Loss of SATB1 induces p21-dependent cellular senescence in post-mitotic dopaminergic neurons (original) (raw)
. Author manuscript; available in PMC: 2020 Sep 16.
Published in final edited form as: Cell Stem Cell. 2019 Sep 19;25(4):514–530.e8. doi: 10.1016/j.stem.2019.08.013
Abstract
Summary
Cellular senescence is a mechanism used by mitotic cells to prevent uncontrolled cell division. As senescent cells persist in tissues, they cause local inflammation and are harmful to surrounding cells, contributing to aging. Generally, neurodegenerative diseases, such as Parkinson’s, are disorders of aging. The contribution of cellular senescence to neurodegeneration is still unclear. SATB1 is a DNA binding protein associated with Parkinson’s disease. We report that SATB1 prevents cellular senescence in post-mitotic dopaminergic neurons. Loss of SATB1 causes activation of a cellular senescence transcriptional program in dopamine neurons, both in human stem cell-derived dopaminergic neurons and in mice. We observed phenotypes which are central to cellular senescence in SATB1 knockout dopamine neurons in vitro and in vivo. Moreover, we found that SATB1 directly represses expression of the pro-senescence factor, p21, in dopaminergic neurons. Our data implicate senescence of dopamine neurons as a contributing factor in the pathology of Parkinson’s disease.
eTOC:
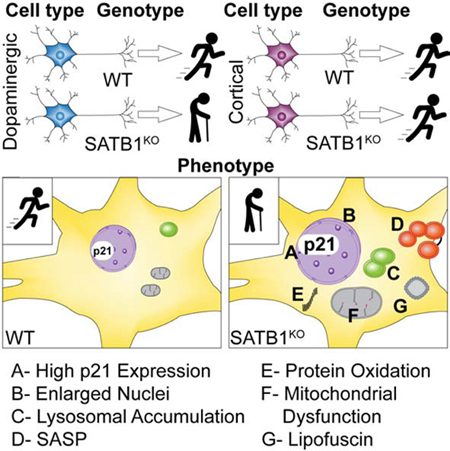
SATB1 is a genetic risk factor for Parkinson’s disease. Riessland and colleagues show that loss of SATB1 induces cellular senescence in mouse and human dopaminergic, but not cortical, neurons, and these senescent neurons secrete inflammatory factors that induce an immune response in mice.
Graphical abstract
Introduction
A key pathological hallmark of Parkinson’s disease (PD) is the progressive degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) of the midbrain. The loss of these cells causes the characteristic motor symptoms of the disease. In contrast, non-dopaminergic neuronal populations, such as cortical (CTX) neurons, are generally spared in the disease (Giguere et al., 2018; Kalia and Lang, 2015; Pedersen et al., 2005; Rajput and Rozdilsky, 1976). The molecular basis of PD remains elusive. Identification of causative Parkinson’s genes has shed light on the molecular mechanisms underlying the degeneration of DA neurons. The functions of many PD genes converge upon mitochondrial quality control (PINK1, PRKN) and lysosomal function (GBA) (Mullin and Schapira, 2015). Age, however, is the principal risk factor in both the sporadic and familial forms of the disease (Reeve et al., 2014). In line with this, mitochondrial and lysosomal dysfunction are both linked with aging (Folick et al., 2015; Tauchi and Sato, 1968).
As part of aging, organisms accumulate senescent cells within their tissues (Herbig et al., 2006). Although detrimental later in life, senescence is an important process in both embryogenesis and wound healing (Storer and Keyes, 2014). Senescent cells have lysosomal and mitochondrial abnormalities. They can damage or negatively impact surrounding cells through the senescence associated secretory phenotype (SASP) (reviewed in (Coppe et al., 2010)). Elimination of senescent cells increases both healthspan and lifespan (Baker et al., 2011). In addition to accumulating in healthy aged tissue, senescent cells can abnormally accumulate in disease states. Recent studies reported increased cellular senescence markers in the midbrain of PD patients and Alzheimer’s mouse models (Bussian et al., 2018; Chinta et al., 2018). Given that cellular senescence is characteristic of mitotic cells, the upregulation of senescence markers in the CNS was therefore attributed to the mitotic cells of the brain. There is however growing interest in cellular senescence in post-mitotic cells (Sapieha and Mallette, 2018). Multiple lines of evidence suggest that terminally differentiated neurons may shift to a state similar to senescence (Baker and Petersen, 2018; Jurk et al., 2012; Piechota et al., 2016).
Cellular senescence is a mechanism of cell cycle arrest, to protect from uncontrolled proliferation (Campisi, 2013; Kirkland and Tchkonia, 2017). Both DNA damage and oxidative stress are able to induce cellular senescence. Either of two pathways can initiate and maintain cellular senescence: the p53-p21-pRB and the p16-pRB pathways (Sultana et al., 2018). Induction of either of these converging pathways results in cell cycle arrest. Along with cell cycle arrest, senescent cells show a host of key phenotypes. These include resistance to apoptosis, reduced lysosomal function, increased senescence-associated β-galactosidase (SA-βGal) expression, lipofuscin accumulation, increased nuclear/cytoplasm ratio, loss of laminin B1, HMGB1 relocalization, secretion of damage-associated molecular patterns (DAMPs), damaged mitochondria, increased ROS formation, oxidative protein damage, and the SASP [Reviewed in (Martinez-Zamudio et al., 2017)]. SASP is the secretion of inflammatory cytokines, chemokines, growth factors, and proteases. The activation of SASP causes local inflammation and is harmful to surrounding cells, if the senescent cell is not removed by the immune system cells (reviewed in (Coppe et al., 2010)).
Special AT-Rich Sequence-Binding Protein 1 (SATB1) is a transcriptional regulator which has recently been identified as a risk factor for PD (Chang et al., 2017; Nalls et al., 2018). Previously, we have reported that the activity of SATB1 is reduced in the vulnerable brain region of PD patients (Brichta et al., 2015). SATB1 is a ubiquitously expressed chromatin organizing factor that confers cell type-specific patterns of transcription and mediates a transcriptional response of midbrain DA neurons to toxic insult.
Here we show that the genetic ablation of SATB1 induces a senescence phenotype in hESC derived DA neurons. In DA neurons lacking SATB1, we observed the hallmarks of cellular senescence. We found that SATB1 directly represses the expression of p21, a crucial determinant of cellular senescence. Inhibition of p21 in SATB1 knockout (KO) cells ameliorates the senescence phenotype. Contrastingly, elimination of SATB1 from cortical neurons did not induce senescence, or p21 expression. In vivo, the reduction of SATB1 in dopamine neurons caused p21 elevation and a local immune response. Finally, we found that p21 is actively expressed in the dopamine neurons of the SNpc of sporadic PD patients, rendering these cells prone to enter a state of senescence.
Results
hESC Derived DA Neurons Require SATB1
To produce pure DA neuronal cultures, we used an optimized DA neuron differentiation protocol (Figure 1A) (adapted from (Kriks et al., 2011; Miller et al., 2013; Steinbeck et al., 2015)), with the goal of obtaining high-purity DA neuron cultures. To achieve this high purity, we further generated a NURR1 (a postmitotic marker for DA neurons (Saucedo-Cardenas et al., 1998))-driven GFP expressing hESC line (NURR1-GFP cells). Using a CRISPR/Cas9 based knock-in approach, we replaced the endogenous NURR1-stop codon with P2A-H2B-GFP (Figure S1A). Using a combinatorial PCR strategy, we confirmed that the NURR1-reporter construct was integrated into the correct genomic locus as a single copy (Figure S1A). These NURR1-GFP cell-derived DA neurons co-express GFP with both NURR1 and tyrosine hydroxylase (TH) (Figure S1B). We then used FACS-based isolation of NURR1+ cells at day 25 of differentiation (Figure S1C). This enabled us to isolate a pure population of GFP+ cells with excellent survival in culture (Figure S1C–D). Single cell qRT-PCR data in differentiated NURR1-GFP cells demonstrated that upregulation of DA neuron markers occurred between day 25 and day 40 of differentiation. These data also confirmed that ~98.5% of the resulting NURR1-GFP cell-derived DA neurons express TH (Figure 1A, S1E), confirming that the sorted NURR1+ cells were highly pure and had the molecular characteristics of DA neurons (Figure 1A). To generate an isogenic SATB1 knockout in these NURR1-GFP cells, we used CRISPR/Cas9 to genetically eliminate SATB1. Sanger sequencing confirmed the successful mutagenesis of the SATB1KO clone, leading to the generation of frame shift mediated-premature stop codons (Figure 1B, S1F). To exclude the possibility of off-site targeting of CAS9 by our gRNA, we used CAS-OFFinder (Bae et al., 2014) to predict putative off target sites. Sanger sequencing confirmed no mutations at these sites in the SATB1KO cells (Table S1). SATB1 protein was eliminated from the hESC SATB1KO cells, which retained normal cellular appearance in culture (Figure 1C, D).
Figure 1. hESC Derived DA Neurons Require SATB1 to Maintain their Identity.

(A) Overview of the differentiation protocol to generate highly enriched cultures of mature DA neurons. Distribution of DA neuron population post FACS, as determined by single-cell qRT-PCR (89 cells analyzed). Immunofluorescent staining of the sorted NURR1-GFP+ neurons with anti-TH and anti-MAP2 showing the high yield of TH+ neurons following differentiation. (B) Amino acid sequence of SATB1 after CRISPR/CAS9 mediated SATB1 knockout reveals the introduction of premature stop codons leading to loss of SATB1 expression. (C) Western blot analysis to confirm SATB1KO in hESC on protein level. (n=6) (D) Microscopic bright light image of SATB1KO hESC shows normal appearance. Scale bar: 200 μm. (E) Immunolabeling of DA precursor markers in WT and SATB1KO hESC derived DA neurons after 16 days of differentiation. (F) Depiction of spontaneous action potentials of WT and SATB1KO DA neurons at different time points show that both genotypes differentiate into mature DA neurons between day 35 and 40. Scale bar: 20 mV, 2 s. (G) DA neurons at day 40 show significant differences in maintenance of response to positive current injections. Scale bar: 20 mV, 200 ms. (5–8 cells recorded per dish, data derived from 3 independent experiments) (H) Longitudinal comparison of cell survival of WT vs. SATB1KO DA neurons revealed a significant reduction in SATB1KO survival between day 30 and 40, reaching a plateau at ~50%. n=12, data are represented as mean ± SEM. (I) Quantification of neurite morphology and complexity in WT and SATB1KO DA neurons. n=24, Data are represented as mean ± SEM. (***p<0.001). See also Figure S1 and Table S1.
Using this approach, we differentiated SATB1KO NURR1-GFP cells into mature DA neurons. Following floor plate differentiation, SATB1KO NURR1-GFP cells expressed key markers such as OTX2, FOXA2 and LMX1A. These markers define the cells as midbrain floor plate precursors at day 16 of differentiation (Figure 1E). To track the functional maturation of the hESC-derived DA neurons, we performed electrophysiological recordings at different time points. Neither WT nor SATB1KO neurons showed spontaneous action potentials at day 30 of differentiation. Cells of both genotypes on day 36 of differentiation started to display spontaneous action potentials, evoked action potentials, as well as voltage-dependent Na+ and K+ currents (Figure 1F, S1G). Finally, the frequency of action potentials of both genotypes was equally increased at day 40 of differentiation (Figure 1F). These results show that the differentiated SATB1KO cells exhibited features of mature DA neurons. However, SATB1KO neurons were unable to maintain the response to positive current injections (Figure 1G).
Previously, our lab reported that the knockdown of SATB1 in the midbrain is sufficient to cause degeneration of DA neurons in mice (Brichta et al., 2015). We therefore monitored the survival of SATB1KO DA neurons during maturation. SATB1KO DA neurons showed a significant reduction in cell numbers starting at day 40 of differentiation (Figure 1H). Interestingly, we observed that the cell loss in SATB1KO cultures stabilized by day 40 and was maintained at that level over the 60 days of differentiation. We speculate that the loss of live SATB1KO DA neurons is at least in part a consequence of detachment from the culture dish due to fundamental reorganization of their gross morphology including the cytoskeleton. Consistent with this, we found that surviving SATB1KO DA neurons showed significantly decreased neurite outgrowth and complexity at day 60, following normal development at earlier days of differentiation (Figure 1I).
SATB1 Acts Predominantly as a Gene Repressor in DA Neurons.
To understand the functional role of SATB1 in DA neurons, we performed concurrent RNA-Seq and ChIP-Seq experiments (Figure 2A). We used ChIP-Seq to compare the genome-wide binding profiles of SATB1 in early and mature DA neurons (Figure 2B). We found that SATB1-binding had the highest intensity in mature DA neurons. We confirmed this finding by analysis of the expression profile changes caused by SATB1KO in DA neurons.
Figure 2. SATB1 Plays Discrete Regulatory Roles in early and mature DA Neurons.

(A) Outline of the experimental approach comparing expression, DNA-binding, and regulator profile of SATB1 in DA neurons. (B) Genome-wide heatmaps of SATB1-ChIP-Seq experiments comparing binding patterns in early and mature DA neurons (ChIP-Seq experiments performed in 4 independent experiments). RNA-Seq expression profile comparing WT vs. SATB1KO of early DA neurons (C) (n=4) and mature DA neurons (D) (n=3). Red dots indicate significantly changed genes (FDR < 0.05, > ±2-fold expression change). BETA plots of combined computational analysis of SATB1-ChIP-Seq and RNA-Seq data of early DA neurons (E) and mature DA neurons (F). Black line: static background, red line: repressive function, blue line: activating function. See also Figure S2.
Comparison of WT and SATB1KO DA neurons at an early timepoint (day 30) revealed few changes in gene expression (Figure 2C). At this timepoint, the cells were phenotypically comparable to WT. At day 50 of differentiation, when surviving SATB1KO neurons showed a phenotype, much greater gene expression changes were observed (Figure 2D). The KO of SATB1 has a more dramatic effect in mature DA neurons than in early DA neurons. Next, we used the binding and expression target analysis (BETA) software (Wang et al., 2013) to incorporate the ChIP-Seq and RNA-Seq data. This analysis showed that SATB1 has no significant effects as a gene regulator in early DA neurons (Figure 2E). In mature DA neurons SATB1 acts as a gene repressor (p = 0.000236) (Figure 2F). Interestingly, network analysis of enriched gene ontologies (GO) in DA neurons revealed that the loss of SATB1 activates connected transcriptional programs that underlie cytoskeleton remodeling as seen in (Figure 1I, S2). Surprisingly, in these postmitotic cells, ontologies related to the negative regulation of cell proliferation were enriched (Figure S2).
Loss of SATB1 in Dopamine Neurons Results in a Senescence Phenotype
Amongst the GO pathways enriched in SATBKO versus WT DA neurons, we found the cellular senescence pathway. The DA neuron enrichment was further confirmed by GSEA of the mature SATB1KO DA neuron transcriptome (Figure 3A). Given this, we sought to investigate if SATB1KO DA neurons present the classical features of cellular senescence. First, we observed a dramatic increase in acidic lysosomal senescence associated beta-Galactosidase (SA-βGal) activity, the hallmark senescence biomarker (Figure 3B). Another key feature of senescent cells is the activation of the SASP. To determine if SATB1KO DA neurons present this phenotype, we evaluated the expression of the described key SASP factors (Coppe et al., 2008). We found an upregulation of the majority of the SASP factors at 50 days of differentiation in the SATB1KO DA neurons versus WT neurons (Figure 3C). We confirmed SASP activation by western blotting. In the conditioned media of SATB1KO neurons, we found IGFBP7, which was absent in the media of WT neurons (Figure 3D). In fact, secretion of IGFBP7 alone is capable of inducing cellular senescence in surrounding cells (Severino et al., 2013). Another well described phenotype of cellular senescence is an increase in nucleus diameter correlating with reduced laminin B1 expression. Using automated high-content imaging, we evaluated nucleus size of SATB1KO DA neurons in comparison to controls. We found a consistent, and significant increase in nucleus diameter in the DA neurons lacking SATB1 (Figure 3E). We also found a corresponding significant decrease in lamin B1 expression (Figure 3F). Furthermore, we found electron dense lipofuscin accumulations in the cytoplasm of SATB1KO DA neurons (Figure 3G). Senescence-associated mitochondrial dysfunction leads to the elevation of reactive oxygen species, which in turn leads to oxidative protein damage. Using an OxiSelect™ Protein Carbonyl ELISA assay, we found that SATB1KO DA neurons show increased oxidized proteins compared to WT (Figure 3H). These findings were confirmed using a GSH/GSSG ratio detection assay and OxyBlot (Figure S3).
Figure 3. Loss of SATB1 in Dopamine Neurons Results in a Neuronal Senescence Phenotype.

(A) Diagrammatic representation of gene enrichment significantly correlating with the expression profile of cellular senescence. (B) Representative microscopic overview images of WT and SATB1KO neurons subjected to X-Gal staining. Blue cells are senescence cells. Bar graph shows significant increase of SA-βGal positive cells. n=4, data are represented as mean ± SEM. (C) Heatmap of genes associated with SASP of RNA-Seq data from Figure 2. (D) Western blot of cell culture supernatant of mature DA neurons. The SASP factor IGFBP7 is secreted from SATB1KO neurons. Ponceau S stain serves as loading control (n=3). (E) Representative images from high-quantity imaging of nuclei in WT and SATB1KO DA neurons (n=24). Scale bar: 5 μm. Bar graph shows significant increase of nucleus size in SATB1KO neurons. (F) Representative western blot and quantification of lamin B1 from WT and SATB1KO DA neurons. n=4, data are represented as mean ± SEM. (G) Representative TEM-image of the electron-dense lipofuscin observed in SATB1KO neurons. (H) Quantification of OxiSelect™ Protein Carbonyl ELISA assay, which was performed with protein from WT and SATB1KO DA neurons (n=8). (I) Bar graph shows cell viability of WT and SATB1KO DA neurons following treatment with diverse senolytic compounds (Azi = Azithromycin, D = Dasatinib, Q= Quercetin) (*p<0.05, ***p<0.001). See also Figure S3.
Another key feature of senescent cells is the inherent sensitivity to senolytics drugs. To test whether the SATB1KO DA neurons are susceptible to senolytics, we treated the WT and the KO cells with senolytic drugs that target disparate molecular pathways, azithromycin (Ozsvari et al., 2018), fisetin (Yousefzadeh et al., 2018), ABT-737 (Yosef et al., 2016) and the dasatinib plus quercetin cocktail (Zhu et al., 2015). After verifying the efficacy in senescent fibroblasts, we tested the compounds in DA neurons (Figure S3 B–C). Compared to WT controls, SATB1KO DA neurons showed significantly lower cell viability under azithromycin, fisetin, and the dasatinib plus quercetin treatment. Neurons of both genotypes were affected by ABT-737, implying a toxic effect on DA neurons (Figure 3I). This is in line with previous work demonstrating that Bcl-xL, the pharmacological target of ABT-737, is essential to neuronal survival (Nakamura et al., 2016).
Dopamine Neurons Lacking SATB1 Have Impaired Lysosomal and Mitochondrial Function
SA-β-Gal activity is indicative of altered function and enlargement of lysosomal compartments. Analysis of RNA-Seq data show a drastic change in lysosomal gene expression in SATB1KO DA neurons (Figure 4A). Based upon these molecular alterations, and given the reported connections between lysosomal alterations and senescence, we performed a detailed characterization of lysosomal function. Using LysoTracker dye and automated high-content imaging, we found that SATB1KO DA neurons showed a significant increase in endo/lysosomal content, a well characterized feature of senescent cells (Figure 4B). To confirm the increase in endo/lysosomal content, we quantified the protein level of the endo/lysosomal marker LAMP1. In line with the imaging data, LAMP1 levels were significantly increased in SATB1KO DA neurons (Figure 4C). Given the structural, ultrastructural (Figure S4A–C) and molecular changes in the lysosomes of SATB1KO DA neurons, we evaluated the lysosomal function in these cells. To assess lysosomal degradation capacity, we used two representative activity assays for enzymes that play important roles in lysosomal degradation. Both the cathepsin D and the β-glucosidase activity assays were significantly altered, implicating severe lysosomal dysfunction (Figure 4D). The observed increase of oxidized proteins suggested mitochondrial dysfunction, so we assessed the function of these organelles in SATB1KO DA neurons. We first quantified ultrastructural alterations in the mitochondria of SATB1KO DA neurons by electron microscopy (EM). We found that the mitochondria in these neurons were damaged and had an abnormal appearance (Figure 4E, F). The evaluation of functioning mitochondria using MitoTrackerCMX, and oxygen consumption by Seahorse respirometry, revealed a significant reduction in basal respiration and ATP production in SATB1KO DA neurons (Figure 4G–I). Moreover, the assembly of mitochondrial complexes was greatly impaired in SATB1KO DA neurons. This was also reflected in the total protein level of the complexes which were altered in SATB1KO DA neurons, although to a lesser extent (Figure 4J, S4D, E).
Figure 4. Dopamine Neurons Lacking SATB1 have impaired Lysosomal and Mitochondrial Function.

(A) Heatmap of the RNA expression of lysosomal genes comparing WT and SATB1KO neurons at day 30 of differentiation. (B) Representative images of LysoTracker staining of WT and SATB1KO neurons. Bar graph shows significant increase of lysosomal content in SATB1KO neurons. n=8, scale bar: 100 μm. Data are represented as mean ± SEM. (C) Representative western blot of mature DA neurons. Bar graph shows significantly increased LAMP1 levels in SATB1KO neurons. n=6, data are represented as mean ± SEM. (D) Bar graphs of cathepsin D and GCase assays. n=5, data are represented as mean ± SEM. (E) Representative TEM-images of mitochondria in mature DA neurons. Green arrow marks normal, yellow marks affected and red arrow marks severely affected mitochondria, quantified in (F) (n=203 mitochondria from 20 WT cells and n=222 mitochondria from 32 SATB1KO cells). Analysis of cellular respiration identified reduced basal respiration (G) and ATP production (H) in SATB1KO neurons (n=10). Data are represented as mean ± SEM. (I) Representative images of MitoTracker CMX stain shows decreased membrane potential in SATB1KO neurons. (J) Native-PAGE of mitochondrial complex integrity using anti-NDUFA9 (Complex I), anti-UQRC2 (Complex III) and anti-MTCO2 (Complex IV) antibodies. (*p<0.05, **p<0.01, ***p<0.001). See also Figure S4.
The Senescence Effects Seen in DA Neurons Lacking SATB1 are not Observed in CTX Neurons
To determine the cell type-specificity of the senescent phenotype, we differentiated the same KO hESC clone into CTX neurons. After cortical differentiation, both WT and SATB1KO expressed the relevant differentiation markers of layer IV and V CTX neurons (Figure 5A). We verified the purity of CTX neuronal cultures by expression profiling, confirming the absence of glial marker expression (Figure S5A) and the efficacy of differentiation by the absence of Ki67 positive cells (Figure 5A). During the course of differentiation, survival of SATB1KO CTX neurons was unaffected (Figure S5C). Both neurite outgrowth and complexity showed a slight delay in development, but both were able to achieve the same level (Figure S5D–F). Taken together, these findings suggest that the loss of SATB1 had distinct physiological effects in these neuronal subtypes. We next performed RNA-Seq and ChIP-Seq experiments in CTX neurons, to further understand the distinct function of SATB1 in comparison to DA neurons. CTX neurons showed very few expression changes caused by SATB1KO (Figure 5B). The effect of SATB1-KO on the overall expression profile in CTX neurons was neglectable since we found that there was almost no overlap in gene expression changes between the DA and CTX neurons (Figure S5B). The combination of RNA-Seq and ChIP-Seq (Figure 5C) data, using the BETA software in CTX neurons, revealed that SATB1 has both repressive (p=0.00791) and activating (p=2.97e−05) functions (Figure 5D), although it regulates far fewer genes in comparison to DA neurons (Figure 2B). Since the expression analysis did not reveal any features of cellular senescence in CTX SATB1KO neurons, we assessed all previously described senescence-associated phenotypes that were observed in DA neurons. The expression data did not reveal any SASP activation in SATB1KO CTX neurons (Figure 5E). Remarkably, one of the major hallmarks of cellular senescence, SA-βGal activity, was virtually absent in SATB1KO and WT CTX neurons (Figure 5F). In addition, CTX SATB1KO neurons do not show any increase in nuclear size or changes in lamin B1 expression (Figure S5H). Furthermore, we did not observe any changes in the endo/lysosomal content of SATB1KO CTX neurons (Figure 5G). Finally, KO of SATB1 in CTX neurons did not alter components of the mitochondrial complexes (Figure S5G). This finding was reflected in cellular respiration and ATP production that remained unchanged in SATB1KO CTX neurons (Figure 5H, I).
Figure 5. CTX Neurons lacking SATB1 do not have a Senescence Phenotype.

(A) Triple immunolabeling of cortical shows that both WT as well as SATB1KO neurons express the essential markers for cortical neurons. Scale bar: 50 μm. (B) RNA-Seq expression profile comparing WT vs. SATB1KO CTX neurons (n=4). Red dots indicate significantly changed genes (FDR < 0.05, > ±2-fold expression change). (C) Genome-wide heatmap of SATB1-ChIP-Seq experiment of CTX neurons (ChIP-Seq experiments performed in 4 independent experiments). (D) BETA plot of combined computational analysis of SATB1-ChIP-Seq and RNA-Seq data of CTX neurons. Black line: static background, red line: repressive function, blue line: activating function. (E) Heatmap of genes associated with SASP. (F) Representative microscopic overview images of WT and SATB1KO neurons subjected to X-Gal staining (n=6). Blue cells are senescence cells. Bar graph shows low levels and no change in numbers of SA-βGal positive cells. Data are represented as mean ± SEM. (G) Representative confocal images of LysoTracker staining of WT and SATB1KO CTX neurons. Bar graph shows no significant change of lysosomal content in SATB1KO CTX neurons. n=8, data are represented as mean ± SEM. Analysis of cellular respiration identified no alteration of basal respiration (H) and ATP production (I) in SATB1KO CTX neurons. n=10, data are represented as mean ± SEM. See also Figure S5.
SATB1 Repression of CDKN1A Prevents Senescence in DA Neurons
We next sought to determine the molecular basis of the link between SATB1 and the observed cellular senescence phenotype in SATB1KO DA neurons. Protein-protein interaction network analysis identified CDKN1A as a critical node likely mediating the molecular profile of the DA neurons (Figure S6A, B). This gene encodes for p21, a critical regulator of senescence. Both CDKN1A transcription and p21 protein levels were significantly increased in SATB1KO DA neurons (Figure 6A, B). Using ChIP-Seq in DA neurons, we found that SATB1 binds the regulatory region of CDKN1A (Figure 6F). Taken together, these data indicate a repressive function of SATB1 at this locus. The elimination of SATB1, and the resulting de-repression of CDKN1A, is thus sufficient to elevate cellular p21 levels. In line with these findings, SATB1 has previously been described to regulate cellular senescence in keratinocytes (Lena et al., 2012), and has also been previously described to repress the promoter of CDKN1A (Wang et al., 2014).
Figure 6. SATB1 Repression of CDKN1A Prevents Senescence in DA Neurons.

(A) Bar graph shows significant upregulation of CDKN1A expression in SATB1KO DA neurons. n=3, data are represented as mean ± SEM. (B) Western blot probed for p21, p53, gamma H2AX, and b-actin comparing WT and SATB1KO DA neurons (n=5). See also Figure S6C. Bar graph shows significant upregulation of p21. Data are represented as mean ± SEM (C) Representative western blot and quantification of p21 protein level in lentiviral transduced SATB1KO DA neurons. n=3, data are represented as mean ± SEM. (D) Representative microscopic images of SATB1KO neurons subjected to X-Gal staining with and without p21-shRNA transduction. Bar graph shows significant decrease of percentage of SA-βGal positive SATB1KO neurons subjected to p21 knockdown (n=6). Data are represented as mean ± SEM. (E) Heat maps of ATAC-Seq experiments comparing open chromatin patterns in WT and SATB1KO mature DA neurons (ATAC-seq done in independent triplicates). (F) The CDKN1A gene overlaid with ATAC-Seq enrichment tracks from WT and SATB1KO DA neurons as well as SATB1-ChIP-Seq from WT DA neurons. (**p<0.01, ***p<0.001, n.s., not significant). See also Figure S6.
We next sought to determine the direct role of p21 in inducing the senescent phenotype in SATB1KO DA neurons. To do so, we used a lentiviral approach to specifically downregulate the expression of CDKN1A in SATB1KO DA neurons. Indeed, the CDKN1A shRNA treatment significantly reduced p21 levels in SATB1KO DA neurons (Figure 6C). p21 reduction in SATB1KO DA neurons was able to significantly reduce the number of SA-βGal positive cells (Figure 6D). Importantly, knockdown of CDKN1A in DA neurons did not induce proliferation (Figure S6D). In addition to the viral knockdown, we found that chemical reduction of p21 by UC2288, resulted in a similar rescue (Figure S6E, F). These results suggest that SATB1 loss-mediated elevation of p21 causes the senescent phenotype in SATB1KO DA neurons. To determine if SATB1 directly represses CDKN1A, we performed a luciferase reporter assay, and found that under conditions of SATB1 knockdown, CDKN1A promoter activity was significantly increased (Figure S6J). To further confirm the specificity of the CDKN1A regulation by SATB1, we performed ATAC-Seq experiments in WT and SATB1KO DA neurons. The ATAC-Seq approach revealed that the global open chromatin structure is not changed in SATB1KO cells (Figure 6E), indicating a specific mode of gene regulation by the protein in these cells. Indeed, comparing the CDKN1A locus between WT and SATB1KO DA neurons, the ATAC-Seq shows a more opened chromatin structure which overlaps with the SATB1 ChIP-Seq binding profile (Figure 6F).
While SATB1KO caused an increase of p21 protein level in DA neurons, it did not change CDKN1A transcription or protein levels of p21 in CTX neurons (Figure S6H, I). Importantly, we found that the CDKN1A locus is more open in WT DA neurons compared to CTX neurons, suggesting a DA neuron-specific role for this gene (Figure S6G). This finding implies an additional mechanism of transcriptional regulation of p21. During the process of p21-mediated cellular senescence, transcription factors such as SMAD3 or FOXO1 increase CDKN1A gene expression (Martinez-Zamudio et al., 2017). Both SMAD3 and FOXO1 play crucial regulatory roles in DA neurons (Doan et al., 2016; Tapia-Gonzalez et al., 2011). Correspondingly, their expression is higher in DA compared to CTX neurons (Figure S6K). Our data suggest that CDKN1A gene expression in DA neurons is under constant influence of promoting factors which aim to increase cellular p21 levels, whereas in CTX neurons this is not the case. SATB1 is therefore an important repressor of CDKN1A transcription (Wang et al., 2014) and regulates the p21 level in DA neurons. In these cells, p16 protein level was undetectable (Figure S6L). This suggests that the direct regulation of SATB1 on p21 expression is sufficient to induce the senescence phenotype. This is in line with the observation that p21 expression is indeed sufficient to induce senescence (Capparelli et al., 2012).
Importantly, to validate the in vitro findings generated with the human stem cell-derived CTX and DA cells, we generated another, independent SATB1KO stem cell clone. We found again that, when we differentiated the SATB1KO clone into DA neurons, the cells senesced, whereas CTX neurons did not show any signs of cellular senescence (Figure S7A–C).
In vivo Reduction of SATB1 Induces Senescence of DA Neurons
Next, in order to validate the human in vitro findings, we shifted to an in vivo mouse model. In this model, we sought to investigate whether the reduction of SATB1 triggers p21 expression and subsequent senescence in vivo. Using stereotactic adeno-associated virus 1 injection expressing short hairpin RNA (AAV1-shRNA), we downregulated the endogenous Satb1 in the neurons of the midbrain of mice (Figure 7A). As previously reported, this knockdown of Satb1 results in an elimination of TH+ neurons three to four weeks after injection (Brichta et al., 2015). We investigated the midbrains of these mice two weeks after injection and found a reduction of TH protein level as assayed by bacTRAP and immunostaining in the remaining DA neurons (Figure 7B, S7D). Microscopic analysis of the midbrains further showed reduction of mitochondrial content and accumulation of lipofuscin, similar to what we have observed in the stem cell-derived SATB1KO DA neurons (Figure S7G, H). Analyzing DA neuron-specific bacTRAP expression data (Brichta et al., 2015), we found that Cdkn1a was amongst the highest upregulated genes in Satb1-knockdown mice the second highest upregulated gene to be Cdkn1a and observed an increase in genes related to the SASP (Figure 7C, E). Moreover, when we stained the remaining DA neurons for expression of p21, we found a significant increase in p21 positive DA neurons (Figure 7D). Importantly, we found no expression of p16/INK4 in control as well as Satb1-knockdown DA neurons (Figure S7F). To assess whether SASP in DA neurons triggers an immune response in the midbrain, we stained for the microglial marker Iba-1. Strikingly, confocal microscope analysis of Satb1-knockdown midbrains two weeks after virus injection revealed that numerous microglia co-localized with the TH+ DA neurons, suggesting a release of immune factors to attract immune cells (Figure 7F, S7E). This mechanism is in line with the finding of significantly elevated expression levels of immune factors (Figure 3C, 7E). To confirm that the SASP in these animals activated the surrounding microglia, we adapted a previously reported method to characterize the activation status of microglia based upon their roundness and cell size (Davis et al., 2017). Using this approach, we found a significantly increased percentage of activated microglia in Satb1-knockdown substantia nigra (Figure 7F, S7F), which target Satb1 depleted DA neurons (Figure S7E).
Figure 7. In vivo reduction of SATB1 induces senescence of DA Neurons.

(A) Virus injection strategy. (B) Representative confocal images of TH and Satb1 staining of control-shRNA and _Satb1_-shRNA TH+ neurons in the SNpc, 2 weeks after injection. Scale bar: 50 μm (C) DAT-bacTRAP-based expression profile changes in DA neurons comparing control-shRNA and _Satb1_-shRNA (n=3) (Data from Brichta et al. 2015). Cdkn1a is amongst the highest upregulated transcripts. (D) Representative confocal images of TH and p21 staining of control-shRNA and _Satb1_-shRNA TH+ neurons in the SNpc, 2 weeks after injection. Scale bar: 50 μm (E) Heatmap of mouse genes associated with SASP (n=3) (Data from Brichta et al. 2015). (F) Representative confocal tile scan images of TH and Iba-1 staining of control-shRNA and _Satb1_-shRNA TH+ neurons in the SNpc, 2 weeks after injection. Scale bar: 100 μm. Corresponding bar graphs show the quantification of high activity microglia, and colocalization between Iba-1 and TH signals, in the SNpc, comparing control-shRNA and _Satb1_-shRNA. n=5, data are represented as mean ± SEM. (G) Representative confocal images of TH and p21 staining of human brain slices of the SNpc derived from age-matched control individual or sporadic PD patient. Bar graph shows quantification of intensity of p21 signal in TH+ SNpc neurons of four age-matched control individuals and four PD patients. Scale bar: 50 μm, 5 μm. Data are represented as mean ± SEM. (*p<0.05). See also Figure S7.
Using a regulatory network analysis of expression data from substantia nigra samples of human subjects with incipient PD, we have previously reported that the regulatory function but not the expression level of SATB1 is significantly decreased (Brichta et al., 2015; Zheng et al., 2010). Based on this finding and the above described discoveries, we analyzed the expression of p21 in human sporadic PD patient midbrain slices (obtained from the Harvard Brain Tissue Resource Center). Crucially, we found a significant increase in the intensity of p21 staining within TH+ DA neurons in PD patient brain slices compared to age-matched controls (Figure 7G). This higher level of p21 shows that PD DA neurons are vulnerable to senescence. Additionally, we found a significant decrease in lamin B1 levels in the PD brain samples (Figure S7I). However, we could not detect any significant difference in SAHF (Figure S7J). These findings potentially link aberrant SATB1 regulation of CDKN1A to PD.
Discussion
The general understanding of cellular senescence is tied to its initial discovery in mitotic cells more than half a century ago. Hayflick and Moorhead (1961) discovered that primary cells in culture undergo a limited number of cell divisions. These cells then reach a state of replicative senescence and can no longer regenerate tissue (Hayflick, 1965). Since then, cellular senescence and the associated SASP have been thoroughly investigated, and many additional biological roles have been identified. Senescence has an established function in cancer prevention by triggering growth arrest and by signaling to the immune system to remove incipient cancerous cells (Acosta et al., 2013; Acosta et al., 2008; Georgilis et al., 2018; Kang et al., 2011). In addition, senescence and SASP have been shown to contribute to development and tissue repair (reviewed in (Munoz-Espin and Serrano, 2014)). If immune cells do not remove senescent cells, their constant pro-inflammatory SASP can be toxic to surrounding cells and therefore has been associated with multiple age-related diseases (Franceschi and Campisi, 2014). Recent studies have suggested that not only mitotic, but also postmitotic cells are capable of entering a state of senescence. This has been observed in a number of cell types from the CNS (Jurk et al., 2012), bone (Farr et al., 2017), cardiac (Wang et al., 2016), and adipose tissue (Minamino et al., 2009).
Evidence suggests that neurons can enter a state of cellular senescence (Baker and Petersen, 2018; Tan et al., 2014). A number of pathological features in the brains of Alzheimer’s and Parkinson’s disease patients overlap with phenotypes observed in cellular senescence (Tan et al., 2014). Whether neurons contribute to these disease features has been an open question. Indeed, some studies revealed progressive activity of SA-βGal in cultured cerebellar granular neurons and hippocampal neurons (Bhanu et al., 2010; Bigagli et al., 2016; Green et al., 2017). However, there has been a debate as to whether SA-βGal activity by itself is a sufficient marker for cellular senescence (Severino et al., 2000).
In the present study, we have utilized nearly pure human stem cell-derived DA neurons, a mouse model, and sporadic PD patient brain slices to establish that SATB1 is a regulator of cellular senescence in DA neurons. We show that SATB1KO DA neurons show virtually all established features of cellular senescence. This includes activation of the SASP, SA-βGal activity, lysosomal dysfunction, lipofuscin accumulation, damaged and dysfunctional mitochondria, enlargement of the nucleus, increased oxidative protein damage, and expression pathway changes towards cellular senescence. Interestingly, we did not observe increased DNA damage in the senescent cells. However, we found that SATB1 binds directly to the regulatory region of CDKN1A repressing its expression, which is in accordance with previous findings (Wang et al., 2014). Moreover, inhibition of CDKN1A in SATB1KO DA neurons significantly reduced the number of senescent cells, demonstrating that the direct regulation of p21 levels in SATB1KO DA neurons triggers the senescence phenotype. Strikingly, when the SATB1KO stem cell clone was differentiated into CTX neurons, none of the senescence phenotypes were observed.
The SATB1-dependent repression of CDKN1A transcription seems crucial to DA neuron function. Loss of SATB1 in DA neurons caused a ~300% elevation in CDKN1A transcription, but did not significantly alter levels in CTX neurons. We speculate that the CDKN1A locus is under a constant transcriptional activation pressure in DA neurons compared to CTX neurons, which is reflected by significantly higher expression of _CDKN1A_-promoting factors (Figure S5I, J; (Tinkum et al., 2013)). These findings suggest that the intrinsic properties of DA neurons, be they structural (size, branching), energetic demand, or high stress level (Pacelli et al., 2015) may render these cells vulnerable to cellular senescence.
As previously reported, the transcriptional activity of SATB1 decreases in incipient PD patient brains (Brichta et al., 2015). This suggests that a p21-induced senescence-like phenotype may be present in human PD brains. Indeed, we describe here a significant increase in p21 levels in DA neurons in the SNpc of PD patients in comparison to age-matched controls. There have been a number of studies which revealed phenotypes in PD brains which could be attributed to cellular senescence (Tan et al., 2014): the discovery of elevated inflammatory factors in the CSF of PD patients for example could be linked to SASP (Blum-Degen et al., 1995). Other studies found evidence for dysfunctional lysosomes in the PD brain (Chu et al., 2009; van Dijk et al., 2013). Also, the accumulation of lipofuscin, increased SA-βGal activity, and enlargement of mitochondria have been described in PD (Braak et al., 2003; Trimmer et al., 2000; van Dijk et al., 2013). However, none of the above-mentioned studies have connected these findings to neuronal senescence (Tan et al., 2014). Evidence of a p21-dependent cellular senescence in PD is supported by genetic mouse models of the disease. Neural stem cells from Prkn−/− mice show an increase of p21 protein levels (Park et al., 2017). In line with this finding, LRRK2 mutant animals show robust increases in Cdkn1a transcript levels (Nikonova et al., 2012). These findings further link familial forms of PD to the p21 pathway.
PD is an age-related disorder. Both sporadic and most familial cases of PD will usually develop the disease later in life. Age is therefore considered the most important contributing risk factor for PD. Critically, the accumulation of senescent cells is also clearly linked to increased age (Soto-Gamez and Demaria, 2017). In young individuals, senescent cells which show a SASP will be detected and eventually removed by the immune system. Yet, with age, as immune surveillance decreases, senescent cells can escape the immune system, a phenomenon directly linked to aging (van Deursen, 2014). The prolonged presence of senescent cells in the tissue is problematic since the SASP secretes pro-inflammatory factors which can trigger local inflammation and spread the senescence phenotype in a paracrine fashion (Acosta et al., 2013). Should cellular senescence be a step preceding the loss of SNpc cells during the development of PD, this could explain why disease symptoms occur before the loss of soma in the midbrain. DA neurons which enter senescence would lose function but would remain in the midbrain until microglia remove them. This would be similar to what we observed in our Satb1-knockdown mouse model (Figure 7F). In aged PD patients, it is possible to envision that senescent DA neurons would not be removed. Instead, they would cause a local inflammation and spreading of senescence, resulting in the expression of the respective markers, which indeed have been identified in PD brains (Tan et al., 2014).
The ultimate confirmation that cellular senescence contributes to aging and age-related disorders came from animal studies which showed that removal of senescent cells has a beneficial effect on the disease phenotype and lifespan (Baker et al., 2016; Baker et al., 2011; Xu et al., 2018). These discoveries are driving the field of the development of drugs which reduce aging by removal of senescent cells. These drugs, termed senolytics, have already been able to improve age-related phenotypes in mice (Xu et al., 2018; Zhu et al., 2015). Based on our finding that SATB1 regulates cellular senescence in DA neurons, which respond to senolytic-treatment (Figure 3I), we hypothesize that SATB1 itself could be a promising target for novel senolytics. Moreover, therapeutic strategies targeting SATB1 or p21 in PD may be a beneficial route to intervention.
STAR Methods
Lead Contact and Materials Availability
Requests for reagent and resource sharing should be addressed to the Lead Contact, Markus Riessland (mriessland@rockefeller.edu) who will fulfil requests. Engineered cell lines are available by contacting the SKI (stem cell research facility at MSKCC, stemcells.mskcc.org) or Lorenz Studer (studerl@mskcc.org).
Experimental Model and Subject Details
hESC culture and differentiation
The human embryonic stem cells [ESCs; wild-type H9 (WA-09), SATB1KO, NURR1-GFP reporter line] were maintained using E8-essential medium (Fisher Scientific) on Vitronectin coated dishes (VTN-N; Fisher Scientific) under feeder-free conditions and passaged every 4–5 days by EDTA. Cortical differentiation (Maroof et al., 2013; Qi et al., 2017) and midbrain dopamine (mDA) differentiation (Chambers et al., 2009; Kriks et al., 2011) from hESCs were carried out following optimized versions of our previously published protocols (Kriks et al., 2011; Qi et al., 2017). Briefly, hESCs were dissociated into single cells using Accutase, and plated at high density on Matrigel (Corning). For cortical neuron patterning, timed exposure to LDN193189, SB431542, XAV939 was used (until day 7) to trigger FOXG1+/PAX6+ precursors for cortical neuron differentiation while timed exposure to LDN193189, SB431542, SHH C25II, and CHIR99021 was used (until day 10) to induce FOXA2+/LMX1A+ midbrain floor plate precursors for mDA neuron differentiation. For mDA neuron induction, floor plate precursors were maintained in mDA differentiation media containing Neurobasal (Life Technologies)/B27(Life Technologies)/L-glutamine supplemented with BDNF (brain-derived neurotrophic factor; R&D), ascorbic acid (Sigma), GDNF (glial cell line-derived neurotrophic factor; Peprotech), TGFβ3 (transforming growth factor type β3; R&D), dibutyryl cAMP (Sigma), and DAPT (R&D) as described previously (Kriks et al., 2011). For cortical neuron induction, precursors were maintained in the same, mDA differentiation media, but in the absence of DAPT and TGFβ3. Both mDA neurons and cortical neurons were cultured on polyornithine (PO)/laminin (L)/fibronectin (FN) coated dish until the desired maturation stage for the further experiments.
Generation of a NR4A2 (NURR1)-driven GFP expressing hESC line
The NR4A2 gene was targeted with CAS9 (Addgene) and gRNA (ATTATTTGTCCAAACTGTTGGGG in gRNA expression vector (Addgene)). The donor plasmid was cloned as 5’arm-P2A-H2B-EGFP-PgkPuro-3’arm in a pug19 vector (Clonetech) designed as an EGFP expression cassette that can replace the stop codon of NR4A2 by gene targeting. 5’arm and 3’arm sequences are provided in Supplementary Table S2. H9 hESCs were transfected with 1μg Cas9 plasmid, 1μg gRNA and/or 2μg DNA donor plasmid by Nucleofection (Lonza) as described (Mali et al., 2013). 1 μg/ml Puromycin (Thermo) was used for selection during 15 days after transfection. At day 15, individual colonies were isolated and cultured as sub-clones.
Flow Cytometry associated cell sorting
NURR1-driven GFP expressing DA neuronal cells at day 25 of differentiation from hESC were sorted using a BDFACS Aria6 cell sorter in Flow Cytometry Core Facility of MSKCC. The GFP positive and negative sorted cells were in vitro cultured for subsequent experiments until use.
Generation of SATB1 Knockout hESC line
The SATB1 CRISPR/Cas9 KO Plasmid Pool (Santa Cruz, sc-401321) was transfected into the NURR1 reporter-hESC using Nucleofector (Lonza, B-016 program). These plasmids contain specific 20 nt guide RNA sequences derived from the GeCKO (v2) library, which target SATB1. 48h after transfection, single hESC clones were picked, expanded, and the knockout of SATB1 was confirmed by Sanger sequencing.
Cell Culture
HeLa cells were maintained in DMEM supplemented with 10% FBS and 1% Pen/Strep. SK-N-MC cells were maintained in EMEM supplemented with 10% FBS and 1% Pen/Strep. Human primary fibroblasts were maintained in DMEM supplemented with 15% FBS and 1% Pen/Strep. In order to generate senescent human dermal fibroblasts, we used a BrdU protocol as previously described (Ozsvari et al., 2018). Briefly, human dermal fibroblasts were seeded on a 96 well plate. Cells were then treated with BrdU (100 μM) for 8 days. Control cells were treated with the same amount of DMSO for the same duration. Cells were then tested for their sensitivity to senolytics.
Mouse strains
Male wild-type C57BL/6 (Jackson Laboratories) were used for all experiments at 10 weeks of age. All animal experiments were approved by the Rockefeller University Institutional Animal Care and Use Committee. All described procedures were performed according to the guidelines described in the US National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the ARRIVE guidelines. Mice were housed in rooms on a 12 h dark/light cycle at 22 °C. Mice feeding was based on rodent diet (Picolab) and water available ad libitum. Mice were housed in groups of up to five animals except for mice that underwent stereotaxic surgery, which were housed singly to ensure recovery and avoid fighting.
Stereotaxic surgery
The stereotaxic injections were carried out as previously described (Brichta et al., 2015). In brief, we used an Angle Two stereotaxic frame for mouse with motorized nanoinjector (Leica) and injected 10-week-old male C57BL/6 WT mice (Charles River Laboratories) which were anesthetized with ketamine and xylazine prior to the procedure. The experimental viruses (total injection volume 0.5 μl) were stereotaxically injected targeting the ipsilateral SNpc (AP: −3.0 mm; ML: −1.2 mm; DV: −4.3 mm) and the control viruses were injected into the contralateral SNpc (AP: −3.0 mm; ML: +1.2 mm; DV: −4.3 mm). The injection rate was 0.05 μl/min using Hamilton syringes (30 gauge). Surgery wounds were sutured and recovery was monitored after the injections.
Method Details
RNA extraction and Real-time qPCR
Total RNA was isolated using TRIzol (Qiagen), and 1ug of RNA was reversed transcribed into cDNA using iScript (BioRad). Real-time qPCR was performed using the SSoFAST EvaGreen Mix (BioRad) in a BioRad CFX96 Thermal Cycler. All reactions were performed according to the manufactured protocol.
Immunocytochemistry
For immunocytochemistry (ICC), cells were grown on sterile glass coverslips. Cells were fixed in 4% PFA in DPBS and incubated at 37oC for 30 minutes. Cells were subsequently washed with serum-free media and then DPBS. Samples were first permeabilized with 0.5% Triton X100 and blocked with 10% Normal Donkey Serum (NDS, Jackson ImmunoResearch #017–000-121). After the blocking step, the samples were incubated overnight at 4oC with primary antibody. The next day, the samples were incubated in secondary antibody diluted in DPBS (1:250) for 1 hour at room temperature. Cells were then mounted onto Superfrost Plus microscope slides (Fisher Scientific) using Prolong Gold Antifade reagent with DAPI mounting medium (Invitrogen #P36931) for subsequent imaging.
RNA-Seq
Total RNA from differentiated neurons was isolated using the Qiagen RNEasy Plus kit according to the manufacturer’s instructions. 300ng of RNA were then used for library preparation using the TruSeq RNA Library Preparation Kit v2, Set B (Illumina). Quality and concentration of the libraries were assessed using a TapeStation (Agilent Technologies). Identical molarities of each library was then multiplexed and sequenced on an Illumina NextSeq High Output under 75 single read and multiplexed conditions.
Chromatin Immunoprecipitation Sequencing (ChIP-seq)
To crosslink the chromatin, differentiated cells were fixed for exactly 8 minutes in fixation buffer containing 1% fresh formaldehyde (Thermofisher). After washing in ice-cold PBS (containing protease inhibitors, Roche) cells were sonicated using a chromatin shearing protocol on a Covaris sonicator to generate chromatin fragments of 100–400 bp in size. Since the reproducibility of the shearing step is crucial for the remaining ChIP experiment, correct chromatin shearing has been successfully confirmed using the same DA neurons. After the fragmentation of chromatin, chromatin immunoprecipitation (ChIP) using a SATB1 antibody and an unspecific antibody (ctrl. IgG, LifeTechnologies) as negative/background control was performed. ChIP was performed and DNA was purified with a magnetic bead-based purification method according to the manufacturer’s protocol (MAGnify Chromatin Immunoprecipitation System-Kit, LifeTechnologies). Subsequently, the sheared DNA was used to generate ChIP-sequencing libraries by employing Ovation Ultralow System V2 Kit (NuGEN). DNA libraries were then checked for quality and concentration using both a Bioanalyzer (High Sensitivity DNA Chip (Agilent Technologies)) and the Tape Station (Agilent Technologies). Afterwards, the exact same molarities of each DNA library was multiplexed and sequenced on Illumina HiSeq 2500 sequencers using 100 single read and multiplexing conditions. The sequencing data was then analyzed using a well-established bioinformatics pipeline.
Fluorescent Dyes for Imaging Mitochondria and Lysosomes
For mitochondrial imaging, MitoTracker™ Red CMXRos (ThermoFisher #M7512) was used. A 500nM solution of the dye was prepared in serum free media. Cells were washed three times with serum free media and then incubated for 15 minutes at 37°C, and then washed again three times with serum free media. For lysosome imaging, LysoTracker™ Deep Red (ThermoFisher #L12492) was used. A 1 μM solution of the dye was prepared in serum free media. Cells were washed three times with serum free media and then incubated for 15 minutes at 37°C, an d then washed again three times with serum free media. Samples where then fixed and mounted to slides as described above.
Fluorescence Microscopy
Fluorescence images were collected using a Zeiss LSM 710 confocal microscope, using 10X, 40X and 63X objectives. Subsequent quantification and analysis of the images were performed in ImageJ. Minimal adjustment of contrast and brightness was performed as well in ImageJ for the optimal representation of the data.
High Content Imaging
High content imaging was performed using a Molecular Devices ImageXpress Micro Fluorescence Microscopic Imaging system, using 10X and 20X objectives. Determination of live cells was done using Calcein AM and dead cells were detected using Propidium Iodine. Neurite morphology of the cells was analyzed using the Calcein AM images, in order to ensure only the neurites of live cells were detected. This analysis was done using the Neurite Outgrowth Application Module of the MetaXpress software (Molecular Devices). For lysosome imaging, LysoTracker™ Deep Red (ThermoFisher #L12492) was used. A 1μM solution of the dye was prepared in serum free media. Cells were washed three times with serum free media and then incubated for 15 minutes at 37°C, and then washed again three times with serum free media.
Electron Microscopy
Cells grown on ACLAR film were fixed with 4% formaldehyde and 2% glutaraldehyde in 0.1M sodium cacodylate buffer (pH 7.4), and post-fixed with 2.5% glutaraldehyde and 0.25% tannic acid in 0.1M sodium cacodylate buffer for 15 minutes. They were then fixed with 2.5% glutaraldehyde in 0.1M sodium cacodylate buffer for 15 minutes and with 1% osmium tetra-oxide in sodium cacodylate buffer for 30 min on ice. The cells were subsequently washed three times in 0.1M sodium cacodylate buffer (pH 7.4) for 5 min and rinsed three times with water. They were then stained with 1% uranyl acetate for 30 min at room temperature followed by 12 hours on ice. The cells were dehydrated in increasing concentrations of ethanol; 50%, 70%, 90%, 100%, and 100% using Pelco Biowave Pro microwave automatic protocol (TedPella, Inc.). They were then infiltrated with Epon812 resin, using an increasing concentration of resin in acetone; 50%, 100%, and 100% using Pelco Biowave Pro microwave automatic protocol (TedPella, Inc.). The cells were infiltrated with Epon812 for 24 hours on a rotating rack, and the resin was replaced three times before polymerization for 48 hours at 60°C. Areas of interest were then selected under the light microscope and trimmed for re-mounting and microtome sectioning. 70 nm sections were cut and collected on electron microscope grids, and samples were counter-stained using 1% uranyl acetate and Sato’s lead stain (Proc. XIth Int. Cong. on Electron Microscopy, Kyoto. 1986, pp. 2181–2182). The sections were then imaged with 1000–10000x magnification using 120kV operated Jeol 1400 plus TEM.
Western blotting
Western blots were performed as previously described (Brichta et al., 2015). In brief, protein samples were isolated from respective cells by lysis in RIPA buffer (#89900, Thermo Scientific) containing protease and phosphatase inhibitors (#11836170001, Roche). Subsequently, a BCA assay (Thermo Scientific) was used to determine the protein concentrations. Equal amounts of protein were boiled in NuPAGE LDS sample buffer (Invitrogen) at 95 °C for 5 min and separate d using either: 8–16% Tris-HCl (Bio-Rad, #345–0038), or 4–20% Tris-glycine (Life Technologies, #EC6025). A wet blotting method was used to transfer the proteins to nitrocellulose membranes (BioRad), blots were blocked in 5% BSA for 2 h at room temperature and incubated with the respective primary antibody at 4 °C overnight. The following p rimary antibodies were used: rabbit monoclonal LAMP1 (CST #D2D11, 1:3000), rabbit monoclonal anti-SATB1 antibody (Abcam, #ab92307; 1:3000), rabbit monoclonal anti-Histone H3 (CST #D1H2, 1:5000), rabbit monoclonal anti-beta Actin (CST #D6A8, 1:15000), goat anti-IGFBP7 (R&D Technologies, AF-1334, 1:3000), rabbit monoclonal anti-p21 (CST, # 2947, 1:3000). Primary antibodies were detected using HRP-linked donkey anti–rabbit IgG (GE Healthcare, #NA934V; 1:10,000), HRP-linked anti-Goat IgG (H+L) Secondary Antibody (Invitrogen, #A15999, 1:10,000) or HRP-linked sheep anti–mouse IgG (GE Healthcare, #NA931V, 1:10,000) together with Western Lightning Plus-ECL (Perkin Elmer, #NEL105001EA). Bands were quantified with ImageJ software and normalized to the corresponding to β-Actin bands.
Senolytic Sensitivity Assay
To test the sensitivity of cells to senolytics, we used four different, well characterized compounds, which are known to act as senolytics and which act on different cellular pathways, namely fisetin, azithromycin, ABT-737, and a dasatinib plus quercetin cocktail. Doses were chosen based on previous literature. Cells were treated for various time courses (Fisetin 48 hours, ABT-737 24 hours, dasatinib plus quercetin 24 hours, and azithromycin 72 hrs). Following treatment, cell viability was determined using the CCK8 cytotoxicity assay (Dojindo).
GBA enzymatic activity assay
The enzymatic activity was measured with the GBA enzymatic activity assay (Novus bio) according to the manufacturer’s protocol. This method utilizes p-nitrophenyl-β-D-glucopyranoside which was hydrolyzed specifically by GBA into a yellow colored product. After an incubation with the substrate, a colorimetric measurement was used to determine the rate of the reaction which is directly proportional to the enzyme activity. Values were normalized to protein content using a BCA assay
Cathepsin D Enzymatic Activity Assay
Cathepsin D enzymatic activity was assessed using the Cathepsin D Activity Fluorometric Assay Kit (BioVision) according to the manufacturers protocol. Values were normalized to protein content using a BCA assay.
ATAC-seq
ATAC-seq was performed according to the protocol published by Buenrostro et al. (Buenrostro et al., 2015). In brief, some 50,000 cells (dopamine neurons, or cortical neurons, respectively) were collected by centrifugation at 1000x g at 4°C. After lysis, nuclei were pelleted and resuspended in transposition reaction mix. After transposase reaction, DNA was purified and transposed fragments were amplified in a PCR reaction using individually barcoded primers. To normalize the amplification rate of the PCR, amplified DNA fragments were quantified using qPCR. After determining the additional PCR cycle numbers to normalize the sequencing libraries, the remaining PCR reaction (before qPCR), was further amplified. The generated libraries were purified using the Monarch PCR & DNA cleanup kit (NEB) and the quality and concentration was assessed using a TapeStation device. Finally, equal molarities of each library was pooled for sequencing (75bp reads, paired end) on a NextSeq sequencer (Illumina).
Viruses
The viruses used for stereotaxic injections were obtained from Vector Biolabs. The virus AAV1-EGFP-U6-shRNA served as a control virus (scrambled shRNA and EGFP, #7040) and as previously reported, the shRNA virus for the silencing of Satb1 was custom-made (Brichta et al., 2015). The Satb1 shRNA construct used was designed to target bp 2329–2349 of Satb1 mRNA (reference sequence: BC011132.1) and included the coding sequence for EGFP. The control virus injected was AAV1-hSYN1-mCherry-WPRE. Viruses were packaged into a solution and injected at a concentration of 1 × 1013 genome copies per ml.
Histology
Two- to three-weeks post injection, the mice were anesthetized with pentobarbital and transcardially perfused using PBS (pH 7.4), followed by 4% paraformaldehyde in PBS. Brains were postfixed in 4% paraformaldehyde in PBS at room temperature for 1 hour and then cryopreserved using a gradient of 5%, 15% and 30% sucrose. The brains were embedded in Neg-50 (Thermo Scientific), frozen and stored at −80 °C and cut into 14-μm-thick coronal sections using a Microm cryostat and thaw-mounted onto Superfrost Plus microscope slides (Fisher Scientific). For indirect IF staining of TH, p21, IBA-1, SATB1, brain sections were washed in PBS and permeabilized with 0.2% Triton X-100 in PBS, followed by blocking with 2% donkey serum and 0.1% fish gelatin in 0.2% Triton X-100 in PBS. Sections were then incubated with rabbit anti-SATB1 (AbCam, #ab189847), rabbit anti-p21 antibody (AbCam, # ab188224), chicken anti-TH (Millipore, #ab9706), or goat anti-IBA-1 (AbCam, # ab5076) all at a concentration of 1:250 overnight. The next day, slides were washed with PBS and incubated with Alexa Fluor appropriate secondary antibodies at a 1:250 dilution. After another washing step, slides were mounted with ProLong Diamond Antifade with DAPI (Life Technologies, #P36962) coverslipped and stored at room temperature in the dark until imaging.
Human Substantia Nigra Histology
Human Substantia Nigra tissue was obtained from the Harvard Brain Tissue Resource Center, and subsequently processed by the Banner Sun Health Research Institute. For IF staining of TH and p21, brain sections were washed in PBS and permeabilized with 0.2% Triton X-100 in PBS, followed by blocking with 2% donkey serum and 0.1% fish gelatin in 0.2% Triton X-100 in PBS. Sections were then incubated with rabbit polyclonal anti-TH antibody (Millipore, #AB152), and anti-p21 (CST, #2947 at a concentration of 1:250 overnight. The next day, slides were washed with PBS and incubated with Alexa Fluor 488 goat anti-rabbit IgG, and Alexa Fluor 546 goat anti-mouse IgG (Life Technologies). Following incubation of secondary antibody, a tissue sections were washed with a Sudan Black 0.1% solution to quench autofluorescence. After another washing step, slides were mounted with Mowiol (Sigma Aldrich) mounting media supplemented with DAPI (Sigma Aldrich) (1:5000 dilution) and then coverslipped and stored at room temperature in the dark until imaging. Samples were coded, and images were acquired as described above. They were then quantified by a different individual who was also blind to the sample identities.
Blue Native Gel Electrophoresis and Western Blotting
For the assessment of mitochondrial complexes, cell lysates were prepared using the NativePAGE Sample Prep Kit (Life Technologies) and solubilized with 1% digitonin. A BCA assay (Thermo Scientific) was used to determine the protein concentrations. Equal amounts of protein were loaded in a NativePAGE Novex 4–16% Bis-Tris Protein Gels (Life Technologies). A wet blotting method was used to transfer the proteins onto a PVDF membrane (Life Technologies, 0.45 μm), the membrane was then incubated with 8% acetic acid for 15min and washed with methanol and dh2o before being blocked with 5% BSA in 20 mM Tris, 150 mM NaCl, and 0.1% (w/v) Tween 20, pH 7.5, for 2 hr. Subsequently after blocking, the membrane was immunoblotted with the respective primary antibody at 4 °C overnight. The primary ant ibodies used were NDUFA9 (a CI Subunit) (1/2500 by vol; ab14713: Abcam), UQCRC2 (a CIII Subunit) (1/2500 by vol; ab203832: Abcam), or MT-CO2 (a CIV Subunit) (1/2500 by vol; ab110258: Abcam). Primary antibodies were detected using either HRP-linked donkey anti–rabbit IgG (GE Healthcare, #NA934V; 1:10,000) or HRP-linked sheep anti–mouse IgG (GE Healthcare, #NA931V, 1:10,000) together with Western Lightning Plus-ECL (Perkin Elmer, #NEL105001EA).
Measurements of cellular respiration
In order to measure cellular respiration which reflects mitochondrial activity in cells, we applied the XF mito stress kit using the Seahorse XFe96 Analyzer (Agilent) according to the manufacturer’s protocol. In brief, we plated out some 40,000 SK-N-MC cells per well, on a 96-well format plate, one day before the measurement and incubated cells in 5% CO2 at 37° C overnight. On the day of measurement, cells were washed with XF cell mito stress test assay medium (Agilent) and incubated in the medium for one hour prior to the measurement in a CO2-free incubator at 37oC. During the measurement program, cells were challenged with 1.0 μM Oligomycin (Port A), 0.5 μM FCCP (Port B) and 0.5 μM Rotenone/antimycin A (Port C). Results of the measurement were subsequently analyzed using the Wave software (Agilent). In order to measure mitochondrial activity in hESC-derived DA neurons, some 40,000 neurons which express NURR1::GFP were plated out at day 25 of differentiation into DA neurons. Then the cells were treated in the same way as described above for SK-N-MC cells, except that 1.5 μM FCCP (Port B) was used.
Luciferase Reporter Assay
The luciferase plasmid LightSwitch™ Promoter Reporter, containing the CDKN1A promoter (Switchgear Genomics, S718663) was co-transfected with renilla luciferase containing plasmid pGL4.82 (Promega) and either a control siRNA or one targeting SATB1 (Invitrogen) into HeLa cells. Transfection was performed by Lipofectamine 3000 (Lonza) using manufacturer’s instructions. Three days after transfection analysis of luciferase expression was performed using the Dual-Glo Luciferase Assay System (Promega). Firefly luciferase activity was normalized to cell number using a CCK8 assay (Dojindo).
Protein Oxidation Assays
Both the OxiSelect™ Protein Carbonyl ELISA (Cell BioLabs) assays well as the luciferase-based GSH/GSSG ratio detection assay (Promega) were performed according to the manufacturers’ protocols. For both assays protein lysates from DA neurons at 40days of differentiation were used.
Quantification and Statistical Analysis
Differential Gene Expression Analysis
Sequences were then aligned using Bowtie (Langmead et al., 2009). Differential gene expression analysis was then performed using edgeR (Robinson et al., 2011).
ChIP-seq Analysis
All sequencing reads were tested for quality using the FastQC online software (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). For further downstream analysis the sequences were trimmed using Trimmomatic (Bolger et al., 2014). For alignment to the human genome and model-based analysis of the results we used a combined approach using Bowtie (Langmead et al., 2009), Presq (http://smithlabresearch.org/software/preseq) and MACS2 (Zhang et al., 2008). Finally, statistical significance was calculated and data were analyzed. ChIP-sequencing data were visualized using the open source software Integrative Genomics Viewer (Robinson et al., 2011).
Binding and expression target analysis (BETA)
We have used the BETA analysis tool to unravel the mode of action of SATB1. In brief, Binding and Expression Target Analysis (BETA) is a software package that is able to integrate ChIP-seq of transcriptional regulators with differential gene expression data to infer direct target genes. Here we used it to predict if the genetic regulator SATB1 has activating or repressive functions. The software package was run by python version 2.7.13. As input files, we used the expression files (.csv) from the RNA-seq analyses (comparing WT vs. SATB1KO) formatted according to the LIMMA standard and the BED files from the SATB1 ChIP-seq analyses that contained the called peaks. We chose a p-value cut-off of 0.01 (parameter-c) and the human genome version hg38 (parameter - g). In summary, the analysis was performed following the protocol described in (Wang et al., 2013) using default parameters, if not mentioned otherwise above (e.g. hg38, p-value).
ATAC-seq Analysis
The resulting sequencing data was thoroughly analyzed using a well-established bioinformatics pipeline. In brief, we used the alignment tool bowtie2 (version 2.3.3.1) (Langmead and Salzberg, 2012; Langmead et al., 2009), post-alignment processing was performed using samtools (version 1.5) (Li, 2011; Li et al., 2009) and picardtools (version 2.9.5) ( http://broadinstitute.github.io/picard/). Peak calling was performed using macs2 (version 2.1.1.20160309) (Zhang et al., 2008). For the graphical presentation of the ATAC-seq, we generated a coverage plot using ngsplotr (version 2.63) (Shen et al., 2014).
Data Analysis
Data analysis for protein quantification of Western blots was performed by using Fiji (Schindelin et al., 2012), Microsoft Excel 16.11 and GraphPad Prism 7. For the analysis and graphical presentation of ChIP-seq and ATAC-seq data, the Integrative Genomics Viewer (IGV) software was used (Robinson et al., 2011; Thorvaldsdottir et al., 2013). For pathway analysis of RNA-Seq data, the GeneTrail2 transcriptomics tool was used (Stockel et al., 2016). Gene Set Enrichment Analysis (GSEA) was performed using 3.0 (Mootha et al., 2003; Subramanian et al., 2005). Gene ontology enrichment and interactome analysis was performed using metascape (Tripathi et al., 2015). Analysis of microglia morphology was performed as described in Davies et al., 2017.
Data and Code Availability
All sequencing data were deposited to the EMBL-EBI arrayexpress server (https://www.ebi.ac.uk/arrayexpress/), ascension number E-MTAB-5965.
Supplementary Material
1
2
Highlights:
- FACS of NURR1::GFP dopaminergic (DA) precursors enriches DA neuron culture purity
- SATB1KO DA neurons show hallmarks of cellular senescence including SASP
- SATB1 is required to repress CDKN1A in DA neurons
- Senescence induced by SATB1 reduction in vivo induces microglial activation
Acknowledgments
We thank the Genomics Resource Center and the Electron Microscopy Resource Center (EMRC) at Rockefeller University for technical support. All mitochondrial respiration measurements were performed using the Agilent Seahorse XFe96 analyzer, as well as High-Content Imaging on the ImageXpress system with generous support from the Rockefeller University High-Throughput Screening and Spectroscopy Resource Center (HTSRC). We thank Elisabeth Griggs for help with graphic design. Human brain tissue was provided by the Harvard Brain Tissue Resource Center, which is supported in part by PHS grant number R24 MH068855. This work was supported by the United States Army Medical Research and Material Command (USAMRMC) under Award No. W81XWH-10–1-0640 (LB), W81XWH-12–1-0039 (MR) and the JPB Foundation, Award #475 (PG), and by NYSTEM contract C32598GG to P.G.(and M.R.) and L.S.. T.W.K. is supported by a NYSCF Druckenmiller fellowship and G.C. is supported by a NYSTEM postdoctoral fellowship from the Center for Stem Cell Biology, MSKCC.. Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the sponsors.
Footnotes
Declaration of Interests
L.S. is a scientific co-founder and paid consultant of Bluerock Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018. [DOI] [PubMed] [Google Scholar]
- Bae S, Park J, and Kim JS (2014). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. (2016). Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, and Petersen RC (2018). Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest 128, 1208–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, and van Deursen JM (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanu MU, Mandraju RK, Bhaskar C, and Kondapi AK (2010). Cultured cerebellar granule neurons as an in vitro aging model: topoisomerase IIbeta as an additional biomarker in DNA repair and aging. Toxicol In Vitro 24, 1935–1945. [DOI] [PubMed] [Google Scholar]
- Bigagli E, Luceri C, Scartabelli T, Dolara P, Casamenti F, Pellegrini-Giampietro DE, and Giovannelli L. (2016). Long-term Neuroglial Cocultures as a Brain Aging Model: Hallmarks of Senescence, MicroRNA Expression Profiles, and Comparison With In Vivo Models. J Gerontol A Biol Sci Med Sci 71, 50–60. [DOI] [PubMed] [Google Scholar]
- Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, and Riederer P. (1995). Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett 202, 17–20. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, and Braak E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Brichta L, Shin W, Jackson-Lewis V, Blesa J, Yap EL, Walker Z, Zhang J, Roussarie JP, Alvarez MJ, Califano A, et al. (2015). Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat Neurosci 18, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, and Baker DJ (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. (2013). Aging, cellular senescence, and cancer. Annu Rev Physiol 75, 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, Ando S, Howell A, Martinez-Outschoorn UE, Sotgia F, et al. (2012). CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neoangiogenesis. Cell Cycle 11, 3599–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature biotechnology 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, International Parkinson’s Disease Genomics, C., andMe Research, T., Kerchner GA, Ayalon G, et al. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Woods G, Demaria M, Rane A, Zou Y, McQuade A, Rajagopalan S, Limbad C, Madden DT, Campisi J, et al. (2018). Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep 22, 930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Dodiya H, Aebischer P, Olanow CW, and Kordower JH (2009). Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 35, 385–398. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, and Campisi J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, and Campisi J. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BM, Salinas-Navarro M, Cordeiro MF, Moons L, and De Groef L. (2017). Characterizing microglia activation: a spatial statistics approach to maximize information extraction. Sci Rep 7, 1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan KV, Kinyua AW, Yang DJ, Ko CM, Moh SH, Shong KE, Kim H, Park SK, Kim DH, Kim I, et al. (2016). FoxO1 in dopaminergic neurons regulates energy homeostasis and targets tyrosine hydroxylase. Nat Commun 7, 12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, et al. (2017). Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 23, 1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folick A, Oakley HD, Yu Y, Armstrong EH, Kumari M, Sanor L, Moore DD, Ortlund EA, Zechner R, and Wang MC (2015). Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science 347, 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, and Campisi J. (2014). Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69 Suppl 1, S4–9. [DOI] [PubMed] [Google Scholar]
- Georgilis A, Klotz S, Hanley CJ, Herranz N, Weirich B, Morancho B, Leote AC, D’Artista L, Gallage S, Seehawer M, et al. (2018). PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer Cell 34, 85–102 e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere N, Burke Nanni S, and Trudeau LE (2018). On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front Neurol 9, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H, Zhang X, Tiklova K, Volakakis N, Brodin L, Berg L, Greengard P, Perlmann T, and Svenningsson P. (2017). Alterations of p11 in brain tissue and peripheral blood leukocytes in Parkinson’s disease. Proc Natl Acad Sci U S A 114, 2735–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L. (1965). The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res 37, 614–636. [DOI] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, and Sedivy JM (2006). Cellular senescence in aging primates. Science 311, 1257. [DOI] [PubMed] [Google Scholar]
- Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Saffrey MJ, Cameron K, et al. (2012). Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia LV, and Lang AE (2015). Parkinson’s disease. Lancet 386, 896–912. [DOI] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. (2011). Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551. [DOI] [PubMed] [Google Scholar]
- Kirkland JL, and Tchkonia T. (2017). Cellular Senescence: A Translational Perspective. EBioMedicine 21, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lena AM, Mancini M, Rivetti di Val Cervo P, Saintigny G, Mahe C, Melino G, and Candi E. (2012). MicroRNA-191 triggers keratinocytes senescence by SATB1 and CDK6 downregulation. Biochem Biophys Res Commun 423, 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Esvelt KM, and Church GM (2013). Cas9 as a versatile tool for engineering biology. Nat Methods 10, 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, et al. (2013). Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Zamudio RI, Robinson L, Roux PF, and Bischof O. (2017). SnapShot: Cellular Senescence Pathways. Cell 170, 816–816 e811. [DOI] [PubMed] [Google Scholar]
- Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, et al. (2009). A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med 15, 1082–1087. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Mullin S, and Schapira A. (2015). The genetics of Parkinson’s disease. Br Med Bull 114, 39–52. [DOI] [PubMed] [Google Scholar]
- Munoz-Espin D, and Serrano M. (2014). Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15, 482–496. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Swahari V, Plestant C, Smith I, McCoy E, Smith S, Moy SS, Anton ES, and Deshmukh M. (2016). Bcl-xL Is Essential for the Survival and Function of Differentiated Neurons in the Cortex That Control Complex Behaviors. J Neurosci 36, 5448–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, et al. (2018). Parkinson’s disease genetics: identifying novel risk loci, providing causal insights and improving estimates of heritable risk. bioRxiv. [Google Scholar]
- Nikonova EV, Xiong Y, Tanis KQ, Dawson VL, Vogel RL, Finney EM, Stone DJ, Reynolds IJ, Kern JT, and Dawson TM (2012). Transcriptional responses to loss or gain of function of the leucine-rich repeat kinase 2 (LRRK2) gene uncover biological processes modulated by LRRK2 activity. Hum Mol Genet 21, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsvari B, Nuttall JR, Sotgia F, and Lisanti MP (2018). Azithromycin and Roxithromycin define a new family of “senolytic” drugs that target senescent human fibroblasts. Aging (Albany NY) 10, 3294–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacelli C, Giguere N, Bourque MJ, Levesque M, Slack RS, and Trudeau LE (2015). Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr Biol 25, 2349–2360. [DOI] [PubMed] [Google Scholar]
- Park MH, Lee HJ, Lee HL, Son DJ, Ju JH, Hyun BK, Jung SH, Song JK, Lee DH, Hwang CJ, et al. (2017). Parkin Knockout Inhibits Neuronal Development via Regulation of Proteasomal Degradation of p21. Theranostics 7, 2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen KM, Marner L, Pakkenberg H, and Pakkenberg B. (2005). No global loss of neocortical neurons in Parkinson’s disease: a quantitative stereological study. Mov Disord 20, 164–171. [DOI] [PubMed] [Google Scholar]
- Piechota M, Sunderland P, Wysocka A, Nalberczak M, Sliwinska MA, Radwanska K, and Sikora E. (2016). Is senescence-associated beta-galactosidase a marker of neuronal senescence? Oncotarget 7, 81099–81109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Zhang XJ, Renier N, Wu Z, Atkin T, Sun Z, Ozair MZ, Tchieu J, Zimmer B, Fattahi F, et al. (2017). Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat Biotechnol 35, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajput AH, and Rozdilsky B. (1976). Dysautonomia in Parkinsonism: a clinicopathological study. J Neurol Neurosurg Psychiatry 39, 1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve A, Simcox E, and Turnbull D. (2014). Ageing and Parkinson’s disease: why is advancing age the biggest risk factor? Ageing Res Rev 14, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nature biotechnology 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapieha P, and Mallette FA (2018). Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol 28, 595–607. [DOI] [PubMed] [Google Scholar]
- Saucedo-Cardenas O, Quintana-Hau JD, Le WD, Smidt MP, Cox JJ, De Mayo F, Burbach JP, and Conneely OM (1998). Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc Natl Acad Sci U S A 95, 4013–4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severino J, Allen RG, Balin S, Balin A, and Cristofalo VJ (2000). Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res 257, 162–171. [DOI] [PubMed] [Google Scholar]
- Severino V, Alessio N, Farina A, Sandomenico A, Cipollaro M, Peluso G, Galderisi U, and Chambery A. (2013). Insulin-like growth factor binding proteins 4 and 7 released by senescent cells promote premature senescence in mesenchymal stem cells. Cell Death Dis 4, e911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E. (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Gamez A, and Demaria M. (2017). Therapeutic interventions for aging: the case of cellular senescence. Drug Discov Today 22, 786–795. [DOI] [PubMed] [Google Scholar]
- Steinbeck JA, Choi SJ, Mrejeru A, Ganat Y, Deisseroth K, Sulzer D, Mosharov EV, and Studer L. (2015). Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson’s disease model. Nature biotechnology 33, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockel D, Kehl T, Trampert P, Schneider L, Backes C, Ludwig N, Gerasch A, Kaufmann M, Gessler M, Graf N, et al. (2016). Multi-omics enrichment analysis using the GeneTrail2 web service. Bioinformatics 32, 1502–1508. [DOI] [PubMed] [Google Scholar]
- Storer M, and Keyes WM (2014). Developing senescence to remodel the embryo. Commun Integr Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana Z, Maiti K, Dedman L, and Smith R. (2018). Is there a role for placental senescence in the genesis of obstetric complications and fetal growth restriction? Am J Obstet Gynecol 218, S762–S773. [DOI] [PubMed] [Google Scholar]
- Tan FC, Hutchison ER, Eitan E, and Mattson MP (2014). Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology 15, 643–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Gonzalez S, Giraldez-Perez RM, Cuartero MI, Casarejos MJ, Mena MA, Wang XF, and Sanchez-Capelo A. (2011). Dopamine and alpha-synuclein dysfunction in Smad3 null mice. Mol Neurodegener 6, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi H, and Sato T. (1968). Age changes in size and number of mitochondria of human hepatic cells. J Gerontol 23, 454–461. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinkum KL, White LS, Marpegan L, Herzog E, Piwnica-Worms D, and Piwnica-Worms H. (2013). Forkhead box O1 (FOXO1) protein, but not p53, contributes to robust induction of p21 expression in fasted mice. J Biol Chem 288, 27999–28008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP Jr., Miller SW, Davis RE, and Parker WD Jr. (2000). Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp Neurol 162, 37–50. [DOI] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. (2015). Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM (2014). The role of senescent cells in ageing. Nature 509, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk KD, Persichetti E, Chiasserini D, Eusebi P, Beccari T, Calabresi P, Berendse HW, Parnetti L, and van de Berg WD (2013). Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov Disord 28, 747–754. [DOI] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, and Liu XS (2013). Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 8, 2502–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gu X, Zhang G, Wang L, Wang T, Zhao Y, Zhang X, Zhou Y, Kadin M, and Tu P. (2014). SATB1 overexpression promotes malignant T-cell proliferation in cutaneous CD30+ lymphoproliferative disease by repressing p21. Blood 123, 3452–3461. [DOI] [PubMed] [Google Scholar]
- Wang Z, Rong X, Luo B, Qin S, Lu L, Zhang X, Sun Y, Hu Q, and Zhang C. (2016). A Natural Model of Mouse Cardiac Myocyte Senescence. J Cardiovasc Transl Res 9, 456–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, et al. (2018). Senolytics improve physical function and increase lifespan in old age. Nat Med 24, 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, et al. (2016). Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7, 11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, et al. (2018). Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, et al. (2010). PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2, 52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, et al. (2015). The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2
Data Availability Statement
All sequencing data were deposited to the EMBL-EBI arrayexpress server (https://www.ebi.ac.uk/arrayexpress/), ascension number E-MTAB-5965.