4, 127.12, 127.05, 126.8, 126.7, 126.3, 126.0, 61.8, 61.1; IR _υ_max/cm−1: 1645 (C=O), 1586 (C=C), 1299 (C–O); LRMS (ESI+) m/z: 421 (M + H), HRMS (ESI+) m/z calculated for C28H21O4 (M+H) 421.1440; found 421.1434.
2,5-Dimethoxy-3-(2,4-dimethoxyphenyl)-6-(naphthalene-3-yl)cyclohexa-2,5-diene-1,4-dione (34).
Synthesized according to the general procedure 3 from 2-naphthyl boronic acid (0.20 g, 1.20 mmol) and 2,4-dimethoxyphenyl boronic acid (0.21 g, 1.00 mmol). The title compound was obtained as an orange solid. Yield 0.29 g (69%); m.p. 159–161°C.
1H NMR (acetone-d6) δ 7.95–7.92 (m, 4H), 7.67–7.54 (m, 2H), 7.50 (dd, J = 8.4, 2.4 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 6.66 (d, J = 2.4 Hz, 1H), 6.61 (dd, J = 8.4, 2.4 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.82 (s, 3H), 3.80 (s, 3H); 13C NMR (acetone-d6) δ 184.0, 183.7, 162.6, 159.5, 156.5, 156.0, 134.0, 133.8, 133.0, 131.0, 129.5, 129.3, 129.1, 128.5, 128.1, 127.7, 127.4, 127.0, 125.8, 113.5, 105.5, 99.2, 61.7, 61.1, 56.0, 55.7; IR _υ_max/cm−1: 1665 (C=O), 1606 (C=C), 1300 (C–O); LRMS (ESI+) m/z: 431 (M+H), HRMS (ESI+) m/z calculated for C26H23O6 (M + H) 431.1494; found 431.1489.
2-(4-Butylphenyl)-3,6-dimethoxy-5-(naphthalene-3-yl)cyclohexa-2,5-diene-1,4-dione (35).
Synthesized according to the general procedure 3 from 2-naphthyl boronic acid (0.20 g, 1.20 mmol) and 4-butylphenyl boronic acid (0.21 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.28 g (67%); m.p. 134–136°C.
1H NMR (acetone-d6) δ 7.96–7.92 (m, 4H), 7.57–7.54 (m, 2H), 7.50 (dd, J = 8.2, 1.2 Hz, 1H), 7.33 (d, J = 8.2 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 3.89 (s, 3H), 3.88 (s, 3H), 2.66 (t, J = 7.6 Hz, 2H), 1.66 (m, 2H), 1.43 (m, 2H), 0.94 (t, J = 7.2 Hz, 3H); 13C NMR (acetone-_d_6) δ 184.32, 184.27, 156.1, 155.7, 143.9, 134.0, 133.8, 131.5 (2C), 131.0, 129.4, 129.2, 129.1, 129.0, 128.9, 128.52 (2C), 128.48, 127.8 (2C), 127.4, 127.0, 61.8, 61.7, 36.1, 34.4, 23.1, 14.2; IR _υ_max/cm−1: 1641 (C=O), 1586 (C=C), 1297 (C–O); LRMS (ESI+) m/z: 427 (M+H), HRMS (ESI+) m/z calculated for C28H27O4 (M+H) 427.1909; found 427.1901.
2,5-Dimethoxy-3-(biaryl-2-yl)-6-(naphthalene-3-yl)cyclohexa-2,5-diene-1,4-dione (36).
Synthesized according to the general procedure 3 from 2-naphthyl boronic acid (0.20 g, 1.20 mmol) and 2-biphenylboronic acid (0.23 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.28 g (65%); m.p. 113–115°C.
1H NMR (acetone-d6) δ 7.94–7.90 (m, 3H), 7.83 (d, J = 1.6 Hz, 1H), 7.56–7.44 (m, 5H), 7.43–7.30 (m, 7H), 3.83 (s, 3H), 3.56 (s, 3H); 13C NMR (acetone-d6) δ 183.9, 183.8, 156.4, 155.8, 143.1, 142.7, 133.9, 133.7, 132.1, 131.0, 130.9 (2C), 130.3, 129.62, 129.56, 129.4, 129.2, 129.1, 129.0 (2C), 128.4 (2C), 128.3, 128.0, 127.72, 127.71, 127.4, 127.0, 61.4, 61.3; IR _υ_max/cm−1: 1646 (C=O), 1583 (C=C), 1300 (C–O); LRMS (ESI+) m/z: 447 (M + H), HRMS (ESI+) m/z calculated for C30H23O4 (M + H) 447.1596; found 447.1592.
2,5-Dimethoxy-3-(biaryl-3-yl)-6-(naphthalene-3-yl)cyclohexa-2,5-diene-1,4-dione (37).
Synthesized according to the general procedure 3 from 2-naphthyl boronic acid (0.20 g, 1.20 mmol) and 3-biphenyl boronic acid (0.23 g, 1.20 mmol). The title compound was obtained as an orange solid. Yield 0.28 g (65%); m.p. 119–121°C.
1H NMR (CDCl3) δ 7.92–7.87 (m, 4H), 7.65–7.60 (m, 4H), 7.55–7.51 (m, 3H), 7.48–7.44 (m, 3H), 7.39–7.35 (m, 2H), 3.90 (s, 3H), 3.84 (s, 3H); 13C NMR (CDCl3) δ 183.7, 183.6, 155.1, 155.0, 141.1, 140.9, 133.3, 132.9, 130.8, 130.5, 129.61 (2C), 129.60, 129.0, 128.6, 128.5, 128.1, 127.9, 127.8, 127.62 (2C), 127.59, 127.4 (2C), 126.9, 126.72, 126.67, 126.4, 61.88, 61.87; IR _υ_max/cm−1: 1647 (C=O), 1583 (C=C), 1301 (C–O); LRMS (ESI+) m/z: 447 (M+H), HRMS (ESI+) m/z calculated for C30H23O4 (M+H) 447.1596; found 447.1592.
General procedure 4 for synthesis of aryl-2,5-bisaminocyclohexa-2,5-diene-1,4-dione.
3-Aryl-2,5-dimethoxy-1,4-benzoquinone or 3,5-diaryl-2,5-dimethoxy-1,4-benzoquinone, synthesized following the general procedures 1 and 3 (1.00 mmol), was dissolved in MeOH (20 ml); then the relevant aliphatic amine (2.00 mmol) was added dropwise. The reaction was stirred at room temperature for 1 h, and the precipitate filtered and washed with MeOH (2 × 20 ml). The desired product dried in vacuo as a red to pink solid.
2,5-Bis(propylamino)3-p-tolylcyclohexa-2,5-diene-1,4-dione (38).
Synthesized according to the general procedure 4 from 13 (0.25 g, 1.00 mmol) and _n_-propyl amine (0.17 ml, 2.00 mmol). The title compound was obtained as a pink solid. Yield 0.27 g (95%); m.p. 166–168°C.
1H NMR (DMSO-d6) δ 7.75–7.72 (s, br, NH), 7.36–7.33 (s, br, NH), 7.15 (d, J = 7.6 Hz, 2H), 7.04 (d, J = 7.60 Hz, 2H), 5.34 (s, 1H), 3.15–3.09 (m, 2H), 2.56–2.52 (m, 2H), 2.32 (s, 3H), 1.58–1.53 (m, 2H), 1.24–1.18 (m, 2H), 0.88 (t, J = 7.6 Hz, 3H), 0.47 (t, J = 7.6 Hz, 3H); 13C NMR (DMSO-d6) δ 177.8, 177.2, 150.9, 146.6, 135.8, 132.1, 131.3, 128.1 (2C), 106.8 (2C), 91.4, 45.0, 43.5, 22.3, 20.9, 20.8, 11.3, 10.7; IR _υ_max/cm−1: 3243 (–NH), 1643 (C=O), 1586 (C=C), 1347 (C–O); LRMS (ESI+) m/z: 313 (M + H), HRMS (ESI+) m/z calculated for C19H24N2O2 (M + H) 313.1915; found 313.1910.
3-(Naphthalene-1-yl)-2,5-bis-(propylamino)cyclohexa-2,5-diene-1,4-dione (39).
Synthesized according to the general procedure 4 from 25 (0.30 g, 1.00 mmol) and _n_-propyl amine (0.17 ml, 2.00 mmol). The title compound was obtained as a pink solid. Yield 0.21 g (85%); m.p. 134–137°C.
1H NMR (DMSO-d6) δ 7.95 (d, J = 7.6 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.76–7.73 (m, NH), 7.66 (d, J = 7.6 Hz, 1H), 7.53–7.44 (m, 4H), 7.34 (dd, J = 7.0, 1.0 Hz, 1H), 5.45 (s, 1H), 3.18–3.13 (m, 2H), 2.21–2.14 (m, 2H), 1.63–1.54 (m, 2H), 1.07–0.93 (m, 2H), 0.90 (t, J = 7.6 Hz, 3H), 0.16 (t, J = 7.6 Hz, 3H); 13C NMR (DMSO-d6) δ 177.4, 177.1, 151.1, 147.3, 133.2, 132.94, 132.89, 129.1, 128.2, 127.5, 126.1, 125.7 (2C), 125.3, 104.0, 91.7, 44.4, 43.6, 22.7, 21.0, 114, 10.4; IR _υ_max/cm−1: 3263 (–NH), 1643 (C=O), 1581 (C=C), 1346 (C–O); LRMS (ESI+) m/z: 349 (M + H), HRMS (ESI+) m/z calculated for C22H25N2O2 (M + H) 349.1916; found 349.1909.
2,5-Bis(3-(dimethylamino)propylamino)-3-(naphthalene-1-yl)cyclohexa-2,5-diene-1,4-diene (40).
Synthesized according to the general procedure 4 from 25 (0.30 g, 1.00 mmol) and _N,N_-dimethylpropane-1,3-diamine (0.20 g, 2.00 mmol). The title compound was obtained as a red solid. Yield: 0.17 g (84%); m.p. 210–212°C.
1H NMR (DMSO-d6) δ 8.0 (s, br, NH), 7.94 (d, J = 7.6 Hz, 1H), 7.90 (d, J = 7.6 Hz, 1H), 7.84 (s, br, NH), 7.67 (d, J = 7.6 Hz, 1H), 7.52–7.44 (m, 3H), 7.34 (d, 7.6 Hz, 1H), 5.43 (s, 1H), 3.25–3.20 (m, 2H), 2.36–2.26 (m, 4H), 2.12 (s, 6H), 1.83 (s, 6H), 1.73–1.66 (m, 2H), 1.60–1.48 (m, 2H), 1.16–1.00 (m, 2H); 13C NMR (DMSO-d6) δ 177.4, 177.2, 151.1, 147.3, 133.2, 133.0, 132.9, 129.1, 128.2, 127.5, 126.1, 125.7 (2C), 125.3, 104.0, 91.7, 44.4, 43.6, 30.7 (4C), 22.7, 21.0, 11.4, 10.4; IR _υ_max/cm−1: 3271 (–NH), 1629 (C=O), 1569 (C=C), 1332 (C–O); LRMS (ESI+) m/z: 435 (M + H), HRMS (ESI+) m/z calculated for C26H35N4O2 (M + H) 435.2759; found 435.2752.
3-(Biaryl-2-yl)-2,5-bis-(propylamino)cyclohexa-2,5-diene-1,4-dione (41).
Synthesized according to the general procedure 4 from 29 (0.30 g, 1.00 mmol) and _n_-propyl amine (0.17 ml, 2.00 mmol). The title compound was obtained as a red solid. Yield 0.31 g (85%); m.p. 177–179°C.
1H NMR (acetone-d6) δ 7.44–7.40 (m, 1H), 7.38–7.33 (m, 2H), 7.31–7.21 (m, 6H), 7.04 (bs, NH), 6.76 (bs, NH), 5.25 (s, 1H), 3.21–3.16 (m, 2H), 2.71–2.56 (m, 2H), 1.71–1.62 (m, 2H), 1.34–1.30 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H), 0.64 (t, J = 7.2 Hz, 3H); 13C NMR (acetone-d6) δ 178.8, 151.5, 144.4, 142.8, 134.8, 133.8, 130.1, 129.3, 128.7, 128.6 (2C), 127.7, 127.5, 92.5, 46.0, 45.8, 44.7, 44.5, 22.78, 23.75, 22.15, 22.13, 11.6, 11.2; IR _υ_max/cm−1: 3272 (–NH), 1652 (C=O), 1587 (C=C), 1295 (C–O); LRMS (ESI+) m/z: 375 (M + H), HRMS (ESI+) m/z calculated for C24H27N2O2 (M + H) 375.2072; found 375.2067.
2-(Naphthalene-1-yl)-5-phenyl-3,6-bis-(propylamino)cyclohexa-2,5-diene-1,4-dione (42).
Synthesized according to the general procedure 4 from 32 (0.37 g, 1.00 mmol) and _n_-propyl amine (0.17 ml, 2.00 mmol). The title compound was obtained as a red solid. Yield 0.37 g (88%); m.p. 226–228°C.
1H NMR (DMSO-d6) δ 7.98–7.92 (m, 2H), 7.76–7.74 (m, 1H), 7.56–7.50 (m, 4H), 7.44–7.29 (m, 7H), 2.59–2.54 (m, 2H), 2.26–2.18 (m, 2H), 1.29–1.20 (m, 2H), 1.09–0.94 (m, 2H), 0.46 (t, J = 7.2 Hz, 3H), 0.17 (t, J = 7.2 Hz, 3H); 13C NMR (DMSO-d6) δ 177.7 (2C), 177.4 (2C), 147.0 (2C), 146.3, 135.2, 133.1, 133.0, 131.6, 129.1, 128.3, 127.6, 126.9, 126.2 (2C), 125.8 (2C), 125.3, 106.6, 103.5, 45.2, 44.5, 22.8, 22,4, 10.7, 10.5; IR _υ_max/cm−1: 3245 (–NH), 1640 (C=O), 1560 (C=C), 1303 (C–O); LRMS (ESI+) m/z: 425 (M + H), HRMS (ESI+) m/z calculated for C28H29N2O2 (M + H) 425.2228; found 425.2221.
2-(Naphthalene-1-yl)-5-naphthalen-3-yl)-3,6-bis(propylamino)cyclohexa-2,5-diene-1,4-dione (43).
Synthesized according to the general procedure 4 from 33 (0.42 g, 1.00 mmol) and _n_-propyl amine (0.17 ml, 2.00 mmol). The title compound was obtained as a red solid. Yield 0.79 g (88%); m.p. 210–213°C.
1H NMR (DMSO-d6) δ 7.99–7.91 (m, 5H), 7.83 (bs, NH), 7.79–7.77 (m, 1H), 7.58–7.43 (m, 9H), 2.59–2.54 (m, 2H), 2.23–2.20 (m, 2H), 1.27–1.18 (m, 2H), 1.12–0.93 (m, 2H), 0.31 (t, J = 7.2, 3H), 0.18 (t, J = 7.2, 3H); 13C NMR (DMSO-d6) δ 177.7, 177.6, 147.0, 146.7, 133.1, 132.9, 132.8, 132.6, 131.9 (2C), 130.0 (2C), 129.8, 129.1, 128.3, 127.7, 127.6, 127.5, 126.7, 126.2, 126.1, 125.9, 125.8, 125.3, 106.4, 103.5, 45.3, 44.5, 22.7, 22.2, 10.6, 10.4; IR _υ_max/cm−1: 3243 (–NH), 1640 (C=O), 1557 (C=C), 1295 (C–O); LRMS (ESI+) m/z: 475 (M+H), HRMS (ESI+) m/z calculated for C32H30N2O2 (M+H) 475.2385; found 475.2379.
Supplementary Material
Supporting Data
Data accessibility
The datasets supporting this article have been uploaded as part of the electronic supplementary material.
Authors' contributions
A.M. and J.A.S. conceived this study. A.G., J.R.B. and C.C.R. are responsible for the synthesis of the analogues reported herein. J.G. conducted the biological screening. A.M. and J.A.S. are the primary authors of this manuscript. A.M. is the recipient of project funding.
Competing interests
We declare we have no competing interests.
Funding
A.M. is the recipient of funding from the Australian Cancer Research Foundation, the Ramaciotti Foundation and the National Health and Medical Research Council (Australia). A.G. is the recipient of a University of Newcastle Postgraduate Research Scholarship. J.R.B. is the recipient of an Australian Postgraduate Award.
References
- 1.AIHW & AACR. 2012. Cancer in Australia: an overview. 2012 Cancer series no. 74. Cat. no. CAN 70. Canberra, Australia: AIHW.
- 2.Joshi S, et al. 2010. The dynamin inhibitors MiTMAB and OcTMAB induce cytokinesis failure and inhibit cell proliferation in human cancer cells. Mol. Cancer Ther. 9, 995–2006. (doi:10.1158/1535-7163.MCT-10-0161) [DOI] [PubMed] [Google Scholar]
- 3.Chircop M, et al. 2011. Inhibition of dynamin by dynole 34–2 induces cell death following cytokinesis failure in cancer cells. Mol. Cancer Ther. 10, 1553–1562. (doi:10.1158/1535-7163.MCT-11-0067) [DOI] [PubMed] [Google Scholar]
- 4.Smith CM, Haucke V, McCluskey A, Robinson PJ, Chircop M. 2013. Inhibition of clathrin by Pitstop 2 activates the spindle assembly checkpoint and induces cell death in dividing HeLa cancer cells. Mol. Cancer 12, 4 (doi:10.1186/1476-4598-12-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill TA, Stewart SG, Gordon CP, Ackland SP, Gilbert J, Sauer B, Sakoff JA, McCluskey A. 2008. Norcantharidin analogues: synthesis, anticancer activity and protein phosphatase 1 and 2A inhibition. ChemMedChem. 3, 1878–1892. (doi:10.1002/cmdc.200800192) [DOI] [PubMed] [Google Scholar]
- 6.Hill TA, Stewart SG, Sauer B, Gilbert J, Ackland SP, Sakoff JA, McCluskey A. 2007. Heterocyclic substituted cantharidin and norcantharidin analogues—synthesis, protein phosphatase (1 and 2A) inhibition, and anti-cancer activity. Bioorg. Med. Chem. Lett. 17, 3392–3397. (doi:10.1016/j.bmcl.2007.03.093) [DOI] [PubMed] [Google Scholar]
- 7.Sakoff JA, Howitt IJ, Ackland SP, McCluskey A. 2004. Serine/threonine protein phosphatase inhibition enhances the effect of thymidylate synthase inhibition. Cancer Chemoth. Pharm. 53, 225–232. (doi:10.1007/s00280-003-0730-9) [DOI] [PubMed] [Google Scholar]
- 8.Tarleton M, Gilbert J, Sakoff JA, McCluskey A. 2012. Synthesis and anticancer activity of a series of norcantharidin analogues. Eur. J. Med. Chem. 54, 573–581. (doi:10.1016/j.ejmech.2012.06.010) [DOI] [PubMed] [Google Scholar]
- 9.Tarleton M, Dyson L, Gilbert J, Sakoff JA, McCluskey A. 2013. Focused library development of 2-phenylacrylamides as broad spectrum cytotoxic agents. Bioorg. Med. Chem. 21, 333–347. (doi:10.1016/j.bmc.2012.10.003) [DOI] [PubMed] [Google Scholar]
- 10.Tarleton M, Gilbert J, Sakoff JA, McCluskey A. 2012. Cytotoxic 2-phenyacrylnitriles, the importance of the cyanide moiety and discovery of potent broad spectrum cytotoxic agents. Eur. J. Med. Chem. 57, 65–73. (doi:10.1016/j.ejmech.2012.09.019) [DOI] [PubMed] [Google Scholar]
- 11.Holland IP, McCluskey A, Sakoff JA, Gilbert J, Chau N, Robinson PJ, Motti CA, Wright AD, van Altena IA. 2009. Steroids from an Australian sponge Psammoclema sp. J. Nat. Prod. 72, 102–106. (doi:10.1021/np800688f) [DOI] [PubMed] [Google Scholar]
- 12.Dyson L, Wright AD, Young KA, Sakoff JA, McCluskey A. 2014. Synthesis and anticancer activity of focused compound libraries from the natural product lead, oroidin. Bioorg. Med. Chem. 22, 1690–1699. (doi:10.1016/j.bmc.2014.01.021) [DOI] [PubMed] [Google Scholar]
- 13.Zaleta-Pinet DA, Holland IP, Muñoz-Ochoa M, Murillo-Alvarez JL, Sakoff JA, van Altena IA, McCluskey A. 2014. Cytotoxic compounds from Laurencia pacifica. Org. Med. Chem. Lett. 4, 8, (doi:10.1186/s13588-014-0008-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trinh TN, McLaughlin EA, Abdel-Hamid MK, Gordon CP, Bernstein IR, Pye V, Cossar PJ, Sakoff JA, McCluskey A. 2016. Quinolone-1-(2_H_)-ones as hedgehog signalling pathway inhibitors. Org. Biomol. Chem. 14, 6304–6315. (doi:10.1039/C6OB00606J) [DOI] [PubMed] [Google Scholar]
- 15.Trinh TN, McLaughlin EA, Gordon CP, Bernstein IR, Pye VA, Redgrove KA, McCluskey A. 2017. Small molecule hedgehog pathway antagonists. Org. Biomol. Chem. 15, 3046–3059. (doi:10.1039/C6OB01959E) [DOI] [PubMed] [Google Scholar]
- 16.Swinney DC, Anthony J. 2011. How were new medicines discovered? Nat. Rev. Drug. Discov. 10, 507–519. (doi:10.1038/nrd3480) [DOI] [PubMed] [Google Scholar]
- 17.de Guzman FS, Copp BR, Mayne CL, Concepcion GP, Mangalindan GC, Barrows LR, Ireland CM. 1998. Bolinaquinone: a novel cytotoxic sesquiterpene hydroxyquinone from a Philippine Dysidea sponge. J. Org. Chem. 63, 8042–8044. (doi:10.1021/jo981037t) [Google Scholar]
- 18.Božić T, Novaković I, Gašić MJ, Juranić Z, Stanojković T, Tufegdžić S, Kljajić Z, Sladić D. 2010. Synthesis and biological activity of derivatives of the marine quinone avarone. Eur. J. Med. Chem. 45, 923–929. (doi:10.1016/j.ejmech.2009.11.033) [DOI] [PubMed] [Google Scholar]
- 19.Muller WE, et al. 1985. Potent antileukemic activity of the novel cytostatic agent avarone and its analogues in vitro and in vivo. Cancer Res. 45, 4822–4826. [PubMed] [Google Scholar]
- 20.Swersey JC, Barrows LR, Ireland CM. 1991. Mamanuthaquinone: an antimicrobial and cytotoxic metabolite of Fasciospongia sp. Tetrahedron Lett. 32, 6687–6690. (doi:10.1016/S0040-4039(00)93575-5) [Google Scholar]
- 21.Rodríguez J, Quiñoá E, Riguera R, Peters BM, Abrell LM, Crews P. 1992. The structures and stereochemistry of cytotoxic sesquiterpene quinones from Dactylospongia elegans. Tetrahedron 48, 6667–6680. (doi:10.1016/S0040-4020(01)80012-0) [Google Scholar]
- 22.Shigemori H, Madono T, Sasaki T, Mikami Y, Kobayashi JI. 1994. Nakijiquinones A and B, new antifungal sesquiterpenoid quinones with an amino acid residue from an Okinawan marine sponge. Tetrahedron 50, 8347–8354. (doi:10.1016/S0040-4020(01)85557-5) [Google Scholar]
- 23.Jiao W-H, Xu T-T, Yu H-B, Mu F-R, Li J, Li Y-S, Yang F, Han B-N, Lin H-W. 2014. Dysidaminones A-M, cytotoxic and NF-B inhibitory sesquiterpene aminoquinones from the South China Sea sponge, Dysidea fragilis. RSC. Adv. 4, 9236–9246. (doi:10.1039/C3RA47265E) [Google Scholar]
- 24.Boyle RG, Travers S. 2006. Hypoxia: targeting the tumour. Anticancer Agents Med. Chem. 6, 281–286. (doi:10.2174/187152006777698169) [DOI] [PubMed] [Google Scholar]
- 25.Faig M, Bianchet MA, Winski S, Hargreaves R, Moody CJ, Hudnott AR, Ross D, Amzel LM. 2001. Structure-based development of anticancer drugs: complexes of NAD(P)H: quinone oxidoreductase 1 with chemotherapeutic quinones. Structure 9, 659–667. (doi:10.1016/S0969-2126(01)00636-0) [DOI] [PubMed] [Google Scholar]
- 26.O'Brien PJ. 1991. Molecular mechanisms of quinone cytotoxicity. Chem. Biol. Interact. 80, 1–41. (doi:10.1016/0009-2797(91)90029-7) [DOI] [PubMed] [Google Scholar]
- 27.Tudor G, Gutierrez P, Aguilera-Gutierrez A, Sausville EA. 2003. Cytotoxicity and apoptosis of benzoquinones: redox cycling, cytochrome c release, and BAD protein expression. Biochem. Pharmacol. 65, 1061–1075. (doi:10.1016/S0006-2952(03)00013-3) [DOI] [PubMed] [Google Scholar]
- 28.Senger MR, Fraga CAM, Dantas RF, Silva FP. 2016. Filtering promiscuous compounds in early drug discovery: is it a good idea? Drug Dis. Today 21, 868–872. (doi:10.1016/j.drudis.2016.02.004) [DOI] [PubMed] [Google Scholar]
- 29.Gordaliza M. 2012. Synthetic strategies to terpene quinones/hydroquinones. Mar. Drugs 10, 358–402. (doi:10.3390/md10020358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon T, Danishefsky SJ, de Gala S. 2003. A concise total synthesis of (±)-mamanuthaquinone by using an _exo_-Diels–Alder reaction. Angew. Chem. Int. Ed. 33, 853–855. (doi:10.1002/anie.199408531) [Google Scholar]
- 31.Bernet A, Schröder J, Seifert K. 2003. Total synthesis of the marine sesquiterpene quinones hyatellaquinone and spongiaquinone. Helv. Chim. Acta 86, 2009–2020. (doi:10.1002/hlca.200390158) [Google Scholar]
- 32.Stahl P, Waldmann H. 1999. Asymmetric synthesis of the nakijiquinones-selective inhibitors of the Her-2/Neu protooncogene. Angew. Chem. Int. Ed. 38, 3710–3713. (doi:10.1002/(SICI)1521-3773(19991216)38:24<3710::AID-ANIE3710>3.0.CO;2-H) [PubMed] [Google Scholar]
- 33.Zayas HA, Bowyer MC, Gordon CP, Holdsworth CI, McCluskey A. 2009. Synthesis of biaryl-styrene monomers by microwave-assisted Suzuki coupling. Tetrahedron Lett. 50, 5894–5895. (doi:10.1016/j.tetlet.2009.07.117) [Google Scholar]
- 34.Trinh TN, Hizartzidis L, Lin AYS, McCluskey A, Gordon CP. 2014. An efficient continuous flow approach to furnish furan-based biaryls. Org. Biomol. Chem. 12, 9562–9571. (doi:10.1039/C4OB01641F) [DOI] [PubMed] [Google Scholar]
- 35.Keegstra EMD, van der Mieden V, Zwikker JW, Jenneskens LW, Schouten A, Kooijman H, Veldman N, Spek AL. 1996. Self-organization of 2,5-di-n-alkoxy-1,4-benzoquinones in the solid state: molecular recognition involving intermolecular dipole-dipole, weak C–H···O = C hydrogen bond and van der Waals interactions. Chem. Mater. 8, 1092–1105. (doi:10.1021/cm950526w) [Google Scholar]
- 36.Viault G, Grée D, Das S, Yadav JS, Grée R. 2011. Synthesis of a focused chemical library based on derivatives of Embelin, a natural product with proapoptotic and anticancer properties. Eur. J. Org. Chem. 2011 1233–1241. (doi:10.1002/ejoc.201001627) [Google Scholar]
- 37.Gan X, Jiang W, Wang W, Hu L. 2009. An approach to 3,6-disubstituted 2,5-dioxybenzoquinones via two sequential Suzuki couplings. three-step synthesis of Leucomelone. Org. Lett. 11, 589–592. (doi:10.1021/ol802645f) [DOI] [PubMed] [Google Scholar]
- 38.Lankveld DP, Van Loveren H, Baken KA, Vandebriel RJ. 2010. In vitro testing for direct immunotoxicity: state of the art. Methods. Mol. Biol. 598, 401–423. (doi:10.1007/978-1-60761-401-2_26) [DOI] [PubMed] [Google Scholar]
- 39.McKim JM. 2010. Building a tiered approach to in vitro predictive toxicity screening: a focus on assays with in vivo relevance. Comb. Chem. High Throughput Screening 13, 188–206. (doi:10.2174/138620710790596736) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergman AM, van Haperen VR, Veerman G, Kuiper CM, Peters GJ. 1996. Synergistic interaction between cisplatin and gemcitabine in vitro. Clin. Cancer Res. 2, 521–530. [PubMed] [Google Scholar]
- 41.Sakoff J, Ackland S. 2000. Thymidylate synthase inhibition induces S-phase arrest, biphasic mitochondrial alterations and caspase-dependent apoptosis in leukaemia cells. Cancer Chemother. Pharmacol. 46, 477–487. (doi:10.1007/s002800000164) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Data
Data Availability Statement
The datasets supporting this article have been uploaded as part of the electronic supplementary material.