A phase 3 trial evaluating panitumumab plus best supportive care vs best supportive care in chemorefractory wild-type KRAS or RAS metastatic colorectal cancer (original) (raw)
Main
Panitumumab, a fully human monoclonal antibody targeting the epidermal growth factor receptor (EGFR), is effective as monotherapy and in combination with chemotherapy for treatment of patients with metastatic colorectal cancer (mCRC) (Van Cutsem et al, 2007; Douillard et al, 2013; Poulin-Costello et al, 2013; Peeters et al, 2015). Signalling through the EGFR pathway initiates a number of signalling cascades that regulate cell proliferation (Mendelsohn and Baselga, 2006; Normanno et al, 2006). A key element of signal transduction through several of these cascades is activation of members of the RAS family of small GTP-binding proteins (Downward, 2003; Schubbert et al, 2007). Kirsten rat sarcoma-2 virus oncogene homologue (KRAS) is a member of the RAS gene family (Downward, 2003; Schubbert et al, 2007), and is frequently mutated in patients with CRC (Schubbert et al, 2007). Oncogenic mutations in KRAS are found most frequently in codons 12 and 13 of exon 2 and occur in ∼30–45% of CRC tumours (Lievre et al, 2006; Benvenuti et al, 2007; Di Fiore et al, 2007). These mutations compromise the GTP-binding domain, causing the protein to stall in the GTP-bound confirmation, thereby resulting in a constitutively active protein (Downward, 2003). Given the role of KRAS in EGFR signalling, it was hypothesised that these activating mutations in KRAS exon 2 may result in a lack of response to panitumumab. A retrospective analysis of the randomised, phase 3 20020408 study which evaluated panitumumab plus best supportive care (BSC) vs BSC alone found a significant improvement in patient outcomes in patients with wild-type KRAS exon 2 tumours compared with those who had mutant KRAS exon 2 tumours (Amado et al, 2008). These findings were subsequently confirmed by prospective analyses of the randomised, phase 3 PRIME and 20050181 studies, which evaluated panitumumab plus FOLFOX in the first-line setting or panitumumab plus FOLFIRI in the second-line setting, respectively (Douillard et al, 2010; Peeters et al, 2010).
More recent retrospective analyses have indicated that additional activating mutations in exons 3 and 4 of KRAS and in exons 2, 3, and 4 of NRAS (another member of the RAS family), are also negative predictors for anti-EGFR efficacy (Douillard et al, 2013; Schwartzberg et al, 2014; Bokemeyer et al, 2015; Peeters et al, 2015; Van Cutsem et al, 2015). These mutations have been identified in approximately 15% of wild-type KRAS exon 2 tumours (Douillard et al, 2013; Van Cutsem et al, 2015). Evaluation of outcomes in patients with wild-type RAS tumours (i.e., wild-type for KRAS and NRAS, exons 2, 3, and 4) by extended RAS analysis have shown numerically improved survival relative to patients with wild-type KRAS exon 2 tumours in the first-line (PRIME) (Douillard et al, 2013) and second-line (20050181) (Peeters et al, 2015) settings. In addition, outcomes for progression-free survival (PFS) and objective response rate (ORR) were more favourable in an extended RAS analysis from the 20020408 study (Patterson et al, 2013; Peeters et al, 2013). These analyses have helped guide treatment selection decisions to better refine patients who are likely to respond to anti-EGFR therapies and to exclude patients unlikely to respond, limiting potential exposure to toxicities. However, although these extended RAS analyses were rigorously conducted, they were retrospective in nature and were not prespecified endpoints in the respective study protocols at the time each study was initiated. Therefore, it is possible that potential sources of bias/confounding were not adequately mitigated. At present, RAS as a predictive biomarker for anti-EGFR therapies has yet to be validated in prospective, phase 3, randomised mCRC studies.
Although the primary objective of the phase 3, open-label, randomised 20100007 study (ClinicalTrials.gov, NCT01412957) was to evaluate the effect of panitumumab plus BSC vs BSC alone on overall survival (OS) in patients with chemotherapy-refractory wild-type KRAS exon 2 mCRC, a critical key secondary objective was to prospectively evaluate the treatment effect of panitumumab in patients with wild-type RAS mCRC. This analysis would provide definitive validation for RAS as a predictive biomarker for anti-EGFR therapies.
Patients and methods
Patient population
Eligible patients (⩾18 years) had histologically or cytologically confirmed metastatic adenocarcinoma of the colon/rectum, wild-type KRAS exon 2 (codons 12 and 13) tumour status confirmed by central laboratory (see below), Eastern Cooperative Oncology Group (ECOG) performance status ⩽2, ⩾1 measurable or non-measurable lesion per Response Evaluation Criteria In Solid tumours (RECIST) version 1.1 (Eisenhauer et al, 2009), had previously received a thymidylate synthase inhibitor (e.g., fluorouracil, capecitabine, raltitrexed or fluorouracil-uracil), and had clinical/radiological disease progression or toxicity on prior regimens for metastatic disease containing irinotecan and oxaliplatin. Relapse within 6 months after adjuvant chemotherapy was considered treatment failure for metastatic disease. Patients were excluded if they had symptomatic brain metastases requiring treatment; major surgery ⩽28 days before randomisation; clinically significant cardiovascular disease ⩽6 months before randomisation; previous anti-EGFR therapy (small-molecule or monoclonal antibody); magnesium below lower limit of normal; inadequate renal, hepatic or haematological function; antitumour therapy ⩽21 days before randomisation; and/or radiotherapy ⩽14 days before randomisation. Patients must have recovered from any acute chemotherapy/radiotherapy toxicities. It is important to note that at the time the trial opened for patient enrollment, a statistically significant OS benefit for panitumumab had not been observed nor had non-inferiority with other anti-EGFR therapies been established. Thus the trial was conceived and conducted under clinical equipoise. The study adhered to all country-specific regulatory requirements, the protocol was approved by an independent ethics committee at each of the study centres, and informed consent was obtained from all patients.
Study design and treatment schedule
This was an open-label, randomised, phase 3 study conducted at 66 centres in 16 countries. Patients were randomly assigned 1 : 1 to the treatment arms using an interactive voice-response system to receive panitumumab 6 mg kg−1 intravenously on day 1 of each 14-day cycle plus BSC or BSC alone. Randomisation was stratified according to geographic region (Europe vs Asia vs rest of the world) and ECOG performance status (0 or 1 vs 2). BSC was defined as the best palliative care available, as judged appropriate by the investigator, consistent with institutional guidelines. BSC included antibiotics, analgesics, radiation for pain control (bone metastases only), corticosteroids, transfusions, psychotherapy, growth factors, palliative surgery or any other symptomatic therapy as clinically indicated. Patients were treated until disease progression, withdrawal of consent or panitumumab intolerance (panitumumab plus BSC arm only). Crossover from the BSC arm to panitumumab plus BSC was prohibited on-study.
Assessments
Radiographic tumour assessments were performed at week 4 (+1 week), week 8 (±1 week) and every 8 weeks (±1 week) thereafter, until radiographic or clinical disease progression. Response was evaluated by investigators per RECIST version 1.1 (Eisenhauer et al, 2009). Patients were followed for survival for up to 24 months or until ∼250 deaths were observed, whichever occurred later. Patients had a safety follow-up visit 30–33 days after the last dose of panitumumab in the panitumumab plus BSC arm; and within 33 days of disease progression or the decision to end treatment in the BSC alone arm. Adverse events (AEs) occurring on-study were recorded and graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0; skin- or nail-related AEs were graded using CTCAE version 3.0 with modifications.
RAS mutational analysis
Three central laboratories screened patient tumour KRAS exon 2 status in formalin-fixed, paraffin-embedded tissue sections using validated KRAS assays that identified seven mutations in codons 12 and 13 to determine eligibility for panitumumab treatment (clinical trial assays based on primers from a DxS/Qiagen assay; Venlo, Netherlands). Extended RAS analysis was conducted in a single central laboratory (blinded to patients’ treatment assignments/outcomes) on banked patient tumour specimens characterised as wild-type KRAS exon 2. Analyses of KRAS exon 3 (codons 59, 61) and exon 4 (codons 117, 146) and NRAS exon 2 (codons 12, 13), exon 3 (codons 59, 61) and exon 4 (codons 117, 146) were prespecified in the study protocol and mutation status determined by bidirectional Sanger sequencing before the primary analysis.
Statistical analyses
The primary endpoint was OS (time from randomisation to death) for all patients with wild-type KRAS exon 2 mCRC. Secondary endpoints included PFS (time from randomisation to disease progression or death) and ORR (rate of either a complete response or partial response per RECIST version 1.1) (Eisenhauer et al, 2009) for patients with wild-type KRAS exon 2 status; OS, PFS and ORR for patients with wild-type RAS status; and safety. Assuming an HR (panitumumab plus BSC to BSC alone) of 0.66, to achieve 90% power for a 5% significance level test, a total of 250 OS events and a sample size of 350 patients were required. OS and PFS analyses included all patients randomised to treatment with wild-type KRAS exon 2 status (i.e., the intent-to-treat analysis set). Safety analyses included all randomised patients according to treatment received, which was the same as treatment randomised for all patients. Wild-type RAS analysis sets were similar and included patients with wild-type KRAS and NRAS exons 2, 3, and 4.
The primary analysis was planned when ∼250 deaths had occurred; no interim efficacy analysis was planned. Log-rank tests stratified by the randomisation factors (baseline ECOG performance status and region) were used to compare OS and PFS between treatment groups. Treatment effects on OS and PFS were estimated using stratified Cox proportional hazards models and the Kaplan–Meier method. Sequential testing based on a two-sided 5% significance level was used; if the OS treatment effect in the intent-to-treat population was significant, PFS in the intent-to-treat population would be tested. If PFS in the intent-to-treat population was significant, OS in the wild-type RAS population would be tested. If OS in the wild-type RAS population was significant, PFS in the wild-type RAS population would be tested. ORR was not formally tested.
RAS analyses were prospective in nature. Although treatment was not randomised within the wild-type RAS population, RAS analyses were specified as secondary objectives, RAS exons were defined in the protocol, a rigorous sequential testing methodology was prespecified, and the testing laboratories were blinded to both treatment assignment and clinical outcome.
Results
Patients
Between November 2011 and July 2013, 377 patients with wild-type KRAS exon 2 mCRC were enrolled (panitumumab plus BSC, _n_=189; BSC alone, _n_=188; Figure 1A). Among these patients, 324 patients (86%) were evaluable for RAS analysis; 270 (83%) had tumours that were wild-type in exons 2–4 of KRAS and NRAS (i.e., wild-type RAS), and 54 (17%) had a mutation in KRAS exon 3 or 4 or NRAS exons 2, 3, or 4 (Figure 1B and Supplementary Table 1). Demographics and baseline characteristics were similar between arms in the wild-type KRAS exon 2, wild-type RAS and wild-type KRAS exon 2 but other RAS mutations populations (Table 1).
Figure 1
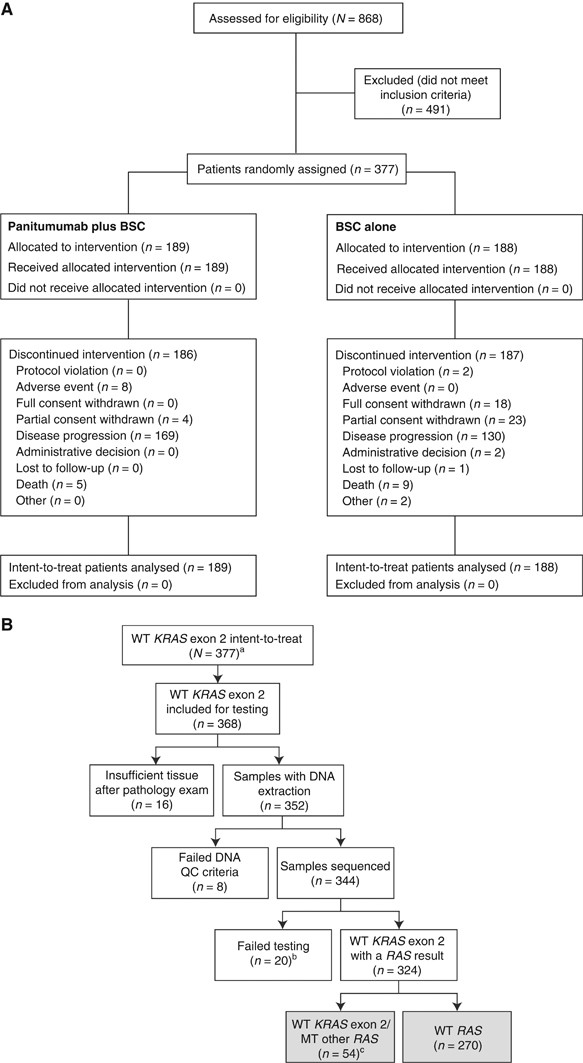
CONSORT diagram (A) and RAS testing (B) for the 20100007 study. a_KRAS_ exon 2 status was based on initial screening test results and wild-type for all 377 patients; mutation status of KRAS exon 3 and 4 and NRAS exons 2, 3, and 4 was determined by Sanger sequencing. bOf 20 samples that failed, four were due to not meeting the analysis criteria and 16 had at least one RAS exon that failed testing and were wild-type in the other exons (see footnote c). cIf a sample had a RAS exon mutation (KRAS exon 3 or 4 or NRAS exons 2, 3 or 4) and one of the other exons failed testing, the sample was characterised as wild-type KRAS exon 2/mutant other RAS. BSC=best supportive care; QC=quality control; WT=wild-type.
Table 1 Baseline demographics and disease characteristics
For patients who received panitumumab plus BSC, median (range) duration of treatment was 16.0 (2.0–80.0) weeks, and median (range) number of infusions was 8.0 (1–40). For patients who received BSC alone, median (range) duration of treatment was 5.1 (0–56.4) weeks. Median follow-up time was 41.0 and 25.5 weeks for panitumumab plus BSC and BSC alone, respectively. At the time of analysis, 99% of patients had discontinued study therapy, most frequently due to disease progression (79%; Figure 1A). After disease progression, 29% of patients in the panitumumab plus BSC arm and 34% of patients in the BSC alone arm received additional antitumour therapy including chemotherapy (panitumumab plus BSC, 26%; BSC alone, 21%), anti-EGFR therapy (1%; 7%) and bevacizumab (2%; 5%).
Efficacy
Panitumumab plus BSC significantly improved OS compared with BSC alone. For patients with wild-type KRAS exon 2 tumours, median (95% CI) OS was 10.0 (8.7–11.4) months in the panitumumab plus BSC arm vs 7.4 (5.8–9.3) months in the BSC alone arm (HR=0.73; 95% CI=0.57–0.93; _P_=0.0096; Figure 2A, Table 2). In OS subgroups defined by baseline characteristics, HRs consistently favoured panitumumab plus BSC over BSC alone including subgroups by ECOG status (0/1 or 2), metastatic sites (1, 2 or ⩾3) and liver limited disease (Supplementary Figure 1A).
Figure 2
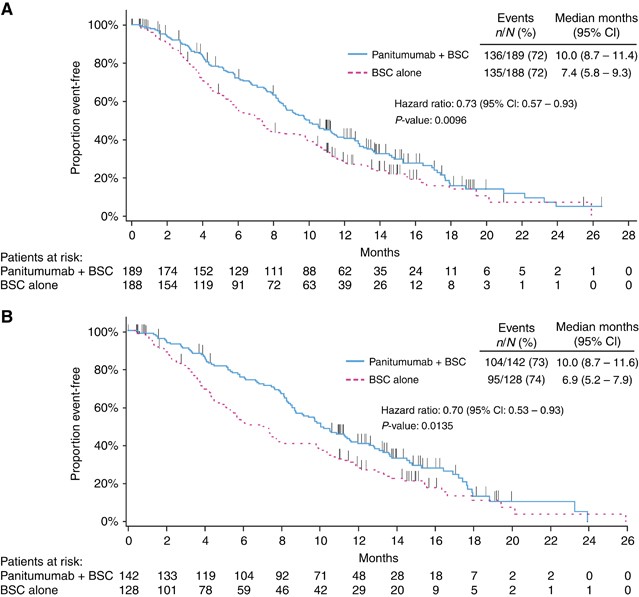
Overall survival in (A) the wild-type KRAS exon 2 group and (B) in the extended wild-type RAS subgroup. BSC=best supportive care.
Table 2 Efficacy results
PFS was also improved among patients in the panitumumab arm vs those in the BSC alone arm. Median (95% CI) PFS was 3.6 (3.4–5.3) months for panitumumab plus BSC vs 1.7 (1.6–1.9) months for BSC alone (HR=0.51; 95% CI=0.41–0.64; P<0.0001; Figure 3A, Table 2). The ORR (95% CI) for panitumumab plus BSC was 27.0% (20.8–33.9) vs 1.6% (0.3–4.6) for BSC alone (odds ratio (OR)=24.9; 95% CI=7.5–123.8; P<0.0001; Table 2).
Figure 3
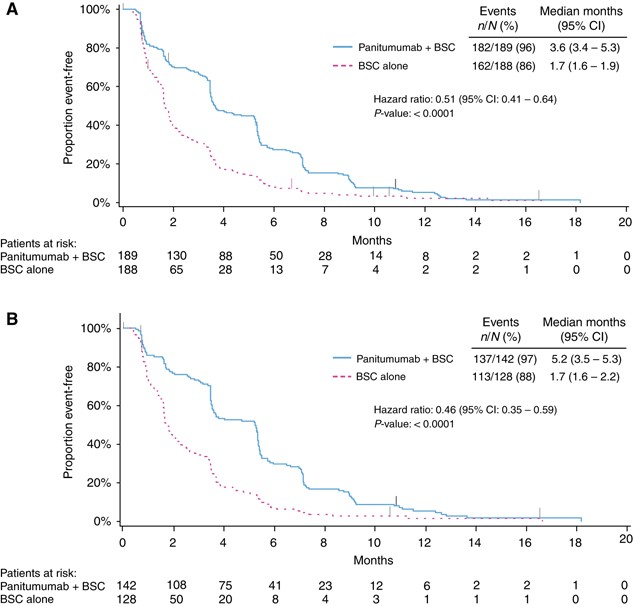
Progression-free survival in (A) the wild-type KRAS exon 2 group and (B) in the extended wild-type RAS subgroup. BSC=best supportive care.
As in the wild-type KRAS exon 2 population, treatment with panitumumab plus BSC improved OS vs BSC alone for patients with wild-type RAS tumours (median (95% CI): 10.0 (8.7–11.6) months vs 6.9 (5.2–7.9) months; HR=0.70; 95% CI=0.53–0.93; _P_=0.0135; Figure 2B, Table 2). In OS subgroups defined by baseline characteristics, results also favoured panitumumab plus BSC over BSC alone (Supplementary Figure 1B). PFS was improved for panitumumab plus BSC (median (95% CI): 5.2 (3.5–5.3) months) vs BSC alone (1.7 (1.6–2.2) months; HR=0.46; 95% CI=0.35–0.59; P<0.0001; Figure 3B, Table 2). In addition, ORR (95% CI) for panitumumab plus BSC was 31.0% (23.5–39.3) vs 2.3% (0.5–6.7) for BSC alone (OR=20.0; 95% CI=5.9–101.6; P<0.0001; Table 2).
Patients with wild-type KRAS exon 2 tumours but with other RAS mutations did not benefit from panitumumab therapy. In this group, the HRs (95% CI) for OS and PFS for panitumumab plus BSC vs BSC alone were 0.99 (0.49–2.00; Supplementary Figure 2A) and 1.03 (0.56–1.90; Supplementary Figure 2B), respectively. Complete or partial responses were not achieved by either group; stable disease was achieved by 42% for panitumumab plus BSC vs 21% for BSC alone (Supplementary Table 2).
Safety
In the wild-type KRAS exon 2 population, 97% of those who received panitumumab plus BSC and 61% of those who received BSC alone experienced an AE of any grade (Table 3). The incidences of grades 3 and 4 AEs were 37% and 9%, respectively, for panitumumab plus BSC and 15% and 3% for BSC alone (Table 3). Eight patients (4%) in the panitumumab plus BSC arm discontinued treatment because of AEs vs 0 patients (0%) in the BSC alone arm. Fatal AEs occurred in 8 (4%) patients who received panitumumab plus BSC (seven due to disease progression and one to gastrointestinal necrosis) vs 15 (8%) patients who received BSC alone (nine due to disease progression and one each to bone marrow toxicity, congestive cardiac failure, abnormal hepatic function, lower gastrointestinal haemorrhage, respiratory distress and sepsis).
Table 3 Summary of adverse events
The incidence of infusion reactions was 1% in the panitumumab plus BSC arm. AEs of any grade with a ⩾5% difference between arms (panitumumab plus BSC, BSC alone) included rash (39%, 1%), dermatitis acneiform (29%, 0%) and hypomagnesemia (28%, 1%; Supplementary Table 3, online only). Incidences of grade 3/4 hypomagnesemia, skin rash, and dermatitis acneiform were 6%, 6%, and 6%, respectively, for panitumumab plus BSC, and 1%, 1%, and 0% for BSC alone (Supplementary Table 3). Similar incidences were observed for the wild-type RAS and the wild-type KRAS exon 2 but mutant other RAS populations (Supplementary Tables 4 and 5).
Discussion
This was the first randomised, phase 3 study to demonstrate a significant OS benefit with panitumumab monotherapy vs BSC in patients with wild-type KRAS exon 2 mCRC. In the 20020408 study of panitumumab plus BSC vs BSC alone, OS benefit was not shown, likely because 76% of patients in the BSC arm opted to cross over and receive panitumumab as allowed in the study protocol (Van Cutsem et al, 2007). Retrospective analyses suggested OS may have been improved if crossover had not occurred (Poulin-Costello et al, 2013). Although caution is warranted when making cross-trial comparisons, median OS in the panitumumab arm in the wild-type KRAS population in this study (10.0 months, 95% CI=8.7–11.4) was moderately longer relative to that in the wild-type KRAS population in the 20020408 study (8.1 months) but was consistent with that reported in the ASPECCT study (panitumumab, 10.4 months, 95% CI=9.4–11.6 months; cetuximab, 10.0 months, 95% CI=9.3–11.0 months; HR=0.97, 95% CI=0.84–1.11) (Price et al, 2014) and in the cetuximab CO.17 study (9.5 months) (Karapetis et al, 2008). The OS HR in this study was 0.73 and was higher than the anticipated HR of 0.66. The assumed OS HR was based on results from the CO.17 study where median OS in the control arm was 4.8 months. The longer median OS for the BSC arm reported in this study (7.4 months) likely impacted the HR and suggests that outcomes with BSC have improved over time. The longer OS time in the BSC arm observed in this study, compared with the 20020408 study, may have resulted from the more extensive use of post-treatment antitumour therapy after disease progression (34% of patients in the BSC arm in 20100007 received post-progression therapy). In the wild-type KRAS exon 2 population, median PFS in the panitumumab arm was also longer than in the BSC arm (3.6 months, 95% CI=3.4–5.3 months vs 1.7 months; 95% CI=1.6–1.9 months). Notably, PFS in the BSC arm was consistent between the 20020408 and 20100007 populations, providing further evidence that the differences in OS times between the studies may have been due to post-progression therapy. No new safety signals were observed in this study. Events occurring more frequently among patients who received panitumumab were consistent with previous studies (Douillard et al, 2010; Price et al, 2014; Peeters et al, 2015), and the rate of infusion reactions with panitumumab therapy was low (1%).
This was also the first phase 3 study to demonstrate significant improvements in OS, PFS and ORR with an anti-EGFR agent in a prospectively defined chemorefractory wild-type RAS population (although, as noted, treatment was not randomised within the wild-type RAS population). These results confirm and extend those from previous retrospective analyses evaluating extended RAS analysis in mCRC (Douillard et al, 2013; Peeters et al, 2013; Bokemeyer et al, 2015; Peeters et al, 2015; Van Cutsem et al, 2015). In the 20020408 study of panitumumab plus BSC vs BSC alone, PFS was significantly improved in patients with wild-type KRAS exon 2 tumours (HR=0.45; 95% CI=0.34–0.59) (Amado et al, 2008), and this treatment effect was greater in retrospective analysis in the wild-type RAS population (HR=0.39; 95% CI=0.27–0.56) (Peeters et al, 2013). In the 20100007 study, improvements in PFS were also seen in both the wild-type KRAS exon 2 population (HR=0.51; 95% CI=0.41–0.64) and the wild-type RAS population (HR=0.46; 95% CI=0.34–0.59). The improvement in outcomes in the wild-type RAS population vs the wild-type KRAS exon 2 population is also consistent with that seen in extended RAS analysis of the PRIME study in the first-line setting (Douillard et al, 2013) and the 20050181 study in the second-line setting (Peeters et al, 2015), indicating the benefit observed in patients with tumours without activating RAS mutations is true across all lines of therapy and for combination treatment as well as monotherapy. In both the 20020408 study and in this study, patients who were wild-type for KRAS exon 2 but had other RAS mutations (i.e., in either KRAS exons 3 or 4 or NRAS exons 2, 3 or 4) were unlikely to benefit from panitumumab. This lack of benefit is consistent with results from other monotherapy studies and studies evaluating combination therapy (Bokemeyer et al, 2009; Douillard et al, 2013; Peeters et al, 2015; Van Cutsem et al, 2015).
The consistency between outcomes in the 20020408 study and the 20100007 study demonstrates the value of rigorously conducted retrospective analyses. The retrospective extended RAS analyses of the randomised, phase 3 20020408, PRIME and 20050181 studies were all conducted following stringent analysis procedures (Douillard et al, 2013; Peeters et al, 2013; Peeters et al, 2015), providing a rigorous framework for analysis that mirrored the procedures followed for the prospective extended RAS analysis of the 20100007 study. First, in both the retrospective and prospective analyses, tissue samples were collected with appropriate informed consent before patients were randomised to treatment arms. Second, the extended RAS analyses conducted for these studies were predefined: the statistical analysis plans were finalised before patient biomarker status information became available. Third, the testing laboratories were all blinded to both treatment assignment and clinical outcome. Finally, these retrospective analyses used data from large, randomised controlled phase 3 trials, and the high RAS ascertainment rates for these studies minimised the potential for ascertainment bias due to missing data on tumour RAS status. RAS ascertainment rates ranged from 75% in the 20020408 study (Patterson et al, 2013), 90% in the PRIME study (Douillard et al, 2013), 85% in the 20050181 study (Peeters et al, 2015), to 86% in this study. In the _RAS_-evaluable populations, reported rates of RAS mutations (KRAS exons 3 or 4 or NRAS exons 2, 3 or 4) found in the wild-type KRAS exon 2 population were between 15 and 20% (Douillard et al, 2013; Peeters et al, 2015), and the 17% frequency observed here in the 20100007 study was consistent with this range. The high ascertainment rate, coupled with the large informative patient populations in the phase 3 trials, provided numerically large patient subgroups for the expanded analyses of activating RAS mutations, thus limiting the potential for influence of outliers on outcomes. Given the rigorous nature of these analyses and the consistency of improvement in outcomes in the wild-type RAS population vs the wild-type KRAS exon 2 population in each study, the studies as a whole provide reassurance that the better predictive value of extended RAS analysis is a genuine finding and not an experimental artefact. Just as the predictive value of KRAS exon 2 mutations was identified in retrospective analysis of the 20020408 study (Amado et al, 2008) and confirmed by prospective analysis of the PRIME and 20050181 studies (Douillard et al, 2010; Peeters et al, 2010), similarly, the predictive value of extended RAS analysis was identified in retrospective analyses from the PRIME and 20050181 studies (Douillard et al, 2013; Peeters et al, 2015) and confirmed by the prospective extended RAS analysis in the 20100007 study. Taken together, these findings indicate that when retrospective analyses are conducted within a predefined rigorous, controlled methodological framework they can serve as an important and valid method for identifying predictive biomarkers, particularly in situations where it may be challenging to conduct a large prospective trial
Although the results from the 20100007 study validate those seen in previous studies, the study did have limitations that may have influenced outcomes. First, although on-study crossover was prohibited, the evaluation of OS may have been confounded by subsequent therapy use. Patients in the BSC alone arm were more likely to receive anti-EGFR therapy or bevacizumab after progression; however, this imbalance would be anticipated to reduce the magnitude of the treatment effect, yet statistically significant improvements were still seen. Second, the extended RAS analysis was performed after randomisation and before the planned primary analysis, and thus the treatment assignment was not randomised within the wild-type RAS subgroup. Although this could potentially have confounded the study results, it must be noted that the distribution of identified RAS activating mutations (KRAS exons 3 or 4 or NRAS exons 2, 3 or 4) was similar between treatment arms and the high ascertainment rate provided a lower probability of ascertainment bias.
The results from the prospective 20100007 trial indicate panitumumab therapy significantly improved OS and PFS, and numerically improved ORR in patients with chemotherapy-refractory wild-type KRAS exon 2 and wild-type RAS mCRC vs BSC alone. RAS analysis from the 20100007 study definitively confirms the negative predictive value of activating RAS mutations for response in patients with mCRC receiving panitumumab monotherapy. Moreover, the concordance observed between the prospective 20100007 study and the retrospective 20020408, PRIME and 20050181 analyses provides strong supporting evidence for the use of prespecified high ascertainment retrospective analyses to effectively identify treatment predictive biomarkers for mCRC outcomes.
References
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26: 1626–1634.
Article CAS Google Scholar - Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S, Bardelli A (2007) Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 67: 2643–2648.
Article CAS Google Scholar - Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P (2009) Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol 27: 663–671.
Article CAS Google Scholar - Bokemeyer C, Kohne CH, Ciardiello F, Lenz HJ, Heinemann V, Klinkhardt U, Beier F, Duecker K, van Krieken JH, Tejpar S (2015) FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur J Cancer 51: 1243–1252.
Article CAS Google Scholar - Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ, Queuniet AM, Paillot B, Sabourin JC, Michot F, Michel P, Frebourg T (2007) Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer 96: 1166–1169.
Article CAS Google Scholar - Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Williams R, Rong A, Wiezorek J, Sidhu R, Patterson SD (2013) Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 369: 1023–1034.
Article CAS Google Scholar - Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Oliner KS, Wolf M, Gansert J (2010) Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol 28: 4697–4705.
Article CAS Google Scholar - Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3: 11–22.
Article CAS Google Scholar - Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228–247.
Article CAS Google Scholar - Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359: 1757–1765.
Article CAS Google Scholar - Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P (2006) KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66: 3992–3995.
Article CAS Google Scholar - Mendelsohn J, Baselga J (2006) Epidermal growth factor receptor targeting in cancer. Semin Oncol 33: 369–385.
Article CAS Google Scholar - Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366: 2–16.
Article CAS Google Scholar - Patterson SD, Peeters M, Siena S, Van Cutsem E, Humblet Y, Van Laethem J-L, Andre T, Tian Y, Sidhu R, Oliner KS (2013) Comprehensive analysis of KRAS and NRAS mutations as predictive biomarkers for single agent panitumumab (pmab) response in a randomized, phase III metastatic colorectal cancer (mCRC) study (20020408). J Clin Oncol 31: abstract 3617.
Google Scholar - Peeters M, Oliner K, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, Ciuleanu TE, Roman L, Van Cutsem E, He P, Yu H, Koukakis R, Terwey JH, Jung AS, Sidhu R, Patterson SD (2015) Analysis of KRAS/NRAS mutations in a phase 3 study of panitumumab with FOLFIRI compared with FOLFIRI alone as second-line treatment for metastatic colorectal cancer. Clin Cancer Res 21: 5469–5479.
Article CAS Google Scholar - Peeters M, Oliner KS, Parker A, Siena S, Van Cutsem E, Huang J, Humblet Y, Van Laethem JL, André T, Wiezorek J, Reese D, Patterson SD (2013) Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res 19: 1902–1912.
Article CAS Google Scholar - Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, Ciuleanu TE, Roman L, Van Cutsem E, Tzekova V, Collins S, Oliner KS, Rong A, Gansert J (2010) Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol 28: 4706–4713.
Article CAS Google Scholar - Poulin-Costello M, Azoulay L, Van Cutsem E, Peeters M, Siena S, Wolf M (2013) An analysis of the treatment effect of panitumumab on overall survival from a phase 3, randomized, controlled, multicenter trial (20020408) in patients with chemotherapy refractory metastatic colorectal cancer. Target Oncol 8: 127–136.
Article Google Scholar - Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, Suresh AS, Thomas A, Tjulandin S, Zhang K, Murugappan S, Sidhu R (2014) Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol 15: 569–579.
Article CAS Google Scholar - Schubbert S, Shannon K, Bollag G (2007) Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7: 295–308.
Article CAS Google Scholar - Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Hecht JR, Yu H, Oliner KS, Go WY (2014) PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 32: 2240–2247.
Article CAS Google Scholar - Van Cutsem E, Lenz HJ, Kohne CH, Heinemann V, Tejpar S, Melezínek I, Beier F, Stroh C, Rougier P, van Krieken JH, Ciardiello F (2015) Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 33: 692–700.
Article CAS Google Scholar - Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG (2007) Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 25: 1658–1664.
Article CAS Google Scholar
Acknowledgements
We thank the patients and families involved in study 20100007 and the study investigators. Medical writing support was provided by Shawn Lee (Amgen Inc.) and Meghan Johnson, PhD (Complete Healthcare Communications, LLC) on behalf of Amgen Inc. This study was funded by Amgen Inc. Trial registration: ClinicalTrials.gov, NCT01412957.
Author information
Authors and Affiliations
- Department of Oncology, Asan Medical Center, University of Ulsan, Seoul, 138-736, South Korea
Tae Won Kim - North Estonia Medical Centre Foundation, Tallinn, 13419, Estonia
Anneli Elme - University Hospital Center ‘Sestre Milosrdnice’, Zagreb, 10000, Croatia
Zvonko Kusic - Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, 135-710, South Korea
Joon Oh Park - MEDISPROF, Cluj-Napoca, 400367, Romania
Anghel Adrian Udrea - National Cancer Center, Goyang-si, Gyeonggi-do, 410-769, South Korea
Sun Young Kim - Yonsei University Health System Severance Hospital, Seoul, 120-752, South Korea
Joong Bae Ahn - Centro Medico Nacional Siglo XXI, Mexico City, 06725, Mexico
Ricardo Villalobos Valencia - Dr Rai Memorial Medical Centre, Chennai, 600018, India
Srinivasan Krishnan - Klinicka bolnica Sveti Duh, Zagreb, 10000, Croatia
Ante Bilic - Clinic for Gastroenterology and Hepatology of Military Medical Academy of Serbia, Belgrade, 11000, Serbia
Nebojsa Manojlovic - Amgen Inc., Thousand Oaks, 91320, CA, USA
Jun Dong, Xuesong Guan, Catherine Lofton-Day & A Scott Jung - Department of Oncology, Center of Oncology, Clinical Hospital Center Split, Split, 21000, Croatia
Eduard Vrdoljak
Authors
- Tae Won Kim
You can also search for this author inPubMed Google Scholar - Anneli Elme
You can also search for this author inPubMed Google Scholar - Zvonko Kusic
You can also search for this author inPubMed Google Scholar - Joon Oh Park
You can also search for this author inPubMed Google Scholar - Anghel Adrian Udrea
You can also search for this author inPubMed Google Scholar - Sun Young Kim
You can also search for this author inPubMed Google Scholar - Joong Bae Ahn
You can also search for this author inPubMed Google Scholar - Ricardo Villalobos Valencia
You can also search for this author inPubMed Google Scholar - Srinivasan Krishnan
You can also search for this author inPubMed Google Scholar - Ante Bilic
You can also search for this author inPubMed Google Scholar - Nebojsa Manojlovic
You can also search for this author inPubMed Google Scholar - Jun Dong
You can also search for this author inPubMed Google Scholar - Xuesong Guan
You can also search for this author inPubMed Google Scholar - Catherine Lofton-Day
You can also search for this author inPubMed Google Scholar - A Scott Jung
You can also search for this author inPubMed Google Scholar - Eduard Vrdoljak
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toTae Won Kim.
Ethics declarations
Competing interests
TWK has received honoraria from Merck Serono, has served as a consultant for Amgen Inc. and Taiho, and has received research funding from Merck Sharp & Dohme and Roche. AE has received honoraria from Roche and Eli Lilly, has served as a consultant for Amgen Inc. and Roche, and has received travel expenses from Amgen Inc. and Roche. JOP has received honoraria from Celgene, has served as a consultant for Celgene and Agios Pharmaceuticals, Inc., and has received research funding from Celgene and AstraZeneca. AAU has served as a consultant for Amgen Inc., Bristol-Myers Squibb, and Teva, and has received travel expenses from Astellas and Teva. SYK has received honoraria from Roche Korea and Ildong Pharmaceutical and has served as a consultant for Bayer Korea. RVV has provided expert testimony on behalf of and received travel expenses from Pfizer, AstraZeneca and Roche. JD is an employee of, has received travel expenses from, and owns stock in Amgen Inc. XG is an employee of and owns stock in Amgen Inc., and has an immediate family member who is an employee of and owns stock in Novartis. CL-D and ASJ are employees of and own stock in Amgen Inc. EV has served as a consultant for Roche, Merck, Amgen Inc., Pfizer, Novartis, Boehringer Ingelheim and AstraZeneca, has served on speakers’ bureaus for Pfizer, Roche, Amgen Inc. and AstraZeneca, and has received travel expenses from Roche and Pfizer. ZK, JBA, SK, AB and NM have nothing to disclose.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article
Cite this article
Kim, T., Elme, A., Kusic, Z. et al. A phase 3 trial evaluating panitumumab plus best supportive care vs best supportive care in chemorefractory wild-type KRAS or RAS metastatic colorectal cancer.Br J Cancer 115, 1206–1214 (2016). https://doi.org/10.1038/bjc.2016.309
- Received: 02 June 2016
- Revised: 11 August 2016
- Accepted: 07 September 2016
- Published: 13 October 2016
- Issue Date: 08 November 2016
- DOI: https://doi.org/10.1038/bjc.2016.309