Cardiomyocyte‐Specific Snrk Prevents Inflammation in the Heart (original) (raw)
Abstract
Background
The SNRK (sucrose‐nonfermenting–related kinase) enzyme is critical for cardiac function. However, the underlying cause for heart failure observed in Snrk cardiac conditional knockout mouse is unknown.
Methods and Results
Previously, 6‐month adult mice knocked out for Snrk in cardiomyocytes (CMs) displayed left ventricular dysfunction. Here, 4‐month adult mice, on angiotensin II (Ang II) infusion, show rapid decline in cardiac systolic function, which leads to heart failure and death in 2 weeks. These mice showed increased expression of nuclear factor κ light chain enhancer of activated B cells (NF‐κB), inflammatory signaling proteins, proinflammatory proteins in the heart, and fibrosis. Interestingly, under Ang II infusion, mice knocked out for Snrk in endothelial cells did not show significant systolic or diastolic dysfunction. Although an NF‐κB inflammation signaling pathway was increased in Snrk knockout endothelial cells, this did not lead to fibrosis or mortality. In hearts of adult mice knocked out for Snrk in CMs, we also observed NF‐κB pathway activation in CMs, and an increased presence of Mac2+ macrophages was observed in basal and Ang II–infused states. In vitro analysis of Snrk knockdown HL‐1 CMs revealed similar upregulation of the NF‐κB signaling proteins and proinflammatory proteins that was exacerbated on Ang II treatment. The Ang II–induced NF‐κB pathway–mediated proinflammatory effects were mediated in part through protein kinase B or AKT, wherein AKT inhibition restored the proinflammatory signaling protein levels to baseline in Snrk knockdown HL‐1 CMs.
Conclusions
During heart failure, SNRK acts as a cardiomyocyte‐specific repressor of cardiac inflammation and fibrosis.
Keywords: cardiac hypertrophy, cardiomyocyte, endothelial cell, fibrosis, heart failure, inflammation, NF‐kB
Subject Categories: Heart Failure, Inflammatory Heart Disease, Cardiomyopathy
Clinical Perspective
What Is New?
- This article reveals the significance of SNRK (sucrose‐nonfermenting–related kinase) in preventing cardiac inflammation during the cardiac remodeling process, which leads to fibrosis and heart failure.
- Angiotensin II–mediated cardiac remodeling induces inflammation and fibrosis in Snrk cardiac‐specific knockout mice leading to death in 2 weeks.
- SNRK influences NF‐κB signaling, which is the key factor in cardiac inflammation and SNRK is a target at the interface of inflammation, fibrosis, and metabolism in cardiomyocytes.
What Are the Clinical Implications?
- Small‐molecule or target‐based therapies directed toward SNRK in cardiomyocytes could improve several of the clinical complications such as inflammation, fibrosis, and metabolic dysfunction that are observed during heart failure.
Heart failure (HF) is 1 of the pressing clinical problems that is a frequent cause of hospitalization.1, 2 Direct cardiac insults such as myocardial ischemia, hypertension, or indirect cardiac insults such as obesity‐ or diabetes mellitus–associated cardiac overload, all eventually result in HF. Significant advances have been made in therapies targeted at both prevention and treatment of HF. However, the patient prognosis once admitted is poor, with 17% to 45% patients dying within 1 year of admission and >50% mortality in 5 years.3 Thus, the mortality and morbidity burden inflicted by HF on society dictate a better understanding of the underlying mechanisms that cause HF. HF is often associated with 2 key pathologies, namely inflammation and fibrosis.4 Inflammation serves as beneficial (reparative) and harmful (persistent) mechanism adopted by the cells associated with the heart (cardiomyocytes [CMs], endothelial cells [ECs], fibroblasts, macrophages, and others) in the context of defense, repair, and regeneration of heart tissue.5, 6 Fibrosis in the heart is associated with preserving tissue architecture, and pathological fibrosis, like inflammation, results in scarring and impairment of heart function.5 Thus, the balance between these 2 key processes is vital for normal heart function.
Cross‐talk between cell types in the heart is critical for cardiac function.7, 8 For example, on angiotensin II (Ang II) exposure, ECs contribute to CM hypertrophy by releasing endothelin‐1 and decreasing nitric oxide release.9 CMs influence long‐term development of coronary arteries by releasing vascular endothelial growth factor.10 Dysfunction of ECs is hypothesized to contribute to cardiac fibrosis and inflammation via expression of adhesion receptors on ECs that recruit immune cells.11, 12 A failing heart thus presumably creates an environment in which dysregulation of signaling pathways and check mechanisms will culminate in a pathological inflammation and a fibrotic state that prevents normal heart function. It is therefore of importance to investigate and identify molecules and associated signaling pathways that function at the interface of cell types12 that can prevent inflammation and fibrosis to restore or improve heart function. Our focus in this work is on ECs and CMs and their role in adult mammalian heart function.
Our laboratory has been studying and characterizing an AMPK (AMP‐activated protein kinase)‐family member, sucrose‐nonfermenting–related kinase (SNRK), and its role in cardiovascular development. SNRK is essential for cardiac metabolism13 and cardiac function14 and is implicated in a cardioprotective function by improving cardiac mitochondrial efficiency and decreasing mitochondrial uncoupling.15 In relation to inflammation, SNRK has been implicated as a repressor of adipose tissue inflammation16, 17 and kidney tissue inflammation.18 In relation to vasculature, SNRK participates with a dual specific phosphatase‐5 to regulate embryonic zebrafish vascular development,19, 20 and in mammals SNRK was shown to be critical for promoting angiogenesis in vivo.21 To date, however, SNRK's role in cardiac inflammation is not known.
In HF, circulating and tissue Ang II concentrations are increased, which results in systolic and diastolic dysfunction.22 Ang II, a renin‐angiotensin system effector molecule, affects cardiac function through both systemic and local actions and plays a key role in cardiac remodeling and dysfunction in the failing heart.23, 24 Ang II initiates injury in the heart by promoting hypertension and stimulating immune and inflammatory signaling, which is independent of blood pressure changes. One prominent inflammatory pathway responsive to Ang II is the translocation of nuclear factor κ light chain enhancer of activated B cells (NF‐κB) to the nucleus, where it drives transcription of a broad array of proinflammatory mediators such as tumor necrosis factor (TNF)‐α, interleukin (IL)‐6, pro‐IL‐1β, and pro‐IL‐18 in the heart.25 These cytokines (TNF‐α and IL‐6) are not constitutively expressed in the normal heart but are produced and upregulated during cardiac remodeling.
Recent work showed that SNRK directly interacts with the phosphorylated p65 (p‐p65) subunit of NF‐κB to prevent inflammation and fibrosis in kidney glomerular ECs.18 Based on prior evidence that Snrk in CMs is essential for cardiac function,14, 15 and its well‐characterized role as a repressor of inflammation in adipocytes16, 17 and kidney ECs,18 in this study we investigated the hypothesis that Snrk represses inflammation in CMs during Ang II–induced cardiac remodeling.
Materials and Methods
All data and supporting materials have been provided with the published article.
Mouse Experiments
The mice were housed in the Medical College of Wisconsin Biological Resource Center, and all experiments were performed in accordance with an approved IACUC animal protocol 1022. The Snrk cmcKO (cardiac myocyte knockout) adult mice and Snrk ecKO (endothelial cell knockout) adult mice were generated from MYH6CRE‐positive Snrk LoxP/wild‐type (WT) males and TIE2CRE positive Snrk LoxP/WT males mated to Snrk LoxP/LoxP females, respectively. The genotyping and characterization information for these alleles are available from our previous publication.13 The in vivo experiments included the following conditions: Snrk WT, Snrk cmcKO, Snrk WT with Ang II, and Snrk cmcKO with Ang II.
HL‐1 Mouse Cardiomyocyte System and Transfection
Mouse HL‐1 atrial cardiomyocytes cell line was cultured in Claycomb media (Sigma‐Aldrich, St. Louis, MO) containing 10% FBS, 0.1 mmol/L norepinephrine, 2 mmol/L l‐glutamine, and penicillin/streptomycin. The culture plates were precoated with gelatin/fibronectin overnight at 37°C. Knockdown in HL‐1 cells was accomplished using small interfering RNA (siRNA) for Snrk and control siRNA (Dharmacon, Lafayette, CO), which were transfected using Lipofectamine2000 reagent. After 48 hours of transfection, HL‐1 cells were subjected to stimulation with and without Ang II (1 μmol/L) or AKT inhibitor, LY294002 (10 μmol/L) for 24 hours. Untreated cells, DMSO‐treated cells, or control siRNA–treated cells were used as controls.
ECHO and Image Analysis
Transthoracic echocardiography (ECHO) was performed in anesthetized (2% isoflurane) 4‐ to 5‐month‐old litter‐matched mice. The Ang II dose used in the study was 1000 ng/kg per minute. The investigator (L.M.H.) who performed the measurements and data analyses was blinded to the study groups. A comprehensive 2‐dimensional transthoracic ECHO with Doppler was performed on animals with a commercially available ECHO system (Vivid 7, General Electric, Wauwatosa, WI), with an 11‐MHz M12‐L linear array transducer. Details of ECHO analysis are provided in our previous publication.13 ECHOs were performed with 2 to 5 mice per group, containing both male and female mice. In the ECHO data (Figure 1 and Figure S1), we have only included data from male mice, and n=3 for WT and conditional knockout groups. The ages of the mice for the Snrk cmcKO group range from 3 months, 5 days (youngest) to 4 months, 18 days (oldest). For the Snrk ecKO group, the age range from 4 months, 21 days (youngest) to 5 months 11 days (oldest). All data sets were normalized with body weight measurements. ECHO experiments were performed multiple times.
Figure 1.
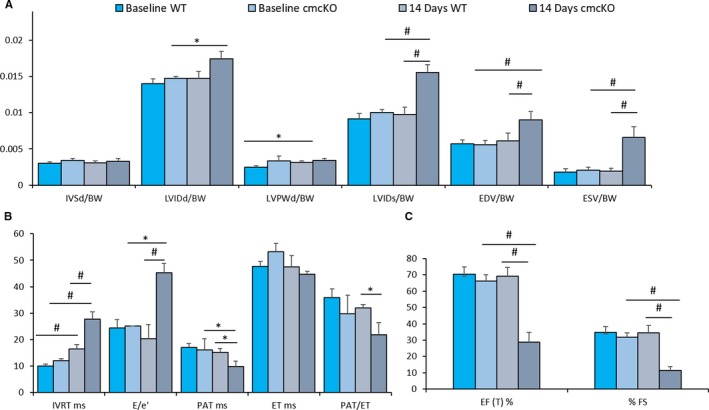
Angiotensin II (Ang II) induces cardiac failure in 2 weeks in Snrk cmcKO mice. A through C, Echocardiogram results for baseline 4‐month‐old Snrk wild‐type (WT) mice and cardiac‐specific knockout (Snrk cmcKO) mice with Ang II infused for 14 days into Snrk WT mice and 14 days into cardiac‐specific knockout (Snrk cmcKO) mice as described before.15 All data were normalized to body weight (BW) and presented as such. The parameters analyzed are interventricular septum thickness at end‐diastole (IVSd/BW), left ventricular internal dimension at end‐diastole (LVIDd/BW), left ventricular posterior wall thickness at end‐diastole (LVPWd/BW), left ventricular internal dimension at end‐systole (LVIDs/BW), end‐diastole volume (EDV/BW), end‐systolic volume (ESV/BW), isovolumic relaxation time (IVRT), peak velocity of early diastolic transmitral flow (E), early diastolic mitral annular velocity (e′), pulmonary acceleration rate (PAT), ejection time (ET), ejection fraction (EF), fractional shortening (FS). Results are presented as mean±SEM (*P<0.05, # P<0.01). The statistical comparison for P value was done by comparing Snrk WT vs Snrk cmcKO, Snrk WT vs Snrk WT‐Ang II, Snrk cmcKO vs Snrk cmcKO‐Ang II, and Snrk WT‐Ang II vs Snrk cmcKO‐Ang II (n=6 for the WT group and n=3 for Snrk cmcKO and the Ang II–induced experimental group).
Tissue Sectioning, Staining, and Image Analysis
Adult mouse hearts were fixed in 4% paraformaldehyde/PBS for 1 to 2 days at 4°C. After fixation, tissues were washed extensively in PBS and embedded in paraffin or sucrose/Optimal Cutting Temperature compound as previously described.15 The microscopic images of the entire tissue section were scanned at ×40 magnification using the Nanozoomer 2.0‐HT (Hamamatsu, Hamamatsu City, Shizuoka, Japan) digital slide scanning system (Children's Research Institute's Imaging Core). Sirus red staining was performed to determine fibrosis of the heart sections. The amount of fibrosis was quantified using a software‐assisted, unbiased microimage quantification method. The scanned images were imported into Visiopharm software (Hørsholm, Denmark), and using the imager module, 3 ×20 region‐of‐interest images were extracted from the left, center, and right regions of the heart. All original images were processed with this preset threshold and linear Bayesian classification to generate a processed image. Total collagen‐positive area per region of interest was measured in micrometers and represented as a percentage of the total tissue area.
Immunohistochemistry
Formalin‐fixed hearts collected from Snrk WT and Snrk cmcKO mice treated independently with vehicle and Ang II were dehydrated, embedded in paraffin, and sectioned at 5 μm. Slides were warmed at 60°C for 1 hour, dewaxed in xylene, and rehydrated. For assessing macrophage infiltration in hearts, anti‐mouse/human Mac2 (Galectin‐3) antibody (Cedarlane, Burlington, ON, Canada; catalog No. CL8942AP) was used. Pretreatment with target retrieval solution (Dako, Santa Clara, CA; S1699) was performed with heating (95°C) over a period of 40 minutes and then cooling at room temperature for 15 minutes. Tissue slices were incubated overnight at 4°C, and stained sections were viewed using light microscopy. All analyses of histological data quantification were done by scanned images that were imported to Visiopharm software (Hørsholm, Denmark) and using the imager module; 10 ×20 region‐of‐interest images were extracted from left, center, and right regions of the heart. All original images were processed with this preset threshold and linear Bayesian classification to generate the processed image.
Fluorescence‐Activated Cell‐Sorting Analysis
HL‐1 CM cells were seeded at a density of 1×105 cells/mL in 35‐mm dishes, and for a knockdown experiment, Snrk siRNA or control siRNA (Dharmacon) were transfected using Lipofectamine2000 reagent. Transfected cells were stimulated with and without Ang II (1 μmol/L) condition for 24 hours and were harvested using absolute methanol for 30 minutes, in 4°C to fix and permeabilize the cells. Cells were washed 3 times with fluorescence‐activated cell‐sorting buffer (1× PBS with 5% FBS and 0.1% NaN3) at 300_g_ for 10 minutes and were subsequently incubated with anti–NF‐κB primary antibody (Cell Signaling Technologies, Danvers, MA; catalog No. 3033) per the manufacturer's protocol. After staining with primary antibody, the cells were washed 3 times and incubated with PE‐conjugated anti‐rabbit secondary antibody (1:500 dilution, Biolegend, San Diego, CA; catalog No. 406421) for 30 minutes at 4°C. Stained cells were washed 3 times and ran on a flow cytometer (BD LSR Fortessa, San Jose, CA). Sample acquisition was done using fluorescence‐activated cell‐sorting DIVA software (BD) and subsequently analyzed on FlowJo software (FlowJo, Ashland, OR). Secondary antibody controls were included for each corresponding experimental sample.
Reporter Gene Assay
HL‐1 CM cells were coinfected with reporter lentivirus (Cignal Lenti NF‐κB GFP [green fluorescent protein] Reporter; Qiagen, Hilden, Germany, catalog No. CLS‐013G‐8) and constitutively active positive control lentivirus (Cignal Lenti RFP [red fluorescent protein] Positive Control; Qiagen, Cat # CLS‐PCR‐8) using an Multiplicity of Infection of 100 and polybrene (5 μg/mL; Santa Cruz Biotechnology, Dallas, TX, catalog No. sc‐134220) in complete growth media. Cells were incubated with viral transduction medium for 24 hours and replaced with complete growth medium. After cell expansion in puromycin (1 μg/mL), infected cells were transfected with Snrk siRNA or control siRNA (Dharmacon) as described above. Transfected cells were stimulated with and without Ang II (1 μmol/L), and the cells were further stimulated with TNF‐α (50 ng/mL) for 24 hours. To quantify NF‐κB GFP reporter activity, medium was aspirated, washed with live cell–imaging solution, and then replaced with fresh warm live cell imaging solution. Fluorescence was quantified with SpectraMax i3x microplate reader (Molecular Devices, San Jose, CA) and reported as GFP (482/510 nm) relative to RFP positive control (555/584 nm). After quantification, fluorescence was directly visualized with Keyence (Osaka, Japan) BZ‐X700 fluorescent microscope using a GFP filter cube (OP‐87763, Keyence) and a Texas Red filter cube (OP‐87765, Keyence).
Immunofluorescence
HL‐1 CM cells were grown on coverslips until reaching 60% to 70% confluency. Cells were then washed with 1× PBS (Gibco) 3 times and then fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA). Fixed cells were washed again with 1× PBS before permeabilization with 0.1% Triton X‐100 (Bio‐Rad, Hercules, CA) and blocked with 4% BSA in PBS and overnight incubation with primary antibodies of cardiac troponin‐I (Proteintech, Rosemont, IL, catalog No. 66376) and NF κB p‐p65 (Cell Signaling Technologies, catalog No. 3033) at a dilution of 1:100. Cells were washed again with 1× PBS and incubated with secondary antibody (1:250 dilution) for 90 minutes at room temperature. Cells were mounted with VECTASHIELD antifade mounting medium with DAPI (Vector Laboratories Inc, Burlingame, CA) and used for fluorescence imaging with a Keyence digital microscope at a magnification of ×20. The HL‐1 CMs were quantified across triplicate samples per condition. Thus, 3 cells/sample were quantified for a total of 9 cells per condition. The NF‐kB p‐65 fluorescence integrated density was measured using ImageJ software, and data were plotted as graphs.
Western Blot Analysis
Adult heart tissues were collected immediately after euthanization, and the proteins were isolated using homogenization in RIPA buffer (Sigma) with complete mini EDTA‐free protease inhibitor cocktail (Roche, Basel, Switzerland) and PhosSTOP phosphatase inhibitor (Roche) using a Qiagen TissueRuptor (Hilden, Germany). Methodologies related to protein estimation and quantitation have been described.13 The following antibodies were used: anti–extracellular signal–regulated kinase (total ERK [catalog No. 9102] and phospho‐ERK [catalog No. 9101], Cell Signaling Technology); anti–protein kinase B (total AKT [catalog No. 9272] and phospho‐AKT [catalog No. 4060], Cell Signaling Technology); anti‐JNK (total JNK [catalog No. 9252] and phospho‐JNK [catalog No. 4668], Cell Signaling Technology); anti–NF‐κB P65, (total P65 [catalog No. 8242] and phospho NF‐κB P65 [catalog No. 3033], Cell Signaling Technology); anti–IL‐6 (catalog No. 12912; Cell Signaling Technology); anti–TNF‐α ([catalog No. GTX110520], GeneTex, Irvine, CA); anti–IL‐10 (catalog No. 12163, Cell Signaling Technology); anti‐SNRK (catalog No. GTX111380, GeneTex); antirabbit HRP (horseradish peroxidase, catalog No. 7074, Cell Signaling Technology), and antimouse HRP (catalog No. 7076, Cell Signaling Technology). Quantification was done using ImageJ software and plotted against the respective controls.
Enzyme‐Linked Immunosorbent Assay
Briefly, HL‐1 CM cells were grown in a 30‐mm dish, and the cells were transfected with control or Snrk siRNA with and without Ang II; 100 μL of the cell‐free supernatant was used for the assessment of TNF‐α and IL‐10 inflammatory cytokines. The protocol was followed according to the manufacturer's instructions (RayBiotech, Peachtree Corners, GA; Mouse ELM‐IL10‐1, Mouse ELM‐TNFa‐1).
Statistical Methods
A t‐test, one‐way or two‐way analysis of variance (ANOVA) was used to examine the effects of various conditions on the outcomes. Repeated‐measures ANOVA were performed for outcomes measured over time. A P<0.05, with no correction for multiple testing, was considered significant. For some analyses, data were log transformed to improve fit. All statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC) software.
Results
Loss of Snrk in CMs and Not in ECs Leads to Ang II–Induced Cardiac Failure
Previously, we have shown that Snrk is essential for mammalian development and cardiac metabolism.13, 14 We have generated both cardiac‐specific (MYH6‐CRE) Snrk cmcKO and endothelial‐specific (TIE2‐CRE) Snrk ecKO mice.13 Snrk cmcKO mice showed cardiac function deficits, and adult mice die after between 8 and 10 months.13 Transthoracic ECHO analysis on 6‐month‐old adult male Snrk cmcKO mice showed numerous cardiac functional parameter deficits including lower strain rates, ejection time, and lower time velocity integrals.13 Here, we performed ECHO on 4‐month‐old Snrk cmcKO mice (Figure 1A through 1C) in the absence and presence of Ang II. We chose this time point because we rationalized that at 4 months the cardiovascular defects would be minimal compared with 6 months. Ang II was infused via injection or pumps implanted in 4‐month‐old mice, and ECHO was performed 2 to 3 weeks later. Remarkably, most of the Ang II–infused Snrk cmcKO mice died in 2 weeks, and their cardiac ventricle functional parameters (ejection fraction, fractional shortening, and a majority of ventricular dimension parameters) were significantly attenuated (Figure 1). We also performed ECHO at 2 and 4 weeks on Ang II–infused 4‐ to 5‐month‐old Snrk ecKO mice (Figure S1A through S1C). Interestingly, these mice tolerated the Ang II infusion dose (1000 ng/kg per minute) well and did not show much of a deficit in cardiac functional output. The Ang II dose used in this study does elevate blood pressure,26, 27 but we did not measure blood pressure in our Ang II–infused mice. Further, the Ang II–infused Snrk WT or Snrk ecKO mice in our studies did not die in the 2‐ to 4‐week period of the experiment as the Ang II–infused Snrk cmcKO mice did, suggesting that Ang II–mediated hypertension is not the only component contributing to HF. These results collectively demonstrate that Snrk in CMs protects the heart from adverse Ang II–induced cardiac remodeling that causes accelerated HF.
Snrk Is Protective Against Inflammatory Condition in the Heart
Inflammation has emerged as a key factor in the development and progression of HF.4 To explore the functional role of Snrk in heart inflammation, we collected heart tissue from vehicle‐treated WT, Snrk cmcKO, and Ang II‐treated WT, Snrk cmcKO (Figure 2A and 2B) and Snrk ecKO mice (Figure 2C and 2D) and assessed for pro‐ and anti‐inflammatory cytokine levels by Western blotting. Proinflammatory cytokines such as IL‐6, TNF‐α, and NF‐κB P65 were increased under basal Snrk cmcKO condition (Figure 2A), and in the Ang II–induced condition the proinflammatory cytokine levels further increased (Figure 2A and 2B). Conversely, the anti‐inflammatory IL‐10 protein expression was decreased in Snrk cmcKO hearts compared with Snrk WT hearts. In contrast, in the Ang II–induced condition, IL‐10 protein expression was further decreased. Hearts from Snrk ecKO mice (Figure 2C and 2D) showed proinflammatory IL‐6, TNF‐α, and NF‐κB P65 protein changes that were not significantly altered in the basal level. In Ang II–induced condition, IL‐6, TNF‐α cytokine levels are attenuated while NF‐κB p‐p65 protein levels increase further. Levels of IL‐10 anti‐inflammatory protein were decreased in Ang II‐induced Snrk ecKO mice hearts compared with vehicle‐treated WT or WT with Ang II‐infused hearts. To confirm the inflammatory effects in CMs, we mimicked the Snrk knockdown in vitro by Lipofectamine‐based silencing of Snrk in the HL‐1 mouse atrial cardiomyocyte cell line using Snrk siRNA (Figure S2A and S2B) with and without Ang II stimulation (Figure 2E and 2F). We found that the inflammatory signaling molecule changes are similar to the in vivo results. Further, we also assessed proinflammatory cytokine TNF‐α and anti‐inflammatory cytokine IL‐10 levels by a second independent ELISA method on HL‐1 cell supernatants (Figure S3) and found increased TNF‐α (Figure S3A) and decreased IL‐10 levels (Figure S3B) in Snrk siRNA HL‐1 cells compared with control siRNA cells. Ang II stimulation on both Snrk siRNA and control siRNA cells showed expected increased and decreased protein levels. Together, these in vivo and in vitro data suggest that Snrk is essential for repressing inflammation in a cell‐autonomous fashion in CMs, an effect that is exacerbated under Ang II–induced conditions.
Figure 2.
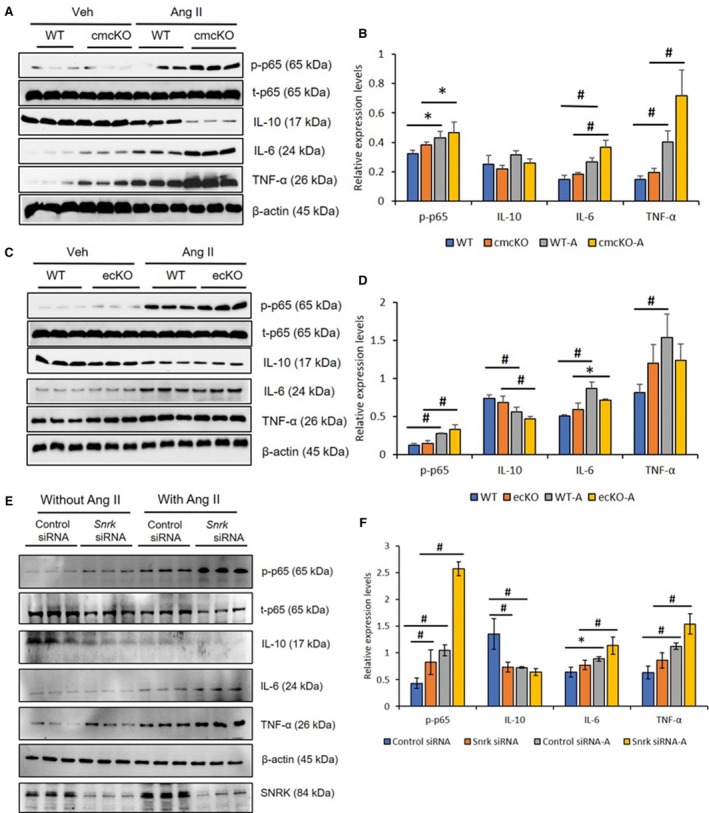
Angiotensin II (Ang II)–infused hearts from Snrk conditional knockout mice and Snrk knockdown cardiomyocytes (CMs) show higher levels of proinflammatory response. A and B, Hearts from Snrk cmcKO and (C and D) Snrk ecKO mice were assessed for pro‐ and anti‐inflammatory signaling by immunoblotting. Both knockout mouse groups were analyzed under vehicle‐treated and Ang II–induced conditions similar to wild‐type (WT) control mice. NF‐κB p‐p65, IL‐6, and TNF‐α were assessed for proinflammatory signaling, and IL‐10 was assessed for anti‐inflammatory signaling. Results are presented as mean±SEM (*P<0.05 and # P<0.01). The statistical comparison for P value was done by comparing Snrk WT vs Snrk cmcKO, Snrk WT vs Snrk WT‐Ang II, Snrk cmcKO vs Snrk cmcKO‐Ang II and Snrk WT‐Ang II vs Snrk cmcKO‐Ang II (n=3 animals in each experimental group). E and F, HL‐1 cardiomyocyte cells were treated with Snrk small interfering (si) RNA with and without Ang II and assessed for pro‐ and anti‐inflammatory signaling. Results are presented as mean±SEM (*P<0.05 and # P<0.01). The statistical comparison for P value was done by comparing control siRNA vs Snrk siRNA, control si RNA vs control siRNA‐Ang II, Snrk siRNA vs Snrk siRNA‐Ang II, and control siRNA‐Ang II vs Snrk siRNA‐Ang II (n=3 in each experimental group). cmcKO indicates cardiomyocyte knockout; ecKO, endothelial cell knockout; SEM, standard error of the mean.
Ang II–Induced SNRK Influences Key Signaling Pathways
We noticed that Ang II–induced SNRK protein expression in HL‐1 cell line (Figure 2F, note control lanes for SNRK). Therefore, to investigate whether Ang II–induced effects may be linked to SNRK in CMs, we focused on key cardiac signaling pathways previously implicated in Ang II–induced cardiac inflammation (pAKT and pERK).28 Snrk siRNA–treated HL‐1 CM cells were treated with and without Ang II and probed for pAKT and pERK signaling proteins. We noticed a significant trend in pAKT protein toward sustained increases in basal Snrk siRNA cells and further increase in Ang II–treated Snrk siRNA cells (Figure 3A and 3B). In heart tissue lysates from Snrk cmcKO (Figure 3C and 3D), under basal conditions, minimal change was noticed in the expression of pAKT. In an Ang II–stimulated condition, pAKT protein level was increased. Interestingly, in Snrk ecKO hearts, under basal conditions, we observed no change in the pAKT protein levels compared with Snrk WT, but with Ang II stimulation, pAKT was found to be decreased (Figure S2C and S2D, *P<0.05). These data collectively suggest that in Ang II conditions, SNRK controls pAKT protein levels in CMs. For pERK, the basal state is high in Snrk cmcKO hearts and low in Snrk ecKO hearts (*P<0.05), which is indicative of a basal stressed state of the heart. Again, under Ang II conditions, high pERK protein levels in Snrk cmcKO hearts became low (Figure 3D), and low pERK protein levels in Snrk ecKO hearts became high (Figure S2C and S2D). These data argue that, as for pAKT protein levels under cardiac stress conditions, SNRK controls pERK protein levels in CMs. Collectively, the HL‐1, Snrk cmcKO heart and Snrk ecKO heart data suggest that under Ang II–induced conditions, in CMs, either directly (Snrk cmcKO) or indirectly (Snrk ecKO), SNRK maintains pAKT and pERK protein levels.
Figure 3.
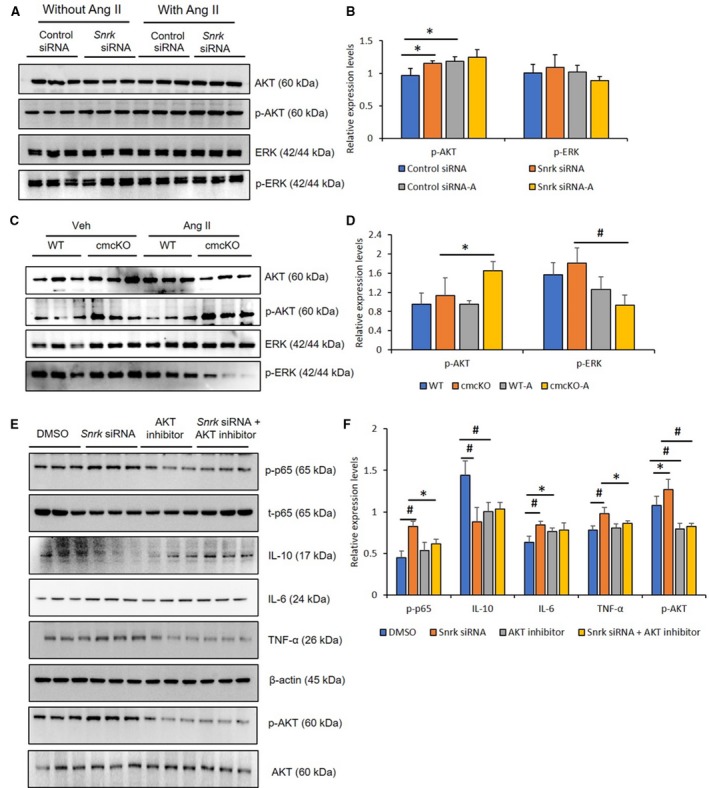
Angiotensin II (Ang II)–infused hearts from Snrk cmcKO and Snrk knockdown cardiomyocytes (CMs) show changes in pAKT and pERK signaling pathways. A and B, Immunoblotting analysis of HL‐1 cardiomyocytes were treated with Snrk small interfering (si) RNA with and without Ang II (1 μmol/L) for 24 hours. Results are presented as mean±SEM (*P<0.05). Statistical comparison for P value was done by comparing control siRNA vs Snrk siRNA, control siRNA vs control siRNA‐Ang II, Snrk siRNA vs Snrk siRNA‐Ang II, and control siRNA‐Ang II vs Snrk siRNA‐Ang II (n=3 in each experimental group). C and D, Snrk cmcKO mouse hearts were assessed for phosphorylated forms of AKT and ERK signaling by immunoblotting, KO mouse hearts were analyzed under vehicle‐treated and Ang II–induced conditions, wild‐type (WT) mice were used as controls for both conditions. Results are presented as mean±SEM (*P<0.05 and # P<0.01). The statistical comparison for P value was done by comparing Snrk WT vs Snrk cmcKO, Snrk WT vs Snrk WT‐Ang II, Snrk cmcKO vs Snrk cmcKO‐Ang II, and Snrk WT‐Ang II vs Snrk cmcKO‐Ang II (n=3 animals in each experimental group). E and F, Briefly, HL‐1 cardiomyocyte cells were subjected to Snrk siRNA transfection and treated with and without AKT inhibitor LY294002 (10 μmol/L) for 24 hours and assessed for phosphorylated AKT and the proinflammatory NF‐κB p‐p65, IL‐6, and TNF‐α and anti‐inflammatory IL‐10 signaling. DMSO‐treated cells served as control. Results are presented as mean±SEM (*P<0.05 and # P<0.01 compared with respective treatment groups; n=3 in each experimental group). cmcKO indicates cardiomyocyte knockout; SEM, standard error of the mean.
Previously, we showed that SNRK regulates CMs metabolic homeostasis via a phosphorylated ACC (pACC)–pAMPK pathway13 that is known to be regulated by AKT.29, 30 Therefore, we hypothesized that a SNRK‐AKT signaling axis in CMs is responsible for controlling inflammation. To investigate this hypothesis, Snrk siRNA–transfected HL‐1 CM cells were treated with and without AKT inhibitor LY294002 and probed for inflammatory proteins (Figure 3E). In their basal state, Snrk siRNA knockdown HL‐1 CM cells showed upregulation of proinflammatory proteins (p‐p65, IL‐6, TNF‐α) and downregulation of anti‐inflammatory protein IL‐10, which was consistent with previous results (Figure 2E). In AKT inhibitor‐treated HL‐1 WT cells, proinflammatory proteins are lower, but little change was observed in the anti‐inflammatory IL‐10 protein level. In Snrk siRNA knockdown HL‐1 cells treated with AKT inhibitor, the basal changes in inflammatory proteins observed in Snrk siRNA‐knockdown HL‐1 cells were rescued (Figure 3E and 3F). These data suggest that in CMs, SNRK controls pAKT, which in turn controls inflammation.
Ang II–Induced Inflammation in CMs Is Repressed by SNRK and Is Mediated Partly by an AKT Pathway
We already have shown that Ang II treatment of Snrk siRNA HL‐1 cells shows higher pAKT protein levels (Figure 3A and 3B), whereas AKT inhibition in Snrk siRNA HL‐1 cells is associated with decreased proinflammation proteins (Figure 3E and 3F). Therefore, we next investigated whether an Ang II–induced SNRK‐AKT signaling axis links to proinflammation signals. We treated control and Snrk siRNA knockdown HL‐1 cells with and without Ang II and/or with and without AKT inhibitor and probed for proinflammatory proteins (Figure 4A and 4B). When compared with control siRNA cells, Snrk siRNA cells showed expected higher amounts of pAKT, p‐p65, IL‐6, and TNF‐α (Figure 4B, compare gray with orange bars). In the presence of Ang II, control siRNA cells (Figure 4B, yellow bars) showed higher levels of all the tested proteins compared to without Ang II (control siRNA [gray bars] or untreated [intermediate blue color bars]). In Ang II alone–treated Snrk siRNA cells (Figure 4B, dark blue color bars), the most pronounced increase was clearly in p‐p65 levels compared with the other proteins, which showed a modest increase (compare with untreated Snrk siRNA cells, Figure 4B, orange bars). This interpretation is also consistent with previous (Figure 3A) results in which Ang II–treated Snrk siRNA cells showed modest upregulation of pAKT. Consistent with this interpretation, in AKT inhibitor alone–treated Snrk siRNA cells (Figure 4B, brown bars), most protein levels did not change compared with untreated Snrk siRNA cells (Figure 4B, orange bars), indicating that a majority of the effect was driven by Ang II treatment. This interpretation is in agreement with the double treatment (Ang II+AKT inhibitor) Snrk siRNA cells (Figure 4B, dark gray bars), where the Ang II exacerbated p‐p65 levels, and other protein levels were reduced. These results collectively suggest that Ang II induces SNRK and pAKT protein levels in HL‐1 cells, which control inflammation. In the absence of SNRK, the inflammatory pathway is upregulated, which can be controlled partly by AKT inhibition. Thus, under increased cardiac remodeling conditions (Ang II), the cell‐autonomous role for SNRK in CMs is protective against cardiac inflammation partly via the AKT signaling axis.
Figure 4.
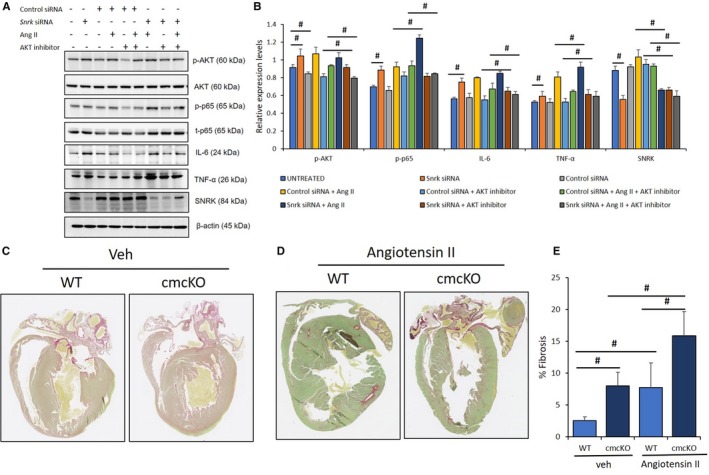
AKT inhibition under angiotensin II (Ang II)–stimulated conditions in cardiomyocytes (CMs) attenuate inflammation. A, HL‐1 CMs were transfected with control and Snrk small interfering (si) RNA, studied with and without Ang II (1 μmol/L) and/or AKT inhibitor LY294002 (10 μmol/L), and assessed for phosphorylated‐AKT (p‐AKT) and proinflammatory markers NF‐κB p‐p65, IL‐6, and TNF‐α. Untreated cells served as control. Results are presented as mean±SEM (*P<0.05 and # P<0.01 vs respective control siRNA‐treated cells; n=3 in each experimental group). B, Quantification of blots from sample in A. C through E, Snrk cmcKO hearts were analyzed for fibrosis using Sirius red staining, which showed an exacerbated fibrotic environment determined by increased accumulation of collagen in the hearts. The mice were treated with vehicle control (C) and Ang II (D). Significant increases in the accumulation of collagen in the Snrk cmcKO mice were observed compared with wild type (WT); on stimulation with Ang II the collagen deposition increases further. Results are presented as mean±SEM (# P<0.01). The statistical comparison for P value was done by comparing Snrk WT vs Snrk cmcKO, Snrk WT vs Snrk WT–Ang II, Snrk cmcKO vs Snrk cmcKO–Ang II, and Snrk WT–Ang II vs Snrk cmcKO–Ang II (n=3 animals in each experimental group). cmcKO indicates cardiomyocyte knockout; SEM, standard error of the mean; veh, vehicle.
Mice Lacking Cardiac Snrk Expression Promote Cardiac Fibrosis Induced by Ang II
Cardiac inflammation is often associated with deposition of collagen leading to fibrosis in the heart.31 Thus, to evaluate the degree of fibrosis in the heart, tissue samples from animals with age‐matched transgenic animals were stained using Sirius red, which stains collagen. As shown (Figure 4C through 4E), tissues from Snrk WT and Snrk cmcKO mice without (Figure 4C) and with (Figure 4D) Ang II–infused hearts were examined. Hearts from Snrk cmcKO mice in both vehicle‐ and Ang II–infused conditions (Figure 4D) showed substantial accumulation of collagen and developed severe fibrosis compared with Snrk WT mice, which did show increased collagen deposition with Ang II infusion (Figure 4C). Hearts from Snrk ecKO mice showed similar collagen deposition without any changes in basal state compared with Snrk WT mice (Figure S4A). In the Ang II–infused condition, both (Snrk WT and Snrk ecKO) mice showed similar increase in collagen deposition with no appreciable difference across the samples (Figure S4B). These data suggest that Snrk is critical for CM health and is protective against inflammation and fibrosis in the heart.
Increased Mac2‐Positive Macrophage Coverage in Hearts of Snrk cmcKO Mice
Infiltration of inflammatory cells such as macrophages in the heart is an early event in Ang II–induced cardiac remodeling.32 Macrophages also contribute to the development of cardiac fibrosis.33 To assess the macrophage numbers in the heart, immunohistochemistry was performed with a Mac2 monoclonal antibody that specifically binds to the mouse Mac2 antigen in the heart tissues. The Mac2 antigen is a 32 000‐Da surface protein expressed on a subpopulation of mouse macrophages.34 As shown (Figure 5A), under basal conditions, vehicle‐treated Snrk cmcKO hearts in comparison to WT mice showed an increased coverage of Mac2‐positive (Mac2+) macrophage in a given surface area in the heart. In the Ang II‐infused condition, both WT and Snrk cmcKO heart showed comparable increase (Figure 5B) in Mac2+ macrophage coverage in the heart. These data suggest that Snrk in CMs is responsible for controlling cardiac inflammation, which partly influences the increased presence of macrophages in the hearts.
Figure 5.
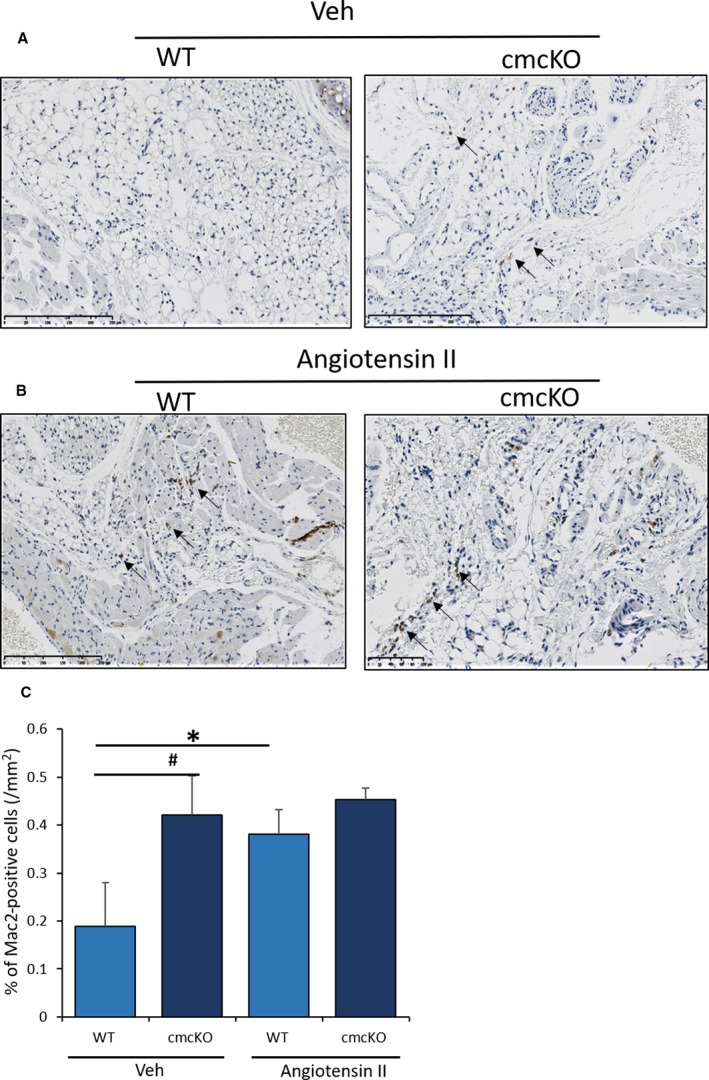
SNRK enhances angiotensin II (Ang II)–induced inflammation in Snrk cmcKO hearts. A, Wild‐type (WT) and Snrk cardiac knockout mice (cmcKO) were treated with vehicle control and Ang II (B). Increased infiltration of macrophages in the Snrk cmcKO mice was observed compared with WT on stimulation with Ang II, and macrophage numbers further increased. Scale bars are 250 μm for Snrk WT, Snrk cmcKO, and Snrk WT‐Ang II and 100 μm for Snrk cmcKO‐Ang II. C, Results are presented as mean±SEM (*P<0.05 and # P<0.01). The statistical comparison for P value was done by comparing Snrk WT vs Snrk cmcKO, Snrk WT vs Snrk WT–Ang II, Snrk cmcKO vs Snrk cmcKO–Ang II, and Snrk WT–Ang II vs Snrk cmcKO–Ang II (n=3 animals in each experimental group). SEM indicates standard error of the mean; veh, vehicle.
Ang II–SNRK–NF‐κB Pathway Mechanism in Inflammatory CMs
Based on recent evidence18 in Ang II–treated glomerular ECs that SNRK interacts with the p‐p65 subunit of nuclear factor κ light‐chain enhancer of the activated B cells (NF‐κB) pathway, we investigated whether this mechanism was responsible for preventing inflammation in CMs. NF‐κB is comprised of homo‐ or heterodimers of the Rel family of DNA‐binding proteins (RelA, p65; RelB, p105 and p100),35 whose activity is regulated by IκB proteins. The phosphorylation of p65 and IκB proteins results in release of NF‐κB from IκB complex and entry into nucleus to transactivate gene targets for proinflammation. We assessed the p‐p65 protein by fluorescence‐activated cell‐sorting analysis in Snrk siRNA or control siRNA knockdown HL‐1 cells in the presence or absence of Ang II stimulation (Figure 6A and 6B). We noticed an increase in the number of cells expressing NF‐κB p‐p65 in Snrk knockdown HL‐1 cells compared with control siRNA HL‐1 cells. In HL‐1 cells treated with Ang II, we observed a higher number of cells expressing p‐p65 (Figure 6A and 6B). To confirm transcriptional activation of the NF‐κB pathway, we performed a GFP reporter gene vector assay, wherein the vector contains NF‐κB response elements in tandem to GFP, which helps to assess the transcriptional activation of the NF‐κB signaling pathway. An RFP‐expressing vector acted as a positive control. Basal Snrk siRNA HL‐1 cells showed increased GFP/RFP ratio compared with control or vector‐alone transfected HL‐1 cells (Figure 6C and 6D). Ang II treatment further increased this ratio in Snrk siRNA HL‐1, indicating a transcriptionally active state of NF‐κB (Figure 6C and 6D). Additionally, we performed Immunofluorescence p‐p65 staining in HL‐1 CMs and noticed a similar increased trend in p‐p65 staining in troponin‐I+ Ang II–treated CMs compared with untreated CMs (Figure 6E and 6F). Akt inhibitor CMs also showed a statistically significant increase in p‐p65 staining (Figure 6E and 6F) compared with untreated CMs. The in vitro HL‐1 p‐p65 results (fluorescence‐activated cell‐sorting analysis, reporter gene assay) correlate with the respective Western blot results for p‐p65 proteins (Figure 2A: Snrk cmcKO heart; Figures 3E and 4A: HL‐1) in vivo heart lysate. Thus, collectively, all these results suggest that SNRK in CMs is a repressor of cardiac inflammation through suppression of NF‐κB signaling, which is attenuated in Ang II–associated cardiac remodeling.
Figure 6.
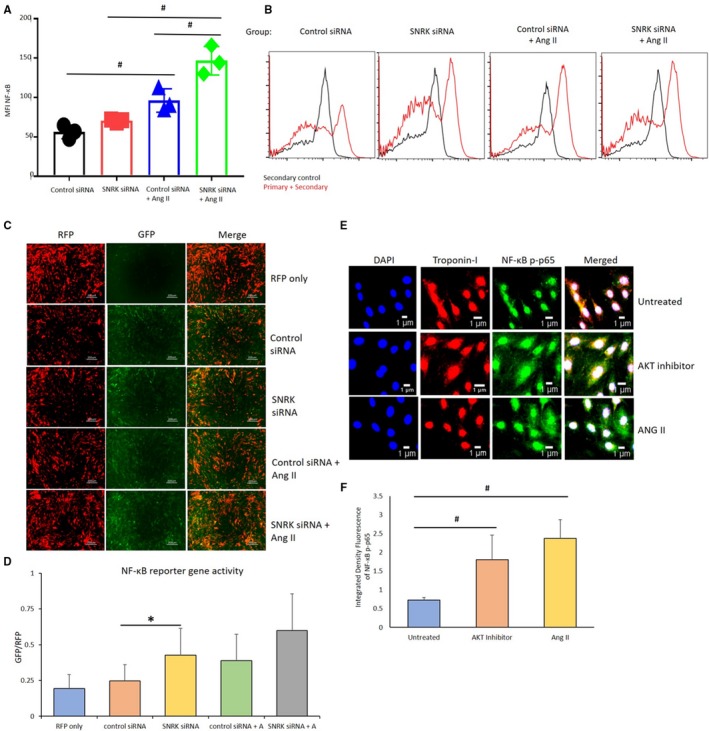
NF‐κB transcription and expression in HL‐1 cardiomyocytes. A and B, Flow cytometry analysis for detection of HL‐1 cells positive for the expression of NF‐κB p‐p65. Results are presented as mean±SEM (# P<0.01). The statistical comparison for P value was done by comparing control small interfering (si) RNA vs Snrk siRNA, control siRNA vs Control siRNA–angiotensin II (Ang II), Snrk siRNA vs Snrk siRNA–Ang II, and control siRNA–Ang II vs Snrk siRNA–Ang II (n=3 per each experimental group). C and D, Reporter vector gene assay for the analysis of transcriptional activation of NF‐κB. NF‐κB vector was tagged with GFP protein, RFP acted as positive control for the assessment of infection efficiency. Scale bars are at 200 μm. Results are presented as mean±SEM (*P<0.05). The statistical comparison for P value was done by comparing control siRNA vs Snrk siRNA, control siRNA vs control siRNA–Ang II, Snrk siRNA vs Snrk siRNA–Ang II, and control siRNA–Ang II vs Snrk siRNA–Ang II (n=12 in each experimental group). E and F, HL‐1 cells were treated with AKT inhibitor/Ang II, where Ang II–treated HL‐1 cells show more NF‐κB p‐p65. The resulting colocalizations were quantified using ImageJ software and represented as graphs. Scale bars are at 1 μm. Results are presented as mean±SEM (# P<0.01 vs untreated control cells; n=3 in each experimental group). GFP indicates green fluorescent protein; MFI, Mean Fluorescence Intensity; RFP, red fluorescent protein; SEM, standard error of the mean.
Discussion
In this study we identified SNRK as a potential suppressor of cardiac inflammation and hypothesize that SNRK in CMs protects against the development of cardiac inflammation and fibrosis commonly observed in HF.5, 6, 31, 32 In our previous studies we observed cardiac functional deficits by ECHO in Snrk cmcKO mice at 6 months, and they succumbed to these defects at ≈9 months.13 To assess how these mice would respond to stress early, we subjected 4‐month‐old Snrk cmcKO mice to Ang II infusion, which is known to increase cardiac load and cause HF.36 Remarkably, the 4‐month‐old Snrk cmcKO succumbed to Ang II infusion within 14 days. The same experiment performed on Snrk ecKO mice did not result in death. Further, fibrosis was only observed in Snrk cmcKO and not in Snrk ecKO mice, and Ang II, exacerbated inflammation and fibrosis in Snrk cmcKO hearts. These results suggest that SNRK in CMs is critical for heart function.
To assess the cause of the death in Ang II–infused Snrk cmcKO mice, we focused on Ang II–induced inflammatory and fibrotic signaling pathways in the heart.23 Ang II acts via NF‐κB to stimulate a proinflammatory environment in the heart,37 and SNRK represses proinflammatory signaling in adipocytes through defective mTOR signaling.17 On Ang II infusion, phosphorylated NF‐κB p65 nuclear subunit protein level becomes higher in the heart tissue lysates and in HL‐1 CMs. Further, more Mac2+ macrophages were noticed in Ang II–infused Snrk cmcKO hearts. However, when these are compared with basal Snrk cmcKO hearts, we do not observe an even greater increase in Mac2+ cells. We postulate that because the Snrk cmcKO mice are on the verge of dying, there is not enough time for sustained Mac2+ cells to reach the heart, and/or Mac2+ cells in the altered environment of Snrk cmcKO hearts may be dying rapidly or preferentially. These hypotheses will need further evaluation. Also, note that anti‐inflammatory IL‐10 levels in Ang II–infused Snrk cmcKO hearts in vivo are different from those in Snrk siRNA HL‐1 cells by Western blot (Figure 2F) or ELISA (Figure S3B) in vitro, which suggests a contribution of an EC compartment or other cell types in the heart to IL‐10 levels. In support of an EC contribution to IL‐10 levels in the heart, Snrk ecKO hearts show lower levels of IL‐10 (Figure 2D). Interestingly, expression of the proinflammatory cytokines IL‐6 and TNF‐α is higher in the hearts of Ang II–infused Snrk ecKO mice (Figure 2C) even though this high level does not seem to influence cardiac function (Figure S1) or mortality in these mice. Thus, the question is how does SNRK repress inflammation in CMs from a mechanism standpoint, and how does this lead to HF? A recent article18 from our collaborators suggests a potential mechanism. They identified that the NF‐κB pathway was a direct target of SNRK in kidney glomerular ECs treated with Ang II. NF‐κB is composed of homo‐ or heterodimers of the Rel family of DNA‐binding proteins (RelA, p65; RelB, p105 and p100), and NF‐κB activity is regulated by proteins called IκB.35, 38 In the canonical mechanism, NF‐κB is complexed with IκB in the cytoplasm, and this complex is in an inactive state. On phosphorylation of IκB, this protein gets targeted for ubiquitinylation, which releases NF‐κB from the IκB‐containing complex, unmasking the nuclear localization signal of NF‐κB. The NF‐κB complex composed of RelA/c‐Rel/p50 enters the nucleus and activates transcription of proinflammatory target genes.35 Further, cardiomyocyte‐specific NF‐κB activation induces reversible inflammatory cardiomyopathy and HF.39 Ang II activates NF‐κB in CMs40 but not in cardiac fibroblasts.41 NF‐κB polymorphisms in the gene promoter are associated with a lower ejection fraction in patients with HF.42 Thus, several lines of evidence suggest a strong association among Ang II, NF‐κB activation, inflammation, and HF.
In this study we have much evidence from heart lysates in vivo and from CMs in vitro that SNRK represses NF‐κB activation. Thus, we hypothesize that in unstimulated CMs, SNRK interacts with p‐p65 (RelA) in the cytoplasm and nucleus. On Ang II stimulation in CMs, we have observed an increase in SNRK protein expression, but whether this increase is cellular compartment specific is unknown. Further, whether there is differential interaction of SNRK with p‐p65 in the nucleus and cytoplasm of CMs is unknown. Thus, in this hypothetical model, the SNRK:p‐p65 interaction of NF‐κB complex will prevent p‐p65 from entering the nucleus to activate proinflammation signaling. This model thus predicts that in the absence of SNRK, the association of p65 to IκB in cytoplasm is likely compromised, thereby facilitating the degradation of IκB and subsequent p‐p65 subunit translocation into the nucleus to activate proinflammatory gene and protein expression.
In terms of fibrosis it is noteworthy that ECs in the heart behave differently than the ECs in the kidney. In our study Snrk ecKO hearts do not show fibrosis in the presence of Ang II, but in the Lu et al study,18 they do show fibrosis in the kidney. These data collectively suggest that the majority of the fibrosis effect in the heart is emerging from CMs. Our work also sheds light on the link between inflammation and fibrosis in HF.5, 6 The Snrk ecKO heart lysates in our study do show upregulation of the inflammatory pathway (Figure 2C), but these hearts do not progress to fibrosis (Figure S3). Thus, we reason that in the heart, SNRK in CMs prevents the progression from inflammation to fibrosis, and such protective fibrotic mechanisms are mediated by ECs in the kidney glomeruli. However, in this hypothesis, the effect of CMs on cardiac fibroblasts is not excluded, and it is possible that signaling from CMs is altered in such a fashion that cardiac fibroblasts become activated and differentiate to myofibroblasts. Myofibroblasts then secrete high levels of proinflammatory and profibrotic paracrine factors. Thus, there are multiple mechanisms for fibrosis to occur in the Snrk cmcKO hearts, but this collectively implies cross‐talk between CMs and cell types in the heart.7 Thus, SNRK's role in protecting against heart inflammation and fibrosis, both of which are cardiomyocyte‐cell autonomous, is one of the salient features of this work.
For downstream signaling mechanisms associated with Ang II–induced cardiac inflammation in Snrk cmcKO hearts, the picture is not yet fully clear, although our work implicates pAKT to a certain extent. Both pAKT and pERK signaling proteins have been implicated previously in cardiac inflammation.43, 44 Although pERK signals were high in Snrk cmcKO hearts, this signal was attenuated in the presence of Ang II. This was not the case for pAKT. Furthermore, pAKT regulates the pACC‐pAMPK pathway, a signaling pathway controlled by SNRK.13 In inflammation, AKT can phosphorylate and activate IKKβ causing translocation of NF‐κB where it promotes the expression of proinflammatory cytokines and accelerates the inflammatory response.45, 46 Snrk knockdown CMs in general showed high levels of basal inflammation, which was exacerbated on Ang II treatment. We found that the basal AKT inhibition in Snrk knockdown CMs modestly influenced inflammation. However, AKT inhibition in Ang II–treated Snrk knockdown CMs blocked proinflammatory protein expression. These data suggest that the majority of the Ang II–induced signals leading to CM inflammation are going through multiple pathways, one of which is pAKT. In this context, SNRK acts to repress Ang II's effect on pAKT‐mediated inflammation in CMs. The association of SNRK, AKT, and Ang II requires further exploration, especially at the level of individual phosphorylation sites in the SNRK and AKT proteins; whether such phosphorylation is negative or positive in their signaling outcome will ultimately clarify the signaling mechanism. Other limitations of this study include the limited sample size for the Ang II ECHO Snrk cmcKO studies, exclusive reliance on loss‐of‐function studies, and lack of thorough follow‐up of in vitro findings in vivo.
The findings in this article suggest the significance of SNRK function in repressing cardiac inflammation during the pathological cardiac remodeling process that leads to HF. Accumulating evidence suggests that chronic inflammation and fibrosis are linked together and play an important role in HF.4, 5, 6, 11, 31 Our work is suggestive that the 2 processes of fibrosis and inflammation can be decoupled in the heart, and the cross‐talk between ECs and CMs where SNRK participates is a key node of intervention. In summary, our data suggest that SNRK serves a critical function in CMs, which is to block heart inflammation that occurs via interfering with an activated NF‐κB pathway. This SNRK function is cell autonomous and exacerbated during the pathological cardiac remodeling process that leads to cardiac functional deficits, fibrosis, and HF.
Sources of Funding
Thirugnanam, Gupta and Ramchandran are supported by NIH grant HL123338 and partly supported by funds from the Department of Pediatrics and Children's Research Institute funds. Ramchandran is also supported by Women's Health Research Program in the Department of Obstetrics and Gynecology at the Medical College of Wisconsin. Strande is supported by NIH grant R01 HL134932. Lu and Zou are supported by NIH grants CA213022, HL080499, HL089220, and HL142287. Chowdhury is supported by Medical College of Wisconsin Cancer Center Pilot Grant, funded through Advancing a Healthier Wisconsin research and education program. Spearman is partially supported by Mend‐a‐Heart Foundation. The funders had no role in the study design or in the collection, analysis, and interpretation of data.
Disclosures
None.
Supporting information
Figure S1. Ang II does not induce cardiac functional deficits in Snrk ecKO mice.
Figure S2. Snrk knockdown and cardiac dysfunctional signaling in endothelial cells.
Figure S3. Expression of pro‐ and anti‐inflammatory cytokine in HL‐1 cells.
Figure S4. Activation of proinflammatory cytokines signals to fibrosis in the heart of Snrk ecKO mice.
(J Am Heart Assoc. 2019;8:e012792 DOI: 10.1161/JAHA.119.012792.)
References
- 1.Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giamouzis G, Kalogeropoulos A, Georgiopoulou V, Laskar S, Smith AL, Dunbar S, Triposkiadis F, Butler J. Hospitalization epidemic in patients with heart failure: risk factors, risk prediction, knowledge gaps, and future directions. J Card Fail. 2011;17:54–75. [DOI] [PubMed] [Google Scholar]
- 3.Ponikowski P, Anker SD, AlHabib KF, Cowie MR, Force TL, Hu S, Jaarsma T, Krum H, Rastogi V, Rohde LE, Samal UC, Shimokawa H, Budi Siswanto B, Sliwa K, Filippatos G. Heart failure: preventing disease and death worldwide. ESC Heart Fail. 2014;1:4–25. [DOI] [PubMed] [Google Scholar]
- 4.Dick SA, Epelman S. Chronic heart failure and inflammation: what do we really know? Circ Res. 2016;119:159–176. [DOI] [PubMed] [Google Scholar]
- 5.Suthahar N, Meijers WC, Sillje HHW, de Boer RA. From inflammation to fibrosis—molecular and cellular mechanisms of myocardial tissue remodelling and perspectives on differential treatment opportunities. Curr Heart Fail Rep. 2017;14:235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Linthout S, Tschope C. Inflammation—cause or consequence of heart failure or both? Curr Heart Fail Rep. 2017;14:251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tirziu D, Giordano FJ, Simons M. Cell communications in the heart. Circulation. 2010;122:928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu C, Zhu JX, Yang Z, Deng W, Tang QZ. Mechanisms contributing to cardiac remodelling. Clin Sci (Lond). 2017;131:2319–2345. [DOI] [PubMed] [Google Scholar]
- 9.Vignon‐Zellweger N, Relle K, Rahnenfuhrer J, Schwab K, Hocher B, Theuring F. Endothelin‐1 overexpression and endothelial nitric oxide synthase knock‐out induce different pathological responses in the heart of male and female mice. Life Sci. 2014;118:219–225. [DOI] [PubMed] [Google Scholar]
- 10.Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, Ruiz‐Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J Jr, Chien KR, Ferrara N. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci USA. 2001;98:5780–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tschope C, Van Linthout S. New insights in (inter)cellular mechanisms by heart failure with preserved ejection fraction. Curr Heart Fail Rep. 2014;11:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leucker TM, Jones SP. Endothelial dysfunction as a nexus for endothelial cell‐cardiomyocyte miscommunication. Front Physiol. 2014;5:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cossette SM, Gastonguay AJ, Bao X, Lerch‐Gaggl A, Zhong L, Harmann LM, Koceja C, Miao RQ, Vakeel P, Chun C, Li K, Foeckler J, Bordas M, Weiler H, Strande J, Palecek SP, Ramchandran R. Sucrose non‐fermenting related kinase enzyme is essential for cardiac metabolism. Biol Open. 2014;4:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cossette SM, Bhute VJ, Bao X, Harmann LM, Horswill MA, Sinha I, Gastonguay A, Pooya S, Bordas M, Kumar SN, Mirza SP, Palecek SP, Strande JL, Ramchandran R. Sucrose nonfermenting‐related kinase enzyme‐mediated rho‐associated kinase signaling is responsible for cardiac function. Circ Cardiovasc Genet. 2016;9:474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rines AK, Chang HC, Wu R, Sato T, Khechaduri A, Kouzu H, Shapiro J, Shang M, Burke MA, Abdelwahid E, Jiang X, Chen C, Rawlings TA, Lopaschuk GD, Schumacker PT, Abel ED, Ardehali H. Snf1‐related kinase improves cardiac mitochondrial efficiency and decreases mitochondrial uncoupling. Nat Commun. 2017;8:14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Feng B, Nie Y, Jiao P, Lin X, Huang M, An R, He Q, Zhou HE, Salomon A, Sigrist KS, Wu Z, Liu S, Xu H. Sucrose nonfermenting‐related kinase regulates both adipose inflammation and energy homeostasis in mice and humans. Diabetes. 2018;67:400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Nie Y, Helou Y, Ding G, Feng B, Xu G, Salomon A, Xu H. Identification of sucrose non‐fermenting‐related kinase (SNRK) as a suppressor of adipocyte inflammation. Diabetes. 2013;62:2396–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu Q, Ma Z, Ding Y, Bedarida T, Chen L, Xie Z, Song P, Zou MH. Circulating miR‐103a‐3p contributes to angiotensin II‐induced renal inflammation and fibrosis via a SNRK/NF‐kappaB/p65 regulatory axis. Nat Commun. 2019;10:2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chun CZ, Kaur S, Samant GV, Wang L, Pramanik K, Garnaas MK, Li K, Field L, Mukhopadhyay D, Ramchandran R. Snrk‐1 is involved in multiple steps of angioblast development and acts via notch signaling pathway in artery‐vein specification in vertebrates. Blood. 2009;113:1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pramanik K, Chun CZ, Garnaas MK, Samant GV, Li K, Horswill MA, North PE, Ramchandran R. Dusp‐5 and Snrk‐1 coordinately function during vascular development and disease. Blood. 2009;113:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Q, Xie Z, Yan C, Ding Y, Ma Z, Wu S, Qiu Y, Cossette SM, Bordas M, Ramchandran R, Zou MH. SNRK (sucrose nonfermenting 1‐related kinase) promotes angiogenesis in vivo. Arterioscler Thromb Vasc Biol. 2018;38:373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldstein S, Sabbah H. Ventricular remodeling and angiotensin‐converting enzyme inhibitors. J Cardiovasc Pharmacol. 1994;24(suppl 3):S27–S31. [PubMed] [Google Scholar]
- 23.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. [DOI] [PubMed] [Google Scholar]
- 24.Saito Y, Berk BC. Angiotensin II‐mediated signal transduction pathways. Curr Hypertens Rep. 2002;4:167–171. [DOI] [PubMed] [Google Scholar]
- 25.Paulus WJ. Cytokines and heart failure. Heart Fail Monit. 2000;1:50–56. [PubMed] [Google Scholar]
- 26.Sparks MA, Stegbauer J, Chen D, Gomez JA, Griffiths RC, Azad HA, Herrera M, Gurley SB, Coffman TM. Vascular type 1A angiotensin II receptors control BP by regulating renal blood flow and urinary sodium excretion. J Am Soc Nephrol. 2015;26:2953–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomolak JR, Didion SP. Angiotensin II‐induced endothelial dysfunction is temporally linked with increases in interleukin‐6 and vascular macrophage accumulation. Front Physiol. 2014;5:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang F, Ren X, Zhao M, Zhou B, Han Y. Angiotensin‐(1‐7) abrogates angiotensin II‐induced proliferation, migration and inflammation in VSMCs through inactivation of ROS‐mediated PI3K/Akt and MAPK/ERK signaling pathways. Sci Rep. 2016;6:34621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP‐activated protein kinase in the heart. J Biol Chem. 2003;278:39422–39427. [DOI] [PubMed] [Google Scholar]
- 30.Soltys CL, Kovacic S, Dyck JR. Activation of cardiac AMP‐activated protein kinase by LKB1 expression or chemical hypoxia is blunted by increased Akt activity. Am J Physiol Heart Circ Physiol. 2006;290:H2472–H2479. [DOI] [PubMed] [Google Scholar]
- 31.Kania G, Blyszczuk P, Eriksson U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc Med. 2009;19:247–252. [DOI] [PubMed] [Google Scholar]
- 32.Brasier AR, Recinos A III, Eledrisi MS. Vascular inflammation and the renin‐angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22:1257–1266. [DOI] [PubMed] [Google Scholar]
- 33.Jia LX, Qi GM, Liu O, Li TT, Yang M, Cui W, Zhang WM, Qi YF, Du J. Inhibition of platelet activation by clopidogrel prevents hypertension‐induced cardiac inflammation and fibrosis. Cardiovasc Drugs Ther. 2013;27:521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong S, Hughes RC. Macrophage surface glycoproteins binding to galectin‐3 (Mac‐2‐antigen). Glycoconj J. 1997;14:267–274. [DOI] [PubMed] [Google Scholar]
- 35.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF‐κB in the heart: to be or not to NF‐κB. Circ Res. 2011;108:1122–1132. [DOI] [PubMed] [Google Scholar]
- 36.Zablocki D, Sadoshima J. Angiotensin II and oxidative stress in the failing heart. Antioxid Redox Signal. 2013;19:1095–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolf G, Wenzel U, Burns KD, Harris RC, Stahl RA, Thaiss F. Angiotensin II activates nuclear transcription factor‐κB through AT1 and AT2 receptors. Kidney Int. 2002;61:1986–1995. [DOI] [PubMed] [Google Scholar]
- 38.Kumar R, Yong QC, Thomas CM. Do multiple nuclear factor kappa B activation mechanisms explain its varied effects in the heart? Ochsner J. 2013;13:157–165. [PMC free article] [PubMed] [Google Scholar]
- 39.Maier HJ, Schips TG, Wietelmann A, Kruger M, Brunner C, Sauter M, Klingel K, Bottger T, Braun T, Wirth T. Cardiomyocyte‐specific IκB kinase (IKK)/NF‐κB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc Natl Acad Sci USA. 2012;109:11794–11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rouet‐Benzineb P, Gontero B, Dreyfus P, Lafuma C. Angiotensin II induces nuclear factor‐κB activation in cultured neonatal rat cardiomyocytes through protein kinase C signaling pathway. J Mol Cell Cardiol. 2000;32:1767–1778. [DOI] [PubMed] [Google Scholar]
- 41.Freund C, Schmidt‐Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, El‐Jamali A, Dietz R, Scheidereit C, Bergmann MW. Requirement of nuclear factor‐κB in angiotensin II– and isoproterenol‐induced cardiac hypertrophy in vivo. Circulation. 2005;111:2319–2325. [DOI] [PubMed] [Google Scholar]
- 42.Santos DG, Resende MF, Mill JG, Mansur AJ, Krieger JE, Pereira AC. Nuclear factor (NF) κB polymorphism is associated with heart function in patients with heart failure. BMC Med Genet. 2010;11:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simoes e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin‐(1‐7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. 2013;169:477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sukumaran V, Veeraveedu PT, Gurusamy N, Yamaguchi K, Lakshmanan AP, Ma M, Suzuki K, Kodama M, Watanabe K. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin‐converting enzyme‐2/angiotensin 1‐7/Mas receptor axis. Int J Biol Sci. 2011;7:1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhingra R, Gang H, Wang Y, Biala AK, Aviv Y, Margulets V, Tee A, Kirshenbaum LA. Bidirectional regulation of nuclear factor‐κB and mammalian target of rapamycin signaling functionally links Bnip3 gene repression and cell survival of ventricular myocytes. Circ Heart Fail. 2013;6:335–343. [DOI] [PubMed] [Google Scholar]
- 46.Shaw J, Yurkova N, Zhang T, Gang H, Aguilar F, Weidman D, Scramstad C, Weisman H, Kirshenbaum LA. Antagonism of E2F‐1 regulated Bnip3 transcription by NF‐κB is essential for basal cell survival. Proc Natl Acad Sci USA. 2008;105:20734–20739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ang II does not induce cardiac functional deficits in Snrk ecKO mice.
Figure S2. Snrk knockdown and cardiac dysfunctional signaling in endothelial cells.
Figure S3. Expression of pro‐ and anti‐inflammatory cytokine in HL‐1 cells.
Figure S4. Activation of proinflammatory cytokines signals to fibrosis in the heart of Snrk ecKO mice.