Arginase I mRNA therapy – a novel approach to rescue arginase 1 enzyme deficiency (original) (raw)
ABSTRACT
Arginase I (ARG1) deficiency is an autosomal recessive urea cycle disorder, caused by deficiency of the enzyme Arginase I, resulting in accumulation of arginine in blood. Current Standard of Care (SOC) for ARG1 deficiency in patients or those having detrimental mutations of ARG1 gene is diet control. Despite diet and drug therapy with nitrogen scavengers, ~25% of patients suffer from severe mental deficits and loss of ambulation. 75% of patients whose symptoms can be managed through diet therapy continue to suffer neuro-cognitive deficits. In our research, we demonstrate in vitro and in vivo that administration of ARG1 mRNA increased ARG1 protein expression and specific activity in relevant cell types, including ARG1-deficient patient cell lines, as well as in wild type mice for up to 4 days. These studies demonstrate that ARG1 mRNA treatment led to increased functional protein expression of ARG1 and subsequently an increase in urea. Hence, ARG1 mRNA therapy could be a potential treatment option to develop for patients.
KEYWORDS: Arginase 1, mRNA therapy, urea cycle disorder
1. Introduction
The urea cycle primarily eliminates nitrogen generated from protein intake in the human body and it is an endogenous source for arginine, citrulline and ornithine synthesis. The urea cycle pathway also metabolizes nitrogen-containing compounds e.g. adenosine monophosphate. An alternate, effective system that clears ammonia or nitrogen from the human body is lacking. Disruption of the urea cycle due to a missing or mutated enzyme therefore causes ammonia accumulation and its related symptoms [1,2].
Urea cycle disorders (UCDs) result from inherited deficiencies in any one of the six enzymes or two transporters of the urea cycle pathway (CPS1, OTC, ASS1, ASL, ARG1, NAGS, ORNT1 or citrin). Deficiency of urea cycle enzymes results in accumulation of ammonia and leads to encephalopathy. Episodes of encephalopathy and associated symptoms are unpredictable and, if untreated, are lethal or produce devastating neurologic sequelae in long-term survivors [1]. Therefore, investigation for hyperammonemia in any infant or child with altered mental status (in the absence of obvious causes, such as trauma, infection, or poisoning) is essential for prompt diagnosis of urea cycle disorders and treatment to avoid brain damage and death [2].
Arginase I (ARG1) is the last enzyme in the urea cycle and it facilitates conversion of arginine to urea and ornithine. Hyperargininemia/Arginase deficiency is an autosomal recessive urea cycle disorder caused by deficiency of the enzyme ARG1, resulting in elevated levels of arginine which can lead to toxicity. The nervous system is especially sensitive to the effects of excess arginine [3]. ARG1 deficient patients can infrequently experience hyperammonemia as well. Symptoms of ARG1 deficiency include increased plasma level of arginine and glutamine, severe spasticity, loss of ambulation, complete loss of bowel and bladder control, seizures, growth deficiency and severe intellectual disability.
Current Standard of Care (SOC) for patients with ARG1 deficiency is diet control. Nitrogen scavenger drugs such as phenyl butyrate, a low-protein diet or ornithine supplementation give symptomatic relief by reduction in arginine levels [4], but fail to prevent neuro-cognitive deficits [5,6]. A protein deficient diet is provided to children with ARG1 deficiency, which in turn results in slow growth and delayed/missed developmental milestones [7,8]. Despite diet and drug therapy, ~25% of patients suffer from severe mental deficits and loss of ambulation. and 75% of patients whose symptoms can be managed through diet therapy continue to suffer from neuro-cognitive deficits [9–11].
ARG1 enzyme replacement therapy normalizes plasma arginine levels in artificial buffers and in whole blood from rabbits [12]. ARG1 loaded erythrocyte therapy has been tested in the past to reduce plasma arginine levels and rescue hyperargininemic episodes, induced by arginine injections, in wild type rats and mice [13,14]. The study showed a small immediate decrease in serum arginine but the decrease was not significant [15]. In a recent study, Burrage et al demonstrated that PEGylated recombinant human ARG1 enzyme appeared to normalize plasma and brain arginine in arginase-deficient mouse models. However, it had no effect in the liver thus failing to rescue the lethal mouse Arg1 knockout (KO) phenotype. In addition, enzyme replacement therapy failed to rescue hyperargininemia [4,9]. An attempt at gene therapy with Shope papilloma virus, to induce virally-encoded arginase was also unsuccessful [16]. However, there have been some promising results in juvenile lethal and inducible _Arg1_-deficient mouse models using adenoviral delivery [5,17,18]. Diet plays a key role in treatment of hyperargininemia. Strict restriction of dietary protein in combination with supplementation with an essential amino acid mixture that is free of arginine can ameliorate arginine levels in plasma and CSF. However, the response to dietary restriction is relatively poor and improvement is unsatisfactory [19,20]. Prasad et al. found clinical improvement in 50%, stabilization in 25%, and progression of the disease in 25% of ARG1-deficient patients despite drug and diet therapy [21]. Liver transplantation in humans cures the disease in the liver but not in other organs including brain, although it may normalize ammonia and arginine in blood [22,23].
Recently, mRNA therapy has emerged as a viable drug modality that can be exploited to deliver a functional protein to diseased cells. Engineering of mRNA molecules to code and transiently express a desired protein can alleviate a diseased condition. Recently, mRNA cancer immunotherapies and vaccines have reached clinical development [24]. Delivery is a primary challenge for developing effective mRNA-based drugs, but lipid nanoparticles (LNPs) are promising for in vivo delivery of nucleotides [24]. To increase stability and translation efficiency of protein from in vitro transcribed mRNA, various engineering approaches have been studied and are currently being tested [25]. Recently, protein engineering and UTR modifications are being explored to synthesize modified mRNA transcripts with improved protein translation and half-life compared to its naturally occurring counterpart. These modifications would result in better efficiency of protein synthesis and increased stability [26].
mRNA therapy is a novel therapeutic approach to effectively replace mutated or deficient proteins. ARG1 is a promising and evolving disease target – in fact, ARG1 deficiency is now included as a part of new born screening tests. The purpose of the present studies is to evaluate ARG1 as a target for mRNA therapy by determining whether delivery of ARG1 mRNA can increase ARG1 protein expression in vitro and in vivo.
2. Results
2.1. In vitro proof of expression studies in multiple relevant cell types
To determine the ability of ARG1 mRNA to encode for protein, in vitro assays were optimized in relevant cell types for studying mRNA uptake and subsequent protein production. HeLa, HepG2 and ARG1 deficient patient fibroblasts (GM00954) were utilized for in vitro proof of expression studies. Transfection conditions were optimized for the three cell types with varied ratios of different lipids to find the optimal condition for transfection. Optimization was performed using enhanced green fluorescent protein (eGFP) mRNA for ease of confirmation through microscopy. Cells were observed for increased intensity of green fluorescence, percent green cells as well as percent viability with each transfection condition to ensure that the lipid or its concentration was not toxic to cells.
mRNA was tested initially in HeLa cells which served as a quick check of ARG1 protein expression. The HeLa cell line was chosen for this experiment due to its low constitutive expression of ARG1 protein which was confirmed by protein expression studies by WES capillary electrophoresis (Figure 1).
Figure 1.
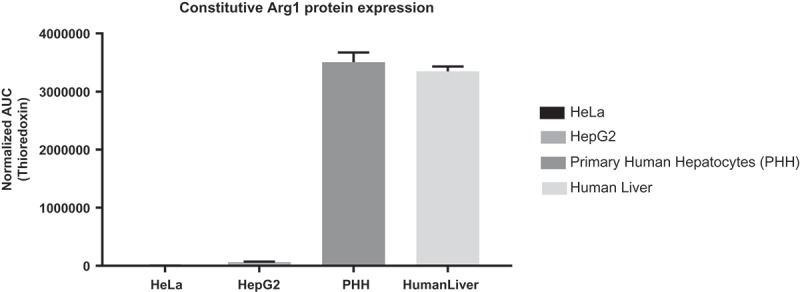
qPCR analysis for constitutive Arg1 protein expression in four different cell lines. HeLa and HepG2 cells express low levels of Arg1 protein, while primary human hepatocytes (PHH) and human liver sample express high levels of Arg1 protein.
In addition to HeLa cells, ARG1 mRNA was also tested in HepG2 and ARG1 deficient fibroblasts. Primary human hepatocytes express significant amounts of ARG1 protein and hence do not serve as an ideal model for increased protein expression studies. HepG2, a liver carcinoma cell line was used as a surrogate to primary human hepatocytes to evaluate protein expression of modified mRNA. HepG2 expresses all other essential enzymes of the urea cycle e.g. CPS1, ASS1 and ASL, at levels comparable to those of primary human hepatocytes [27–29], but is deficient in expressing ARG1 and OTC. Lastly, ARG1 deficient patient fibroblasts, GM00954 [30], demonstrating decreased arginase enzyme activity were also tested.
Two forms of ARG1-encoding mRNA i.e. wild type (WT) and FLAG-tagged were synthesized for the study. Transfection of ARG1 mRNA and its FLAG-tagged version in cell types (HeLa, HepG2, and ARG1 deficient patient fibroblasts) with low levels and deficient activity of ARG1, increased expression of ARG1 protein 24h post transfection. Each transfected cell line data point was compared to the well transfected with mRNA encoding for eGFP protein as the vehicle control, to which all the data was normalized. In parallel, constitutive ARG1 protein expression of the cell line was determined by utilizing the ‘cells only’ well. FLAG-tagged version of mRNA was initially designed to ensure detection of translated recombinant protein. Since a specific antibody to human ARG1 was optimized for use in these experiments, all treatment groups were subsequently evaluated with the optimized anti-human ARG1 antibody, and anti-FLAG antibody was not utilized. ARG1 mRNA transfected cells expressed increased levels of ARG1 protein in HeLa, HepG2 and in ARG1 deficient patient fibroblasts (Figure 2, n = 3 individual experiments).
Figure 2.
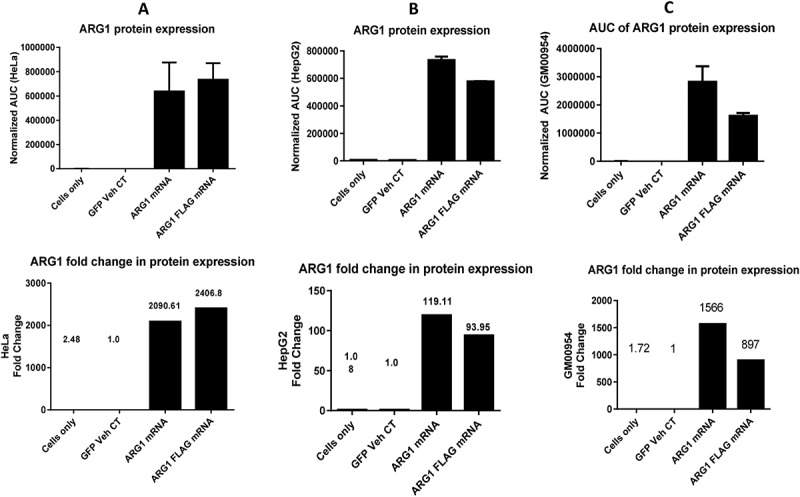
Arg1 protein expression profile across multiple relevant cell types in upper panel and fold change of protein expression when normalized to eGFP vehicle control in lower panel. A. HeLa, B. HepG2 and C. GM00954 (Arg1 deficient patient fibroblasts) show increased protein expression when transfected with Arg1 mRNA (WT and FLAG).
2.2. Bioactivity of ARG1 recombinant protein
To confirm the enzyme activity of recombinant ARG1 protein, a quantitative ARG1 enzyme activity assay was developed. In this assay, known concentration of urea was used to generate a standard curve. A linear range of protein expression was determined using wildtype ARG1 mRNA transfected lysate expressing high amount of ARG1 protein. Two-fold serial dilution of lysate was tested in duplicate in a 96 well format. HepG2 and ARG1 deficient patient fibroblasts, when transfected with ARG1 mRNA, produced more urea compared to eGFP vehicle control group and cells only group for each cell line (Figure 3, n = 3 individual experiments). Serial dilution of HeLa cell lysates demonstrated that increased levels of urea were produced even at very high dilutions. Since, HeLa was only used to validate protein expression of the construct; no further optimization was done with this cell line.
Figure 3.
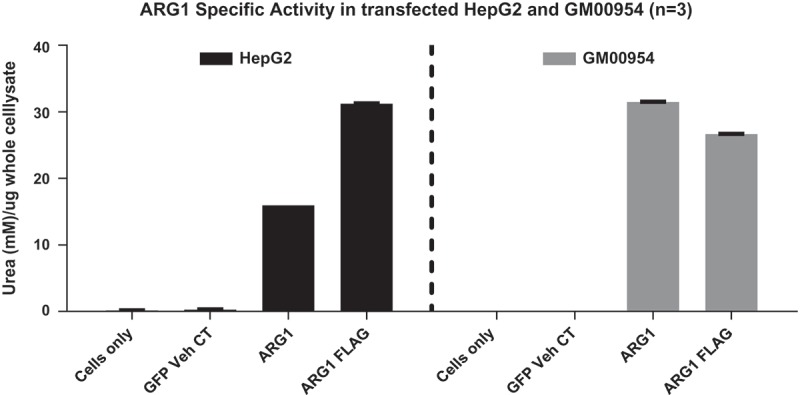
Specific activity of Arg1 measured in terms of urea synthesized in A. HepG2 and B. GM00954 (Arg1 deficient patient fibroblasts). Note cells only and GFP vehicle controls do not show any urea synthesized due to lack of Arg1 enzyme; when cells are transfected with Arg1 mRNA (WT and FLAG), urea synthesis is increased.
2.3. In vivo proof of expression in C57bl/6 mice
We next sought to determine if ARG1 mRNA could increase ARG1 protein expression in vivo. ARG1 mRNA (WT and FLAG-tagged) were formulated in house with proprietary self-assembling lipid mixes from Precision Nanosystems. The in vivo proof of expression study in 8-week-old C57BL/6 mice was designed to monitor expression of human recombinant ARG1 protein. Luciferase mRNA and PBS-treated arms were used as negative controls in this in vivo experiment. Protein expression studies were performed using capillary electrophoresis in the WES machine (Protein Simple) with human ARG1-specific antibody (ab133543). Optimization experiments with multiple ARG1 antibodies from various vendors were carried out to identify an antibody specific for human ARG1 protein. We observed increased ARG1 recombinant protein expression (resolved from endogenous mouse Arg1 protein) at 24h which was sustained at 48h post injection (Figure 4) in both WT-ARG and FLAG-ARG1 mRNA injected groups. When compared to the luciferase mRNA treatment at 24h, ARG1 mRNA treatment increased protein expression by ~ 37.8 fold at 24h and ~12.9 fold at 48h post mRNA injection.
Figure 4.
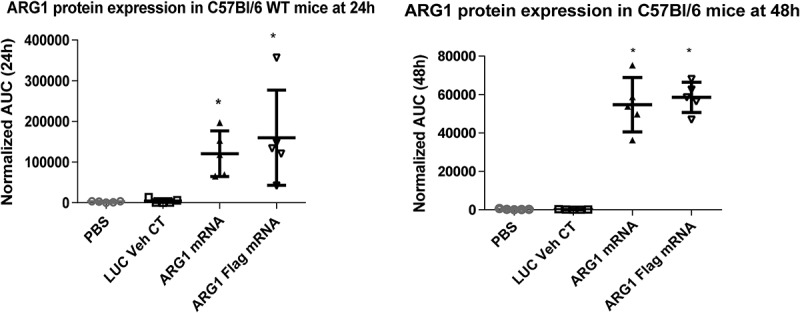
LNP formulated Arg1 mRNA (WT and FLAG) expresses protein in mouse liver. Arg1 protein expression increases statistically in treated animals at 24h (A) and 48h (B).
After confirming liver expression of ARG1 via capillary electrophoresis, we wanted toto confirm intrahepatic protein localization. Immunohistochemistry (IHC) and in situ hybridization (ISH) were performed on FFPE liver sections from treated mice at 24 and 48 h time points. ARG1 antibody, specific for detection of human ARG1 protein, was used for the IHC assay. IHC detected human recombinant protein localized in hepatocyte cytoplasm (Figure 5). The staining intensity was proportional to the amount of protein present. ISH studies on liver sections were performed with a human ARG1 mRNA specific probe set to ensure it does not cross-react with mouse Arg1 sequence. Single red dots were observed indicating single copies of ARG1 mRNA in both 24 and 48 h liver sections from treated mice. Additionally, ARG1 mRNA was distributed in hepatocyte cytoplasm (Figure 6) and these findings were consistent with IHC results.
Figure 5.
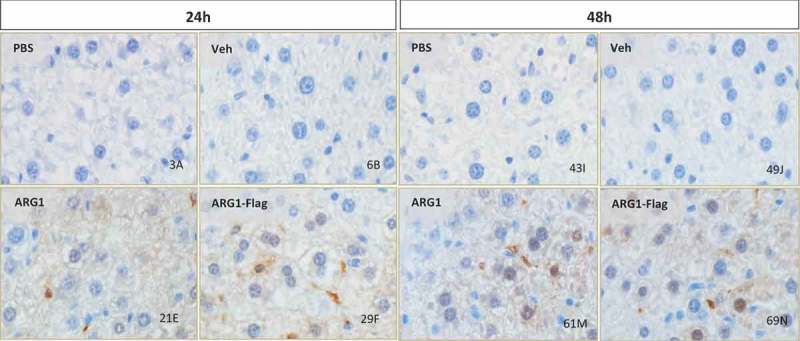
Arg1 immunostaining showed protein biodistribution in hepatocyte cytoplasm, nucleus and sinusoidal spaces at 24h (A) and 48h (B) timepoint in C57Bl/6 wild type mice post Arg1 mRNA treatment (WT and FLAG). 40x magnification.
Figure 6.
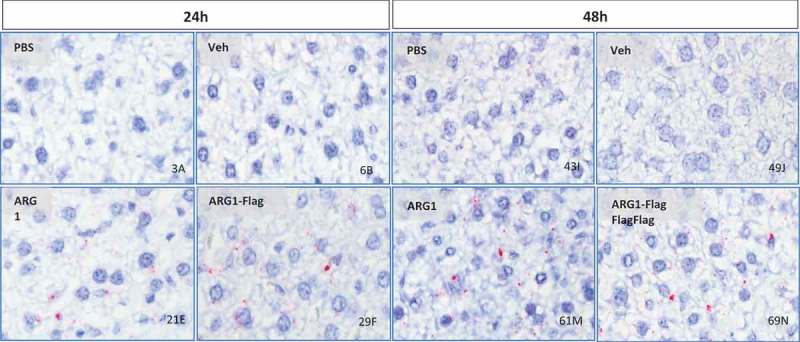
In situ hybridization (ISH) to detect Arg1 mRNA in mouse liver treated with Arg1 WT or FLAG-tagged mRNA. Arg1 mRNA detected in hepatocyte cytoplasm (single red dots) and as mRNA clusters (larger red dots) in sinusoidal spaces (40x magnification).
2.4. Stability of ARG1 protein expression in vivo in C57Bl/6 mice
Since we observed sustained elevated levels of ARG1 protein expression with capillary electrophoresis and IHC, we evaluated the stability of recombinant protein expression encoded by injected mRNA. We evaluated ARG1 protein expression over 168h (7 days) in wild type mice after a single injection of ARG1 mRNA. In this study, mRNA encoding eGFP along with the PBS group served as negative controls. Only ARG1 WT mRNA was administered in this study. ELISA was performed on liver homogenates of in vivo samples to detect ARG1 protein expression in wild type mice. We observed significant protein expression at 24 and 48 h consistent with the findings of proof of expression studies done previously. The study showed ARG1 protein expression lasting up to 168h via single dose of mRNA i.v. (Figure 7). When compared to the eGFP mRNA control group, ARG1 mRNA increased protein expression ~25-fold at 24h and ~6-fold at 168h. To understand this further, mRNA stability was determined by qRT-PCR. Liver tissue was homogenized to extract total RNA from the ARG1 mRNA-treated samples and cDNA prepared for qRT-PCR. We detected ARG1 mRNA expression in the injected samples out to 168h (Figure 8).
Figure 7.
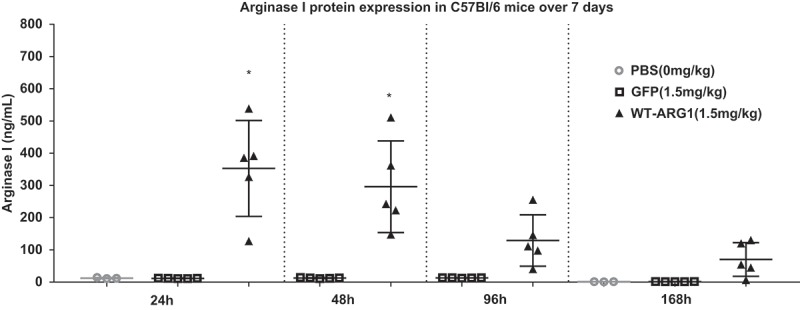
Arg1 mRNA demonstrated extended duration of protein expression in mouse liver. Arg1 protein expression increases in treated animals at 24h and 48h with lasting protein expression seen up to 168h.
Figure 8.
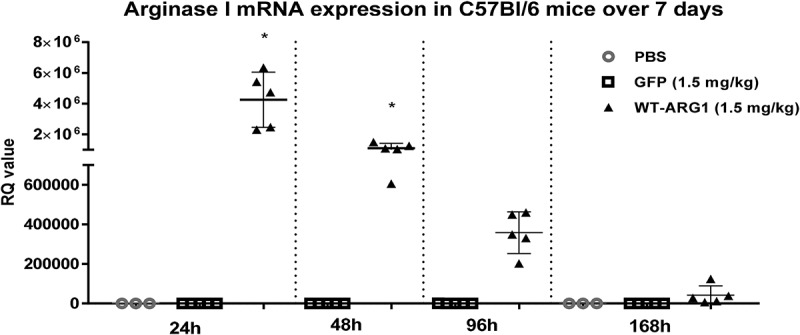
Extended time course of LNP-formulated Arg1 mRNA (WT) in mouse liver. Upon mRNA treatment, Arg1 mRNA level is high at 24h and is detectable until 7d post-injection.
3. Discussion
In this study, we demonstrate that ARG1 protein could be expressed in vitro and in vivo utilizing in vitro transcribed (IVT) ARG1 mRNA, thus postulating that mRNA therapy could serve as a therapeutic for Arginase I deficiency.
In this paper, we focus on exogenous expression of ARG1 and its effects on urea cycle both in vitro and in vivo. Firstly, we showed that ARG1 mRNA significantly increased expression and activity in vitro in relevant cell types. Bioactivity of recombinant protein was confirmed in vitro. Furthermore, we demonstrated that LNP formulated ARG1 mRNA, injected in wildtype mice, increased ARG1 protein and mRNA expression in vivo in liver, confirmed with CE, IHC and ISH. To conclude, an extended time course study showed lasting mRNA and protein expression up to 168h in wildtype animals with a single dose of ARG1 mRNA. The recombinant protein produced by delivered mRNA could metabolize arginine and ammonia in hepatocytes and has the potential to become the standard of care for this disease thus significantly improving quality of life of patients.
To ensure that encoded ARG1 protein is bioactive and can complete the urea cycle to convert arginine to urea and ornithine, a quantitative urea assay was developed which required significant optimization due to the wide variance in ARG1 levels between different cell types. Arginase activity is very low or absent, reduced to 10% of normal or less, in red blood cells of all Arg1-deficient patients in whom it was tested [31]. Interestingly with our mRNA delivery in vitro, protein expression was elevated ~ 20 fold and ~ 30 fold respectively in HepG2 and in GM00954 cells, implying a potential to increase ARG1 enzyme in ARG1-deficient cells.
To validate in vitro findings, proof of expression studies were performed in vivo in wild type mice. We observed significantly increased ARG1 protein expression at 24 and 48 h post mRNA injection. Generally, i.v. administered mRNA levels peak at 6h, but degrade soon after due to nuclease and cellular degradation mechanisms. As opposed to its natural counterparts, these IVT mRNA possess modified nucleotides. These mRNA also have optimized coding sequence with specific 3’ and 5’ UTRs, enzymatically generated 5’cap structure with a long poly(A)-tail rendering them more stable and translatable. At 48h, protein levels appeared to decrease compared to 24h, indicating there was a steady decrease in protein levels in tissue. A probable explanation could be an increased translation of ARG1 protein that lasts for an extended time.
Localization of delivered mRNA and synthesized recombinant protein in the appropriate compartment is quintessential for being therapeutically active. In our study, we observed the delivered ARG1 mRNA and ARG1 recombinant protein localized to hepatocyte cytoplasm in mouse liver tissue. This could likely be due to the size, polydispersity index and composition of LNP formulation used for delivery of these mRNA. Thus, optimization of appropriate delivery LNP is necessary for any mRNA or RNA therapeutic. Different lipids may have different profiles based on the size and charge on the mRNA of interest. An optimized LNP might be able to deliver mRNA to the appropriate compartment with uniform bio distribution which could result in increased protein translation and reduced immune response. It is possible that each potential therapeutic mRNA target might have to be delivered with a unique optimized LNP formulation.
To understand duration of action of single dose of ARG1 mRNA and track half-life of recombinant ARG1 protein, an extended time course study was carried out in wildtype mice. The extended time course study demonstrated repeatability of previous findings. We observed protein expression lasting for 168h in a normal wild type animal. Unfortunately, enzyme assays could not be performed given the elevated levels of endogenous mouse Arg1 in wildtype animals.
While our findings are promising, further research is warranted to assess biomarker changes in terms of levels of ammonia and urea in these mice over extended periods of time. Delivery is another bottle neck in mRNA therapy. With the available lipids, repeat dosing can be toxic and hence single administration was the only feasible choice. While evaluating lipid options for mRNA delivery, strategies to improve mRNA backbone structure and/ or chemistry can possibly stabilize mRNA over long periods of time thus extending its half-life. This in turn would result in stable ARG1 protein expression eliminating the need for frequent dosing. Due to the limitations associated with the current research, studies were performed in wild type mice as opposed to an _Arg1_-deficient disease model. Accurate analysis of the benefits of ARG1 mRNA will require multidose studies in an _Arg1-_deficient mouse model. Thus, to substantiate our findings, future studies are required to understand the pharmacodynamic effect and impact on survival in a disease model.
4. Material and methods
4.1. Cell culture and modified mrna transfection
The human cervical carcinoma cell line (HeLa) and human liver carcinoma (HepG2) cell line were obtained from American Type Culture Collection. The cells were cultured in MEM (ThermoFisher Scientific, 32561037) and supplemented with 10% fetal bovine serum (FBS). Arg1- deficient patient fibroblasts (Coriell Institute, GM00954) are cultured in MEM media supplemented with 15% fetal bovine serum. All cells were maintained at 37°C with 5% CO2 in a humidified atmosphere.
Two synthetic chemically-modified mRNAs were ordered from Trilink; one using native ARG1 protein sequence referred to as ‘wild type’ sequence (NM_000045), and the other mRNA with a FLAG tag referred to as ‘Flag’ sequence. eGFP mRNA coding for eGFP protein was used as an mRNA-loaded vehicle control in this study and as a positive control for transfection.
In vitro proof of expression was performed in HeLa (ATCC, CCL-2) HepG2 (ATCC, HB-8065), and Arg1-deficient patient fibroblasts (GM00954) to study increase in ARG1 protein expression in vitro with the two ARG1 modified mRNA (WT and Flag).
All the steps were performed in a sterile environment in a biosafety cabinet. Transfection optimization was performed for each cell type. Cells were plated in 24 well tissue culture plates (HeLa 50,000 cells per well; HepG2 100,000 cells per well; ARG1-deficient patient fibroblasts 40,000 cells per well) in MEM with 10% FBS (HeLa and HepG2); EMEM with 15% FBS (GM00954) prior to day of transfection. The following day at ~70–80% confluence, cells were transfected with 1.25 µg of modified mRNA with 2 µl of Lipofectamine 2000 (HeLa; ThermoFisher Scientific, 11668019) or 4 µl of Messenger Max (HepG2, ThermoFisher Scientific, LMRNA015) or 2 µl of Messenger Max (GM00954) per manufacturer’s protocol. Appropriate volume of mRNA was dissolved in OptiMEM (ThermoFisher Scientific, 31985062) to make a total volume of 100 µl. Similarly, appropriate volume of lipid was dissolved in OptiMEM to make a total volume of 100 µl. Diluted mRNA was mixed into diluted lipid and the mRNA-lipid complex was incubated for 15 minutes at room temperature before adding to cells. All cells were harvested at 24 hours post transfection and processed for protein analysis.
4.2. Cell lysis and target protein detection
At 24h, cells were observed under the microscope for eGFP positive cells and confirmed for cell health and viability. Once eGFP signal was seen, cells were washed twice with 500 µl PBS (Gibco, ThermoFisher Scientific, 14190094) to remove any residual media. Cells were lysed with 100 µl of freshly prepared RIPA buffer (Sigma Aldrich, R0278) with 1% Halt protease and phosphatase inhibitors (ThermoFisher Scientific, 78430) and collected with a cell scraper. Cell lysates were collected in 0.5 ml tubes and clarified by centrifugation at 18,000 × g for 10 mins at 4°C to pellet cell debris. Clarified cell lysate supernatant was carefully transferred to a new labelled tube and placed on ice for protein quantification with BCA protein detection kit (ThermoFisher Scientific, 23227). BCA assay was performed according to manufacturer’s protocol and total protein concentration was determined. After determining total protein concentration, Arg1 protein was detected using a WES capillary electrophoresis machine (Protein Simple, San Jose, CA). Total protein concentration of 0.2 µg/ml was loaded on the WES and protein detected by human ARG1-specific primary antibody (Abcam, ab133543). Secondary antibody and detection reagents were provided in WES -rabbit master detection kit (Protein Simple, SM-W004). Manufacture’s protocol was followed with 0.2mg/ml of protein loaded in each capillary. Band intensities were quantified by determination of area under curve (AUC) of ARG1 for each sample and the values were normalized to a cytoplasmic housekeeper protein, thioredoxin (Cell Signaling Technology, 2429).
4.3. Bioactivity studies of recombinant ARG1 protein
ARG1 mRNA transfected cell lysates were tested for ARG1 specific activity to ensure that mRNA-encoded ARG1 protein was active. Arginase activity colorimetric assay kit (Sigma-Aldrich, MAK112) was used. In this assay, ARG1enzyme catalyzes conversion of arginine to urea and ornithine. Urea specifically reacts with substrate to generate a colored product, proportional to arginase activity present. A quantitative assay was developed for absolute measurement of urea. In this assay, known concentration of urea was used to generate a standard curve and a linear range of protein expression was determined using native ARG1 mRNA transfected lysate expressing high amount of protein. Optimal linear range of assay was determined with a standard curve using urea standard provided in the kit. After determining optimal working range of assay, manufacturer’s protocol was followed with appropriate working protein concentration for each cell type. Plate was incubated at room temperature for 2 hours and read on Synergy Neo plate reader (BioTek, Winooski VT). Concentration of urea produced in transfected cell lysates was determined with a urea standard curve.
4.4. Animal studies
Alexion IACUC guidelines and regulations were followed while housing, handling and during experimental procedures with rodents and all procedures and protocols were approved by the Alexion IACUC committee. C57Bl/6 male mice (6-week-old) were used in this study. Mice were kept in a temperature-controlled environment with a 12h/12h light-dark cycle, with a standard diet and water ad libitum.
4.5. Formulation of mRNA into lipid nanoparticles and in vivo dosing
mRNA constructs were formulated using a self-assembling ionizable LNP containing ARG1 mRNA or its flag tagged construct. eGFP encoding mRNA was used as an mRNA-loaded vehicle control and PBS was used as a negative control in this experiment. LNPs were prepared by rapid mixing of mRNA in acetate buffer at pH 4.0 with a lipid mix, Hepato9 mRNA kit (Precision Nanosystems), suspended in ethanol at a lipid-to-drug ratio of ~11 (wt/wt). LNPs were immediately filtered through 0.2 μm sterile filter. Animals were injected with a single intravenous dose of 1.5 mg/kg of formulated mRNA through the tail vein. Liver samples were harvested, snap frozen and stored at −80°C until processing (n = 5 per time point) to assay hepatic protein and mRNA expression.
4.6. Liver tissue homogenization and total protein determination
Whole liver tissues were weighed to approximately 50–100 mg and were placed in ceramic bead tubes on ice. 1 ml of RIPA lysis buffer and 1% Halt protease and phosphatase inhibitors were added to each tube. Samples were homogenized using two 15 second pulses at 6,800 RPM using Precellys Evolution tissue homogenizer. On observing a homogenized lysate solution, bead tubes were centrifuged at 18000 x g for 15 minutes at 4°C using Eppendorf 5430R tabletop centrifuge. Supernatants were carefully transferred to new labelled tubes and stored on ice. Tissue lysates were diluted 25-fold and diluted lysate was used to determine total protein concentration in duplicate by BCA assay in a 96 well plate format. Target protein detection was performed by capillary electrophoresis with liver lysates normalized to 0.2 mg/ml protein concentration as described previously.
4.7. Immunohistochemistry (IHC)
After collecting whole liver from treated mice that were euthanized, left lateral lobes were placed flat in tissue cassettes. Cassettes were then placed into a container filled with 10% Neutral Buffered Formalin, NBF (Sigma HT501128-4L) fixative solution at minimum 1:20 ratio of tissue volume vs. the fixative solution. Samples were fixed for a minimum of 24h but no more than 48h at room temperature. After tissue fixing, the cassette was placed into a container filled with PBS for not more than 3 days. After PBS wash, liver tissues were processed and embedded into paraffin blocks to get formalin fixed paraffin embedded (FFPE) 5 µm tissue sections on slides. The slides were then used to perform automated IHC on Bond Rx (Leica Biosystems, Leica Microsystems Inc., Buffalo Grove, IL) according to standard protocol with appropriate positive controls. Human specific rabbit anti ARG1 mAb (Abcam, ab133543) was used to detect target ARG1protein. For the automated staining, epitope retrieval was performed with ER1 citrate buffer, pH 6 (Leica, AR9961), EDTA-Tris, pH 8.0 solution for 20 mins and primary antibody was incubated at 1:400 dilution for 30 mins. After completing the staining protocol, slides were dehydrated in ethanol and xylene and were sealed with a coverslip using Cytoseal (ThermoFisher Scientific, 8310–16). Once the slides were dried, they were imaged on VS120 system Olympus Image scanner at 40X magnification and results were reported.
4.8. In situ hybridization (ISH)
ISH was performed to visualize mRNA biodistribution. Tissues were processed to get FFPE sections on glass slides as described previously. ISH was performed on FFPE liver sections using the automated BondRx (Leica Biosystems, Leica Microsystems Inc., Buffalo Grove IL) using ViewRNA eZ-L Detection Kit (Affymetrix, Santa Clara CA). Custom RNA in situ hybridization probe (Affymetrix, Santa Clara CA) was designed to detect the modified ARG1 mRNA (Human NM_000045). ISH was run following the manufacturer’s protocol for Affymetrix probes. The ViewRNA eZ-L assay was run according to manufacturer’s protocol but briefly, RNA was unmasked with a 10-min incubation at 95°C in Bond Epitope Retrieval Solution 1 (Leica Biosystems, AR9961) followed by 20-min incubation with Proteinase K from the Bond Enzyme Pretreatment Kit at 1:1000 dilution (Leica Biosystems, AR9551). Hybridization was performed with 1:40 probe dilution followed by FastRed detection. To ensure RNA integrity and assay procedure, additional sections were also hybridized with a probe for peptidyl prolyl isomerase B (PPIB), an endogenous housekeeping protein. To control for non-specific staining, control probe against bacterial protein dihydrodipicolinate reductase (dapB) was used as a negative control. Slides were air dried, cleared in xylene and mounted using Cytoseal. Slides were imaged using the VS120 system at 40X magnification (Olympus) and results were reported.
4.9. qRTPCR
Liver punches were collected at their respective time points, stored in RNALater (ThermoFisher Scientific, AM7021), and stored at −20C. Tissues were then weighed to reach an optimal range of 15–35 mg and total RNA was extracted via Maxwell RSC simply RNA Tissue Kit (Promega, AS1610) using the manufacturer’s protocol. RNA concentration was then determined and diluted to 150 ng/ml. cDNA was prepared by generating a 2x RT master mix using High-Capacity cDNA Reverse Transcription kit (ThermoFisher Scientific, 4368813). qRT-PCR was then conducted using Taqman Fast Master Mix (ThermoFisher Scientific, 4400088) on Quant Studio 7 (ThermoFisher Scientific, Waltham MA) using human specific ARG1 Taqman (ThermoFisher Scientific, 4331182) probe. mRNA levels were quantified using the ΔCT method to get a RQ value.
4.10. ELISA
To understand more about stability of protein expression, a 168h (7 days) time course study was performed in vivo in wild type mice with the same dose of ARG1 mRNA. Single dose i.v. of LNP formulated ARG1 mRNA was injected into 8-week old male wild type mice. Liver expression of Arg1 protein was monitored by human ARG1 specific ELISA at four time points after dosing (24, 48, 96 and 168 h). Tissues were processed as described above with total protein quantification done by BCA. Human ARG1 specific ELISA (BioVendor, RD193028000R) was used to determine amount of human recombinant ARG1 protein encoded by mRNA. Linear range of protein detection was determined, and the optimized condition was used for ARG1 protein detection in the samples. Lysates were diluted 10-fold to be able to detect protein across all time points. Significant protein expression was observed at 24 and 48 h groups in extended time course study which compared with the findings of in vivo proof of expression study.
Relevant data and statistical analysis was carried out using Microsoft Excel and GraphPad Prism. All charts or graphs displayed in this manuscript were constructed using GraphPad Prism.
All the procedures for animal studies were approved and performed under Alexion IACUC guidelines and regulations.
Funding Statement
This work was supported by the Alexion Pharmaceuticals.
Acknowledgments
The authors would like to acknowledge Brendan Connolly, Cleo Isaacs, Jeremiah D. Farelli, Jeffrey M. Brown, Mary R. Stahley and Joanna S. deBear, of Alexion Pharmaceuticals, Inc. for their support.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Bk B. Urea cycle disorders. Clin Liver Discov. 2000. November;4(4):815–830. [DOI] [PubMed] [Google Scholar]
- 2.Helman G, Pacheco-Colon I, Gropman AL. The urea cycle disorders. Semin Neurol. 2014;34(3):341–349. [DOI] [PubMed] [Google Scholar]
- 3.Carvalho DR, Brum JM, Speck-Martins CE, et al. Clinical features and neurologic progression of hyperargininemia. Pediatr Neurol. 2012;46(6):369–374. [DOI] [PubMed] [Google Scholar]
- 4.Burrage LC, Sun Q, Elsea SH, et al. Human recombinant arginase enzyme reduces plasma arginine in mouse models of arginase deficiency. Hum Mol Genet. 2015;24(22):6417–6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ballantyne LL, Sin YY, St. Amand T, et al. Strategies to rescue the consequences of inducible arginase-1 deficiency in mice. PLoS One. 2015;10(5):e0125967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Segawa Y, Matsufuji M, Itokazu N, et al. A long-term survival case of arginase deficiency with severe multicystic white matter and compound mutations. Brain Dev. 2011;33(1):45–48. [DOI] [PubMed] [Google Scholar]
- 7.Scheuerle AE, McVie R, Beaudet AL, et al. Arginase deficiency presenting as cerebral palsy. Pediatrics. 1993;91(5):995–996. [PubMed] [Google Scholar]
- 8.Harrington JW, Stiefel M, Gianos E. Arginase deficiency presenting with cerebral oedema and failure to thrive. J Inherit Metab Dis. 2000;23(5):517–518. [DOI] [PubMed] [Google Scholar]
- 9.Sin YY, Baron G, Schulze A, et al. Arginase-1 deficiency. J Mol Med (Berl). 2015;93(12):1287–1296. [DOI] [PubMed] [Google Scholar]
- 10.Wong D, Cederbaum S, Crombez EA. Arginase deficiency In: Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993–2018 [PubMed] [Google Scholar]
- 11.Therrell BL, Currier R, Lapidus D, et al. Newborn screening for hyperargininemia due to arginase 1 deficiency. Mol Genet Metab. 2017;121(4):308–313. [DOI] [PubMed] [Google Scholar]
- 12.Kanalas JJ, Spector EB, Cederbaum SD. Hollow-fiber reactors containing mammalian arginase: an approach to enzyme replacement therapy. Biochem Med. 1982;27(1):46–55. [DOI] [PubMed] [Google Scholar]
- 13.Carvalho DR, Brand GD, Brum JM, et al. Analysis of novel ARG1 mutations causing hyperargininemia and correlation with arginase I activity in erythrocytes. Gene. 2012;509(1):124–130. [DOI] [PubMed] [Google Scholar]
- 14.Adriaenssens K, Karcher D, Marescau B, et al. Hyperargininemia: the rat as a model for the human disease and the comparative response to enzyme replacement therapy with free arginase and arginase-loaded erythrocytes in vivo. Int J Biochem. 1984;16(7):779–786. [DOI] [PubMed] [Google Scholar]
- 15.Sakiyama T, Nakabayashi H, Shimizu H, et al. A successful trial of enzyme replacement therapy in a case of argininemia. Tohoku J Exp Med. 1984;142(3):239–248. [DOI] [PubMed] [Google Scholar]
- 16.Terheggen HG, Lowenthal A, Lavinha F, et al. Unsuccessful trial of gene replacement in arginase deficiency. Z Kinderheilkd. 1975;119(1):1–3. [DOI] [PubMed] [Google Scholar]
- 17.Lee EK, Hu C, Bhargava R, et al. Long-term survival of the juvenile lethal arginase-deficient mouse with AAV gene therapy. Mol Ther. 2012;20(10):1844–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gau CL, Rosenblatt RA, Cerullo V, et al. Short-term correction of arginase deficiency in a neonatal murine model with a helper-dependent adenoviral vector. Mol Ther. 2009;17(7):1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snyderman SE, Sansaricq C, Chen WJ, et al. Argininemia. J Pediatr. 1977;90(4):563–568. [DOI] [PubMed] [Google Scholar]
- 20.Cederbaum SD, Shaw KN, Spector EB, et al. Hyperargininemia with arginase deficiency. Pediatr Res. 1979;13(7):827–833. [DOI] [PubMed] [Google Scholar]
- 21.Prasad AN, Breen JC, Ampola MG, et al. Argininemia: a treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: case reports and literature review. J Child Neurol. 1997;12(5):301–309. [DOI] [PubMed] [Google Scholar]
- 22.Morioka D, Kasahara M, Takada Y, et al. Current role of liver transplantation for the treatment of urea cycle disorders: a review of the worldwide English literature and 13 cases at Kyoto University. Liver Transpl. 2005;11(11):1332–1342. [DOI] [PubMed] [Google Scholar]
- 23.Whitington PF, Alonso EM, Boyle JT, et al. Liver transplantation for the treatment of urea cycle disorders. J Inherit Metab Dis. 1998;21 Suppl 1:112–118. [DOI] [PubMed] [Google Scholar]
- 24.Sahin U, Kariko K, Tureci O. mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discov. 2014;13(10):759–780. [DOI] [PubMed] [Google Scholar]
- 25.Mignone F, Gissi C, Liuni S, et al. Untranslated regions of mRNAs. Genome Biol. 2002;3(3):REVIEWS0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chatterjee P, Basu S, Zubek J, et al. PDP-CON: prediction of domain/linker residues in protein sequences using a consensus approach. J Mol Model. 2016;22(4):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sauro HM. Control and regulation of pathways via negative feedback. J R Soc Interface. 2017;14(127). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sauro HM. Correction to ‘Control and regulation of pathways via negative feedback’. J R Soc Interface. 2017;14(129). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mavri-Damelin D, Eaton S, Damelin LH, et al. Ornithine transcarbamylase and arginase I deficiency are responsible for diminished urea cycle function in the human hepatoblastoma cell line HepG2. Int J Biochem Cell Biol. 2007;39(3):555–564. [DOI] [PubMed] [Google Scholar]
- 30.Michels VV, Beaudet AL. Arginase deficiency in multiple tissues in argininemia. Clin Genet. 1978;13(1):61–67. [DOI] [PubMed] [Google Scholar]
- 31.Crombez EA, Cederbaum SD. Hyperargininemia due to liver arginase deficiency. Mol Genet Metab. 2005;84(3):243–251. [DOI] [PubMed] [Google Scholar]