Intracellular re-routing of prion protein prevents propagation of PrPSc and delays onset of prion disease (original) (raw)
Abstract
Prion diseases are fatal and transmissible neurodegenerative disorders linked to an aberrant conformation of the cellular prion protein (PrPc). We show that the chemical compound Suramin induced aggregation of PrP in a post-ER/Golgi compartment and prevented further trafficking of PrPc to the outer leaflet of the plasma membrane. Instead, misfolded PrP was efficiently re-routed to acidic compartments for intracellular degradation. In contrast to PrPSc in prion-infected cells, PrP aggregates formed in the presence of Suramin did not accumulate, were entirely sensitive to proteolytic digestion, had distinct biophysical properties, and were not infectious. The prophylactic potential of Suramin-induced intracellular re-routing was tested in mice. After intraperitoneal infection with scrapie prions, peripheral application of Suramin around the time of inoculation significantly delayed onset of prion disease. Our data reveal a novel quality control mechanism for misfolded PrP isoforms and introduce a new molecular mechanism for anti-prion compounds.
Keywords: aggregation/prion protein/quality control/ re-routing/Suramin
Introduction
Prion diseases are fatal, neurodegenerative disorders in humans and animals, and include Creutzfeldt–Jakob disease (CJD) in humans, scrapie in sheep, and bovine spongiform encephalopathy (BSE) in cattle. All of these disorders are characterized by the accumulation of an abnormally folded isoform of the cellular prion protein (PrPc), denoted PrPSc, which represents the major component of infectious scrapie prions (Prusiner, 1998; Weissmann et al., 2001). The formation of PrPSc is accompanied by profound changes in PrPc structure and biochemical properties. PrPc, which is rich in α-helical regions, is converted into a molecule with mainly β-sheet structure and becomes partially resistant to proteolytic digestion (Cohen et al., 1994; Prusiner, 1998; Clarke et al., 2001). During biogenesis, PrPc transits through the secretory pathway and is modified by the attachment of two N-linked carbohydrate chains and a glycolipid anchor. The conversion of PrPc into PrPSc is thought to occur after PrPc has reached the plasma membrane or is re-internalized for degradation (Caughey and Raymond, 1991; Borchelt et al., 1992; Taraboulos et al., 1995). The molecular mechanisms of the conversion reaction remain enigmatic. It is known that PrPc biosynthesis is a pre-requisite for PrPSc formation, and studies in transgenic animals favor a model in which PrPc and PrPSc interact directly, possibly in a complex with auxiliary factors (Prusiner, 1998).
The low frequency of prion diseases in man indicates that the spontaneous formation of PrPSc is a rare process. However, through exogenous infection with PrPSc, the disease can be efficiently transmitted to susceptible organisms. The fact that, even in a scrapie-infected cultured cell, only 1–2% of newly synthesized PrPc is converted into PrPSc (Borchelt et al., 1992) raises the possibility that only a subfraction of PrPc is eligible for PrPSc formation. After PrPSc is formed it accumulates in brains of infected organisms (Prusiner, 1998). The inhibitory potential of chemical chaperones (Tatzelt et al., 1996) on the propagation of PrPSc in scrapie-infected N2a cells adds to the notion that a conformational change of PrPc features in the formation of PrPSc.
A variety of cellular quality control mechanisms ensures detection and rapid degradation of misfolded proteins. In the secretory pathway of eukaryotic cells the most important control system is located in the ER, although additional post-ER/Golgi systems have been described (for review see Ellgaard et al., 1999). Indeed, some mutated prion proteins were found subjected to ER-based quality controls (Rogers et al., 1990; DeArmond et al., 1997; Muramoto et al., 1997; Singh et al., 1997; Zanusso et al., 1999). But overall, the impact of quality control mechanisms and how cells subject misfolded prion proteins to either lysosomal or cytosolic degradation are poorly understood.
We used the chemical compound Suramin to modulate PrP folding in cultured cells and in vitro. Suramin, a bis-hexasulfonated naphthylurea, was initially developed to treat trypanosomiasis (Dressel and Oesper, 1961). Suramin has previously been tested in a hamster scrapie model for anti-prion effects (Ladogana et al., 1992). The compound is known to down-regulate surface proteins and to interfere with the oligomerization state of certain proteins (for review see Stein et al., 1995). Here we report that in the presence of Suramin, aggregates of fully matured PrP are re-routed directly from the Golgi/_trans_-Golgi network (TGN) to lysosomal degradation, bypassing the plasma membrane. We show further that this mechanism interferes with PrPSc propagation in cell culture and scrapie-infected animals.
Results
Suramin interferes with the folding of PrPc and induces the formation of detergent-insoluble PrP
Based on previous observations that Suramin modulated the expression of certain cell surface proteins, we incubated N2a and ScN2a cells in the presence of Suramin and examined the relative amount of PrP by immunoblotting. To separate PrPc and PrPSc we used the ‘classical’ detergent assay, namely solubilization of cellular proteins in a buffer containing 1% sarcosyl and centrifugation at 100 000 g for 1 h. Without Suramin only detergent-soluble PrPc was present in N2a cells (Figure 1A, lanes 1 and 5), whereas ScN2a cells contained significant amounts of detergent-insoluble PrPSc (lanes 2 and 6). One should note that detergent-insoluble PrPSc composed of N-terminally truncated PrP molecules (∼aa 90–231; double-, mono- or un-glycosylated) is found in murine ScN2a cells without exogenous proteinase K (PK) digestion. After cultivation for 7 days in the presence of Suramin, the amount of detergent-soluble PrPc was significantly reduced in both cell lines (lanes 7 and 8). In addition, the amount of PrPSc in ScN2a cells was highly reduced (lane 4). Instead, Suramin-treated N2a and ScN2a cells now harbored a fraction of putatively full-length PrP that partitioned into the detergent-insoluble phase (lanes 3 and 4).
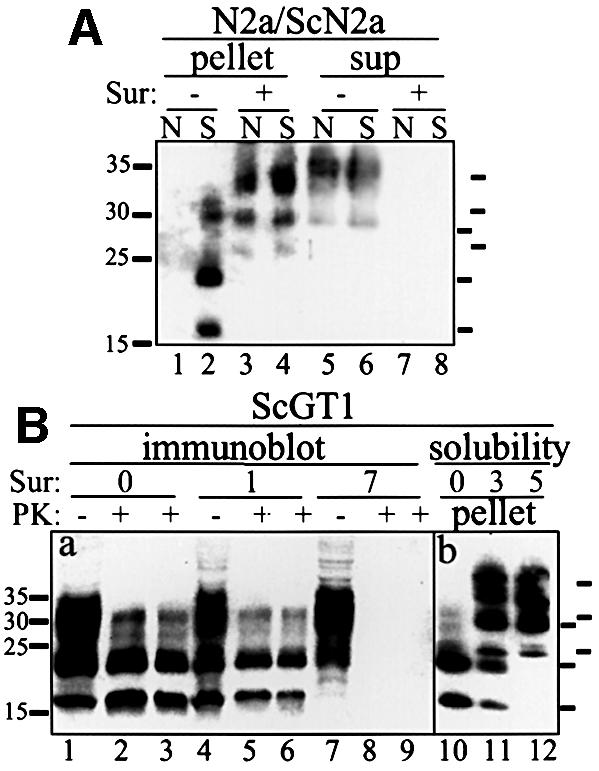
Fig. 1. Induction of insoluble PrP aggregates and reduction of PrPSc by Suramin. (A) Induction of insoluble PrP in N2a and ScN2a cells and reduction of PrPSc determined using a solubility assay. N2a (N; lanes 1, 3, 5 and 7) and ScN2a (S; lanes 2, 4, 6 and 8) cells were treated with Suramin for 7 days (200 µg/ml; lanes 3, 4, 7 and 8) or mock treated (lanes 1, 2, 5 and 6). Cells were lysed and postnuclear lysates were subjected to a solubility assay. PrP was analyzed by immunoblotting using the anti-PrP antibody SAF 70. Detergent-insoluble fractions are shown on the left (pellet; lanes 1–4), soluble fractions on the right (sup; lanes 5–8). Molecular size markers are depicted on the left (kDa); bars on the right indicate the six PrP-specific bands (full-length PrP and N-terminally truncated PrPSc). (B) Reduction of PrPSc and induction of insoluble PrP in ScGT1 cells. Panel (a) shows an immunoblot analysis. ScGT1 cells were treated with Suramin for 1 or 7 days (200 µg/ml; lanes 4–6 and 7–9, respectively) or mock treated (lanes 1–3). Cells were lysed, postnuclear lysates were divided into two parts, and either subjected to digestion with PK (lanes 2, 5 and 8: 20 µg/ml; lanes 3, 6 and 9: 40 µg/ml for 30 min) or proceeded further without PK treatment (lanes 1, 4 and 7). PrP was analyzed by immunoblotting using SAF 70. Panel (b) shows a solubility assay (lanes 10–12). ScGT1 cells were either mock treated (lane 10) or treated for 3 or 5 days with Suramin (200 µg/ml; lanes 11 and 12, respectively). Cells were lysed and postnuclear lysates subjected to a solubility assay. Detergent-insoluble fractions are shown. Bars on the right indicate PrP-specific bands.
To corroborate these findings we used other prion-infected cell culture systems. ScGT1 cells derived from the central nervous system (Schätzl et al., 1997) were treated with Suramin, lysates digested with PK as indicated and analyzed in immunoblot (Figure 1B). Whereas after 24 h of Suramin treatment only a slight reduction of PrPSc was evident, after 7 days of treatment no more PrPSc signal was detectable. We then looked for induction of detergent-insoluble PrP by Suramin in ScGT1 cells (lanes 10–12). After 5 days of Suramin treatment PrPSc was undetectable and a detergent-insoluble PrP of apparently full length was generated (lane 12). As was the case in ScN2a cells, the level of PrPSc was decreased by Suramin treatment in a dose- and time-dependent manner, and the generation of detergent-insoluble PrP was induced.
In summary, we show that Suramin decreases the steady state level of soluble PrPc and induces the intracellular formation of detergent-insoluble PrP.
PrP aggregates formed in the presence of Suramin are sensitive to proteolytic digestion, do not accumulate, and are not infectious
Insolubility in non-ionic detergents and partial resistance to proteolytic digestion are biochemical markers of infectious prions. Consequently, we examined the biochemical features of PrP aggregates formed in the presence of Suramin. 3F4-ScN2a cells (overexpressing 3F4-tagged PrP) were treated for 7 days with Suramin and aliquots of the lysates subjected to a limited proteolytic digestion before analyzing them in a solubility assay (Figure 2A). Without subsequent PK digestion (lanes 1 and 5) both Suramin- and mock-treated 3F4-ScN2a cells harbored detergent-insoluble PrP. However, PrP aggregates formed in the presence of Suramin were completely degraded by PK (lanes 2–4). These assays indicated that Suramin induced a detergent-insoluble fraction of PrP that differed from PrPSc in persistently prion-infected cells.
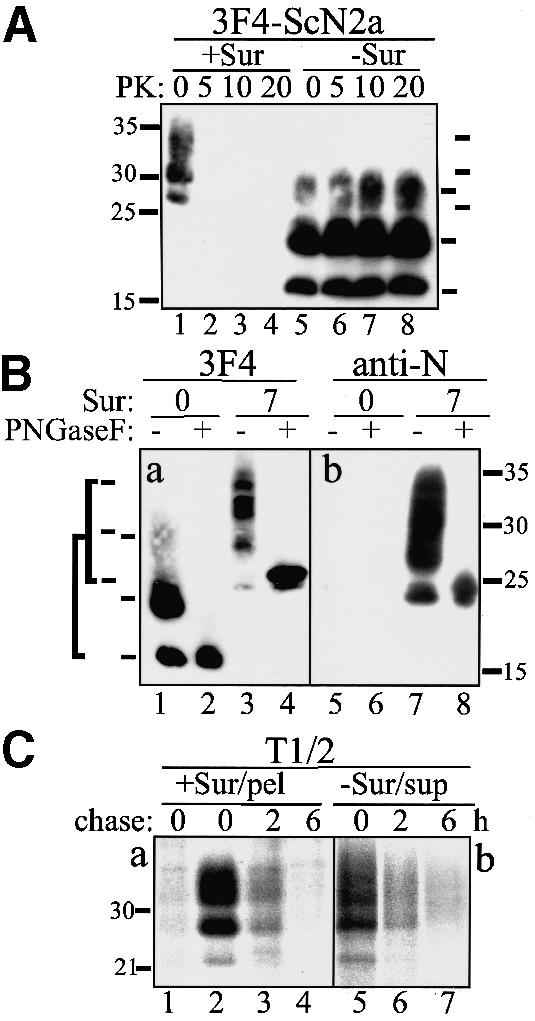
Fig. 2. Detergent-insoluble PrP induced by Suramin is not PK resistant, nor is it N-terminally truncated, and it has a half-life time comparable to PrPc. (A) PrP aggregates formed in the presence of Suramin are completely sensitive to proteolytic digestion. 3F4-ScN2a cells incubated with Suramin (200 µg/ml) for 7 days (+Sur; lanes 1–4) were compared with mock-treated cells (lanes 5–8). Cells were lysed and postnuclear lysates subjected to proteolytic digestion with various amounts of PK as indicated. PK digestion was terminated by the addition of proteinase inhibitors, lysates were adjusted to 1% sarcosyl and centrifuged for 1 h at 100 000 g. An immunoblot analysis of the remaining PrP present in the detergent-insoluble fraction is shown using the anti-PrP antibody 3F4. Molecular size markers are shown on the left (kDa); bars on the right indicate PrP-specific bands. (B) Suramin induces aggregation of full-length glycosylated PrP. 3F4-ScN2a cells were mock treated or incubated in the presence of 200 µg/ml Suramin for 7 days. Detergent-insoluble fractions were prepared as described in (A), and incubated with PNGase F or left untreated. PrP was analyzed by immunoblotting using 3F4 (3F4; lanes 1–4) or the anti-PrP antibody 4F2 (anti-N; lanes 5–8), directed to the N-terminus of PrP. Bars on the left indicate the six PrP-specific bands [glycoforms of full-length PrP (aa 23–231) and N-terminally truncated PrPSc (aa ∼90–231)]. (C) Detergent-insoluble PrP does not accumulate in Suramin-treated cells. 3F4-N2a cells were mock treated [panel (b); –Sur/sup] or treated for 1 day with Suramin [panel (a); +Sur/pel] and metabolically labeled for 2 h with [35S]Met (+/– Suramin). One plate was harvested directly (chase: 0); the other plates were incubated further in [35S]Met-free medium for 2 and 6 h, respectively. Cells were harvested and detergent-soluble (panel b) and -insoluble fractions (panel a) prepared as described. PrP was immunoprecipitated using 3F4. Lane 1 is the pellet fraction of non-Suramin-treated cells. Molecular size markers are shown on the left (kDa).
To characterize further the PrP aggregates induced by Suramin we performed a PNGase F digestion (Figure 2B). PrP 27–30, present in the detergent-insoluble fraction of untreated 3F4-ScN2a cells, was converted into unglycosylated PrP of an apparent mass of ∼19 kDa (panel a). In contrast, detergent-insoluble PrP isolated from Suramin-treated 3F4-ScN2a cells was converted into a molecule with a molecular mass of ∼24–25 kDa (PrP23–231). Using an antibody directed against the N-terminus (panel b), we showed that this molecule (lanes 7 and 8), in contrast to PrP 27–30 (lanes 5 and 6), contained the N-terminal segment (i.e. aa 23–90).
Next we analyzed the half-life time of Suramin-induced PrP aggregates by metabolic labeling experiments. 3F4-N2a cells cultivated without Suramin (panel b) or in the presence of Suramin for 1 day (panel a) were radiolabeled and either harvested directly or cultivated further in [35S]Met-free medium for 2 and 6 h (Figure 2C). PrP present in the detergent-soluble (panel b) or insoluble fraction (panel a) was analyzed by immunoprecipitation. The detergent-insoluble PrP formed in the presence of Suramin showed a half-life time similar to that of soluble PrP in untreated cells with ∼3–4 h under our experimental conditions. This is much shorter than the half-life time of PrPSc and similar to those previously reported for PrPc (Borchelt et al., 1992). To confirm that the detergent-insoluble PrP fraction present in Suramin-treated cells was not PrPSc, we performed bioassays. In mice intracerebrally inoculated with Suramin-treated ScN2a or ScGT1 cells a significant prolongation of incubation time was observed compared with untreated cells (data not shown).
The performed analysis reveals that Suramin interferes with the propagation of PrPSc, and that PrP aggregates formed in the presence of Suramin do not contain infectious prions. Instead, they might represent an aggregated population of PK-sensitive and full-length PrP molecules that can be efficiently degraded by the cell.
Suramin induces aggregation of PrP in the Golgi/TGN and re-routing to acidic compartments
The experiments described above established that Suramin induces a population of detergent-insoluble PrP that is distinct from PrPSc. The following experiments were designed to localize the subcellular compartments where Suramin interferes with the proper folding of PrPc. Using endo H and brefeldin A we demonstrated that fully glycosylated PrP aggregated in post-ER compartments (data not shown).
To corroborate our findings we performed surface fluorescence-activated cell sorting (FACS) studies (Figure 3A). Suramin treatment resulted in a significant down-regulation of surface PrPc (left panel). When the total amount of PrPc was measured by intracellular FACS with permeabilized cells, no significant effect of Suramin on PrPc was detectable (right panel). We then performed biochemical studies where we analyzed the amounts of phosphatidylinositol-specific phospholipase C (PIPLC)-releasable PrPc. PIPLC, when added to the medium, results in a continuous release of surface-located PrPc into the medium, and subsequently, in a decrease in the cell lysate fraction. Whereas PIPLC could efficiently release surface PrPc in non-Suramin-treated cells (Figure 3B, lanes 1 and 2), the amount of PrPc in Suramin-treated cells was unaffected by PIPLC digestion (lanes 3 and 4). Our studies indicate that PrP had no access to the outer leaflet of the plasma membrane in the presence of Suramin. This effect was found in all cell types that we analyzed (e.g. epithelial, neuronal, blood-derived cells, fibroblasts; data not shown).

Fig. 3. Suramin induces intracellular re-routing of PrP from post-ER compartments to acidic vesicles. (A) PrP is not detectable by surface FACS analysis in Suramin-treated cells. The left panel shows the surface FACS analysis of 3F4-N2a cells [the bold line is PrP reactivity without Suramin; the broken line is in the presence of Suramin (200 µg/ml for 24 h)]. The _y_-axis denotes the number of cells, the _x_-axis the fluorescence intensity (lg-scale). The polyclonal rabbit anti-PrP antibody A4 was used. The right panel shows the identical analysis with permeabilized cells (intracellular FACS). (B) No release of surface PrP by PIPLC in cells treated with Suramin. 3F4-N2a cells were incubated for 3 days with Suramin (200 µg/ml medium), treated with PIPLC, and the cellular lysate preparations analyzed in immunoblot (3F4). In non-Suramin-treated cells, PIPLC treatment results in the expected reduction of PrP in the cellular lysate fraction (lane 2); in the presence of Suramin, PIPLC treatment has no effect (lane 4). (C) Suramin induces intracellular retention of PrP in the Golgi/TGN and re-routing to acidic compartments. 3F4-N2a cells were mock treated (panel a) or incubated in the presence of Suramin (100µg/ml for 24 h) [panels (b) and (c)] and analyzed in indirect immunofluorescence experiments using confocal microscopy. (Panel a) PrP is excluded from the cell surface of Suramin-treated cells. Cell surface proteins of fixed and non-permeabilized 3F4-N2a cells were unspecifically surface-labeled with biotin; at the same time PrP present at the cell surface was specifically labeled with the antibody 3F4. The left panel shows surface biotinylated proteins (biotin), the middle panel cell surface PrP (3F4), and the right panel the overlay of both signals in yellowish color (merge). The upper right cell does not express detectable levels of 3F4-PrP, which indicates the specificity of the PrP antibody used. (Panel b) Suramin induces the retention of PrP in a Golgi/TGN compartment. 3F4-N2a cells were treated with Suramin, fixed, permeabilized and incubated with a polyclonal anti-mannosidase II antiserum to localize the medial to _trans_-Golgi compartments (left panel, manII). In parallel, PrP was analyzed with 3F4 (middle panel). In the right panel the overlay of both signals indicates co-localization (merge). (Panel c) PrP is re-routed to acidic compartments in Suramin-treated cells. Cells and treatment as in panel (b), with the exception that leupeptin was added. To stain for acidic endosomal/lysosomal compartments, an antibody against the acidotropic amine DAMP was used. The left panel shows the DAMP staining, the middle panel is PrP (3F4), and the right panel denotes the overlay of both signals (merge).
The experiments described above indicated that Suramin interacted with fully matured PrP and prevented the further trafficking of PrP to the outer cell surface. To identify the intracellular compartments that harbored the detergent-insoluble PrP we used confocal laser scanning microscopy. In mock-treated control cells we found the expected cell surface localization of PrPc (Figure 3C, panel a). In the presence of Suramin, PrP was no longer detectable on the plasma membrane; however, it was present in intracellular compartments (panels b and c). Co-localization analysis with mannosidase II indicated that PrP in Suramin-treated cells was present in a compartment corresponding to the middle and late Golgi/TGN (panel b). In addition, using a marker for endosomal/lysosomal compartments [i.e. the acidotropic amine 3-(2,4-dinitroanilino) 3′-amino-_N_-methyl dipropylamine (DAMP)] and treatment with the lysosomal inhibitor leupeptin, we could show that a fraction of PrP was present in acidic compartments, presumably in lysosomal vesicles (panel c).
Using biochemical and immunological assays as well as confocal laser scanning microscopy we could show that Suramin induces the formation of detergent-insoluble PrP aggregates in a post-ER compartment. Suramin prevents further targeting to the plasma membrane, and instead re-routes PrP to acidic compartments for lysosomal degradation.
Suramin induces in vitro aggregation of PrP, but does not induce a conformational shift from α-helix to β-sheet
Having found that Suramin induces PrP aggregates within living cells, we asked whether Suramin can also induce PrP aggregates in crude cell lysates. We lysed N2a cells either with lysis buffer containing detergent [0.5% Triton X-100 and 0.5% sodium deoxycholate (DOC), or by scraping the cells into lysis buffer without detergent and disrupting them by freeze–thawing. The lysates were then incubated for 16 h with Suramin, adjusted to 0.5% DOC] and Triton X-100 each, and subjected to a solubility assay (Winklhofer and Tatzelt, 2000) (Figure 4A). When the cells were lysed in detergent buffer, Suramin did not alter the solubility profile of PrP (lanes 3 and 4). In contrast, in crude lysates prepared by freeze–thawing and lacking any detergents, Suramin efficiently induced insoluble PrP and reduced the soluble PrP fraction (lanes 1 and 2).

Fig. 4. In vitro aggregation of PrP in the presence of Suramin without inducing a conformational shift in PrP from α-helix to β-sheet. (A) Suramin induces PrP aggregation in crude cellular lysates. 3F4-N2a cells were scraped off the cell culture dish and either lysed in lysis buffer (0.5% Triton X-100 and 0.5% DOC in PBS; lanes 3 and 4) or resuspended in PBS and disrupted by freeze–thawing (lanes 1 and 2). The crude lysates were incubated for 16 h at 4°C with (lanes 2 and 4) or without Suramin (lanes 1 and 3). All samples were adjusted to 0.5% Triton X-100 and DOC each, and centrifuged at 15 000 g for 20 min at 4°C. PrP present in the detergent-soluble (sup) and -insoluble phase (pellet) was analyzed by immunoblotting using the anti-PrP antiserum A4. (B) Suramin induces aggregation of purified recombinant PrP. In panels (a) and (b), studies with CD spectroscopy of recombinant PrP and re-aggregated PrP 27–30 are shown. Recombinant Syrian hamster PrP 29–231 (panel a) and PrP 90–231 (panel b) were either mock treated (bold line) or incubated with Suramin (7.5 µM shown; broken line) in sodium phosphate buffer pH 7 and directly analyzed by CD. The molar ellipticity is plotted against the wave length. In panel (b), the CD signal obtained for re-aggregated Syrian hamster PrP 27–30 is overlaid (dotted line; taken from Post et al., 1998, Figure 2). Aggregation of PrP is in good agreement with the quenching of Trp fluorescence (panel c). Recombinant PrP 90–231 was incubated with various concentrations of Suramin (0–10 µM), and the fluorescence emission was measured between 340 and 500 nm using an excitation wavelength of 280 nm.
To gain insight into structural differences between PrPSc and aggregates formed in the presence of Suramin we employed an in vitro aggregation model. Incubation of recombinantly expressed and purified PrP with increasing concentrations of Suramin yielded insoluble aggregates as analyzed by differential centrifugation (data not shown). We then determined the secondary structure of the Suramin-induced aggregates by circular dichroism (CD) and compared it with the secondary structure of re-aggregated PrP 27–30 prepared from the brains of terminally scrapie-sick Syrian hamsters (Post et al., 1998). Whereas PrP 27–30 after solubilization in 0.2% SDS and re-aggregation by removal of the SDS showed the well known β-sheet-containing conformation (Figure 4B, panel b; dotted line), Suramin-induced aggregates still showed a predominantly α-helical conformation, although decreased in the amplitude of CD spectra (panels a and b; broken lines). Since spectra from soluble and insoluble conformations of PrP are compared, they are not evaluated quantitatively in terms of α-helix and β-sheet content. However, they show unambiguously that the Suramin-induced aggregation is very similar to the predominantly α-helical conformation and quite different from the β-sheet-containing conformation, which is typical for a PrPSc-like structure. Of note, recombinant hamster PrPs 29–231 and 90–231 provided a very similar behavior (panels a and b). The analysis of the Trp fluorescence is in very good agreement with a Suramin-induced aggregation of recombinant PrP leading to a nearly total Trp quenching (panel c). The findings are characteristic for the cellular conditions of neutral or nearly neutral pH. In contrast, at acidic pH (pH 4.0–5.5) the binding mode of Suramin to PrP was quite different. Binding of Suramin and aggregation of PrP were concomitant with acquisition of β-sheet structure (data not shown).
In summary, our in vitro aggregation studies revealed that Suramin induces PrP aggregates in the absence of cellular metabolism and without inducing a conformational shift in PrP from α-helix to β-sheet, when assayed at pH conditions present in the cell. The profound biophysical differences between PrPSc and PrP aggregates induced by Suramin explained the differences in cellular life time and resistance to PK.
Suramin-induced re-routing of PrP and prophylaxis against prion diseases
Finally, we addressed in vivo the potential prophylactic use of Suramin in the case of prion inoculation from peripheral sites. In initial in vivo studies we have found that Suramin applied intravenously (i.v.) to mice significantly reduced PrP surface expression of peripheral blood leukocytes (PBL) in a dose- and time-dependent manner (our unpublished results). We then chose a combination of prion inoculation via intraperitoneal (i.p.) route and application of Suramin using the identical route. Suramin was applied 7 days prior to, simultaneously with (on the other side of the peritoneum), and 7 days after prion inoculation, yielding a total of three applications (two groups of mice with 2 and 5 mg of Suramin, respectively; Table I). Compared with mock-treated control mice, the incubation time to terminal prion disease was dose-dependently prolonged, with a difference of ∼12 days for the 2 mg group, and of ∼28 days for the 5 mg group of mice (Figure 5). This represents a delay in the onset of prion disease of 15.4 and 35.3%, respectively, when compared with the incubation time of untreated animals.
Table I. Prophylactic use of Suramin significantly prolongs prion disease incubation timea,b.
| Treatment | Mice (scrapie sick/total) | Mean incubation time to illness (days, ± SD) | Delay | Significancec p value (_t_-test) | |
|---|---|---|---|---|---|
| Daysd | %e | ||||
| No treatment | 18/18 | 78.5 (SD ± 5.3) | NA | NA | NA |
| 2 mg Suraminb | 17/17 | 90.6 (SD ± 3.9) | 12.1 | 15.4 | <0.001 |
| 5 mg Suraminb | 10/10 | 106.2 (SD ± 7.3) | 27.7 | 35.3 | <0.001 |

Fig. 5. Suramin delays onset of prion disease in scrapie-infected mice. Days to onset of terminal prion disorder of mock-treated Tg20 mice (control; n = 18), compared with mice treated with 2 mg (n = 17), and mice treated with 5 mg of Suramin (n = 10), respectively. Individual data points represent the percentage of mice with symptoms of the total number of mice for that group, and may include more than one mouse.
In summary, we could demonstrate that Suramin significantly delayed the onset of terminal prion disease when the drug was applied around the time of prion inoculation. These data demonstrate the prophylactic potential of Suramin and might indicate possible post-expositional and therapeutic strategies against prion infections.
Discussion
A profound conformational change is thought to be a decisive step in the conversion of PrPc into infectious prions. To decipher molecular events in the pathogenesis of prion diseases it is important not only to understand the nature of this conversion reaction but also to characterize cellular control mechanisms counteracting the formation of PrPSc. We could show that the compound Suramin interfered with the folding of PrPc in a post-ER compartment and that misfolded PrP was efficiently re-routed to an acidic compartment for intracellular degradation. In contrast to PrPSc, Suramin-induced PrP aggregates were sensitive to proteolytic digestion and were not infectious, explainable by the distinct biophysical properties. Moreover, we demonstrated that the Suramin-induced intracellular re-routing can be used for prophylactic approaches against prion diseases in vivo.
Suramin induces aggregation of PrPc and prevents propagation of PrPSc
The initial experiments indicated that Suramin decreased the steady state level of detergent-soluble PrP in a dose- and time-dependent manner. The apparent loss of PrP was not due to an impaired biosynthesis of PrP but rather to the formation of misfolded and aggregated PrP molecules fractionating into the detergent-insoluble phase. The effect of Suramin on folding of PrPc had a profound impact on the propagation of prions in prion-infected cells. Under Suramin treatment, ScN2a and ScGT1 cells lost PK-resistant PrPSc as well as infectious prions. Unexpectedly, Suramin-treated cells still harbored significant amounts of detergent-insoluble PrP. This Suramin-induced insoluble PrP consisted of full-length PrP, whereas the PrPSc found in murine prion-infected cells typically was already N-terminally truncated. Further analysis indicated that PrP aggregates induced by Suramin and aggregated PrPSc also possessed significantly different biochemical properties. In contrast to the highly extended half-life time of PrPSc, PrP aggregates induced by Suramin had a similar half-life time to soluble PrPc. Most notably, the PK-resistant phenotype of PrPSc was not found for PrP aggregates induced by Suramin. Using spectroscopic studies of aggregated recombinant PrPs and comparing them with re-aggregated solubilized PrP 27–30 we could show that, in fact, Suramin-induced aggregates do not possess a defined β-sheet conformation, explaining the obvious sensitivity to proteolytic digestion. The obtained results also indicate the presence of a variety of PrP isoforms that are detergent insoluble but completely unrelated to infectious PrPSc.
Our data demonstrate that insolubility of PrP in non-ionic detergent, relative PK resistance and the formation of infectious PrPSc are clearly distinct entities, and moreover, can even co-exist within a scrapie-infected cell.
Intracellular retention and re-routing of PrP: a post-ER quality control step?
We found that the detergent-insoluble PrP fraction induced by Suramin represents an aggregated population of mature and complex glycosylated PrP. This added to the assumption that Suramin interfered late with PrP folding and trafficking, probably in the mid/_trans_-Golgi or TGN. When analyzing the subcellular localization and trafficking there was no indication that PrP was present on the outer leaflet of the plasma membrane in Suramin-treated cells. Whereas sulfated glycans might stimulate the endocytosis of PrPc (Shyng et al., 1995), Suramin obviously completely prevented the plasma membrane localization of PrP. Our co-localization studies finally demonstrated that in Suramin-treated cells PrP aggregated in the Golgi/TGN and was re-routed directly to acidic compartments. This scenario is schematically depicted in Figure 6.

Fig. 6. Model of Suramin effects on PrP biogenesis and propagation of PrPSc. The biosynthesis of PrPc in the exocytotic pathway and its recycling and degradation in the endocytotic pathway are depicted (–Sur; PrPc shown by shaded circles). The putative subcellular compartment of conversion of PrPc into PrPSc in the early endocytotic pathway is marked (rafts or caveolae; Taraboulos et al., 1995). PrPSc, which is not efficiently degraded in lysosomes, is indicated by black boxes. The induction of detergent-insoluble PrP aggregates by Suramin in post-ER compartments and the direct re-routing to acidic vesicles are indicated (+Sur; red arrow; PrP depicted by red boxes). As a result of this re-routing, the plasma membrane localization and the putative compartment of prion conversion are bypassed.
Based on previous studies, Suramin was thought to act in the endocytotic pathway (Stein et al., 1995). Our report indicates that Suramin can also modulate folding of proteins in the exocytotic pathway. How does Suramin reach these compartments? It was shown before that Suramin enters endosomal compartments (Baghidiguian et al., 1996). These compartments are known to communicate with the TGN, allowing reversible cycling and cross-talk of proteins between TGN and acidic vesicles. Indeed, the entry of Suramin from endosomes to the TGN was reported for NIH 3T3 fibroblasts (Huang et al., 1997). This gives rise to a scenario where Suramin enters Golgi/TGN compartments and interacts there with already fully folded and glycosylated PrP. This interaction changes the folding state of PrP, induces aggregation and might thereby activate the cellular quality control present in Golgi/TGN compartments (Ellgaard et al., 1999). Now labeled as ‘misfolded’, PrP is marked for lysosomal degradation (Figure 6). Within the acidic vesicles, detergent-insoluble PrP is then efficiently degraded.
To our knowledge, intracellular retention in the late Golgi/TGN and subsequent re-routing is a novel quality control mechanism for the mammalian prion protein. Similar phenomena have been observed in studies with mutated prion proteins, with the important difference, however, that these PrP mutants were subjected to an ER-based quality control, probably resulting in cytosolic degradation by the proteasome (Rogers et al., 1990; DeArmond et al., 1997; Muramoto et al., 1997; Singh et al., 1997; Zanusso et al., 1999). Suramin induced aggregation of fully matured wild-type PrP in Golgi/TGN compartments, and misfolded PrP was re-routed to a lysosomal degradation pathway. This represents an example of a post-ER quality control mechanism in mammalian cells, previously described in yeast.
Folding, quality control and implications for prion disease
The formation of different PrP conformations is linked to prion diseases (Prusiner, 1998; Clarke et al., 2001; Weissmann et al., 2001). In this respect, scrapie prions can be regarded as misfolded proteins that should be subjected to cellular quality control mechanisms. How an aberrant conformation is formed and which structure exactly determines the pathological phenotype remains elusive. Studies in cell culture and animal models favor a model where scrapie propagation proceeds at the cell surface or in endosomal vesicles of infected cells. This would place the locale decisive in the formation of PrPSc outside of any known cellular quality control mechanism. In the secretory pathway of eukaryotic cells two main systems are established. In the ER, misfolded or mis-matured proteins are recognized and targeted for cytosolic degradation by the proteasome. Further distal, a second system seems to exist in the Golgi/TGN that re-routes misfolded proteins for lysosomal degradation. This system is common in yeast and further studies should establish its role in mammalian cells. Our study now demonstrates that matured PrP present in Golgi/TGN can efficiently be targeted for lysosomal degradation, if misfolding occurs. It also provides evidence that the PrP precursor required for a productive PrPSc propagation was not formed in Suramin-treated prion-infected cells. Whether the correct PrP conformation promoting conversion into PrPSc was missing, or whether PrP was excluded from the correct com partment, can not be distinguished at present. Interest ingly, PrP aggregates in Suramin-treated ScN2a cells ended up in lysosomal vesicles, a compartment where in fectious PrPSc was found in untreated ScN2a cells, yet these aggregates were not converted into scrapie prions.
Our studies emphasize a protective role of cellular quality control in the formation of infectious prions. This might be of special interest for the pathogenesis of sporadic prion diseases, and might also be relevant to the aggregation of other cellular proteins that are hallmarks in the pathogenesis of a variety of neurodegenerative disorders.
Suramin and prophylaxis against prion diseases
After elucidating in vitro the molecular mechanisms underlying the observed effects exerted by Suramin we asked whether this is of practical use in in vivo situations. The reduction of the operational prion titers of persistently prion-infected cells treated with Suramin already demonstrated that Suramin is able to interfere with prion biogenesis in living cells. A final question was whether Suramin-induced removal of surface PrP is of importance in prion neuroinvasion in vivo. Regrafting studies into PrP knockout mice combined with peripheral prion inoculation demonstrated that peripheral PrP expression is an absolute requirement for prion neuroinvasion (Blättler et al., 1997). B cells might be crucial players, although PrPc expression on B cells seems to be dispensable (Klein et al., 1997, 1998). Prophylactic effects have been found using Pentosan polysulphate (Dealler, 1998), porphyrin compounds (Priola et al., 2000) and soluble lymphotoxin beta receptor (Montrasio et al., 2000). To ultimately test our hypothesis that drug-induced removal of surface PrP is of importance in neuroinvasion, we performed prophylactic studies in mice. Treatment of mice around the time of peripheral prion inoculation with Suramin significantly delayed the onset of terminal prion disease in a dose-dependent manner (Table I). Taking all of our studies into account we showed evidence for the prophylactic and eventually therapeutic potential of Suramin in prion disease scenarios in vivo. The high and persistent blood concentrations that can be achieved by Suramin application might result in a transient and therefore conditioned knockout of peripheral surface PrPc, thereby eventually preventing the replication and/or the transport of pathological prions to the central nervous system. On the other hand, the underlying mechanism in vivo may not necessarily be related to the effects observed in cultured cells. Further studies tracking the fate of inoculated PrPSc will be necessary to address this issue. Detailed dose- and application time-finding studies are needed to use the ‘time window’ in initial prion infection—reducing the replication potential and increasing therewith the chance for clearance of prion infectivity. The combination of effective doses of Suramin with other anti-prion compounds might result in additive or even multiplied anti-prion effects combined with tolerable side effects. This raises the possibility that Suramin alone or in combination with other drugs could be used with prophylactic intentions and especially in cases of unintended or accidental exposure to prions.
Materials and methods
Reagents
The polyclonal anti-PrP antibodies kan72 and A4 and the monoclonal anti-PrP antibodies 3F4, 4F2 and SAF70 have previously been described (Hölscher et al., 1998; Winklhofer and Tatzelt, 2000). Kan72 was a gift of Dr Bürkle, Newcastle, UK; 4F2 of Dr Hunsmann, Göttingen, Germany; and SAF70 of Dr Grassi, Gif sur Yvette, France.
Cell culture and expression of 3F4-tagged PrP
The mouse neuroblastoma cell lines N2a and ScN2a and the hypothalamic lines GT1/ScGT1 have been described (Tatzelt et al., 1995; Schätzl et al., 1997). Cells were maintained in Dulbecco’s modified Eagle’s (DMEM) or Opti-MEM medium containing 10% fetal calf serum, antibiotics and glutamin. Suramin (Bayer) was dissolved in NaCl (0.9%) at a stock concentration of 200 mg/ml (light protected at 4°C). 3F4-N2a and 3F4-ScN2a cells represent stably transfected clones of N2a/ScN2a cells that overexpress 3F4-epitope-tagged murine PrP. Residues 109 and 112 (numbering according to Schätzl et al., 1995; Wopfner et al., 1999) of murine PrP were replaced by methionine. For generation of stable transfectants we used the vector pcDNA3.1/Zeo (Invitrogen, Leek, The Netherlands). Lipofection of cells with recombinant plasmids was carried out using standard procedures and recombinant clones were selected by addition of 300 µg of Zeocin/ml medium.
Immunoblot and PK analysis
Confluent cell cultures were lysed in cold lysis buffer (10 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM EDTA, 0.5% Triton X-100, 0.5% DOC). Postnuclear lysates were split between those with and without PK digestion. Samples without PK digestion were supplemented with proteinase inhibitors (5 mM phenylmethylsulfonyl fluoride, 0.5 mM Pefabloc, and aprotinin) and directly precipitated with ethanol. Samples for PK digestion were incubated with 20 µg/ml PK for 30 min at 37°C; digestion was stopped with proteinase inhibitors, and samples were ethanol precipitated. After centrifuging for 30 min at 2500 g, the pellets were redissolved in TNE buffer and gel loading buffer was then added. After boiling for 5 min an aliquot was analyzed on 12.5% PAGE. For western blot analysis, the proteins were electrotransferred to polyvinylene difluoride membranes. The membrane was blocked with 5% non-fat dry milk in TBST (0.05% Tween 20, 100 mM NaCl, 10 mM Tris–HCl pH 7.8), incubated overnight with the primary antibody at 4°C, and stained using the enhanced chemiluminescence blotting kit from Amersham Corporation.
Detergent solubility assay
Cells were lysed as described for immunoblot analysis. Postnuclear cell lysates were supplemented with proteinase inhibitors and _N_-lauryl sarcosine to 1%, and ultracentrifuged in a Beckman TL-100 ultracentrifuge for 1 h at 100 000 g, 4°C. Supernatant fractions (soluble fraction) were precipitated with ethanol; pellet fractions (insoluble fraction) were resuspended in 50 µl of TNE (50 mM Tris–HCI pH 7.5, 150 mM NaCl, 5 mM EDTA) and analyzed in immunoblot or RIPA assays.
Metabolic radiolabeling and immunoprecipitation assay (RIPA)
Confluent cells were washed twice with phosphate-buffered saline (PBS) and incubated for 1 h in RPMI without methionine/cysteine containing 1% fetal calf serum (FCS) (+/– Suramin). The medium was supplemented with 400 µCi/ml [35S]l-Met/Cys (Amersham) for 2 h (label; +/– Suramin) and cells were chased for various periods as indicated (+/– Suramin). After incubation, cells were washed twice in cold PBS and lysed in cold lysis buffer (100 mM NaCl, 10 mM Tris–HCl pH 7.8, 10 mM EDTA, 0.5% Triton X-100, 0.5% DOC). Postnuclear lysates were subjected to ultracentrifugation (1 h at 100 000 g) in the presence of 1% sarcosyl. Pellets were resuspended in 100 µl of RIPA buffer (0.5% Triton X-100, 0.5% DOC, in PBS) with 1% SDS, and supernatant and pellet fractions were boiled for 10 min. The pellet fraction was diluted with 900 µl of RIPA buffer (supplemented with 1% sarcosyl). The primary antibody was incubated overnight at 4°C; Protein A–Sepharose beads were added for 90 min at 4°C. The immuno-adsorbed proteins were washed in RIPA buffer supplemented with 1% SDS and analyzed on 12.5% SDS–PAGE followed by autoradiography.
PIPLC release and PNGase F treatment
For PIPLC treatment, cells were washed with PBS; 400 mU of PIPLC (Boehringer Mannheim) were added in serum-free DMEM and incubated for 4 h at 37°C. Total postnuclear cell lysates were tested directly by immunoblotting. For PNGase F analysis the pellet fraction after ultracentrifugation was resuspended in 500 µl of lysis buffer. After heating for 10 min at 94°C, an aliquot was supplemented with 5 U of PNGase F (Boehringer Mannheim) and incubated for 24 h at 37°C. The reaction was stopped by ethanol precipitation and the pellet analyzed by immunoblotting.
FACS analysis
For surface protein analysis, cells were suspended in PBS containing 1 mM EDTA, centrifuged, resuspended in FACS buffer (PBS with 2.5% FCS and 0.05% Na-acid) and incubated for 5 min on ice. Primary antibodies were incubated in FACS buffer for 45 min on ice, washed three times, and the secondary antibody (FITC-labeled) incubated for another 45 min. After the last wash, cells were resuspended in FACS buffer with propidium iodide (2 µg/ml). For intracellular protein detection, saponin buffer was used (0.1% saponin in FACS buffer) instead of FACS buffer; cells were fixed in 4% paraformaldehyde (PFA) and quenched. The FACS analysis was performed in a Becton Dickinson FACSCalibur apparatus.
Confocal laser scanning microscopy
Cells were plated at low density 1–3 days prior to staining. Suramin and leupeptin (Boehringer Mannheim) were applied at 100 µg/ml (overnight and for 6 h, respectively). Cells were washed twice in cold PBS and fixed in 4% PFA for 30 min at room temperature. After sequential treatment with NH4Cl (50 mM in 20 mM glycine), Triton X-100 (0.3%) and gelatine (0.2%) for 10 min each, the first antisera were added in PBS and incubated for 30 min. After three washes in PBS, FITC- and Rhodamine-conjugated secondary antisera were used and immunostaining was accomplished according to standard procedures. For staining of surface proteins, cells were incubated for 30 min on ice with a cell membrane-impermeable biotinylation compound for surface biotinylation. FITC-conjugated streptavidin was used for completion of staining. For staining of lysosomes, 30 µM DAMP was added to the cell medium (of cells pretreated with leupeptin) for 30 min, cells were washed, and fresh medium was added for another 30 min. Staining was completed using FITC-conjugated anti-DAMP antibody (Molecular Probes, Eugene, OR) at 1:70 in PBS. Slides were mounted in anti-fading solution (5% propyl gallate in 70% glycerol, 100 mM Tris–HCl pH 9.0) and kept dry at –20°C. Confocal laser scanning microscopy was carried out using a Leica TCSNT/DMIRB Confocal System (Heerbrugg, Switzerland).
In vitro aggregation
Cells were washed twice, scraped off the cell culture dish in a small volume of PBS buffer and disrupted by freeze–thawing. Suramin was added and the aliquots were incubated for 16 h at 4°C. The solution was adjusted to 0.5% DOC and Triton X-100 each, and analyzed by the detergent solubility assay as described (Winklhofer and Tatzelt, 2000).
Recombinant PrP and spectroscopic studies
Lyophilized recombinant Syrian hamster PrP 29–231 and PrP 90–231, and infectious Syrian hamster PrP 27–30 were kindly provided by Prof. S.B.Prusiner, San Francisco, CA. Denaturation and refolding of lyophilized recombinant hamster PrPs as well as re-aggregation of solubilized PrP 27–30 were carried out as described (Post et al., 1998). CD measurement and fluorescence measurement of recombinant PrPs were performed in the presence of various concentrations of Suramin (ranging from 0–10 µM; the latter represents a molar ratio of ∼3:1 in relation to recombinant PrP). The preparations contained 50 ng/µl rec ha PrP and various concentrations of Suramin, as indicated, in 1 mM sodium phosphate buffer pH 7.0. Each sample was prepared freshly and measured straight away after addition of Suramin. CD spectra were recorded with a Jasco J715 spectropolarimeter (Jasco Labor- und Datentechnik GmbH, Germany), from 188 nm to 260 nm. Fluorescence emission of Suramin was scanned from 340 nm to 500 nm using an excitation wavelength of 315 nm.
Prophylactic use of Suramin in vivo
For studies addressing prophylaxis, Tg20 mice, which overexpress murine PrP, were inoculated with a 10% homogenate of brain prepared from terminally sick CD-1 mice infected with the RML scrapie strain. The mean titer of this inoculum has been tested by end-point titrations. One hundred microliters were used for i.p. injection. Mice were monitored every second day, and scrapie was diagnosed according to standard clinical criteria. For Suramin prophylaxis, three doses of Suramin (2 or 5 mg each) were administered intraperitoneally into mice (in 100 µl). Suramin was applied 7 days prior to prion inoculation, simultaneously with prion inoculation (into different sides of the peritoneum), and 7 days post inoculation.
Acknowledgments
Acknowledgements
We are grateful to U.Koszinowski for his continuous support; to S.B.Prusiner, G.Hunsmann, J.Grassi and A.Bürkle for providing antisera and other materials; to E.Wolf for performing Suramin in vivo studies; and to A.Weber for performing the bioassays. This work was supported in part by grants from the Volkswagen-Foundation (I/72765), the Friedrich-Baur Foundation (21/97), Munich, Germany, the DFG (SCHA 594/3-3; TA 167/2), the BMBF of Germany (01KI9758), and by the EU BIOMED (contracts BMH4-CT98–6040; BMH4-CT98–6050) program.
References
- Baghidiguian S., Boudier,J.A., Boudier,J.L. and Fantini,J. (1996) Co-localization of suramin and serum albumin in lysosomes of suramin-treated human colon cancer cells. Cancer Lett., 101, 179–184. [DOI] [PubMed] [Google Scholar]
- Blättler T., Brandner,S., Raeber,A.J., Klein,M.A., Voigtlander,T., Weissmann,C. and Aguzzi,A. (1997) PrP-expressing tissue required for transfer of scrapie infectivity from spleen to brain. Nature, 389, 69–73. [DOI] [PubMed] [Google Scholar]
- Borchelt D.R., Taraboulos,A. and Prusiner,S.B. (1992) Evidence of synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem., 267, 16188–16199. [PubMed] [Google Scholar]
- Caughey B. and Raymond,G.J. (1991) The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem., 266, 18217–18223. [PubMed] [Google Scholar]
- Cohen F.E., Pan,K.-M., Huang,Z., Baldwin,M., Fletterick,R.J. and Prusiner,S.B. (1994) Structural clues to prion replication. Science, 264, 530–531. [DOI] [PubMed] [Google Scholar]
- Clarke A.R., Jackson,G.S. and Collinge,J. (2001) The molecular biology of prion propagation. Philos. Trans. R. Soc. Lond. B Biol. Sci., 356, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dealler S. (1998) Post-exposure prophylaxis after accidental prion inoculation. Lancet, 351, 600. [DOI] [PubMed] [Google Scholar]
- DeArmond S.J. et al. (1997) Selective neuronal targeting in prion disease. Neuron, 19, 1337–1348. [DOI] [PubMed] [Google Scholar]
- Dressel J. and Oesper,R.E. (1961) The discovery of Germanin by Oskar Dressel and Richard Kothe. J. Chem. Educ., 38, 620–621. [Google Scholar]
- Ellgaard L., Molinari,M. and Helenius,A. (1999) Setting the standards: quality control in the secretory pathway. Science, 286, 1882–1888. [DOI] [PubMed] [Google Scholar]
- Hölscher C., Delius,H. and Bürkle,A. (1998) Overexpression of nonconvertible PrPcΔ114-121 in scrapie-infected neuroblastoma cells leads to trans-dominant inhibition of wild-type PrPSc accumulation. J. Virol., 72, 1153–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.S., Koh,H.A. and Huang,J.S. (1997) Suramin enters and accumulates in low pH intracellular compartments of v-sis-transformed NIH 3T3 cells. FEBS Lett., 416, 297–301. [DOI] [PubMed] [Google Scholar]
- Klein M.A. et al. (1997) A crucial role for B cells in neuroinvasive scrapie. Nature, 390, 687–690. [DOI] [PubMed] [Google Scholar]
- Klein M.A., Frigg,R., Raeber,A.J., Flechsig,E., Hegyi,I., Zinkernagel, R.M., Weissmann,C. and Aguzzi,A. (1998) PrP expression in B lymphocytes is not required for prion neuroinvasion. Nature Med., 4, 1429–1433. [DOI] [PubMed] [Google Scholar]
- Ladogana A., Casaccia,P., Ingrosso,L., Cibati,M., Salvatore,M., Xi,Y.G., Masullo,C. and Pocchiari,M. (1992) Sulphate polyanions prolong the incubation period of scrapie-infected hamsters. J. Gen. Virol., 73, 661–665. [DOI] [PubMed] [Google Scholar]
- Montrasio F., Frigg,R., Glatzel,M., Klein,M.A., Mackay,F., Aguzzi,A. and Weissmann,C. (2000) Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science, 288, 1257–1259. [DOI] [PubMed] [Google Scholar]
- Muramoto T, DeArmond,S.J., Scott,M., Telling,G.C., Cohen,F.E. and Prusiner,S.B. (1997) Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an α-helix. Nature Med., 3, 750–755. [DOI] [PubMed] [Google Scholar]
- Post K. et al. (1998) Rapid acquisition of β-sheet structure in the prion protein prior to multimer formation. Biol. Chem., 379, 1307–1317. [DOI] [PubMed] [Google Scholar]
- Priola S., Raines,A. and Caughey,W.S. (2000) Porphyrin and phthalocyanine antiscrapie compounds. Science, 287, 1503–1506. [DOI] [PubMed] [Google Scholar]
- Prusiner S.B. (1998) Prions. Proc. Natl Acad. Sci. USA, 95, 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers M., Taraboulos,A., Scott,M., Groth,D. and Prusiner,S.B. (1990) Intracellular accumulation of the cellular prion protein after mutagenesis of its Asn-linked glycosylation sites. Glycobiology, 1, 101–109. [DOI] [PubMed] [Google Scholar]
- Schätzl H., DaCosta,M., Taylor,L., Cohen,F. and Prusiner,S.B. (1995) Prion protein gene variation among primates. J. Mol. Biol., 245, 362–374. [DOI] [PubMed] [Google Scholar]
- Schätzl H., Laszlo,L., Holtzman,D.M., Tatzelt,J., Weiner,R.I., Mobley, W. and Prusiner,S.B. (1997) A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J. Virol., 71, 8821–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S.-L., Lehmann,S., Moulder,K.L. and Harris,D.A. (1995) Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPc, in cultured cells. J. Biol. Chem., 270, 30221–30229. [DOI] [PubMed] [Google Scholar]
- Singh N., Zanusso,G., Chen,S.G., Fujioka,H., Richardson,S., Gambetti,P. and Petersen,R.B. (1997) Prion protein aggregation reverted by low temperature in transfected cells carrying a prion protein gene mutation. J. Biol. Chem., 272, 28461–28470. [DOI] [PubMed] [Google Scholar]
- Stein C.A., Khan,T.M., Khaled,Z. and Tonkinson,J.L. (1995) Cell surface binding and cellular internalization properties of suramin, a novel antineoplastic agent. Clin. Cancer Res., 1, 509–517. [PubMed] [Google Scholar]
- Taraboulos A., Scott,M., Semenow,A., Avrahami,A., Laszlo,L., Prusiner,S.B. and Avraham,D. (1995) Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol., 129, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatzelt J., Zuo,J., Voellmy,R., Scott,M., Hartl,U., Prusiner,S.B. and Welch,W.J. (1995) Scrapie prions selectively modify the stress response in neuroblastoma cells. Proc. Natl Acad. Sci. USA, 92, 2944–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatzelt J., Prusiner,S.B. and Welch,W.J. (1996) Chemical chaperones interfere with the formation of scrapie prion protein. EMBO J., 15, 6363–6373. [PMC free article] [PubMed] [Google Scholar]
- Weissmann C., Raeber,A.J., Montrasio,F., Hegyi,I., Frigg,R., Klein,M.A. and Aguzzi.A. (2001) Prions and the lymphoreticular system. Philos. Trans. R. Soc. Lond. B Biol. Sci., 356, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklhofer K.F. and Tatzelt,J. (2000) Cationic lipopolyamines induce degradation of PrPSc in scrapie-infected mouse neuroblastoma cells. Biol. Chem., 381, 463–469. [DOI] [PubMed] [Google Scholar]
- Wopfner F., Weidenhöfer,G., Schneider,R., Gilch,S., von Brunn,A., Schwarz,T.F., Werner,T. and Schätzl,H.M. (1999) Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J. Mol. Biol., 289, 1163–1178. [DOI] [PubMed] [Google Scholar]
- Zanusso G., Petersen,R.B., Jin,T., Jing,Y., Kanoush,R., Ferrari,S., Gambetti,P. and Singh,N. (1999) Proteasomal degradation and N-terminal protease resistance of the codon 145 mutant prion protein. J. Biol. Chem., 274, 23396–23404. [DOI] [PubMed] [Google Scholar]