Immunogenicity and safety of an investigational tetravalent recombinant subunit vaccine for dengue: results of a Phase I randomized clinical trial in flavivirus-naïve adults (original) (raw)
ABSTRACT
There is an unmet medical need for vaccines to prevent dengue. V180 is an investigational recombinant subunit vaccine that consists of truncated dengue envelope proteins (DEN-80E) for all 4 serotypes. Three dosage levels of the tetravalent DEN-80E antigens were assessed in a randomized, placebo-controlled, Phase I dose-escalation, first-in-human proof-of-principle trial in healthy, flavivirus-naïve adults in Australia (NCT01477580). The 9 V180 formulations that were assessed included either ISCOMATRIX™ adjuvant (2 dosage levels), aluminum-hydroxide adjuvant, or were unadjuvanted, and were compared to phosphate-buffered saline placebo. Volunteers received 3 injections of assigned product on a 0, 1, 2 month schedule, and were followed for safety through 1 year after the last injection. Antibody levels were assessed at 6 time-points: enrollment, 28 days after each injection, and 6 and 12 months Postdose 3 (PD3). Of the 98 randomized participants, 90 (92%) received all 3 injections; 83 (85%) completed 1-year follow-up. Immunogenicity was measured by a qualified Focus Reduction Neutralization Test with a 50% neutralization cutoff (FRNT50). All 6 V180 formulations with ISCOMATRIX™ adjuvant showed robust immunogenicity, while the 1 aluminum-adjuvanted and 2 unadjuvanted formulations were poorly immunogenic. Geometric mean antibody titers generally declined at 6 months and 1 year PD3. All 9 V180 formulations were generally well tolerated. Formulations with ISCOMATRIX™ adjuvant were associated with more adverse events than aluminum-adjuvanted or unadjuvanted formulations.
Keywords: dengue vaccine, recombinant, subunit, immunogenicity, safety
Introduction
Dengue is an important cause of human morbidity and mortality. Over 140 countries have epidemic or endemic dengue transmission. Of the estimated 3.97 billion people living in tropical or subtropical areas where transmission occurs, dengue viruses are estimated to infect ~390 million annually, resulting in ~96 million symptomatic cases, including ~500,000 with severe disease and ~22,000 deaths.1–3
Dengue infection may be caused by any of 4 virus serotypes (DENV-1, DENV-2, DENV-3, and DENV-4), leading to a range of clinical findings: inapparent or mild febrile illness, classic dengue fever (DF), and the life-threatening conditions of dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS).1,2 DF, although less severe than DHF or DSS, is associated with a substantial burden of illness.1,2 Treatment of dengue is limited to supportive therapy.
Currently, one dengue vaccine, Dengvaxia™ (Sanofi Pasteur, Lyon, France), has been licensed in some countries for the prevention of dengue disease caused by any of the 4 serotypes among persons 9 to 45 years old.4 Recently reported data showed that Dengvaxia™ performs differently by baseline serostatus: compared with baseline seropositive individuals, those who were seronegative at baseline had both a lower vaccine efficacy against virologically confirmed symptomatic dengue in the 25 months after the first vaccine dose, and an increased risk of hospitalized dengue and severe dengue beginning about 30 months after the first dose.4 Based on these findings, the World Health Organization Strategic Advisory Group of Experts has recommended that countries considering vaccination with Dengvaxia™ as part of their dengue-control program implement a pre-vaccination screening strategy, in order to limit vaccination to dengue-seropositive persons.4 To address the remaining unmet medical need, a number of candidate dengue vaccines with the potential for broader use are in development.
V180 is a candidate vaccine comprised of recombinant subunit proteins utilizing 80% of the envelope glycoprotein (DEN-80E) of the 4 dengue virus strains.5 Immunogenicity and efficacy of the 4 DEN-80E proteins were demonstrated in preclinical models with a variety of adjuvants, including aluminum hydroxide Alhydrogel™ (Brenntag Biosector, Frederikssund, Denmark) and the investigational saponin-based ISCOMATRIX™ adjuvant (CSL Behring, King of Prussia, PA, USA).5–8
Results
Participant demographics and accounting
Among the 98 adults who were randomized into the trial, 57 (58%) were female, 92 (94%) were white, and the mean age was 27 years (range, 18 to 48 years). The gender, race/ethnicity, and age distributions were generally consistent across the treatment groups (data not shown).
The first, second, and third injections of trial product were received by 98 (100%), 94 (96%), and 90 (92%) of randomized participants, respectively (Figure 1). Overall, 83 (85%) participants completed the trial. Participants who received < 3 injections or who discontinued during long-term follow-up were distributed across the V180 and placebo groups, with no preponderance in any group (data not shown).
Figure 1.
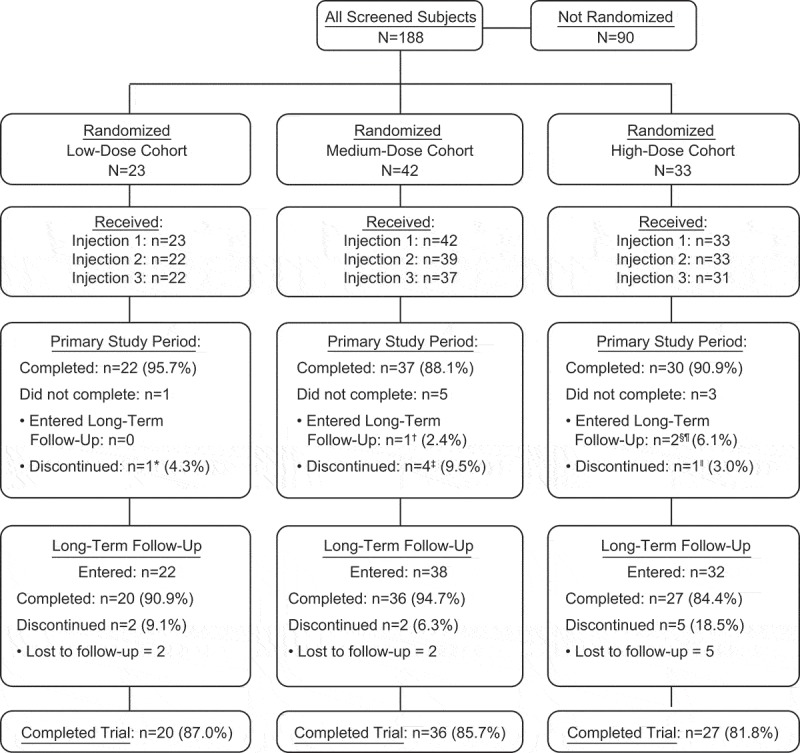
Participant disposition.
Primary study period extended from receipt of the first injection through 28 days after the last injection.Completed primary study period means participant received all 3 injections and completed 28 days of safety and immunogenicity follow-up after each injection.Long-term follow-up period extended from the end of the primary study period through 1 year after the last injection. Participants who entered long-term follow-up included those who completed the primary study period, and those who did not receive all 3 injections during the primary study period and agreed to participate in long-term follow-up.Completed long-term follow-up means participant completed the long-term follow-up visits at 6 months and 1 year following the last injection.Completed trial means completed long-term follow-up.* Participant withdrew consent for participation for personal reasons after receiving 1 injection.† Investigator discontinued participant’s dosing after 1 injection due to adverse events of arthralgia (left knee and ankle) and injection-site swelling; participant completed long-term follow-up and the trial.‡4 participants withdrew consent for participation for personal reasons: 2 after receiving 1 injection, and 2 after receiving 2 injections.§ Participant received 3 injections but did not complete the primary study period prior to completing 28 days of primary safety follow-up due to relocating internationally. Participant completed long-term follow-up via telephone contacts, and thus completed the trial.¶ Participant received 2 injections; the third injection could not be administered due to receipt of medically-necessary vaccines outside of the protocol. The participant agreed to enter long-term follow-up, but was lost during this period.‖ Participant was lost to follow up after receiving 2 injections.
Immunogenicity
Virus-neutralizing antibody
Immunogenicity was measured by a qualified Focus Reduction Neutralization Test with a 50% neutralization cutoff (FRNT50). The protocol-specified criterion for a positive immune response, applied to each formulation, was that at 28 days PD3, at least 3 serotypes would have an FRNT50 seroconversion rate (SCR) of ≥75%. Serotype-specific seroconversion rates (SCRs, the proportion of participants with an FRNT50 titer ≥10) and geometric mean titers (GMTs) for each treatment group were summarized at 28 Days Postdose 3 (Table 1). Each of the 6 V180 formulations containing ISCOMATRIX™ adjuvant met the pre-specified definition of a positive immune response, with seroconversion rates of ≥85.7% for all 4 dengue serotypes; GMTs ranged from 73 to 1344. Within each V180 dose level, GMTs were slightly higher (within 2-fold) for formulations with 60 ISCO™ units than formulations with 30 ISCO™ units. In contrast, for a given dose level of ISCOMATRIX™ adjuvant, GMTs did not increase with increasing doses of V180 antigen. The medium-dose V180 formulation with Alhydrogel™ and the unadjuvanted high-dose V180 formulation did not meet the pre-specified definition of a positive immune response, but showed limited evidence of immunogenicity for all 4 serotypes: SCRs ranged from 14.3 to 62.5%, while GMTs ranged from <10 to 20. There was no detectable immune response in the unadjuvanted medium-dose V180 group or the placebo group. In each formulation with an immune response, the lowest GMTs were observed for DENV4.
Table 1.
Neutralizing antibody (FRNT50) SCRs and GMTs at 28 days postdose 3, by treatment group (per-protocol immunogenicity population, all cohorts).
| Antigen Dose Level (µg) | Adjuvant | N (per protocol) | Postdose 3 FRNT50 SCR† and GMT‡ | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DENV1 | DENV2 | DENV3 | DENV4 | |||||||
| SCR | GMT | SCR | GMT | SCR | GMT | SCR | GMT | |||
| Low(3/3/3/6) | 30 ISCO™ units | 8 | 100 | 197 | 100 | 259 | 100 | 891 | 100 | 127 |
| 60 ISCO™ units | 8 | 100 | 345 | 100 | 589 | 100 | 1,344 | 100 | 180 | |
| Medium(10/10/10/20) | 30 ISCO™ units | 8 | 100 | 121 | 100 | 281 | 100 | 503 | 87.5 | 75 |
| 60 ISCO™ units | 9 | 100 | 153 | 100 | 494 | 100 | 659 | 100 | 111 | |
| Alhydrogel | 8 | 37.5 | 11 | 50 | 17 | 62.5 | 20 | 25 | <10 | |
| None | 7 | 0 | <10 | 0 | <10 | 0 | <10 | 00 | <10 | |
| High(50/50/50/100) | 30 ISCO™ units | 9 | 100 | 111 | 100 | 129 | 100 | 488 | 100 | 73 |
| 60 ISCO™ units | 9 | 100 | 207 | 100 | 198 | 100 | 534 | 85.7 | 83 | |
| None | 7 | 14.3 | <10 | 42.9 | 13 | 42.9 | 15 | 14.3 | <10 | |
| Placebo | None | 17 | 0 | <10 | 0 | <10 | 0 | <10 | 0 | <10 |
Within each V180 dose cohort, formulation-specific GMTs were plotted by time (Figure 2A, B, and C). In accordance with the protocol, all participants were seronegative at baseline. All 6 V180 formulations with ISCOMATRIX™ adjuvant had similar profiles: GMTs increased by Month 2 (28 Days Postdose 2), increased further by Month 3 (28 Days Postdose 3), and then declined over time through Month 14 (1 Year Postdose 3), remaining generally above baseline for DENV1, DENV2, and DENV3, and generally returning to baseline for DENV4. During long-term follow-up, GMTs generally remained higher in the 60 ISCO™ unit group than the 30 ISCO™ unit group for the low-dose V180 cohort, but tended to converge in the medium-dose and high-dose V180 cohorts. For the medium-dose V180 formulation with Alhydrogel™ and the unadjuvanted high-dose V180 formulation, GMTs for some serotypes increased by Month 3, and then returned to baseline by Month 8 (6 Months Postdose 3).
Figure 2.
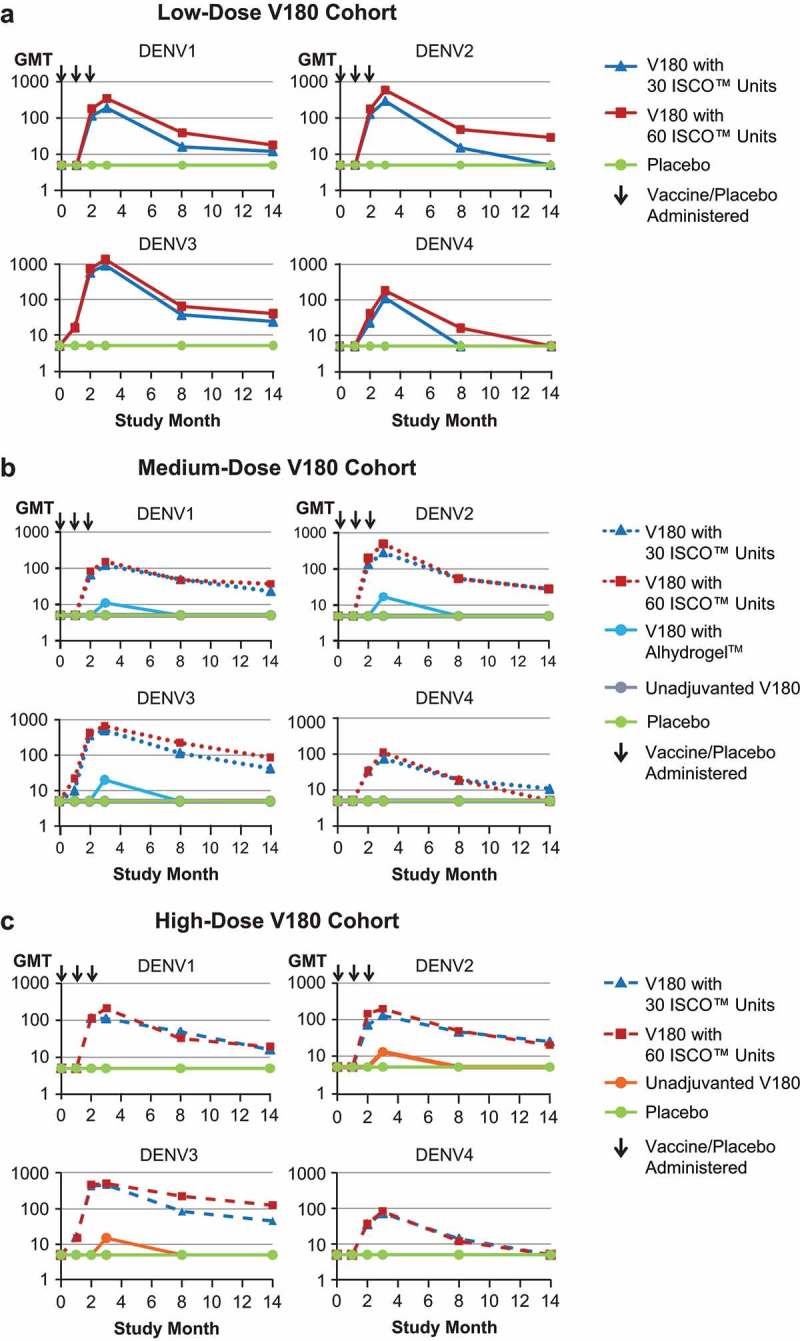
Neutralizing antibody kinetics: GMTs through 1 Year postdose 3 (per-protocol population).
DENV = Dengue Virus; GMT = Geometric Mean Titer; Panel A: Low-Dose V180 Cohort; Note: The arrows indicate when the study product (V180/placebo) was administered. Some data points for unadjuvanted V180 are < 10, and hidden by the placebo arm. Study Month 3 is 28 days Postdose 3; Panel B: Medium-Dose V180 Cohort; Note: The arrows indicate when the study product (V180/placebo) was administered. All data points for unadjuvanted V180 and some data points for the V180 with Alhydrogel are < 10, and hidden by the placebo arm. Study Month 3 is 28 days Postdose 3; Panel C: High-Dose V180 Cohort; Note: The arrows indicate when the study product (V180/placebo) was administered. Some data points for unadjuvanted V180 are < 10, and hidden by the placebo arm. Study Month 3 is 28 days Postdose 3
The proportions of participants exhibiting a tetravalent (FRNT50 titers ≥10 for all 4 dengue serotypes) or a ≥trivalent (FRNT50 titers ≥10 for 3 or 4 dengue serotypes) response are shown by time in Figure 3A and B, respectively. Among the ISCOMATRIX™ adjuvant groups, tetravalent responses were observed in 71 to 88% at Month 2, 86 to 100% at Month 3, 43 to 72% at Month 8, and 0 to 43% at Month 14, while ≥trivalent responses were observed in 100% of recipients at Months 2 and 3, 57 to 100% at Month 8, and 29 to 86% at Month 14. Tetravalent or ≥trivalent responses were exhibited by lower proportions of recipients of medium-dose V180 with Alhydrogel or high-dose unadjuvanted V180, and by no recipients of medium-dose unadjuvanted V180. In the majority of instances when participants had FRNT50 titers ≥10 for only 3 seotypes, DENV4 was the serotype with a titer <10.
Figure 3B.
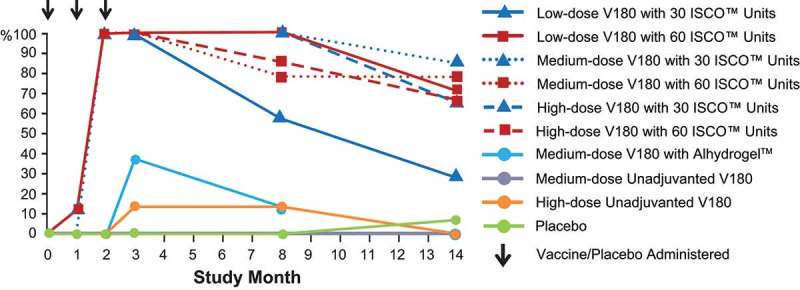
Proportions of participants with a tetravalent response, by study month (per-protocol population).
Note: The arrows indicate when the study product (V180/placebo) was administered. The 0% data points for groups other than placebo are hidden by the placebo group. Tetravalent Response = FRNT50 titers ≥10 for all 4 DENV serotypes.Study Month 3 is 28 days Postdose 3.
Figure 3A.
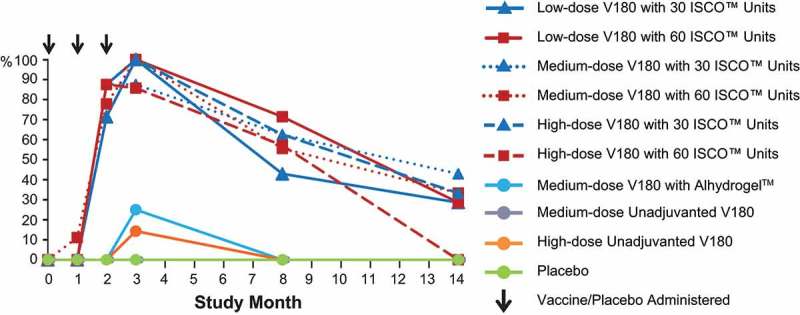
Proportions of participants with a tetravalent response, by study month (per-protocol population).
Note: The arrows indicate when the study product (V180/placebo) was administered. The 0% data points for groups other than placebo are hidden by the placebo group. Tetravalent Response = FRNT50 titers ≥10 for all 4 DENV serotypes.Study Month 3 is 28 days Postdose 3.
Figure 4.
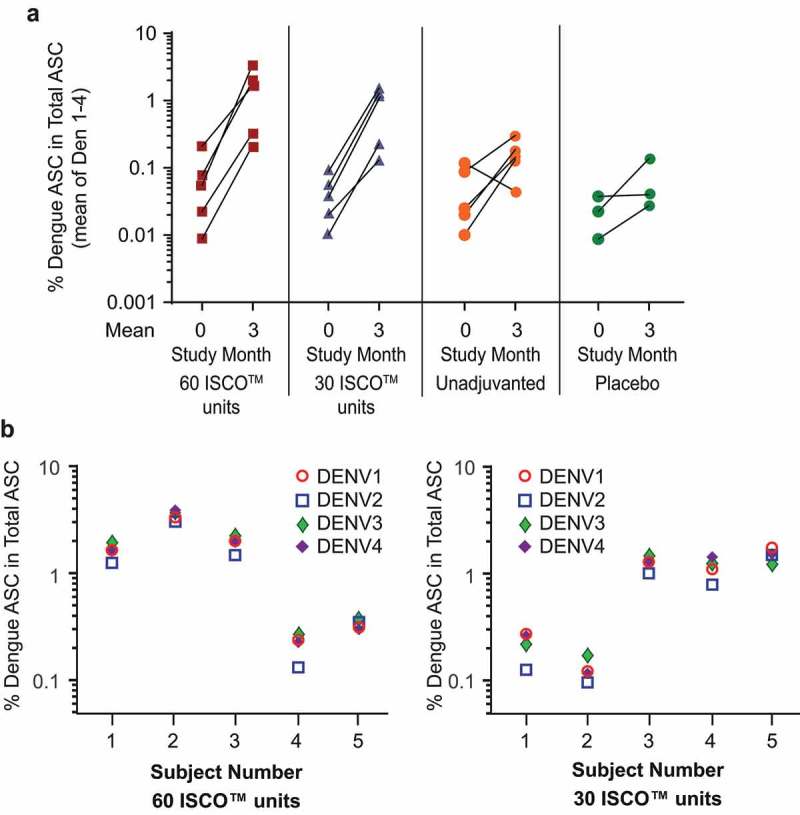
Induction of B-cell memory to each of the four DENV serotypes in a subset of 18 high-dose cohort subjects who received 3 injections of study product (vaccine/placebo).
Blood samples to assess B-cell memory were collected at 2 time points (at Study Month 0 immediately before the first injection, and at Study Month 3 [28 days Postdose 3]) in 18 high-dose cohort participants: 5 recipients of V180 with 30 ISCO™ units of ISCOMATRIX™ adjuvant, 5 recipients of V180 with 60 ISCO™ units of ISCOMATRIX™ adjuvant, 5 recipients of unadjuvanted V180, and 3 placebo recipients.Panel A: Change in Proportion of Dengue-specific memory B cells from Study Month 0 (prevaccination baseline) to Study Month 3 (28 days Postdose 3)Note: Each line represents the data for 1 participantPanel B: Dengue-specific memory B cell frequencies at Study Month 3 (28 days Postdose 3)Note: ASC = antibody secreting cells
Memory B-cell responses
Induction of B-cell memory to each of the four DENV serotypes was observed in peripheral blood mononuclear cells among all participants who received 3 injections of high-dose V180 with ISCOMATRIX™ adjuvant (30 or 60 ISCO™ units) at 28 Days Postdose 3, the mean number of dengue-specific memory B cells in these recipients had increased in frequency by 1 to 2 logs over the pre-vaccination baseline (Figure 4A). In contrast, the mean change ranged from a 0.4-log decrease to 1-log increase among recipients of unadjuvanted high-dose V180, and generally increased <1 log among placebo recipients. Among recipients of 3 injections of V180 with ISCOMATRIX™ adjuvant, at 28 Days Postdose 3, dengue-specific memory B cells comprised 0.1 to 3% of total IgG secreting cells (Figure 4A), and the frequencies of dengue-specific memory B cells appeared generally balanced across the four dengue serotypes (Figure 4B).
Safety
All 9 V180 formulations were generally well tolerated. Participants assessed the majority of adverse events (AEs) to be of mild or moderate intensity. There were no serious AEs (SAEs) or deaths during the trial.
The AE data for the individual V180 treatment groups revealed clear trends by adjuvant type, but not by dose level of V180 antigen, dose level of ISCOMATRIX™ adjuvant, or among the 3 placebo groups. For this reason, AE data are summarized by treatment type: V180 with ISCOMATRIX™ adjuvant (6 treatment groups combined), V180 with Alhydrogel™ (1 treatment group), unadjuvanted V180 (2 treatment groups combined), and placebo (3 treatment groups combined). For all 4 treatment types, pain/tenderness was the most frequent injection-site AE, followed by erythema and swelling (Table 2). V180 with ISCOMATRIX™ adjuvant was associated with a higher frequency of injection-site AEs overall, injection-site AEs of erythema or swelling that were ≥5 cm or ≥10 cm, and a higher frequency of injection-site pain/tenderness that participants assessed as severe (defined in the protocol as the inability to do work or usual activities). V180 with ISCOMATRIX™ adjuvant was also associated with higher frequencies of systemic AEs overall, and those assessed by the investigator as related to study product (Table 3). Fever (temperature ≥38.0°C [100.4°F]) was reported in 5 (9%) ISCOMATRIX™ adjuvant recipients, 0 (0%) Alhydrogel™ recipients, 0 (0%) unadujuvanted recipients, and 1 (6%) placebo recipients.
Table 2.
Incidence of injection-site adverse events days 1 through 5 after any injection, by treatment type.
| Adverse Event | V180 with ISCOMATRIX™ adjuvant (30 or 60 ISCO™ units)N = 53n (%) | V180 with Alhydrogel™ (225 µg aluminum)N = 9n (%) | Unadjuvanted V180N = 18n (%) | PlaceboN = 18n (%) |
|---|---|---|---|---|
| Injection Site, Any | 52 (98%) | 6 (67%) | 7 (39%) | 5 (28%) |
| Pain/tenderness†, AnyPain/tenderness†, Severe‡ | 52 (98%)5 (9%) | 6 (67%)0 (0%) | 7 (39%)0 (0%) | 3 (17%)0 (0%) |
| Erythema, AnyErythema ≥5 cm‡Erythema >10 cm‡ | 37 (70%)21 (40%)7 (13%) | 3 (33%)0 (0%)0 (0%) | 3 (17%)1 (6%)1 (6%) | 1 (6%)0 (0%)0 (0%) |
| Swelling, AnySwelling ≥5 cm‡Swelling > 10 cm† | 31 (58%)13 (25%)5 (9%) | 2 (22%)0 (0%)0 (0%) | 2 (11%)0 (0%)0 (0%) | 1 (6%)0 (0%)0 (0%) |
Table 3.
Incidence of adverse events days 1 through 14 after any injection, by treatment type.
| Adverse Event | V180 with ISCOMATRIX™ adjuvant (30 or 60 ISCO™ units)N = 53n (%) | V180 with Alhydrogel™ (225 µg aluminum)N = 9n (%) | Unadjuvanted V180N = 18n (%) | PlaceboN = 18n (%) |
|---|---|---|---|---|
| Injection Site, Any | 52 (98%) | 6 (67%) | 7/18 (39%) | 5/18 (28%) |
| Systemic, Any | 50 (94%) | 7 (78%) | 12 (67%) | 11 (61%) |
| Systemic, Related to study product† | 44 (83%) | 7 (78%) | 9 (50%) | 10 (56%) |
| Toxicity Grade 3‡, Any | 11 (21%) | 0 (0%) | 1 (6%) | 1 (6%) |
| Toxicity Grade 3‡, Related to study product† | 11 (21%) | 0 (0%) | 1 (6%) | 0 (0%) |
The majority of AEs were assessed by the investigator as Toxicity Grade 1 (mild) or 2 (moderate). Eleven (21%) of recipients of V180 with ISCOMATRIX™ adjuvant experienced a total of 18 Toxicity Grade 3 (severe) events (Table 3), including 15 injection-site AEs (8 events of erythema >10 cm, 6 events of swelling >10 cm, and 1 event of pain/tenderness) and 3 other events (1 each of fatigue, fever, and headache) (See Supplemental Material for the definitions of the Toxicity Grade 3 events). Of the 18 Grade 3 events among recipients of V180 with ISCOMATRIX™ adjuvant, 9 events in 6 participants occurred Postdose 2, and 9 events in 6 participants occurred Postdose 3. No Grade 3 events occurred among recipients of V180 with Alhydrogel™. One recipient (6%) of unadjuvanted medium-dose V180 experienced 1 Grade 3 event of injection-site erythema >10 cm. All of the Grade 3 events experienced by V180 recipients were assessed by the investigator as related to study product. One placebo recipient (6%) had a Grade 3 event of headache, which the investigator assessed as not related to study product. All Grade 3 events resolved completely.
The investigator discontinued dosing of 1 participant after the first injection of medium-dose V180 with 30 ISCO™ units of ISCOMATRIX™ adjuvant. This participant, who had a medical history of left knee injury, experienced vaccine-related Toxicity Grade 2 AEs of arthralgia (left knee and ankle) and injection-site swelling.
Discussion
This trial assessed the magnitude and durability of the immune response, and the number of dengue serotypes to which an immune response was induced, following a 3-injection series of 3 dose levels of V180 formulated with or without adjuvant. The data suggest that an adjuvant is needed for the immunogenicity of V180 when used as a stand-alone vaccine, and that Alhydrogel™ with 225 µg elemental aluminum may not be sufficient for this purpose. These findings are as expected for soluble subunit antigens, and are consistent with the modest and short-lived immunogenicity observed in the published clinical trial of monovalent DEN1-80E containing 1.25 mg of elemental aluminum as Alhydrogel™.5 The lower aluminum dose selected for the present study aligned with licensed Alhydrogel™-adjuvanted vaccines.9
All 6 V180 formulations with ISCOMATRIX™ adjuvant met the protocol-specified criterion for a positive immune response. The magnitude and persistence of the immune response appeared to be driven more by the presence of ISCOMATRIX™ adjuvant than by the dose level of V180 or ISCOMATRIX™ adjuvant. The finding that, within each dose level of ISCOMATRIX™ adjuvant, GMTs did not tend to increase with higher V180 doses, is consistent with preclinical data.5,8,10 The memory B-cell responses are consistent with the neutralizing antibody results observed in these groups.
All V180 formulations in the present study included twice the amount of the DEN4-80E component than the other 3 components. Despite this increased dosage, the neutralizing antibody titers induced for DENV4 were generally lower and less durable than for the other serotypes, although the memory B-cell responses were balanced across serotypes.
All 9 V180 formulations tested were generally well tolerated. No SAEs, Toxicity Grade 4 (life-threatening) events, or deaths were reported. Evaluation of the safety profiles for each of the treatment groups demonstrated clear differences by adjuvant type (ISCOMATRIX™ adjuvant, Alhydrogel™, or none), but not by the dosage level of V180 or ISCOMATRIX™ adjuvant. Consistent with previous reports, formulations with ISCOMATRIX™ adjuvant were associated with an increased frequency of injection-site AEs, systemic AEs, and fevers.11,12 ISCOMATRIX™ adjuvant recipients also experienced more frequent Toxicity Grade 3 (severe) events, which all occurred either Postdose 2 or Postdose 3.
The immunogenicity assessments conducted during this trial are limited by several factors. First, no correlate of protection has been established for dengue. However, measuring virus neutralizing antibody provides a functional assay that may predict the potential for protection.13,14 Second, the V180 formulations were tested as a series of 3 injections administered 1 month apart. Preclinical data suggest that a longer vaccination schedule might induce higher and more durable antibody levels;5,8 assessing this possibility would require further study. Third, only flavivirus-naïve persons were eligible to participate in this trial; the need for an adjuvant may be less in flavivirus-experienced populations. Finally, the exploratory assessment of immune memory was conducted in a small number of high-dose cohort participants, and limited to B cells; more definitive conclusions will require larger numbers of participants and a wider battery of tests, such as memory T-cell assays, B- and T-cell subset analyses, and assessment of antibody affinity.
The safety assessments conducted in this trial are primarily limited by the small sample size. Larger sample sizes will be needed to better understand the safety profile of any V180 formulations studied in the future.
The V180 vaccine construct includes a limited number of T-cell epitopes, which reduces any potential contribution of these epitopes to protection. Because partial and waning immunity represent a theoretical risk for post-vaccination enhancement of dengue disease, V180 as a stand-alone vaccine may not represent an optimal approach for protecting against dengue disease. In a subsequent prime-boost study (NCT02450838), adults who had previously received 1 or 2 doses of an investigational live-attenuated tetravalent dengue vaccine and responded to at least 3 of the 4 vaccine serotypes were administered a single dose of V180 (either unajuvanted or adjuvanted with Alhydrogel™) as a booster. The V180 booster doses were immunogenic and generally well tolerated in these dengue-experienced participants.15
In summary, in a population of flavivirus-naïve adults who received 3 injections of V180 at 1-month intervals, V180 formulations with ISCOMATRIX™ adjuvant met the protocol-specified criterion for a positive immune response; antibody levels declined over time but generally remained above baseline through 1 year after the third injection. Aluminum-adjuvanted and unadjuvanted formulations were poorly immunogenic, and did not meet the pre-specified criterion of a positive immune response; titers generally declined to baseline by 6 months Postdose 3.
All tested formulations were generally well tolerated. Formulations with ISCOMATRIX™ adjuvant were more often associated with AEs than aluminum-adjuvanted and non-adjuvanted formulations.
Methods
Population
Flavivirus-naïve, generally healthy adults 18 to 49 years old were eligible for enrollment. In accordance with the protocol (see Supplemental Material for additional details), prior to receiving any study-related injections, all potential participants were screened by medical history, physical examination, and laboratory testing to ensure seronegativity for flaviviruses at baseline (See Supplemental Material). Exclusion criteria included pregnancy, known allergy to a trial product component, known or suspected immunocompromising condition, pre-specified laboratory abnormalities, hospitalization for acute illness ≤ 3 months before enrollment, and febrile illness ≤ 72 hours before enrollment.
The protocol was conducted in accordance with principles of Good Clinical Practice (GCP), including obtaining written informed consent from each participant prior to study entry, and was approved by the human studies committees of each study site.
Vaccines
V180 is comprised of recombinant DEN-80E proteins for the 4 dengue serotypes (DEN1-80E, DEN2-80E, DEN3-80E, and DEN4-80E) (see Supplemental Material). Three V180 dose levels were studied: low-dose (3, 3, 3, and 6 μg, respectively), medium-dose (10, 10, 10, and 20 μg, respectively), and high-dose (50, 50, 50, and 100 μg, respectively) (Table 4). All V180 formulations included twice the amount of the DEN4-80E antigen than the other antigens, in an effort to overcome the preclinical observation that DEN4-80E tended to induce lower immune responses than the other components.5,8 Two adjuvants were tested: Alhydrogel™ (aluminum hydroxide), at a dose of 225 µg elemental aluminum, and ISCOMATRIX™ adjuvant (containing ISCOPREP™ saponin, cholesterol, and phospholipids) at a dose of 30 or 60 ISCO™ units (see Supplemental Material). Nine adjuvanted or unadjuvanted V180 formulations were compared with phosphate-buffered saline placebo (Table 4).
Table 4.
Number randomized by treatment group.
| V180 Antigen Dose Level(µg of DEN1-80E/DEN280E/DEN3-80E/DEN4-80) | ISCOMATRIX™ adjuvant | Alhydrogel™(225 µg aluminum) | Unadjuvanted | Placebo (phosphate-buffered saline) | Total | |
|---|---|---|---|---|---|---|
| 30 ISCO™ units | 60 ISCO™ units | |||||
| Low (3/3/3/6) | 8 | 9 | – | – | 6 | 23 |
| Medium (10/10/10/20) | 9 | 9 | 9 | 9 | 6 | 42 |
| High (50/50/50/100) | 9 | 9 | – | 9 | 6 | 33 |
| All Combined | 26 | 27 | 9 | 18 | 18 | 98 |
All products were prepared, packaged, and labeled in accordance with Good Manufacturing Practice, guidelines for GCP from The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, and applicable local laws and regulations. Vaccine supplies were shipped, stored, and distributed in accordance with the trial protocol.
Trial design
This Phase I randomized, double-blind, placebo-controlled trial (V180-001; NCT01477580) was conducted at 2 sites in Australia between July 2012 and December 2014. Participants received 3 injections of the allocated product at 1-month intervals. The original sample size was 8 for all active treatment groups, and 6 for all placebo groups (N = 90). After 1 participant in the low-dose cohort elected to discontinue participation following the first injection, that participant was replaced, and the sample size of all subsequent active treatment groups was increased to 9 as a contingency against further discontinuations. A total of 98 participants were randomized (Table 4).
The safety of this trial was monitored and advised at pre-specified time points by an unblinded independent external Data Monitoring Committee (eDMC), comprised of experts in clinical research, pharmacovigilance, and/or dengue. This dose-escalation trial was enrolled into successive cohorts defined by V180 antigen level, following eDMC approval to continue. No safety concerns were identified by the eDMC during the trial.
Immunogenicity assessments
Blood samples for quantifying neutralizing antibodies were to be collected from all participants at 6 time points: prevaccination, 28 days after each injection, and 6 months and 1 year after the last injection. At each time point, virus-neutralizing antibody responses were measured using a Focus Reduction Neutralization Test with a 50% neutralization cutoff (FRNT50) (see Supplemental Material).16,17
Blood samples to assess B-cell memory were collected at 2 time points (immediately before the first injection and at 28 days Postdose 3) in a subset of 18 high-dose cohort participants (5 recipients each of V180 with 30 ISCO™ units of ISCOMATRIX™ adjuvant, V180 with 60 ISCO™ units of ISCOMATRIX™ adjuvant, and unadjuvanted V180, and 3 placebo recipients). Levels of dengue serotype-specific memory B cells were assessed in these samples by an optimized memory B ELISPOT assay (see Supplemental Material).
Immunogenicity data analysis
No specific immunogenicity hypotheses were tested. The primary objective of the trial was to evaluate if, at 28 days Postdose 3, at least one V180 formulation induced neutralizing antibodies to dengue serotypes. For each formulation, the statistical criterion for a positive immune response was that the seroconversion rate (SCR, the proportion of participants with a titer ≥10) at Day 28 Postdose 3 was ≥75% for at least 3 of the 4 serotypes. An exploratory objective was to describe neutralizing antibody levels for each formulation at each time point by computing serotype-specific SCRs, geometric mean titers (GMTs), and proportions of participants with a tetravalent response (FRNT50 titers ≥10 for all 4 dengue serotypes) or a ≥trivalent response (FRNT50 titers ≥10 for 3 or 4 dengue serotypes). Titers < 10 were designated as 5 when computing GMTs.
The per-protocol population, which served as the primary population for the analysis of neutralizing-antibody data, excluded participants with major protocol deviations that may have substantially interfered with the immune response. Excluded participants were identified before trial unblinding according to pre-specified criteria
Evaluation of memory B-cell immune responses in a subset of high-dose cohort participants was an exploratory objective. For each participant, the percentage of antigen-specific antibody-secreting cells was computed for each DENV serotype at baseline and 28 days Postdose 3, and described within and across formulations.
Safety assessments
A secondary study objective was to assess safety and tolerability of the tested products. All participants, including those who did not complete the 3-injection series, were to be followed for serious adverse events (SAEs) (including death) from informed consent through 1 year following the last injection. All participants who received ≥1 injection recorded all post-vaccination adverse events (AEs) from Day 1 (day of injection) through Day 14 following each injection on a Vaccination Report Card (VRC). In addition to unsolicited AEs, the VRC prespecified the following solicited AEs: injection-site AEs of pain/tenderness (captured on the VRC as “pain or tenderness”), erythema, and swelling from Days 1 through 5; and systemic AEs of fatigue, headache, and myalgia from Days 1 through 14. Additionally, a daily measurement of oral temperature from Days 1 through 14 was recorded. Laboratory testing was performed prior to the first injection and approximately every 2 weeks through 28 days following the last injection.
Participants recorded the maximum size for any injection-site erythema or swelling, and the maximum intensity (“mild” [awareness of symptom, but easily tolerated]; “moderate” [discomfort enough to cause interference with usual activities], or “severe” [inability to do work or usual activities]) for other AEs. Investigators scored AEs according to an FDA toxicity scale17 (see Supplemental Material) as Grade 1 (“mild”), Grade 2 (“moderate”), Grade 3 (“severe”), or Grade 4 (“potentially life-threatening”).18 All injection-site AEs were considered to be related to the trial product. While the study was blinded, investigators assessed the likelihood that each non-injection-site AE was related to the study product.
Safety data analysis
All participants who received ≥1 injection were included in the safety analyses. Safety data were summarized for individual treatment groups, and overall by adjuvant type. No formal safety hypotheses were tested.
Funding Statement
Funding for this research was provided by Merck & Co., Inc., Kenilworth, NJ, USA.
Supplementary Material
Supplemental data for this article can be accessed here
Supplemental Material
References
- 1.World Health Organization Dengue and severe dengue. Geneva (Switzerland) 2016. [accessed 2017 April 14]. http://www.who.int/mediacentre/factsheets/fs117/en/. [Google Scholar]
- 2.Editorial: dengue–an infectious disease of staggering proportions. Lancet. 2013;381(9884):2136. doi: 10.1016/S0140-6736(13)61423-3. [DOI] [PubMed] [Google Scholar]
- 3.Screaton G, Mongkolsapaya J, Yacoub S, Roberts C.. New insights into the immunopathology and control of dengue virus infection. Nat Rev Immunol. 2015;15(12):745–759. doi: 10.1038/nri3916. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization Revised SAGE recommendation on use of dengue vaccine – 19 April 2018. Weekly Epidemiologic Rec. No 23, 2018. June 8, 93, 337–340. [accessed 2017 Jun 14]. http://apps.who.int/iris/bitstream/handle/10665/272782/WER9323.pdf?ua=1 [Google Scholar]
- 5.Manoff SB, George SL, Bett AJ, Yelmene ML, Dhanasekaran G, Eggemeyer L, Sausser ML, Dubey SA, Casimiro DR, Clements DE, et al. Preclinical and clinical development of a dengue recombinant subunit vaccine. Vaccine. 2015;33(50):7126–7134. doi: 10.1016/j.vaccine.2015.09.101. [DOI] [PubMed] [Google Scholar]
- 6.Harris JR, Soliakov A, Lewis RJ, Depoix F, Watkinson A, Lakey JH. Alhydrogel® adjuvant, ultrasonic dispersion and protein binding: a TEM and analytical study. Micron. 2012;43(2–3):192–200. doi: 10.1016/j.micron.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 7.Morelli AB, Becher D, Koernig S, Silva A, Drane D, Maraskovsky E. ISCOMATRIX: a novel adjuvant for use in prophylactic and therapeutic vaccines against infectious diseases. J Med Microbiol. 2012;61(Pt 7):935–943. doi: 10.1099/jmm.0.040857-0. [DOI] [PubMed] [Google Scholar]
- 8.Govindarajan D, Meschino S, Guan L, Clements DE, Ter Meulen JH, Casimiro DR, Coller BA, Bett AJ. Preclinical development of a dengue tetravalent recombinant subunit vaccine: immunogenicity and protective efficacy in nonhuman primates. Vaccine. 2015. August 7;33(33):4105–4116. doi: 10.1016/j.vaccine.2015.06.067. [DOI] [PubMed] [Google Scholar]
- 9.Lindblad EB. Aluminium adjuvants–in retrospect and prospect. Vaccine. 2004;22(27–28):3658–3668. doi: 10.1016/j.vaccine.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 10.Clements DE, Coller BA, Lieberman MM, Ogata S, Wang G, Harada KE, Putnak JR, Ivy JM, McDonell M, Bignami GS, et al. Development of a recombinant tetravalent dengue virus vaccine: immunogenicity and efficacy studies in mice and monkeys. Vaccine. 2010; 28(15): 2705–2715. doi: 10.1016/j.vaccine.2010.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenzie A, Watt M, Gittleson C. ISCOMATRIX vaccines: safety in human clinical studies. Hum Vaccin. 2010;6(3):pii: 10754 PMID: 20595811. [DOI] [PubMed] [Google Scholar]
- 12.Bigaeva E, Doorn EV, Liu H, Hak E. Meta-analysis on randomized controlled trials of vaccines with QS-21 or ISCOMATRIX adjuvant: safety and tolerability. PLoS One. 2016;11(5):e0154757. doi: 10.1371/journal.pone.0154757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.World Health Organization Dengue guidelines for diagnosis, treatment, prevention and control: new edition. 2009. Geneva (Switzerland) [accessed 2018 June 14]. http://www.who.int/tdr/publications/documents/dengue-diagnosis.pdf. [PubMed] [Google Scholar]
- 14.Crill WD, Hughes HR, Delorey MJ, Chang GJ. Humoral immune responses of dengue fever patients using epitope-specific serotype-2 virus-like particle antigens. PLoS One. 2009;4(4):e4991. doi: 10.1371/journal.pone.0004991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coller BAG, Durbin A, Kirkpatrick B, Pierce K, Grier P, Sabundayo B, Larsson C, He H, Sausser M, Russell A, et al. A phase I clinical trial evaluating the impact of tetravalent recombinant subunit dengue vaccine boost administered to subjects who have previously been vaccinated with a live-attenuated tetravalent dengue vaccine. Presented at the 2016 Annual Meeting of the American Society of Tropical Medicine and Hygiene, Abstract #59; http://www.astmh.org/ASTMH/media/Documents/Abstracts-1-500-ASTMH-2016-Annual-Meeting-Abstract-Book.pdf.
- 16.World Health Organization Guidelines for plaque reduction neutralizing testing of human antibodies to dengue viruses. Geneva (Switzerland) 2007. [accessed 2018 June 14]. http://apps.who.int/iris/bitstream/handle/10665/69687/who_ivb_07.07_eng.pdf;jsessionid=026253819A4C9E43950ADDF81F87A8B8?sequence=1. [Google Scholar]
- 17.Roehrig JT, Hombach J, Barrett AD. Guidelines for plaque-reduction neutralization testing of human antibodies to dengue viruses. Viral Immunol. 2008;21:123–132. doi: 10.1089/vim.2008.0007. [DOI] [PubMed] [Google Scholar]
- 18.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research Guidance for industry: toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials. September 2007. [accessed 2018 Apr 14]. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Vaccines/ucm091977.pdf.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material