Prokaryotic and Eukaryotic Genomes Submission Guide (original) (raw)
Both WGS and non-WGS genomes, including gapless complete bacterial chromosomes, can be submitted via the Submission Portal. You will be asked to choose whether the genome being submitted is considered WGS or not. The differences for GenBank purposes are:
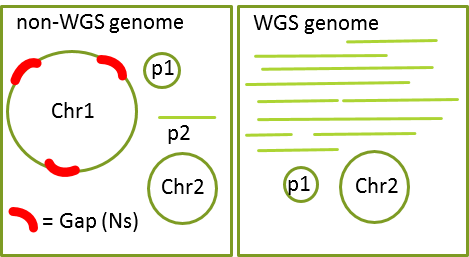
non-WGS
- Each chromosome is in a single sequence and there are no extra sequences
- Each sequence in the genome must be assigned to a chromosome or plasmid or organelle
- Plasmids and organelles can still be in multiple pieces.
WGS
- One or more chromosomes are in multiple pieces and/or some sequences are not assembled into chromosomes
In both cases
- There can still be gaps within the sequences; you will supply that information in the submission
- Plasmids and organelles can still be in multiple pieces.
- Internal sequences must be arranged in the correct order and orientation.
- Sequences concatenated in unknown order are not allowed.
Table of Contents
- Type of submission
- Events
- Submission files : fasta , .sqn , AGP , Genome Info
- Common metadata for all genomes
- Submitting genomes
- Requesting Prokaryotic Genome Annotation Pipeline (PGAP) annotation of prokaryotic genomes
- Running PGAP yourself
Type of submission
- No annotation or requesting PGAP annotation? Submit the fasta sequences in single or Batch mode
- Annotated genome? Create a .sqn file in ASN format and submit it in single or Batch mode
- Lots of genomes in the same BioProject? Use the Batch mode
- Individual assemblies of the haplotypes of a diploid or polyploid genome? See Submitting Multiple Haplotype Assemblies
Submit a single genome
This is the simplest submission route because you just fill in a web form in the Submission Portal and upload fasta (or sqn) files of the genome sequences. You will need to:
- Provide the BioProject created for this research effort, e.g., during submission of the reads to SRA OR register a new BioProject during the genome submission.
- Provide the BioSample created during submission of the reads to SRA OR register a BioSample during the genome submission
- Assert whether this is a WGS or non-wgs genome assembly
- Upload fasta sequences of the genome (or .sqn, file if the genome is annotated)
- Upload optional AGP file(s) to assemble scaffolds (unplaced or unlocalized) and/or chromosomes from the submitted sequences. This is for WGS only. Remember that you can submit the gapped scaffolds themselves instead of submitting contigs plus an AGP file
- Provide information in response to prompts during the genome submission (see the common metada section):
- Genome Assembly Data and other information about this genome assembly
- Gap Information (What the Ns represent)
- Chromosome and plasmid assignments. Every sequence in a non-wgs genome must have a chromosome or plasmid assignment and every chromosome must be submitted as a single sequence.
- Authors and a title (for fasta submissions)
- Release date (immediately after processing OR a specific date. Release will be on that date or upon publication, whichever is first)
- Optional request for annotation of prokaryotic genomes by PGAP
Submit a batch of genomes
This submission route allows you to submit as many as 400 WGS or non-wgs genomes in a single batch submission. In this route you choose Batch/multiple in the Genome Submission Portal , fill in the web form, upload a Genome Info file with genome metadata, and upload or preload fasta files (or sqn files if there is annotation) of the genome sequences. All the genomes within a batch must:
- Be part of the same BioProject
- Be either WGS or non-wgs, not a mix of both types
- Have the same (initial) release date
- Have the same gap/Ns information
- Contain either fasta files or ASN ( .sqn ) files, not a mix of file types. We recommend submitting fasta files unless the submission needs to include annotation or the Genome-Assembly-Data structured comment
- Have a single file for each genome, including any plasmid or organelle sequences
- Have a separate file for each genome, not all the genomes together
- Request PGAP annotation or not (only relevant for prokaryotic genomes)
- Be just a single layer (= no AGP file(s))
You will need to:
- Provide the BioProject created for this research effort, e.g., during submission of the reads to SRA OR register a new BioProject during the genome submission.
- Provide the BioSamples that were preregistered, eg during submission of the reads to SRA OR register BioSamples during the genome submission
- Include assignment (ie, chromosome, plasmid or organelle) information about the sequence in the fasta files (see the Additional requirements for batch submissions section)
- Upload or preload fasta sequences (or sqn files for annotated genomes) of the genomes. Each genome is in a separate single file, uniquely named, but the files can be archived together
- An option for batch submission is to preload the files of genome assemblies before beginning the submission, rather than uploading them in the browser during the submission. You can preload using Aspera, the FTP protocol or Filezilla. Detailed instructions for using the preload option for genome submissions are at How to preload files.
- Upload a Genome Info table with information specific to each genome
- Provide this information in response to prompts on the web pages during the genome submission (see the common metadata section):
- Gap Information (What the Ns represent)
- Authors and a title (for fasta submissions)
- Release date (immediately after processing OR a specific date. Release will be on that date or upon publication, whichever is first)
- Optional request for annotation of prokaryotic genomes by PGAP
Events
- Only if you will be submitting a genome with annotation and you have not yet registered a BioProject and BioSample for this genome, then you will register the genome sequencing project with the BioProject and BioSample databases so that a locus_tag prefix will be assigned to the BioProject:BioSample pair. If you have already registered a BioProject and BioSample for this genome, eg when submitting the reads to SRA, then a locus_tag prefix should have already been assigned. A file of the locus_tag prefix(es) for the BioSamples within a BioProject is linked to the BioProject submission. Write to genomes@ncbi.nlm.nih.gov if you did not receive a locus_tag prefix. Do not register a duplicate BioProject or BioSample for the same genome. Provide these preregistered BioProject and BioSample accessions in the genome submission. Remember that annotation is optional for genome submissions. If you are submitting a genome without annotation, even if you will be requesting PGAP annotation, then you'll create the BioSample (and BioProject, if necessary) during the genome submission. Genomes sequenced as part of the same research effort can belong to a single BioProject, so it's common to create a BioProject during the submission of one genome and then include that BioProject during the submission of additional genomes.
- Make the genome assembly data files.
- Unannotated genomes just need fasta files
- Annotated genomes need to make .sqn file submissions by running the command line program table2asn (the replacement of tbl2asn), and then fixing Errors and Fatals that are indicated in the .val and .dr files. Failure to do this will cause serious delays in processing.
- If you have higher-level assembly information, scaffolds and/or chromosomes, then generate an AGP file to build those objects from the wgs-contigs.
- If you are submitting a batch of genomes (maximum of 400 per batch), then create a Genome Info file. Note that for batch submissions all chromosome and plasmid assignment information must be included in the header of the relevant fasta sequence, as described in the 'see details' section of the Additional requirements for batch submissions
- Submit via the Genome Submission Portal.
- What happens after submission
Submission Files
Fasta files
Put the sequences file into fasta format
- These files have the suffix .fsa.
- Each sequence has a definition line beginning with a '>' and a unique identifier (SeqID), eg contig001, contig002. The SeqIDs must:
- Be <50 characters
- Can only include letters, digits, hyphens (-), underscores (_), periods (.), colons (:), asterisks (*), and number signs (#).
- Be unique within a genome
- Include in the definition line the organism and the relevant strain, breed, cultivar or isolate, if one exists for the sequenced organism. Any additional source qualifiers will be added from the registered BioSample to the genome during processing.
- Remove any Ns from the beginning or end of each sequence.
- Contigs should be >199nt, unless they are part of multi-component scaffolds in an AGP file
IMPORTANT Additional requirements for batch submissions
[1] All the sequences of single genome must be in one file
[2] The chromosome, plasmid, and organelle assignment information must be encoded in the input files of a batch submission, as described in these details:
- To indicate that a single gapped or ungapped sequence represents the chromosome, include [location=chromosome] in the fasta definition line. This must be true for at least one sequence when "option 1" (non-wgs genome) is selected.
- Sequences that are a complete circular chromosome or plasmid need to have the circular topology and the completeness included. If a gapped or nongapped circular chromosome has a gap at the end or has not been circularized, please also include that information unbracketed. Examples:
- [topology=circular] [completeness=complete]
- [topology=circular] gap at end, not circularized
- Sequences that are part of a plasmid, or an organellar chromosome, or specific nuclear chromosomes need to have that information included in the fasta definition line, in these formats:
- [plasmid-name=pBR322]
- [plasmid-name=unnamed] (when the plasmid name is not known. However, be sure that each plasmid has a unique name, eg unnamed1 and unnamed2. )
- [location=mitochondrion]
- [location=chloroplast]
- [chromosome=2]
- Follow the Plasmid and chromosome names rules
- Here is an example of the definition line for the complete plasmid of a bacterial submission (all the text must be in a single line):
>contig02 [organism=Clostridium difficile] [strain=ABDC] [plasmid-name=pABDC1] [topology=circular] [completeness=complete] - Here is an example of a gapped sequence that represents chromosome 2 of a eukaryotic genome (all the text must be in a single line), so both the chromosome location and chromosome name are included:
>Seq001 [organism=Puma concolor] [isolate=ABDC] [location=chromosome] [chromosome=2] - Here is an example of sequences that belong to chromosome 5 so only the chromosome name is included (all the text must be in a single line):
>Seq001 [organism=Puma concolor] [isolate=ABDC] [chromosome=5]
>Seq002 [organism=Puma concolor] [isolate=ABDC] [chromosome=5]
.sqn files
These are generally required only when the submitter wants to include annotation. Annotation is optional for GenBank genome submissions.
see details
Prepare a .sqn file for submission using table2asn. table2asn reads a template file along with the fasta sequence and annotation table files, and outputs an ASN (.sqn) file for submission to GenBank. Follow these three steps:
- Prepare data files
Prepare fasta files as above, with one file per genome.
Prepare these additional files:
- a template file with submitter and publication information.
- annotation files. These correspond to and have the same basenames as the .fsa files. There are two different file formats:
- 5-column feature table files that have the suffix .tbl. Be sure to read the annotation requirements in the appropriate annotation guidelines:
* Prokaryotic Annotation Guidelines
* Eukaryotic Annotation Guidelines - GFF files in GenBank-specific format that have the suffix .gff. Be sure to read the instructions at Genome Annotation with GFF or GTF files.
- 5-column feature table files that have the suffix .tbl. Be sure to read the annotation requirements in the appropriate annotation guidelines:
- Genome-Assembly-Data Structured Comment. This information can be provided during the genome submission, but if many genomes are being submitted it could be simpler to include this in the .sqn file itself. To do that, use the Genome-Assembly-Data Structured Comment Template to create the file and then have it included with
-w genasm.cmtin the tbl2asn commandline, below. - quality scores of the sequences. These files correspond to and have the same basenames as the .fsa files, but have the suffix .qvl. The quality scores are optional.
- Run table2asn
A. Annotation is in GenBank-specific GFF files: follow the instructions for GFF files.
B. Annotation is in .tbl files: follow these instructions. Note that a few of the arguments in table2asn have changed relative to tbl2asn, eg -indir instead of -p. The table2asn page provides more details. Here are the instructions for creating annotated genome files when the annotation is in .tbl files:
Sample command line when the sequences are contigs (overlapping reads with no Ns representing gaps) is
table2asn -indir path_to_files -t template -M n -Z
If the sequences contain Ns that represent gaps, then run the appropriate table2asn command line with the -l and -gaps-min arguments, as described in the Gapped Genome Submission page. The command line for the most common situation (runs of 10 or more Ns represent a gap, and there are no gaps of completely unknown size, and the evidence for linkage across the gaps is "paired-ends") is:
table2asn -indir path_to_files -t template -M n -Z -gaps-min 10 -l paired-ends
For either case you can include the source information in the definition line of each contig, as described in the fasta defline components section, above. Alternatively, the organism and strain (or breed or isolate) can be included with -j in the table2asn command line. The additional source qualifiers will be obtained from the registered BioSample. However, chromosome, plasmid & organelle assignment information must be included in the fasta definition lines. In addition, if the submission is an annnotated prokaryotic genome, then include the genetic code with -j in the commandline, for example:
table2asn -indir path_to_files -t template -M n -Z -j "[organism=Clostridium difficile ABDC] [strain=ABDC] [gcode=11]"
Here are some commonly used arguments when there is no annotation or when the annotation input is .tbl file:
| Option | Description |
|---|---|
| -M n | To run genome-specific functions and validator and to fix some known product name problems |
| -Z | Runs the sequence discrepancy report, which looks for subtle inconsistencies within a set of related records, and outputs a file with the .dr suffix. See the Discrepancy Report page for information about its output. NOTE: this argument is changed from tbl2asn because it no longer requires (or accepts) an output file name. |
| -t template.sbt | Specifies the template file (.sbt), which can be be created at GenBank Submission Template. If the .sbt file is in a different directory the full path must be specified. |
| -j | Allows the addition of source qualifiers that are the same for every sequence in every fasta file being read. Examples:-j "[organism=Mus musculus] [tissue-type=liver]"-j "[organism=Escherichia coli] [strain=ABC1] [gcode=11]" |
| -V b | Generate GenBank Flatfile with a .gbf suffix. This file is only for viewing; it is not for submission. Adding this could slow table2asn so you may choose to include it only for the first run to make sure that the annotation looks as expected. |
| -c s | Add exception to every CDS with an intron shorter than 11bp. Adds /artificial_location="low-quality sequence region" to the CDS, allowing the CDS to pass the ShortIntron error, and causes the protein definition line to be prefaced with "LOW QUALITY PROTEIN:". This option should only be used if you are confident that the protein translation is correct. Do not use short introns to force a translation containing frameshifts or large deletions. |
| -Y File_name | Import a file that is a text comment |
| -w assembly.cmt | Import Structured Comment Table. This is optional, but can be helpful when there are multiple genomes, because there will be less information to supply on the web form during submission. This file can be created at Structured Comment Template |
- Check the output of the validation and discrepancy report and fix problems
A. Check the .stats file for the number, severity and type of errors that are present in the .val files. All Errors and Rejects need to be fixed. The presence of errors will slow processing. See the genome validation errors for guidance. Contact genomes@ncbi.nlm.nih.gov with any questions about the validation output. During processing there may be some questions about other aspects of the submission.
B. Check the .dr file for the results of the discrepancy report. Categories prefaced with FATAL are nearly always unacceptable and must be fixed. (The exceptions are FATALs about bacteria when the genome is not bacterial.) Some of the categories are informational, for example PROTEIN_NAMES: All proteins have same name "hypothetical protein". Reports that are not flagged as fatal should be examined to determine if they represent annotation artifacts that need to be corrected or if they are acceptable due to the biology of the genome. See the discrepancy report examples and explanations and common discrepancy reports for guidance. Write to genomes@ncbi.nlm.nih.gov and send the .dr file with questions about this report.
Some common discrepancy reports of which to be aware:
- NO_ANNOTATION and LONG_NO_ANNOTATION. If either of these is expected, that is fine. However, if not expected, then check that the IDs in the .tbl file definition lines match the SeqIDs of the sequences in the fasta file. When you submit, please let us know when the sequences in the LONG_NO_ANNOTATION report are expected to be unannotated, so that we know to ignore this report.
- PROTEIN_NAMES: All proteins have same name "hypothetical protein". If this is expected, that is fine.
- FATAL: BACTERIAL_PARTIAL_NONEXTENDABLE_PROBLEMS. If this is a eukaryotic genome, you can ignore this error. If this is a prokaryotic genome, then every CDS must begin and end with valid start and stop codons, respectively, or be partial and either extend to the end of the sequence or abut a gap within the scaffold sequence. However, in the .tbl file you should annotate with pseudo any genes that are 'broken' but are not thought to be pseudogenes. These are genes that do not encode the expected translation, for example because of internal stop codons or missing start or stop codons, and are often caused by problems with the sequence and/or assembly.
C. Make any necessary fixes to the input .fsa and/or .tbl files and run table2asn again.
AGP file (optional)
AGP files provide the ordering and orientation information to construct scaffolds from contigs, or to construct chromosomes from scaffolds and/or contigs. However, remember that we do accept the gapped scaffolds themselves as the basic sequences of the genome. If you choose to submit a multi-layer submission with and AGP file, then know that the AGP file defines these genome assemblies, so be sure to include all wgs-contigs that are considered to be part of the genome in the AGP file. However, if the sequences in the fasta (or .sqn) files are already the scaffolds or chromosomes, then do not make an AGP file.
see details
See this page for the AGP format.
There are 3 types of AGP files:
- Unplaced scaffolds = scaffolds without chromosome or plasmid assignments
- Chromosome = the objects built in the AGP file represent the nuclear or organellar chromosomes or plasmids
- Unlocalized scaffolds = scaffolds that are known to belong to a nuclear or organellar chromosome or plasmid but are not part of the assembly to build those chromosomes or plasmids
- NOTE: assignments will need to be provided for the objects being assembled in the Chromosome and Unlocalized AGP files, not the sequences in the fasta/sqn files
Some specific requests are:
- Encode the type of object in the object names in column 1 like this:
- scaffold01, scaffold02, etc for scaffolds
- chr (for bacteria) OR chr1, chr2, etc for eukaryotic chromosomes
- the plasmid names for plasmids (eg pBR322). If the name is not known, then use 'unnamed'.
- MT for the mitochondrial genome. MT_scaf01, MT_scaf02, etc for mitochondrial scaffolds.
- Use "100" as the length and U as the component-type for gaps of unknown size, as that is the GenBank convention. These will appear as gap(unk100) in the flatfile view of the GenBank record.
- Use the same contig identifiers in column 6 (the component-id) that you used in the .fsa files. If the components have already been assigned accession numbers, then you need to use the accession.version numbers as the component identifiers; do not use just the accession number.
- Use different identifiers for the object in column 1 and the component in column 6, even if the object consists of a single component.
You can validate the basic format of your AGP file at http://www.ncbi.nlm.nih.gov/projects/genome/assembly/agp/agp_validate.cgi. In addition, the standalone commandline program, agp_validate is available by anonymous FTP to validate the AGP file more extensively yourself. The -help option details the arguments and command line format.
Genome Info table
The Genome Info table is required for batch submissions and is used to provide the Genome Assembly Data of each. You can either fill in the table during the genome submission or prepare the file ahead of time and upload it during the submission. To prepare it ahead of time, download the Genome Info file template. The instructions are on the first tab of this file and the template is on the second tab. Complete the second tab (Genome_Data), then save the worksheet as a Text (Tab-delimited) file -- (use 'File, Save as, Save as type: Text (Tab-delimited)' ).
see details of the required and optional information
Each row in the template represents a genome. The required fields are:
- Biosample accession OR sample_name
- Assembly method
- Assembly method version
- Genome coverage
- Sequencing technology
- File name
Optional fields:
- Assembly date
- Assembly name
- Reference genome
- Update (update_for)
- bacteria_available_from
Definitions of these fields are in the Genome Assembly Data section and also as comments in the template itself. Instructions:
- If you created BioSamples previously, provide accessions in the form of SAMN# in the column biosample_accession. See the Example of Genome Info file using BioSample Accessions.
- If you are creating samples during this genome submission, provide the names of samples that you just created in the column 'sample_name'. Please note that sample names must be unique within your entire account. See the Example of Genome Info file using BioSample Names. Do not include both sample_name and biosample_accession for a genome.
- If you archived your files by tar utility, you must list file names that are contained in the archive not the name of the tarred file.
- Provide exact file names (including extensions) in the filename column.
- Supported extensions for compressed files: tar, tar.gz, gz, bz2 (do not use zip!).
- File names must be unique.
Metadata required for all genome submissions
BioProject
The BioProject contains the description of the research effort, relevant grant(s), and has links to the public data for the proejct. Each genome must belong to a BioProject, and genomes sequenced as part of the same research effort can belong to a single BioProject. Use the same BioProject for the sequence reads and genome assembly made from those reads; do not create duplicate BioProjects. If a new BioProject is necessary for unannotated (or PGAP-annotated) genomes, then registering during the genome submission process is simplest. However, genomes submitted with annotation will need to be pre-registered so that a locus_tag prefix can be assigned to the BioProject/BioSample pair and used to identify each gene within that genome uniquely. A file of the locus_tag prefix(es) for the BioSamples within a BioProject is linked to the BioProject submission. Write to genomes@ncbi.nlm.nih.gov if you did not receive a locus_tag prefix after preregistering a BioSample for your BioProject.
BioSample
The BioSample contains the source information of the sample that was sequenced. Use the same BioSample for the sequence reads and genome assembly made from those reads; do not create duplicate BioSamples. Registering a new BioSample can be done during the genome submission process for unannotated (or PGAP-annotated) genomes; however, genomes submitted with annotation will need to be pre-registered to get a locus_tag prefix. Include the registered BioProject when you register the BioSample so that a locus_tag prefix is assigned to the pair. You'll find the locus_tag assignment(s) in a file linked to the BioProject submission.
During processing of the genome the relevant information from the genome and BioSample will be merged so that they are in agreement. If the genome and BioSample have a conflict in the value of an attribute, we will stop and ask the submitter to clarify what the correct value is. There is some extra validation in GenBank compared to BioSample, eg ‘altitude’ is defined as being in meters in BioSample but it must have ‘m’ present in the GenBank genome. We will fix simple issues like this.
Genome Assembly Data and other information about a genome assembly
- Assembly method : Name of the assembly algorithm(s)
- Assembly method version or date : version of the algorithm or date it was run
- Genome coverage : The estimated base coverage across the genome, eg 12x.
- Sequencing technology : sequencing platform(s) used
- Assembly date : Optional. Year, month or day the assembly was made. Date formats: YYYY-MM-DD; YYYY-MM; YYYY
- Assembly name : Optional and not usually relevant for prokaryotes. This is a short name suitable for display that does not include the organism name eg, LoxAfr_3.0 for a Loxodonta africana assembly, version 3.0
- Full or Partial Genome in the sample : the answer is nearly always "yes, Full". Choose "no, partial" only if a subset of the sample was deliberately selected, eg just exomes or a single chromosome of a eukaryote or only the non-repetitive regions of the genome
- Reference genome : If this is NOT a de novo assembly, you will need to provide the accession.version and/or the assembly name of the genome assembly that was used as the reference guide for this assembly
- Update : accession of the genome being updated, when appropriate
- bacteria_available_from : Optional. For prokaryotes provide a name and physical address (not email) of the lab or PI, or a culture collection identifier where scientists could obtain this bacterial culture
Gap Information: What the Ns represent
- The minimum number of consecutive Ns that represents a gap (must be 10 or less). Be aware that the assembly statistics are always calculated using 10 or more Ns as a gap, regardless of the presence/absence of gaps in the final genome sequence.
- The number of Ns that represents a gap of completely unknown length (usually 0; sometimes 100 or another value)
- The evidence used to assert that the sequence on either side of the gap is linked (usually paired-ends)
- This information is collected in the submission form for individual and batch submissions. Default answers are those that have been most commonly submitted. Be sure to select the correct answer when the defaults are incorrect for the genome(s) being submitted.
Chromosome and plasmid assignments
- Indicate any sequences that are chromosomes or plasmids, or that belong to chromosomes or plasmids.
- Follow the chromosome and plasmid names rules
- Every sequence in a non-wgs genome must have a chromosome or plasmid assignment and every chromosome must be submitted as a single sequence.
Plasmid and chromosome names rules
see details
Chromosome and plasmid names can only digits, dots, underscores, and ASCII characters in plain text in the standard English alphabet. In addition, there are rules specific for each.
Chromosome names
- Can contain only digits, dots, underscores, and ASCII characters in plain text in the standard English alphabet.
- Cannot include "chr" or "chromosome". However, linkage group names should include "LG" as part of the name.
- Are limited to 33 characters
- Cannot include these words: unknown, Un, Unk, 0 (= zero as the full name). This restriction is because each unplaced sequence should be separate, not concatenated with others, and without an assignment.
Plasmid names
- Can contain only digits, dots, underscores, and ASCII characters in plain text in the standard English alphabet.
- Should start with lower case 'p' UNLESS the plasmid name is not known. In that case use 'unnamed', or "unnamed1" & "unnamed2" for distinct unnamed plasmids.
- Cannot include the word 'plasmid'
- Are limited to 20 characters
Submit the genomes to the Genome Submission Portal
All files must be submitted via the Genome Submission Portal. Choose "Single genome" or "Batch/multiple genomes". Answer the questions and upload the necessary files Review the summary page and click the "Submit" button. The submission will be given a 'SUB' temporary identifier which you can use in correspondence before an accession number is assigned to the genome submission.
What happens next
Once we receive your genome submission, several automated validations are run and a member of our staff conducts an initial review. If no significant issues are found, the genome will be assigned an accession number.
If there are problems
The submitted files will be marked in the submission portal as "Error" and you will receive an email with details of the problems. Errors found in the automated validations are automatically reported back to the submission portal and an email is automatically sent to the submitter with instructions on how to proceed. In addition, a member of our staff conducts an initial review of each submission and reviews several additional validations. The problems, including those described in the Fix problems section, could be:
- Any Error-level errors and some Warning-level errors from the validation. For .sqn file submissions you would see these in the .val file(s) generated by table2asn.
- Any FATAL or problem categories from the discrepancy report, the .dr file generated by table2asn.
- Sequence contamination in the genome sequences
- Bad format of the AGP files or inconsistencies between the AGP and fasta/.sqn files.
- If the genome size is not within the expected range of the median size of the genomes of that species already in GenBank. Because this test uses the genomes that are already in GenBank, genomes could pass it simply because there are not yet enough genomes of that species in GenBank. Note that you can run this Genome Size Check test yourself before submitting.
- If ANI analysis of a prokaryotic genome indicates that the genome is misidentified. This test uses the genomes that are already in GenBank as the reference, so genomes could pass this test simply because there are not yet enough genomes of that species in GenBank.
Once you have made the fixes, log back into theGenome Submission Portal, retrieve that submission by its 'SUB' identifier and click the "FIX" button of that submission. You will be back in the original submission and will need to delete the files that are marked as having errors, and then upload new files in their place.
Once your submission is assigned an accession number it undergoes a thorough review by our staff. This review is critical because we are striving to present genome annotation in an accurate and consistent manner so that database users can make maximum use of the data. If we encounter problems during this review, we will contact you by email.
Submission statuses in the submission portal
- Queued : the submission is waiting for initial review
- Error : one or more genomes has errors in its files, so needs to be resubmitted. Use the same file name when resubmitting batch submissions.
- Processing, and no accession number : all of the genomes have passed the initial automated validations and are waiting for additional review.
- Processing, and accession number : genome accessions have been assigned and the genomes will be processed by NCBI staff. Genomes will remain at this status until they are released. We will contact you during processing if the submission has issues that require additional information.
- Processed : the genome has been publicly released.
If you elected to hold your genome until a particular date (or publication, whichever is first), we ask that you provide us with the expected publication date and also notify us in a timely manner of the upcoming publication and the relevant citation details. This will allow us to coordinate the release of your genome with the appearance of the paper. Please provide at least two weeks' notice of any upcoming publication.
NOTE: As of January 2017, genomes will be released on their release date without additional communication, as is the normal GenBank policy. Be sure to request an extension of the release date if the genome is not yet published and you wish to continue to keep it confidential.
Requesting PGAP annotation of prokaryotic genomes
Requests for annotation by theProkaryotic Genomes Annotation Pipelineis a step during submission of the genome to GenBank. Prepare a regular GenBank genome submission and request PGAP annotation during the submission process by clicking on the box "Annotate this prokaryotic genome in the NCBI Prokaryotic Annotation Pipeline before being released". The annotated genome will be posted back to the Submission Portal for your review. You may edit the file and resubmit that to GenBank; however, this is not required and is generally not recommended, as it will slow processing and may introduce problems that you would need to fix.
Running PGAP yourself
If you would like to annotate your prokaryotic genome with the NCBI Prokaryotic Genomes Annotation Pipeline (PGAP) before or without submitting your data to GenBank, NCBI has made an external version available for you to download and run. It will generate a GenBank-compliant annotated genome that is submission-ready. If you are interested in running PGAP yourself, please see the NCBI Insights announcementand find more details at github, or see this short video.
After your genome is annotated using external PGAP, you may choose to submit it to GenBank:
- Submit the ASN output file of your PGAP run to GenBank via the usual Genome Submission Portal.
- Information for single submission.
- Information for batch submission.
- As usual during the genome submission, you will be asked to provide the BioProject and BioSample that you may have already created for that genome or its sequencing reads, or to create them if they do not already exist. Please do not create duplicates.
- Any locus_tag prefix can be used to run PGAP. However, during GenBank processing that value will be automatically changed to the officially registered locus_tag prefix for the BioProject:BioSample pair of that genome. Be sure to include the official locus_tags in your publication.
- You have the option to obtain the official locus_tag prefix before running PGAP, but that is not necessary. To do that, you would register the BioProject, then register the BioSample(s), then look for the locus_tag_prefix.txt file posted to the BioProject submission. A separate BioSample is required for each strain, but multiple genomes can, and usually do, belong to a single BioProject.
- The submitted genomes will undergo the standard validations, as described above including being screened for foreign contaminants and vector sequences and also being analyzed to check the organism identification. Any annotated assemblies that do not pass these validations may need to be modified.
- Genomes that pass the validations will be assigned accession numbers and made public on the release date you have selected during submission. Be sure to use the official GenBank-assigned accessions when you publish.