Neurons Promote Macrophage Proliferation by Producing Transforming Growth Factor-β2 (original) (raw)
Abstract
The infiltration of bone marrow-derived macrophages into the CNS contributes to growth and reactions of microglia during development or after brain injury. The proliferation of microglial cells is stimulated by colony-stimulating factor 1 (CSF-1), an astrocyte-produced growth factor that acts on mononuclear phagocytes. In the present study, we have shown, using an in vitro model system, that rodent neurons obtained from the developing cerebral cortex produce a soluble factor that strongly enhances the proliferation of macrophages cultured in the presence of CSF-1. Both macrophages isolated from the developing brain and those from the adult bone marrow were stimulated. Kinetic analyses of [3H]thymidine incorporation into macrophages indicated that their response to the neuron-derived factor involved a shortening of the cycle of proliferating cells. The effect of neurons on macrophages was blocked in the presence of antibodies neutralizing transforming growth factor-β2 (TGF-β2), whereas recombinant TGF-β2 stimulated macrophage proliferation in the presence of CSF-1. Neuronal secretion of TGF-β2 was confirmed by reverse transcription-PCR detection of TGF-β2 transcripts and immunodetection of the protein within neurons and in their culture medium. In situ hybridization and immunohistochemical experiments showed neuronal expression of TGF-β2 in sections of cerebral cortex obtained from 6-d-old rats, an age at which extensive developmental recruitment of macrophages occurs in this cerebral region. Altogether, our results provide direct evidence that neurons have the capacity to promote brain macrophage proliferation and demonstrate the role of TGF-β2 in this neuronal function.
Keywords: microglia, bone marrow-derived macrophage, neuron, colony-stimulating factor 1, transforming growth factor-β, rat
The infiltration of the CNS by bone marrow-derived monocytes contributes to the establishment and turnover of microglia. In the developing CNS, invading monocytes proliferate and are transformed into ameboid microglia, also called brain macrophages (BMs) (Ling and Wong, 1993). Maturation of the CNS is associated with a progressive differentiation of BMs into resting, ramified microglia. However, several pathological states lead to the reappearance of proliferating BMs as a consequence of increased infiltration of monocytes or activation and transformation of ramified microglial cells into BMs (Perry et al., 1994). Accumulating evidence indicates that BMs produce trophic as well as toxic compounds that can act on different CNS lineages and promote inflammatory reactions by presenting antigen to T-cells (Mallat and Chamak, 1994; Gehrmann et al., 1995). Hence, the regulation of intracerebral proliferation of macrophages is a key issue in the understanding of physiological and pathophysiological remodeling of CNS tissue.
As with other tissue macrophages or their precursors, the proliferation of BMs can be stimulated by colony-stimulating factors (CSFs), such as CSF-1 (also called macrophage CSF) and granulocyte macrophage CSF (GM-CSF), which are encoded by distinct genes (Giulian and Ingeman, 1988; Metcalf, 1989; Hao et al., 1990; Roth and Stanley, 1992;Théry and Mallat, 1993). The expression of CSF-1 and CSF-1 mRNA has been detected in extracts of developing and adult rodent CNS (Théry et al., 1990, Hulkower et al., 1993, Chang et al., 1994,Roth and Stanley, 1996). Recent analyses of mice genetically deficient in CSF-1 indicate a primary role of CSF-1 in the recruitment of BMs. Indeed, despite the presence of resting, ramified microglia, the proliferation of activated microglia and the occurrence of BM phenotypes appeared dramatically reduced in the mutant brain after ischemic or mechanical injury (Raivich et al., 1994; Berezovskaya et al., 1995).
Cellular sources of CSF-1 were studied in cultures derived from human or rodent CNS; astrocytes were found to be the main source and to produce this factor constitutively (Hao et al., 1990; Frei et al., 1992; Théry et al., 1992; Lee et al., 1993; Théry and Mallat, 1993). In addition, astrocytes stimulated with inflammatory cytokines synthesized GM-CSF (Malipiero et al., 1990; Aloisi et al., 1992), indicating that these cells can promote macrophage growth by producing at least two different mitogenic agents. In contrast, the influence of neurons on macrophage proliferation is poorly defined. CSF-1 has been detected in neuronal cultures derived from mouse cerebellum but not in those obtained from cerebral cortex (Théry et al., 1990; Nohava et al., 1992). This prompted us to set up an_in vitro_ coculture system to study the influence of neurons on the growth of macrophages isolated from bone marrow or from CNS. We observed that rat neurons stimulated the proliferation of macrophages by producing a factor enhancing the activity of CSF-1. This neuron-derived factor was identified as a member of the transforming growth factor-β (TGF-β) family (Massagué et al., 1994), TGF-β2.
MATERIALS AND METHODS
Reagents for cell cultures
Recombinant human CSF-1 (rhCSF-1; specific activity, 2–5 × 106 U/mg protein), rhTGF-β1, rhTGF-β3, and purified goat polyclonal antibodies neutralizing TGF-β2, TGF-β3, interleukin 3 (IL-3), GM-CSF, or tumor necrosis factor-α (TNF-α) were obtained from R & D Systems (Abingdon, UK); rhTGF-β2, mouse monoclonal antibodies (mAbs) neutralizing TGF-β1, -β2, and -β3 (IgG1), TGF-β2 and -β3 (IgG2b), or IL-7 came from Genzyme (Le Perray en Yvelines, France). Affinity-purified rabbit polyclonal anti-TGF-β2 antibodies raised against a peptide fragment of mature human TGF-β2 (residues 50–75) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Methyl-[3H]thymidine (specific activity, 46 Ci/mmmol) was purchased from Amersham (Les Ulis, France). Insulin, iron-free transferrin, progesterone, putrescin, and selenium came from Sigma (L’isle d’Abeau, France). All other components of the culture media were from Life Technology (Eragny, France). Contamination of the culture media with lipopolysaccharide was <0.01 ng/ml, as indicated by the limulus amebocyte assay from Sigma. Nonpurified murine CSF-1 was obtained as a conditioned medium from L929 cells cultured at 2 × 105 cells/ml for 7 d in DMEM supplemented with 10% fetal calf serum (FCS) (Stanley and Heard, 1977). All cell types were cultured in plastic dishes from Nunc (Naperville, IL).
Cell populations
Bone marrow-derived macrophages. Bone marrow-derived macrophages (BMMs) were obtained from cultures of bone marrow collected from male Sprague Dawley rat femurs (Charles River, St. Aubin les Elbeuf, France) as described by Tushinski et al. (1982). Bone marrow cells were flushed out from the femur with a 25 gauge needle into ice-cold PBS containing 33 mm glucose. The marrow plugs were dispersed and incubated for 10 min at 4°C in Gey’s solution to lyse the red cells and then washed in DMEM (250 gm, 10 min at 4°C). Cells were seeded into Petri dishes (1.5 × 106cells/ml) in 10 ml of DMEM supplemented with 10% FCS and 20% L929-conditioned medium (L9CM). After a 3 d incubation (37°C, 5% CO2), the culture medium containing the nonadherent cells was transferred into new plates, and 5 ml of fresh complete medium was added. After 3 d of culture, adherent cells were discarded, whereas nonadherent cells were collected and washed twice before being used as BMMs in the experiments. The purity of isolated BMMs was checked by immunocytochemical detection of specific macrophage markers. Virtually all the cells bore ED1 macrophage antigen as well as complement receptor type 3.
Brain macrophages. BMs (>99% pure) were isolated from 2-week-old primary glial cultures derived from the cerebral cortex and striatum of embryonic day 17 (E17) OFA rats (IFFA Credo) and grown in DMEM supplemented with 10% FCS, as described previously (Théry et al., 1991). Cells were washed in DMEM before use.
Neurons. Neuronal primary cultures were derived from E17 rat cerebral cortex or from E14 rat mesencephalon as described previously by Théry et al. (1991). Briefly, dissociated cells were seeded on plastic or on 14 mm glass coverslips coated with polyornithine (15 μg/ml; 4 × 105 cells/glass coverslip) and cultured in a chemically defined medium (CDM) consisting of DMEM/F12 nutrients (1:1) supplemented with 33 mmd-glucose, 2 mml-glutamine, 9 mm NaHCO3, 5 mm HEPES, 25 μg/ml insulin, 100 μg/ml iron-free transferrin, 20 nmprogesterone, 60 μm putrescin, and 30 nmselenium. In these culture conditions, >95% of the cells bore neuronal markers such as microtubule-associated protein 2 (MAP2) and neurofilament subunits, whereas <5% of the cells expressed glial fibrillary acidic protein (GFAP) or macrophagic ED1 antigen. Neurons from cerebellum were obtained from 6-d-old postnatal OFA rats as described by Van Vliet et al. (1989) and cultured in CDM supplemented with 25 mm K+.
Coculture system
The influence of neurons on macrophages was studied in coculture allowing exchange of soluble factors between the two cell populations. Macrophages were seeded at low density into 16 mm wells (8000–20,000 cells/well) in 600 μl of CDM (described above) supplemented with 20% L9CM or 2% FCS and rhCSF-1. Twenty-four hours after macrophage seeding, neurons cultured on top of 14 mm glass coverslips were introduced with their glass substrate into the wells. Glass coverslips were mounted on 3 mm paraffin feet allowing space between the macrophages and the cell free bottom sides of the slides introduced into the wells. Controls were performed by adding glass coverslips devoid of cells in wells containing the macrophages. At the end of the coculture, the glass coverslips were removed, and the macrophages were fixed for immunocytochemical analysis and cell count. The lack of any contaminating cells derived from the neuronal compartment in the bottom of the wells was checked by introducing glass coverslips coated with neurons in wells devoid of macrophages.
Cell number determination
For routine cell counting macrophages were fixed by a 10 min incubation at 4°C in 2.5% glutaraldehyde, which was added to the 16 mm wells without removing the culture medium. Cells were then washed extensively in PBS and incubated 10–15 min with 0.05% toluidine blue in 2% Na2CO3. Stained cells were washed with distilled water, air dried, and examined with a Nikon optical microscope. Cell growth was determined in each well by counting the number of macrophages in 15 high-power fields (magnification, 200×) covering 1/40 of the surface of the well. The percentage of cells recovered 24 hr after plating was estimated from the number of cells counted (mean from four sister wells) using the formula: percentage of cells = 100 × (number of cells counted × 40/number of cells plated).
[3H]Thymidine incorporation and autoradiography
Cultured BMMs were incubated for different times in the presence of 1 μCi/ml methyl-[3H]thymidine. At the end of the labeling period, BMMs were fixed with 2.5% glutaraldehyde (10 min at 4°C), and the wells were washed six times with PBS before air drying. The wells were then coated with K5 emulsion in gel form (Ilford Scientific Products) and stored at 4°C for 3 d before developing. Cultures were then counterstained with 0.05% toluidine blue in 2% Na2CO3, and both [3H]thymidine-labeled and unlabeled cells were counted under an optical microscope as described above.
ELISA detection of TGF-β2
A commercially available immunoassay (ELISA) set up to detect biologically active human TGF-β2 (R & D Systems) was used to quantify this cytokine in the medium of cultures according to the instructions of the manufacturer. Latent TGF-β2 was also assayed after activation by transient acidification of the culture media: pH 2 for 20 min by adding 100 mm HCl to the media, followed by neutralization with an equal amount of NaOH and addition of 20 mm HEPES. Controls were performed using the culture medium incubated free of cells.
Reverse transcription-PCR and Southern blot procedure
Total RNA was isolated from cells lysed in guanidinium isothiocyanate as described by Chomczynski and Sacchi (1987). Two micrograms of RNA were reverse-transcribed (avian myeloblastosis virus, 42°C) using a kit from Promega (Charbonnières, France) and TGF-β1–β3 cDNAs were amplified by PCR (30 cycles) using specific primers and the following conditions for each cycle: 30 sec at 94°C, 1 min at the annealing temperature (58°C for TGF-β1, 62°C for TGF-β2, and 60°C for TGF-β3), and 1 min elongation at 72°C. The final extension was allowed to continue for 10 min. Amplified products were size-fractioned by electrophoresis through a 1.5% agarose gel. Gels were then denatured for 30 min in a solution of 0.5 mNaOH and 1.5 m NaCl, neutralized for 20 min in 0.5m Tris buffer, pH 8, containing 1.5 m NaCl, and blotted onto nitrocellulose filters (Bioprobe Systems, Montreuil-sous-Bois, France). Filters were hybridized overnight at 65°C with 32P-labeled cDNA probes in 5× SSC, 50 mm PBS, pH 6.5, 5× Denhardt’s solution, 100 μg/ml salmon sperm DNA, and 0.4% SDS. Blots were washed at 65°C in 1× SSC and 0.1% SDS.
Rat TGF-β1 sense and antisense primers were 5′-GAAGTCACCCGCGTGCTAAT-3′ and 5′-TTGCGACCCACGTAGTAGAC-3′, giving a product of 800 bp (Qian et al., 1990); mouse TGF-β2 sense and antisense primers were 5′-CTCCTGCATCTGGTCCCGGT-3′ and 5′-GCACAGCGTCTGTCACGTCG-3′, giving a product of 592 bp (Proetzel et al. 1995); and mouse TGF-β3 sense and antisense primers were 5′-GAAGATGACCATGGCCGTGG-3′ and 5′-GCTGGCCTCAGCTGCACTTA-3′, giving a product of 470 bp (Proetzel et al., 1995). The TGF-β1 probe was a 730 bp _Sac_I–_Pvu_II fragment of porcine TGF-β1 cDNA (Kondaiah et al., 1988). The TGF-β2 probe was a 2.35 kb simian cDNA (Hanks et al., 1988). For the TGF-β3 probe, a 339 bp_Nco_I–_Sal_I fragment of murine TGF-β3 cDNA (Schmid et al., 1991) was used. Positive controls for TGF-β expression were provided with cDNA obtained from adult mouse brain, rat epithelial liver cells (REL cell line), and peripheral nerve ganglions from adult rat (Unsicker et al., 1991; Mercier et al., 1995). PCR controls performed with the non-reverse-transcribed RNA samples were negative.
Immunocytochemistry
Immunoperoxidase detection of neuronal, astrocytic, or macrophagic markers in cultures was performed according to a procedure described previously (Chamak et al., 1994). Primary mAbs applied for 1 hr at room temperature were 1/300 IgG1 anti-MAP2 from BioMakor (Rehovot, Israel), 1/1000 SMI 31 IgG1 anti-phosphorylated neurofilament H from Sternberger Monoclonal Inc. (Baltimore, Maryland), 1/200 IgG1 anti-GFAP, from Amersham, and 1/100 IgG1 ED1 or 1/100 IgG2a OX42 anti-complement receptor type 3 from Serotec (Bicester, UK). mAbs were detected by peroxidase-conjugated goat anti-mouse IgG antibodies from Byosis (Compiègne, France) or by the biotin–avidin–peroxidase method using the Vectastain Elite kit from Vector Laboratories (Peterborough, UK) and diaminobenzidine hydrochloride (Sigma) as the chromogen.
For double immunofluorescent staining, fixed cells (4% paraformaldehyde) permeabilized with 0.1% Triton X-100 (Chamak et al., 1994) were saturated with 20% normal donkey serum (Jackson ImmunoResearch, West Grove, PA) diluted in PBS for 1 hr at room temperature. Cells were then incubated in PBS containing 2% normal donkey serum (1 hr at room temperature followed by washes) with sequentially 1/2000 rabbit polyclonal anti-TGF-β2 (Santa Cruz Biotechnology), 1/100 fluorescein isothiocyanate (FITC)-conjugated donkey F(ab′)2 anti-rabbit IgG (Jackson ImmunoResearch), and 1/300 anti-MAP2 mAb and 1/100 tetramethyl rhodamine isothiocyanate (TRITC)-conjugated donkey F(ab′)2 anti-mouse IgG.
For in situ detection of TGF-β2 and MAP2, postnatal day 6 (P6) rats were deeply anesthetized with diethyl ether and then perfused transcardially with PBS followed by 2% periodate–lysine–paraformaldehyde fixative containing 2% paraformaldehyde. Brains were post-fixed in the same fixative (2 hr at 4°C), cryoprotected by overnight (4°C) immersion in 10% sucrose in PBS, and rapidly frozen. Cryostat cut coronal sections (20 μm thick) were mounted onto poly-l-lysine-coated slides, air dried, and stored at −20°C before use. Double immunocytochemical detection of TGF-β2 and MAP2 was performed as described above with minor modifications; incubations and washes were all performed in the presence of 0.1% Triton X-100, and anti-TGF-β2 antibodies were applied overnight at 4°C.
Controls were performed by substituting primary mAbs with unrelated mouse IgG1 or IgG2a and by incubating anti-TGF-β2 polyclonal antibodies with a 20-fold excess of the synthetic peptides used to generate the antisera (supplied by Santa Cruz Biotechnology).
In situ hybridization
Sample preparation and hybridization were performed according to a reported procedure (Gautron et al., 1992). Frozen tissue sections fixed as described above were thawed, dehydrated in ethanol, and air dried before post-fixation in 2% paraformaldehyde for 20 min at room temperature and washes in PBS. Samples were treated for 8 min at room temperature with 20 μg/ml proteinase K (Boehringer Mannheim, Mannheim, Germany) diluted in 50 mm Tris and 5 mm EDTA, pH 8, washed in PBS, fixed again with 2% paraformaldehyde (5 min, room temperature), and rinsed in PBS and H2O. Sections were then acetylated, washed, and dehydrated with solutions of increasing ethanol concentration before air drying.
Sense and antisense TGF-β2 RNA probes were labeled with35S-uridine triphosphate (UTP, >1000 Ci/mmol; Amersham) to a specific activity of >109 cpm/μg using SP6 or T7 RNA polymerase according to the instructions of the manufacturer (Stratagene, La Jolla, CA). The riboprobe templates were 339-nucleotide-long fragments, subcloned into pGEM5 (Promega), and corresponded to the cDNA sequence encoding the mature form (including the stop codon) of murine TGF-β2 (Schmid et al., 1991). Labeled probes were fragmented by alkaline digestion. Hybridization was performed overnight at 52°C in 50% formamide, 10% dextran sulfate, 0.3 m NaCl, 20 mm Tris-HCl, pH 7.5, 10 mm dithiothreitol, 5 mm EDTA, 1× Denhardt’s reagent, and 0.5 mg/ml yeast tRNA (Sigma) supplemented with a 5 × 104 cpm/μl 35S-UTP-labeled RNA probe. Slides were washed as follows: 5× SSC and 10 mmdithiothreitol (DTT), 30 min at 50°C; 2× SSC, 50% formamide, and 10 mm DTT, 20 min at 60°C; and twice in washing solution containing 0.3 m NaCl, 20 mm Tris, and 5 mm EDTA, 10 min at 37°C. Sections were treated with 20 μg/ml RNase A and 2 μg/ml RNase T1 (Boehringer Mannheim) in the washing solution for 30 min at 37°C, followed by washes in washing solution (5 min, 37°C), 2× SSC, and finally 0,1 × SSC (15 min, 37°C each). Slides were dehydrated, air dried, and coated with Ilford K5 photo emulsion before exposure for 2 weeks at 4°C. After development, slides were dehydrated and stained with 0.05% toluidine blue.
RESULTS
Influence of CSF-1 on the growth of cultured BMMs
Macrophages directly harvested from bone marrow culture (BMMs) provide a highly homogeneous cell population and are much easier to obtain than circulating monocytes from rodent blood. Therefore, BMMs were used to investigate the early influence of neurons on mononuclear phagocytes that infiltrate the CNS parenchyma. Consistent with previous reports (Tushinski et al., 1982; Tushinski and Stanley, 1985), in vitro growth of isolated BMMs required addition of CSF-1 to the culture medium (Fig. 1). The concentration of CSF-1 for optimal stimulation has been shown to depend on the macrophage density in cultures, because binding of CSF-1 to BMMs leads to internalization and degradation of the cytokine (Tushinski et al., 1982). In our assay, using a low cell density, growth of BMMs estimated 4 d after cell plating was found to be optimal at a rhCSF-1 concentration of 2 ng/ml. Similar growth was obtained using 20% L9CM as a source for CSF-1 (Fig.1). In these optimal conditions, quantitation of BMMs 1 d after plating showed that according to the experiments, a proportion of 15–50% of the cells seeded was rescued from death, and these cells were adhering to the plastic substrate. Figure 2 shows that beyond the first day after BMM isolation, surviving cells stimulated with CSF-1 underwent proliferation, as indicated by the progressive increase in culture well BMM number. In cultures without CSF-1, a drop in BMM number was already obvious between 2 and 24 hr after cell isolation, indicating that the survival of freshly harvested BMMs was strongly dependent on the presence of CSF-1 (Fig. 2).
Fig. 1.

Influence of neurons and CSF-1 on BMM growth. BMMs were seeded at a density of 10,000 cells/well in CDM supplemented with L9CM or rhCSF-1 and 2% FCS. Cells were cultured for 96 hr with neurons (hatched bars) or without neurons (open bars) added for the last 72 hr. At the end of the culture, the number of BMM was determined by counts in 15 fields for each well as described in Materials and Methods. Each value is the mean number of cells ± SEM from four wells in one experiment representative of three independent experiments. Comparison between numbers of BMMs in the presence and absence of neurons was performed for each CSF-1 concentration using Student’s t test (*p < 0.05).
Fig. 2.

Effect of CSF-1 and TGF-β2 on the time course of BMM growth. BMMs were seeded at a density of 10,000 cells/well in CDM supplemented with 2% FCS with or without 10 ng/ml rhCSF-1 or 0.1 ng/ml rhTGF-β2. The number of BMMs was determined at different times after cell plating. Values are mean number of cells ± SEM from four wells in one experiment representative of two independent experiments.Open circles, Culture without recombinant cytokines;filled squares, culture with rhCSF-1; open triangles, culture with rhTGF-β2. In the presence of CSF-1, the estimated proportion of BMM recovered 24 hr after cell plating was 50%; from this time point, the number of BMMs increased significantly within 24 hr (p < 0.05). Statistical analyses were performed by one-way ANOVA followed by Dunnett’s test.
Influence of CNS neurons on BMM proliferation
The paracrine effect of neurons on BMM growth was investigated in a coculture system that allowed the exposure of BMMs to soluble factors released by neurons. Figure 1 illustrates that in the absence of CSF-1, the addition of cultured neurons (1 d in vitro) from E17 cerebral cortex to wells seeded with BMMs failed to rescue BMMs from degeneration. This result is consistent with the lack of CSF-1 production by these neurons (Théry et al., 1990) (A. Dobbertin and M. Mallat, unpublished results). However, at optimal concentration of CSF-1 (2 or 10 ng/ml rhCSF-1 or 20% L9CM), the presence of neurons led to a twofold increase in the number of BMMs after 3 d of coculture (Fig. 1). Increased BMM numbers were observed when starting the cocultures with highly pure neuronal populations grown on glass coverslips in CDM. The purity of the neuronal population was also checked at the end of the coculture. More than 95% of the cells displayed the typical shape of cultured neurons and expressed specific neuronal markers such as MAP2 or the 200 kDa neurofilament subunit. Immunocytochemical detection of the ED1 antigen and GFAP confirmed that contaminating brain macrophages and astrocytes within the neuronal culture remained <5% of the total cell population after a 3 d exposure to low FCS (2%) and rhCSF-1 or L9CM.
The number of BMMs was also significantly increased, although to a lower level, when medium conditioned by neurons was added to BMM culture wells instead of living neurons. Thus, neurons do not require stimulation by BMMs to release growth factors acting on the macrophages (data not shown).
These results indicate that neuron-derived factors are ineffective by themselves to promote macrophage survival and proliferation but enhance the mitogenic activity of CSF-1. This enhancement of proliferation was further characterized by kinetic analyses of DNA synthesis using BMMs that had entered a quiescent state before exposure to neurons. It has been shown that removal of CSF-1 from the culture induces proliferating BMMs to enter a quiescent (G0/G1) state and that the cells can reenter the growth cycle if CSF-1 is re-added soon enough to avoid BMM death (Tushinski and Stanley, 1985). In our model, preliminary studies monitoring cell death and DNA synthesis showed that BMM quiescence was fully achieved after a 24 hr deprivation of CSF-1. Kinetics of [3H]thymidine nuclear incorporation indicated that these BMMs were able to synthesize DNA after re-addition of CSF-1 (Fig. 3). During the first 20 hr, the number of BMMs entering the S phase of the cell cycle was significantly increased when the cells were stimulated with both neurons and CSF-1 compared with CSF-1 alone. However, after 24 hr, the same proportion of cells (70%) was labeled under both conditions, indicating that neurons act by shortening the cell cycle of BMMs stimulated by CSF-1 rather than by inducing proliferation of BMMs unresponsive to CSF-1 alone (Fig. 3).
Fig. 3.

Effects of neurons and CSF-1 on the time course of DNA synthesis in quiescent BMMs. Isolated BMMs were cultured for 2 d in the presence of CSF-1 (20% L9CM) and washed, and synchronization of the cultures in a growth-arrested phase was obtained by a 24 hr incubation in CDM free of CSF-1. Cells were then incubated in CDM supplemented with L9CM and 1 μCi/ml [3H]thymidine with or without neurons. At indicated times, [3H]thymidine incorporation was stopped, and macrophages were processed for autoradiography. Results are from one experiment representative of three independent experiments. Values are presented as percentage of labeled nuclei (mean ± SEM) determined in four wells for each condition.Filled squares, Percentage of labeled macrophage nuclei after coculture with neurons; open circles, control without neurons. Comparison of the proportions of labeled BMMs cultured with or without neurons was performed at different times using Student’s t test (16 hr, p = 0.0002; 20 hr, p = 0.06).
Stimulation of BMM proliferation in the presence of CSF-1 was also observed using neuronal cultures derived from E17 cerebral cortex, E14 mesencephalon, or P6 cerebellum (Fig. 4). Comparison between neurons derived from E17 cortex and grown in CDM for 1 or 7 d before coculture indicated that the _in vitro_neuronal maturation did not alter their ability to increase BMM proliferation (Fig. 4). Similar results were obtained using mouse-derived BMM and neurons from embryonic mouse cerebral cortex or mesencephalon (data not shown).
Fig. 4.

Effect of different neuronal populations on BMM growth. Neurons from different CNS regions were seeded on glass coverslips (4 × 105 cells/coverslip) and grown for different times before coculture with BMM in CDM supplemented with 20% L9CM. Cx 1DIV, Cx 7DIV, Neurons from E17 cerebral cortex 1 or 7 d in vitro before coculture;Mes, mesencephalic neurons from E14 rat embryos 1 d_in vitro_ before coculture; Cb, P6 cerebellar neurons 3 d in vitro before coculture. The numbers of BMMs counted after 72 hr of coculture are expressed as percentages of the mean control value set as 100% (BMMs in wells without added neurons). Each value is the mean ± SEM from counts in four wells in one experiment representative of three independent experiments. The actual number of BMMs counted in control conditions was 126 ± 23. Coculture and control values were compared using Student’s t test (*p < 0.01).
TGF-β2 is responsible for the increased proliferation of macrophages in the presence of neurons
The possible involvement of cytokines suggested to modulate CSF-1 activity (Chen et al., 1988; Celada and Maki, 1992; Guilbert et al., 1993; Jacobsen et al., 1994) was investigated in our model by adding different blocking antibodies to the coculture medium. As shown in Figure 5A, neuronal stimulation was not significantly affected in the presence of antibodies neutralizing GM-CSF, IL-3, TNF-α, or IL-7. In contrast, the effect of neurons on BMM proliferation was fully prevented in the presence of two different monoclonal antibodies directed against TGF-β and neutralizing three (β1–β3) or two (β2 and β3) of the TGF-β isoforms (Fig.5A). A primary role for TGF-β2 in BMM proliferation was further demonstrated by the complete blockade of the neuronal stimulation with monospecific antibodies directed against whole TGF-β2 (goat antibodies from R & D Systems) or a TGF-β2 fragment (rabbit antibodies from Santa Cruz Biotechnology). In contrast, anti-TGF-β3 antibodies did not affect stimulation of BMMs by neurons (Fig. 5A) when used at a concentration neutralizing up to 0.1 ng/ml rhTGF-β3.
Fig. 5.

Neutralization of neuronal stimulation by different anti-TGF-β antibodies. Cocultures were all performed in CDM supplemented with 2% FCS and 10 ng/ml rhCSF-1. Antibodies were added together with neurons into wells previously seeded with BMM (A) or BM (B). Final concentrations of added antibodies were 20 μg/ml for polyclonal anti-TNF-α antibodies, anti-IL7 mAb, anti-TGF-β2 and -β3 mAbs, and control unrelated IgG2b or 10 μg/ml for other mAbs or polyclonal antibodies. For each antibody, the results of one experiment representative of at least two independent experiments are shown.Hatched bar, Control number (set as 100%) of BMMs or BMs cultured without neurons. Open bar, Number (percentage of control) of BMMs (A) or BMs (B) counted at the end of 72 hr cocultures in the presence or absence of antibodies. Values are mean ± SEM from counts in four wells. The actual numbers of cells in the seven (A) and two (B) controls were A, 68 ± 10, 222 ± 14, 104 ± 7, 42 ± 2, 420 ± 12, 64 ± 2, 308 ± 20;B, 123 ± 10 and 63 ± 8 (from_top_ to bottom). Neuronal stimulation without antibodies compared with control values (without neurons) were all significant (p < 0.01;p < 0.05 for the experiment performed with anti-TGF-β2 and -β3 antibodies). Addition of unrelated mAbs, rabbit IgGs, or monospecific antibodies directed against GM-CSF, IL-3, IL-7, TNF-α, or TGF-β3 did not significantly alter neuronal stimulation. Comparisons between anti-TGF-β antibodies (mAbs neutralizing two or three isoforms or rabbit monospecific anti-TGF-β2) and unrelated IgGs of matched isotype or species confirmed directly the specificity of blocking effects (in all cases p < 0.05). Significant differences were also checked between goat monospecific anti-TGF-β2 and anti-TGF-β3 in cocultures with BMMs (p < 0.05) or BMs (p < 0.01). Statistical analyses were performed using one-way ANOVA followed by multiple comparison Bonferroni’s test.
The above-mentioned effects were observed with cultured macrophages derived from bone marrow, used as a model for phagocyte precursors infiltrating the CNS. In another series of experiments, we investigated how neurons influence the in vitro growth of resident macrophages belonging to the microglial population (BMs) and obtained from developing CNS. Figure 5B shows that neurons also increased proliferation of BMs cultured in the presence of CSF-1, and that stimulation of BM proliferation was inhibited by anti-TGF-β2 antibodies. Thus, both BMs and BMMs exhibited the same biological response to anti-TGF-β antibodies.
None of the anti-TGF-β antibodies tested interfered with neuron survival or with CSF-1-stimulated macrophage proliferation when BMs and BMMs were cultured in the absence of neuronal cells. The specificity and efficiency of the TGF-β antibodies was checked by Western blot or the Mink lung assay test using recombinant isoforms (data not shown).
rhTGF-β isoforms 1–3 increase BMM and BM growth in the presence of CSF-1
The ability of TGF-β to increase CSF-1-dependent proliferation of BMMs and BMs was investigated directly by adding recombinant β1–β3 isoforms of TGF-β to the medium of macrophage cultures. Figure 6 shows that each of the three isoforms displayed the biological effects characterizing neuron-derived factors in cocultures. In the case of BMMs (Fig. 6A), significant stimulations were observed from concentrations of 10 pg/ml (rhTGF-β1) or 100 pg/ml (rhTGF-β2 and -β3). For each of the three isoforms, optimal stimulations reached levels similar to those observed in cocultures with neurons. As for BMs (Fig. 6B), concentrations required for significant stimulation were 10 pg/ml (rhTGF-β3) and 100 pg/ml (rhTGF-β1 and -β2). The magnitudes of optimal stimulation were similar for the three isoforms but were lower than those obtained with BMMs. In the absence of CSF-1, none of the TGF-β isoforms allowed macrophages to survive to 96 hr (number of BMs or BMMs less than five for each determination in cultures supplemented with TGF-β1–β3 at concentrations up to 1 ng/ml). Kinetic analyses performed with BMM cultures failed to reveal any short-term improvement in cell survival when TGF-β2 was added to cultures free of CSF-1 (Fig. 2).
Fig. 6.
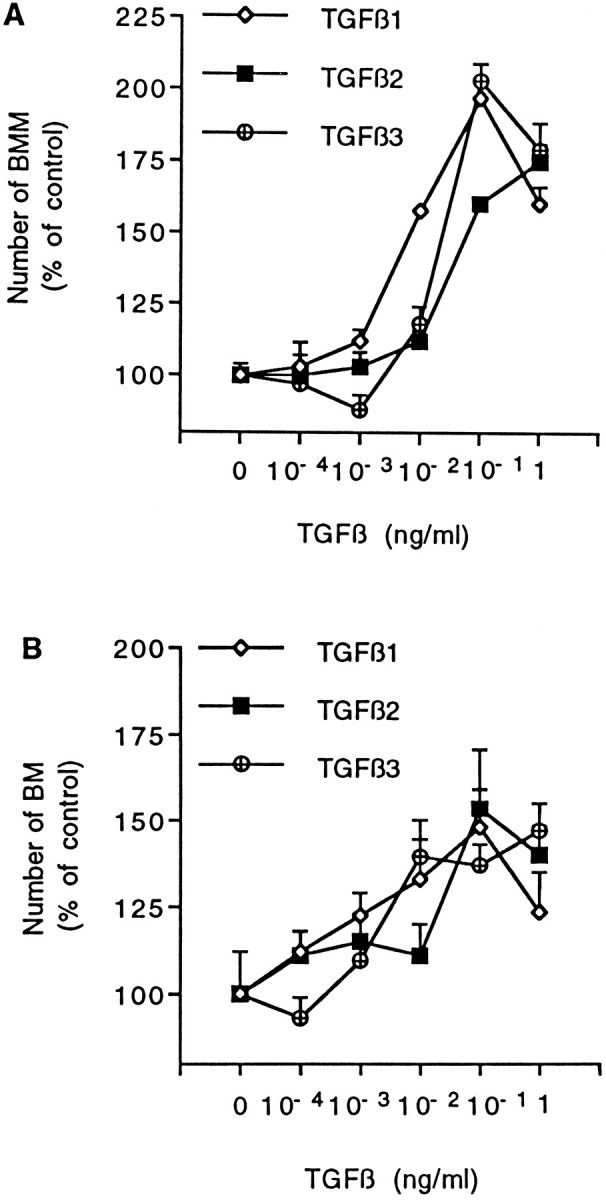
Dose–response effect of recombinant TGF-βs on BMM (A) and BM (B) growth. Cells were cultured for 96 hr in CDM supplemented with 2% FCS and 10 ng/ml rhCSF-1. TGF-β isoforms were added daily to culture at indicated final concentrations for the last 72 hr. The number of macrophages in each condition is given as percentage of mean control value obtained from wells without TGF-β (set as 100%). Data are mean ± SEM of five determinations from one experiment representative of five (BMM) or three (BM) independent experiments. Actual control numbers of BMs and BMMs were 54 ± 6 and 222 ± 9, respectively. Significant increases in macrophage number compared with control values were determined by ANOVA followed by Dunnett’s test. BMM cultures (A), p < 0.01 from 0.01 ng/ml for TGF-β1 and from 0.1 ng/ml for TGF-β2 and -β3. BM cultures (B), p < 0.05 from 0.1 ng/ml for TGF-β1, and p < 0.01 from 0.1 ng/ml for TGF-β2 and from 0.01 ng/ml for TGF-β3.
Synthesis of TGF-β by neurons of the developing cerebral cortex
The expression of TGF-β genes in our neuronal and macrophage cultures was directly investigated by reverse transcription-PCR and Southern blot analyses. Transcripts of TGF-β2 and TGF-β3 but not TGF-β1 genes were detected in neuronal cultures with or without addition of 10 ng/ml rhCSF-1 and 2% FCS to the medium (Fig.7, lanes 2, 3). In contrast, BMs expressed high levels of TGF-β1 but not TGF-β2 or TGF-β3 transcripts (Fig.7, lane 5), a result expected from previous studies with cultured mouse BMs (Constam et al., 1992). TGF-β1 transcripts were also detected in BMMs, although the level of expression was lower (Fig.7, lane 4).
Fig. 7.

Reverse transcription-PCR and Southern blot analysis of TGF-β mRNA in neurons and macrophages. Total RNA was extracted from 4-d-old cultures of BMs, BMMs, or neurons derived from E17 cerebral cortex. Autoradiograms were obtained by Southern hybridization of amplified fragments as described in Materials and Methods. Lane 1, Controls (mouse brain for TGF-β1, rat epithelial liver cell line for TGF-β2, and peripheral nerve ganglions mRNA for TGF-β3); lanes 2 and 3, neurons cultured in CDM without (lane 2) or with (lane 3) 10 ng/ml rhCSF-1 and 2% FCS added for the last 72 hr; lane 4, BMMs; lane 5, BMs. Macrophages were cultured in CDM supplemented with 10 ng/ml rhCSF-1 and 2% FCS.
Considering the role of TGF-β2 in our cocultures, neuronal expression of this cytokine was further studied at the cellular level by immunocytochemical detection of the protein. We took advantage of monospecific anti-TGF-β2 antibodies against human TGF-β2, which was previously used to label this cytokine in various mammalian tissues (Anderson et al., 1995; Stewart et al., 1995; Frank et al., 1996). Double immunostaining using anti-MAP2 antibodies revealed that >80% of neurons in cultures derived from E17 cerebral cortex displayed intracellular TGF-β2 immunoreactivities (Fig.8G,H). Staining with anti-TGF-β2 was restricted to a perinuclear area, which is likely to be the Golgi apparatus (Fig. 8G); such a pattern is reminiscent of that previously reported for cultured peripheral nerves (Stewart et al., 1995).
Fig. 8.
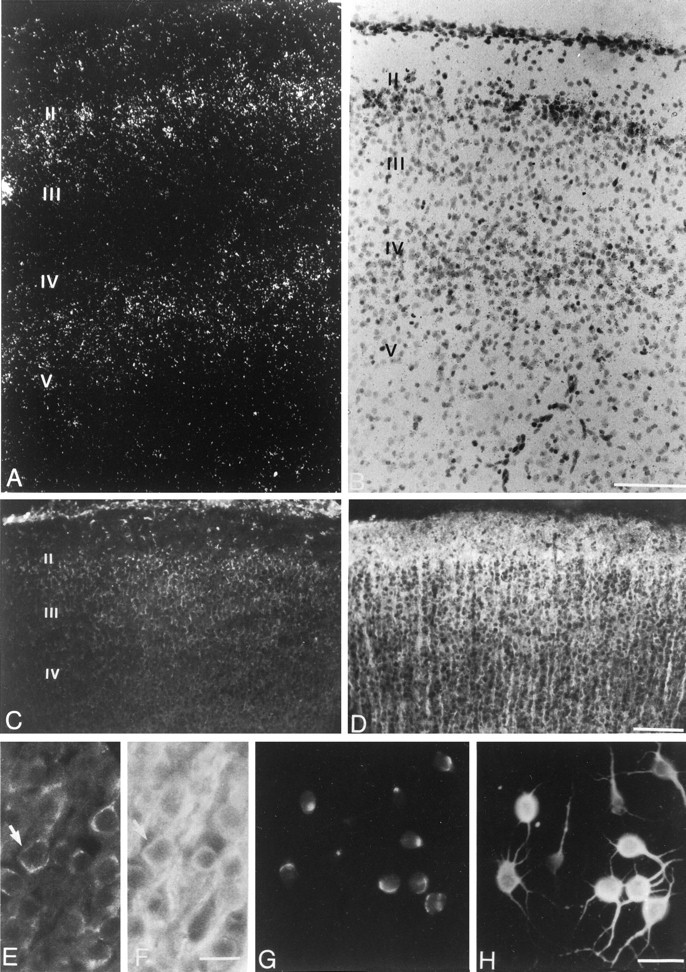
Expression of TGF-β2 by neurons of the cerebral cortex in vivo and in vitro. A, B, In situ hybridization dark-field (A) and bright-field (B) views of two different sections through the frontal part of the rat cerebral cortex at postnatal day 6. Cortical layers_II–V_ are marked. The dark-field view shows accumulation of TGF-β2 mRNA in cortical layers II and IV. Scale bar, 100 μm.C–F, Double immunofluorescent staining of P6 frontal cortex. C, E, anti-TGF-β2 staining (FITC); D, F, same fields stained with anti-MAP2 (TRITC). Scale bars:C, D, 100 μm; E, F, 25 μm.Arrows in E and F mark a cell body double-stained with anti-MAP2 and anti-TGF-β2. G, H, Double staining of neuronal cultures (4 d in vitro) derived from E17 cerebral cortex with anti-TGF-β2 mAb (FITC; G) and anti-MAP2 mAb (TRITC,H), Scale bar, 20 μm.
TGF-β is generally secreted as a latent TGF-β complex unable to bind to TGF-β receptors and requiring extracellular activation for biological activity (Flaumenhaft et al., 1993). In our coculture model, the presence of TGF-β2 activity indicates that cultured neurons produce active forms of TGF-β2 or that activation of neuron-derived latent TGF-β2 occurs in the presence of macrophages. Using an ELISA specific for active human TGF-β2, we failed to detect accumulation of mature TGF-β isoforms in the medium assayed at the end of the cocultures (threshold for detection, 2 pg/ml using rhTGF-β2). Mature TGF-β2 was detectable by ELISA in the medium of a pure neuronal culture when cell densities were higher than those routinely used to perform cocultures. Consistent with reverse transcription-PCR results, TGF-β2 was not detected in BM or BMM cultures (data not shown). However, latent TGF-β2 was repeatedly detected in the media assayed at the end of cocultures and control neuronal cultures, after transient acidification of the media, at mean concentrations ranging between 5 and 10 pg/ml (means from three wells for each culture; SD, <1 pg/ml).
The synthesis of TGF-β2 by neurons of the cerebral cortex was also investigated in tissue sections from developing brain. We chose to focus our observations on postnatal day 6, a developmental stage that corresponds to both infiltration and active proliferation of macrophages in this CNS region (Ling and Wong, 1993). In situ hybridization revealed TGF-β2 mRNA expression in frontal, parietal, and cingular cortices (Fig. 8). Hybridization signals obtained with a TGF-β2 antisense riboprobe displayed a laminar pattern corresponding mostly to cortical layers II and IV (Fig.8A,B). Controls performed using the corresponding sense probe provided a weak and uniformly distributed nonspecific background signal (not shown). TGF-β2 immunoreactivity was detected in adjacent tissue sections across all cortical layers but was more intense in layer II (Fig. 8C). Double immunostaining with anti-MAP2 antibodies allowed unambiguous localization of TGF-β2 immunoreactivity to perinuclear regions of neuronal cell bodies (Fig.8E,F). Weak staining with anti-TGF-β2 was also observed in cells scattered in the corpus callosum. Control experiments for immunocytochemistry including peptide adsorption of the anti-TGF-β2 antibodies were negative (not shown).
DISCUSSION
Infiltration of the CNS by bone marrow-derived phagocytes and the proliferation of macrophages are seminal events of microglial growth and pathological reactions (Hickey and Kimura 1988; Perry et al., 1994). This recruitment of macrophages is supported by CSF-1, a growth factor that is produced by astrocytes (Giulian and Ingeman, 1988; Hao et al., 1990; Théry et al., 1990; Roth and Stanley, 1992, 1996; Raivich et al., 1994).
The present study describes a novel biological mechanism by which neurons regulate macrophage proliferation within the CNS. Using a coculture assay, we show that neurons release compounds that increase mitogenic responses of BMs and BMMs to CSF-1. This capacity is shared by rat and mouse neurons of different brain regions such as the cerebral cortex, cerebellum, and mesencephalon.
TGF-β1–β3 are highly conserved 25 kDa homodimers encoded by distinct genes that present substantial amino acid sequence homologies (70–80%). These peptides modulate both functional properties and growth of mesenchymal and neuroepithelial cells (for review, see Sporn and Roberts, 1992; Massagué et al., 1994; Krieglstein et al., 1995a). Focusing on neurons of the cerebral cortex, TGF-β2 produced by these cells seems to be the primary, if not the unique, molecular effector of macrophage proliferation. Indeed, the effect of neurons was completely blocked in the presence of antibodies neutralizing TGF-β2. Purified TGF-β2, added to BM and BMM cultures, was found to enhance the mitogenic activity of CSF-1, and the secretion of TGF-β2 by cerebrocortical neurons was demonstrated by detection of the transcripts and the protein. Although we did not attempt to confirm the role of TGF-β2 in coculture performed with neurons of cerebellum or mesencephalon, our analysis is consistent with detection of TGF-β2 gene expression in neuronal cultures derived from these two CNS regions (Krieglstein et al., 1995b; De Luca et al., 1996).
TGF-β is generally secreted as an inactive form resulting primarily from its noncovalent association to the N-terminal cleavage product of the TGF-β precursor. Latent TGF-β complex can be activated by different means, including acid treatment and exposure to glycosidases and proteases (Flaumenhaft et al., 1993). ELISA determination confirmed the production of latent TGF-β2 by neurons from the cerebral cortex, but this method did not allow detection of the mature form in our coculture conditions. The quantitation of TGF-β2 in our cultures using an ELISA optimized for detection of human TGF-β2 might suffer from a reduced cross-reactivity with the rat-derived isoform. However, our results support evidence indicating that latent TGF-β can be activated at the surface of different target cells by mechanisms that can prevent the release of mature TGF-β in a form detectable in a conventional bioassay (Lucas et al., 1990; Saad et al., 1991; Arrick et al., 1992; Flaumenhaft et al., 1993). In our experiments, generation of mature TGF-β2 at the macrophage surface could involve proteases or extracellular matrix proteins such as plasmin and thrombospondin-1. In fact, these compounds are known to activate TGF-β, and they can be produced by macrophages, including BMs (Nakajima et al., 1992a,b;Flaumenhaft et al., 1993; Schultz-Cherry et al.,1993; Chamak et al., 1994).
Previous studies focusing on TGF-β1 have shown that this isoform increases or reduces the mitogenic effect of CSF-1 depending on the species, the tissue of origin, and the stage of differentiation of cells from the macrophage lineage (Ohta et al., 1987; Celada and Maki, 1992; Suzumura et al., 1993; Rosenfeld, 1994). Here we found that rhTGF-β1–β3 on their own do not support survival of rat BMs or BMMs but markedly enhance the CSF-1-induced proliferation of these phagocytes. This result is in line with TGF-β1 stimulations reported for adhering mouse BMMs (Celada and Maki, 1992), but our data are in contrast with those of Suzumura et al. (1993), suggesting that TGF-β1 reduces mitogenic effects of CSF-1 on mouse BMs. The common effect of the various TGF-β isoforms in our model is in agreement with the fact that the various isoforms share promiscuous receptors displaying serine threonine kinase activity (Massagué et al., 1994). TGF-β is known to be a potent chemoattractant acting on BMs and blood monocytes (Wahl et al., 1987; Yao et al., 1990). In addition to cell attraction, our results suggest that this cytokine also favors local expansion of macrophages in the CNS by enhancing the effect of CSF-1. The molecular mechanism of this synergy remains to be investigated. TGF-β could modulate the macrophage expression of the CSF-1 receptor and/or act downstream at the level of intracellular transduction processes.
We have also observed that cultured neurons from the cerebral cortex express TGF-β2 and TGF-β3 but not TGF-β1 transcripts. However, although rhTGF-β3 is at least as potent as rhTGF-β2 in promoting macrophage proliferation in the presence of CSF-1, we found no evidence that TGF-β3 is involved in the neuronal effect. Indeed, the influence of neurons on macrophages was abrogated by antibodies neutralizing TGF-β2 but not TGF-β3. This indicates that the neuron-derived TGF-β3 did not reach sufficient concentrations in the cocultures to stimulate macrophage growth. Any marginal contribution of this isoform was further ruled out by the fact that monospecific antibodies blocking TGF-β3 did not modify the neuron effect on macrophage proliferation. Previous studies have also failed to detect any production of TGF-β3 in cultures of mouse astrocytes or cerebellar granule cells, despite the expression of a TGF-β3 transcript in these cultures (Constam et al., 1992, 1994). In line with these observations, a post-transcriptional downregulation of TGF-β3 synthesis was documented by identification of a 5′ noncoding region of the TGF-β3 mRNA which exerts a potent inhibitory effect on translational efficiency in cell lines (Arrick et al., 1991).
The production of TGF-β isoforms in the developing CNS has been investigated in vivo (for review, see Krieglstein et al., 1995a). Except for the mouse cerebellum, in which expression of TGF-β2 mRNA was studied during postnatal periods of development (Constam et al., 1994), the expression of TGF-β genes in the developing mammalian CNS has been studied mostly during prenatal stages (Gatherer et al., 1990; Flanders et al., 1991; Millan et al., 1991;Pelton et al., 1991; Schmid et al., 1991). TGF-β2 and -β3 mRNA were also observed in the cerebral cortex of 1-d-old rats (Poulsen et al., 1994). Our own in situ detection of TGF-β2 extends these previous analyses to a postnatal stage when both the infiltration of bone marrow-derived monocytes and intracerebral proliferation of macrophages account for a marked expansion of microglia in the cerebral cortex (Ling and Wong, 1993). In addition, we have demonstrated the localization of TGF-β2 in neuronal cell bodies by double staining with antibodies raised against a neuronal marker. The in situ detection of transcripts confirms the expression of the TGF-β2 gene in this developing region. High levels of both transcripts and proteins were localized to layer II. However, the protein appeared homogeneously distributed in the cortical layers underneath, whereas TGF-β2 transcript accumulated in layer IV (Fig.8). Such a discrepancy in the amount of TGF-β2 transcript and translation products has previously been emphasized when proteins and transcripts were compared in different regions of the peripheral nervous system and the CNS (Pelton et al., 1991; Unsicker et al., 1991;Stewart et al., 1995). The apparent lack of TGF-β2 transcripts in cells immunoreactive for TGF-β2 could reflect technical limits for the detection of low levels of mRNA using _in situ_hybridization. Immunoreactivity of some cells could also stem from the uptake of TGF-β2 secreted in their vicinity.
Considering the expression of CSF-1 in the developing CNS (Théry et al., 1990; Chang et al., 1994; Roth and Stanley, 1996), the present study strongly suggests that neurons support microglial growth during development by secreting TGF-β2, which stimulates the proliferation of BMs and their precursors infiltrating the CNS tissue. Furthermore, our results provide a new functional significance for the intracerebral synthesis of TGF-β2. Beyond developmental topics, TGF-β2 and CSF-1 expression has been detected in adult CNS, and studies performed with CSF-1-deficient mice indicate that CSF-1 is required for the occurrence of macrophages in the injured brain (Hulkower et al., 1991; Unsicker et al., 1991; Raivich et al., 1994; Berezovskaya et al., 1995). Thus, the synergic effect of CSF-1 and TGF-β cytokines could contribute to the macrophage reaction, which has been demonstrated in both experimental lesions and a variety of human pathologies, including acquired immunodeficiency syndrome and neurodegenerative diseases (Dickson et al., 1993; McGeer et al., 1993; Perry et al., 1994). Noteworthy, different types of CNS injuries are associated with an induction of TGF-β1 synthesis localized to reactive astroglial cells and macrophages (Krieglstein et al., 1995a). Although the neuronal expression of TGF-β1 transcripts has also been observed as a consequence of ischemia or cranial nerve transection (Lefebvre et al., 1992; Knuckey et al., 1996), modulation of other neuronal TGF-β isoforms remains little studied. In this respect, the regulation of neuronal secretion of TGF-β2 and its biological effect under pathological conditions deserves further investigation.
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale and a grant from Agence Nationale de Recherche sur le SIDA. We thank Dr. Annette Koulakoff for participation in this work, Dr. Seillan-Heberden for providing the liver epithelial cell line, and Dr. Charles Félix Calvo for critical reading of this manuscript.
Correspondence should be addressed to Dr. Michel Mallat, Institut National de la Santé et de la Recherche Médicale U 114, Chaire de Neuropharmacologie, Collège de France, 11 Place Marcelin Berthelot, 75231 Paris Cedex 05, France.
REFERENCES
- 1.Aloisi F, Care A, Borsellino G, Gallo P, Rosa S, Bassani A, Cabibbo A, Testa U, Levi G, Peschle C. Production of hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1β and tumor necrosis factor-α. J Immunol. 1992;149:2358–2366. [PubMed] [Google Scholar]
- 2.Anderson DH, Guerin CJ, Hageman GS, Pfeffer BA, Flanders KC. Distribution of transforming growth factor-β isoforms in the mammalian retina. J Neurosci Res. 1995;42:63–79. doi: 10.1002/jnr.490420108. [DOI] [PubMed] [Google Scholar]
- 3.Arrick BA, Lee AL, Grendell RL, Derynck R. Inhibition of translation of transforming growth factor-β3 mRNA by its 5′ untranslated region. Mol Cell Biol. 1991;11:4306–4313. doi: 10.1128/mcb.11.9.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arrick BA, Lopez AR, Elfman F, Ebner R, Damsky CH, Derynck R. Altered metabolic and adhesive properties and increased tumorigenesis associated with increased expression of transforming growth factor-β1. J Cell Biol. 1992;118:715–726. doi: 10.1083/jcb.118.3.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berezovskaya O, Maysinger D, Fedoroff S. The hematopoietic cytokine colony-stimulating factor in the CNS: congenital absence of CSF-1 in mice results in abnormal microglial response and increased neuron vulnerability to injury. Int J Dev Neurosci. 1995;13:285–299. doi: 10.1016/0736-5748(95)00013-7. [DOI] [PubMed] [Google Scholar]
- 6.Celada A, Maki RA. Transforming growth factor-β enhances the M-CSF and GM-CSF-stimulated proliferation of macrophages. J Immunol. 1992;148:1102–1105. [PubMed] [Google Scholar]
- 7.Chamak B, Morandi V, Mallat M. Brain macrophages stimulate neurite growth and regeneration by secreting thrombospondin. J Neurosci Res. 1994;38:221–233. doi: 10.1002/jnr.490380213. [DOI] [PubMed] [Google Scholar]
- 8.Chang Y, Albright S, Lee F. Cytokines in the central nervous system: expression of macrophage colony stimulating factor and its receptor during development. J Neuroimmunol. 1994;52:9–17. doi: 10.1016/0165-5728(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 9.Chen BDM, Clark C, Chou T. Granulocyte/macrophage colony-stimulating factor stimulates monocyte and tissue macrophage proliferation and enhances their responsiveness to macrophage colony-stimulating factor. Blood. 1988;71:997–1002. [PubMed] [Google Scholar]
- 10.Chomczynski P, Sacchi N. Single step method of RNA isolation by acid guanidium thiocyanate phenol chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Constam DB, Philipp J, Malipiero UV, Ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-β1, -β2, and -β3 by glioblastoma cells, astrocytes, and microglia. J Immunol. 1992;148:1404–1410. [PubMed] [Google Scholar]
- 12.Constam DB, Schmid P, Aguzzi A, Schachner M, Fontana A. Transient production of TGF-β2 by postnatal cerebellar neurons and its effect on neuroblast proliferation. Eur J Immunol. 1994;6:766–778. doi: 10.1111/j.1460-9568.1994.tb00988.x. [DOI] [PubMed] [Google Scholar]
- 13.De Luca A, Weller M, Fontana A. TGF-β-induced apoptosis of cerebellar granule neurons is prevented by depolarization. J Neurosci. 1996;16:4174–4185. doi: 10.1523/JNEUROSCI.16-13-04174.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickson DW, Lee SC, Mattiace LA, Yen SC, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 15.Flanders KC, Lüdecke G, Engels S, Cissel DS, Roberts AB, Kondaiah P, Lafyatis R, Sporn MB, Unsicker K. Localization and actions of transforming growth factor-βs in the embryonic nervous system. Development (Camb) 1991;113:183–191. doi: 10.1242/dev.113.1.183. [DOI] [PubMed] [Google Scholar]
- 16.Flaumenhaft R, Kojima S, Abe M, Rifkin DB. Activation of latent transforming growth factor β. Adv Pharmacol. 1993;24:51–76. doi: 10.1016/s1054-3589(08)60933-3. [DOI] [PubMed] [Google Scholar]
- 17.Frank S, Madlener M, Werner S. Transforming growth factors β1, β2, and β3 and their receptors are differentially regulated during normal and impaired wound healing. J Biol Chem. 1996;271:10188–10193. doi: 10.1074/jbc.271.17.10188. [DOI] [PubMed] [Google Scholar]
- 18.Frei K, Noahava K, Malipiero UV, Schwerdel C, Fontana A. Production of colony-stimulating factor by astrocytes and brain macrophages. J Neuroimmunol. 1992;40:189–195. doi: 10.1016/0165-5728(92)90133-6. [DOI] [PubMed] [Google Scholar]
- 19.Gatherer D, ten Dijke P, Baird DT, Akhurst RJ. Expression of TGF-β isoforms during first trimester human embryogenesis. Development (Camb) 1990;110:445–460. doi: 10.1242/dev.110.2.445. [DOI] [PubMed] [Google Scholar]
- 20.Gautron S, Dos Santos G, Pinto-Henrique D, Koulakoff A, Gros F, Berwald-Netter Y. The glial voltage-gated sodium channel: cell- and tissue-specific mRNA expression. Proc Natl Acad Sci USA. 1992;89:7272–7276. doi: 10.1073/pnas.89.15.7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 22.Giulian D, Ingeman J. Colony-stimulating factors as promoters of ameboid microglia. J Neurosci. 1988;8:4707–4717. doi: 10.1523/JNEUROSCI.08-12-04707.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guilbert LJ, Winkler-Lowen B, Smith A, Branch DR, Lloret MG. Analysis of the synergistic stimulation of mouse macrophage proliferation by macrophage colony-stimulating factor (CSF-1) and tumor necrosis factor alpha (TNFα). J Leukoc Biol. 1993;54:65–72. doi: 10.1002/jlb.54.1.65. [DOI] [PubMed] [Google Scholar]
- 24.Hanks SK, Armour R, Baldwin JH, Maldonado F, Spiess J, Holley RW. Amino acid sequence of the BSC-1 cell growth inhibitor (polyergin) deduced from the nucleotide sequence of the cDNA. Proc Natl Acad Sci USA. 1988;85:79–82. doi: 10.1073/pnas.85.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hao C, Guilbert LJ, Fedoroff S. Production of colony-stimulating factor-1 (CSF-1) by mouse astroglia in vitro. J Neurosci Res. 1990;27:314–323. doi: 10.1002/jnr.490270310. [DOI] [PubMed] [Google Scholar]
- 26.Hickey W, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- 27.Hulkower K, Brosnan CF, Aquino DA, Cammer W, Kulshrestha S, Guida MP, Rapoport DA, Berman JW. Expression of CSF-1, c-fms, and MCP-1 in the central nervous system of rats with experimental allergic encephalomyelitis. J Immunol. 1993;6:2525–2533. [PubMed] [Google Scholar]
- 28.Jacobsen FW, Veilby OP, Jacobsen SEW. IL-7 stimulates CSF-induced proliferation of murine bone marrow macrophages and Mac-1+ myeloid progenitors in vitro. J Immunol. 1994;153:270–275. [PubMed] [Google Scholar]
- 29.Kondaiah P, Van Obberghen-Schilling E, Ludwig RL, Dhar R, Sporn MB, Roberts AB. cDNA cloning of porcine transforming growth factor-β1 mRNAs. J Biol Chem. 1988;34:18313–18317. [PubMed] [Google Scholar]
- 30.Knuckey NW, Finch P, Palm DE, Primiano MJ, Johanson CE, Flanders KC, Thompson NL. Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat following transient forebrain ischemia. Mol Brain Res. 1996;40:1–14. doi: 10.1016/0169-328x(96)00016-2. [DOI] [PubMed] [Google Scholar]
- 31.Krieglstein K, Rufer M, Suter-Crazzolara C, Unsicker K. Neural functions of the transforming growth factors β. Int J Dev Neurosci. 1995a;13:301–315. doi: 10.1016/0736-5748(94)00062-8. [DOI] [PubMed] [Google Scholar]
- 32.Krieglstein K, Suter-Crazzolara C, Fischer WH, Unsicker K. TGF-β superfamily members promote survival of midbrain dopaminergic neurons and protect them against MPP+ toxicity. EMBO J. 1995b;14:736–742. doi: 10.1002/j.1460-2075.1995.tb07052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee SC, Liu W, Roth P, Dickson DW, Berman JW, Brosnan CF. Macrophage colony-stimulating factor in fetal astrocytes and microglia: differential regulation by cytokines and lipopolysaccharide and modulation of class II MHC on microglia. J Immunol. 1993;150:594–604. [PubMed] [Google Scholar]
- 34.Lefebvre PP, Martin D, Staecker H, Weber T, Moonen G, Van deWater TR. TGFβ1 expression is initiated in adult auditory neurons by sectioning of the auditory nerve. NeuroReport. 1992;3:295–298. doi: 10.1097/00001756-199204000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Ling EA, Wong WC. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia. 1993;7:9–18. doi: 10.1002/glia.440070105. [DOI] [PubMed] [Google Scholar]
- 36.Lucas C, Bald LN, Fendly BM, Mora-Worms M, Figari IS, Patzer EJ, Palladino MA. The autocrine production of TGF-β1 during lymphocyte activation. J Immunol. 1990;145:1415–1422. [PubMed] [Google Scholar]
- 37.Malipiero UV, Frei K, Fontana A. Production of hemopoietic colony-stimulating factors by astrocytes. J Immunol. 1990;144:3816–3821. [PubMed] [Google Scholar]
- 38.Mallat M, Chamak B. Brain macrophages: neurotoxic or neurotrophic effector cells. J Leukoc Biol. 1994;56:416–422. doi: 10.1002/jlb.56.3.416. [DOI] [PubMed] [Google Scholar]
- 39.Massagué J, Attisano L, Wrana JL. The TGF-β family and its composite receptors. Trends Cell Biol. 1994;4:172–178. doi: 10.1016/0962-8924(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 40.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in degenerative neurological disease. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- 41.Mercier T, Gaillard-Sanchez I, Martel P, Seillan-Heberden C. Constitutive overexpression of c-fos protein in rat liver epithelial cells decreases TGF-β synthesis and increases TGF-β1 receptors. Biochim Biophys Acta. 1995;1266:64–72. doi: 10.1016/0167-4889(94)00240-f. [DOI] [PubMed] [Google Scholar]
- 42.Metcalf D. The molecular control of cell division, differentiation commitment and maturation in haemopoietic cells. Nature. 1989;339:27–30. doi: 10.1038/339027a0. [DOI] [PubMed] [Google Scholar]
- 43.Millan FA, Denhez F, Paturu K, Akhurst RJ. Embryonic gene expression patterns of TGF β1, β2 and β3 suggest different developmental functions in vivo. Development (Camb) 1991;111:131–144. doi: 10.1242/dev.111.1.131. [DOI] [PubMed] [Google Scholar]
- 44.Nakajima K, Tsuzaki N, Nagata K, Takemoto N, Kohsaka S. Production and secretion of plasminogen in cultured rat microglia. FEBS Lett. 1992a;308:179–182. doi: 10.1016/0014-5793(92)81270-v. [DOI] [PubMed] [Google Scholar]
- 45.Nakajima K, Tsuzaki N, Shimojo M, Hamanoue M, Kohsaka S. Microglia isolated from rat brain secrete a urokinase-type plasminogen activator. Brain Res. 1992b;577:285–292. doi: 10.1016/0006-8993(92)90285-h. [DOI] [PubMed] [Google Scholar]
- 46.Nohava K, Malipiero U, Frei K, Fontana A. Neurons and neuroblastoma as a source of macrophage colony-stimulating factor. Eur J Immunol. 1992;22:2539–2545. doi: 10.1002/eji.1830221012. [DOI] [PubMed] [Google Scholar]
- 47.Ohta M, Greenberger JS, Anklesaria P, Bassols A, Massagué J. Two forms of transforming growth factor-β distinguished by multipotential haematopoietic progenitor cells. Nature. 1987;329:539–541. doi: 10.1038/329539a0. [DOI] [PubMed] [Google Scholar]
- 48.Pelton RW, Saxena B, Jones M, Moses HL, Gold LI. Immunohistochemical localization of TGFβ1, TGFβ2, and TGFβ3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol. 1991;115:1091–1105. doi: 10.1083/jcb.115.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perry VH, Lawson LJ, Reid DM. Biology of the mononuclear phagocyte system of the central nervous system and HIV infection. J Leukoc Biol. 1994;56:399–406. doi: 10.1002/jlb.56.3.399. [DOI] [PubMed] [Google Scholar]
- 50.Poulsen KT, Armanini MP, Klein RD, Hynes MA, Phillips HS, Rosenthal A. TGFβ2 and TGFβ3 are potent survival factors for midbrain dopaminergic neurons. Neuron. 1994;13:1245–1252. doi: 10.1016/0896-6273(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 51.Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MWJ, Doetcman T. Transforming growth factor-β3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qian SW, Kondaiah P, Roberts AB, Sporn MB. cDNA cloning by PCR of rat transforming growth factor-β1. Nucleic Acid Res. 1990;18:3059. doi: 10.1093/nar/18.10.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raivich G, Moreno-Flores MT, Möller JC, Kreutzberg GW. Inhibition of posttraumatic microglial proliferation in a genetic model of macrophage colony-stimulating factor deficiency in the mouse. Eur J Neurosci. 1994;6:1615–1618. doi: 10.1111/j.1460-9568.1994.tb00552.x. [DOI] [PubMed] [Google Scholar]
- 54.Rosenfeld CS. Transforming growth factor-β1 augments macrophage-colony stimulating factor activity on human marrow. Stem Cells (Dayt) 1994;12:527–532. doi: 10.1002/stem.5530120509. [DOI] [PubMed] [Google Scholar]
- 55.Roth P, Stanley ER. The biology of CSF-1 and its receptor. Curr Top Microbiol Immunol. 1992;181:141–167. doi: 10.1007/978-3-642-77377-8_5. [DOI] [PubMed] [Google Scholar]
- 56.Roth P, Stanley ER. Colony stimulating factor-1 expression is developmentally regulated in the mouse. J Leukoc Biol. 1996;59:817–823. doi: 10.1002/jlb.59.6.817. [DOI] [PubMed] [Google Scholar]
- 57.Saad B, Constam DB, Ortmann R, Moos M, Fontana A, Schachner M. Astrocyte-derived TGF-β2 and NGF differentially regulate neural recognition molecule expression by cultured astrocytes. J Cell Biol. 1991;115:473–484. doi: 10.1083/jcb.115.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmid P, Cox D, Bilbe G, Maier R, McMaster G. Differential expression of TGFβ1, β2 and β3 genes during mouse embryogenesis. Development (Camb) 1991;111:117–130. doi: 10.1242/dev.111.1.117. [DOI] [PubMed] [Google Scholar]
- 59.Schultz-Cherry S, Murphy-Ullrich JE. Thrombospondin causes activation of latent transforming growth factor-β secreted by endothelial cells by a novel mechanism. J Cell Biol. 1993;122:923–932. doi: 10.1083/jcb.122.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sporn MB, Roberts AB. Transforming growth factor-β: recent progress and new challenges. J Cell Biol. 1992;119:1017–1021. doi: 10.1083/jcb.119.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stanley ER, Heard P. Factors regulating macrophage production and growth. Purification and some properties of the colony-stimulating factor from medium conditioned by mouse L cells. J Biol Chem. 1977;252:4305–4312. [PubMed] [Google Scholar]
- 62.Stewart HJS, Rougon G, Dong Z, Dean C, Jessen KR, Mirsky R. TGF-βs upregulate NCAM and L1 expression in cultured Schwann cells, suppress cyclic AMP-induced expression of O4 and galactocerebroside, and are widely expressed in cells of the Schwann cell lineage in vivo. Glia. 1995;15:419–436. doi: 10.1002/glia.440150406. [DOI] [PubMed] [Google Scholar]
- 63.Suzumura A, Sawada M, Yamamoto H, Marunouchi T. Transforming growth factor-β suppresses activation and proliferation of microglia in vitro. J Immunol. 1993;151:2150–2158. [PubMed] [Google Scholar]
- 64.Théry C, Mallat M. Influence of interleukin-1 and tumor necrosis factor-alpha on the growth of microglial cells in primary cultures of mouse cerebral cortex: involvement of colony-stimulating factor 1. Neurosci Lett. 1993;150:195–199. doi: 10.1016/0304-3940(93)90534-r. [DOI] [PubMed] [Google Scholar]
- 65.Théry C, Hétier E, Evrard C, Mallat M. Expression of macrophage colony-stimulating factor gene in the mouse brain during development. J Neurosci Res. 1990;26:129–133. doi: 10.1002/jnr.490260117. [DOI] [PubMed] [Google Scholar]
- 66.Théry C, Chamak B, Mallat M. Cytotoxic effect of brain macrophages on developing neurons. Eur J Neurosci. 1991;3:1155–1164. doi: 10.1111/j.1460-9568.1991.tb00050.x. [DOI] [PubMed] [Google Scholar]
- 67.Théry C, Stanley ER, Mallat M. Interleukin 1 and tumor necrosis factor-α stimulate the production of colony-stimulating factor 1 by murine astrocytes. J Neurochem. 1992;59:1183–1186. doi: 10.1111/j.1471-4159.1992.tb08366.x. [DOI] [PubMed] [Google Scholar]
- 68.Tushinski RJ, Stanley ER. The regulation of mononuclear phagocyte entry into S phase by colony stimulating factor CSF-1. J Cell Physiol. 1985;122:221–228. doi: 10.1002/jcp.1041220210. [DOI] [PubMed] [Google Scholar]
- 69.Tushinski R, Oliver I, Guilbert L, Tynan P, Warner J, Stanley ER. Survival of mononuclear phagocytes depends on a lineage-specific growth factor that the differentiated cells selectively destroy. Cell. 1982;28:71–81. doi: 10.1016/0092-8674(82)90376-2. [DOI] [PubMed] [Google Scholar]
- 70.Unsicker K, Flanders KC, Cissel DS, Lafyatis R, Sporn MB. Transforming growth factor beta isoforms in the adult rat central and peripheral nervous system. Neuroscience. 1991;44:613–625. doi: 10.1016/0306-4522(91)90082-y. [DOI] [PubMed] [Google Scholar]
- 71.Van-Vliet BJ, Sebben M, Dumuis A, Gabrion J, Bockaert J, Pin J-P. Endogenous amino acid release from cultured cerebellar neuronal cells: effect of tetanus toxin on glutamate release. J Neurochem. 1989;52:1229–1239. doi: 10.1111/j.1471-4159.1989.tb01870.x. [DOI] [PubMed] [Google Scholar]
- 72.Wahl SM, Hunt DA, Wakefield L, Mc Cartney-Francis N, Wahl LM, Roberts AB, Sporn MB. Transforming growth factor beta (TGF-β) induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci USA. 1987;84:5788–5792. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yao J, Harvath L, Guilbert DL, Colton CA. Chemotaxis by a CNS macrophage, the microglia. J Neurosci Res. 1990;27:36–42. doi: 10.1002/jnr.490270106. [DOI] [PubMed] [Google Scholar]