Evidence for cocaine and methylecgonidine stimulation of M2 muscarinic receptors in cultured human embryonic lung cells (original) (raw)
Abstract
- Muscarinic cholinoceptor stimulation leads to an increase in guanylyl cyclase activity and to a decrease in adenylyl cyclase activity. This study examined the effects of cocaine and methylecgonidine (MEG) on muscarinic receptors by measurement of cyclic GMP and cyclic AMP content in cultured human embryonic lung (HEL299) cells which specifically express M2 muscarinic receptors.
- A concentration-dependent increase in cyclic GMP production was observed in HEL299 cells incubated with carbachol, cocaine, or MEG for 24 h. The increase in cyclic GMP content was 3.6 fold for 1 μM carbachol (P<0.01), 3.1 fold for 1 μM cocaine (P<0.01), and 7.8 fold for 1 μM MEG (P<0.001), respectively. This increase in cyclic GMP content was significantly attenuated or abolished by the muscarinic receptor antagonist atropine or the M2 blocker methoctramine.
- In contrast, cocaine, MEG, and carbachol produced a significant inhibition of cyclic AMP production in HEL299 cells. Compared to the control, HEL299 cells treated with 1 μM cocaine decreased cyclic AMP production by 30%. MEG and carbachol at 1 μM decreased cyclic AMP production by 37 and 38%, respectively. Atropine or methoctramine at 1 or 10 μM significantly attenuated or abolished the cocaine-induced decrease in cyclic AMP production. However, the antagonists alone had neither an effect on cyclic GMP nor cyclic AMP production. Pretreatment of HEL299 cells with pertussis toxin prevented the cocaine-induced reduction of cyclic AMP production.
- Western blot analysis showed that HEL299 cells specifically express M2 muscarinic receptors without detectable M1 and M3. Incubation of HEL299 cells with cocaine, carbachol, and atropine did not alter the expression of M2 protein levels. However, the inducible isoform of nitric oxide synthase (iNOS) was induced in the presence of cocaine or carbachol and this induction was significantly attenuated after addition of atropine or methoctramine.
- The present data show that cocaine and MEG significantly affect cyclic GMP and cyclic AMP production in cultured HEL299 cells. Our results also show that these effects result from the drug-induced stimulation of M2 muscarinic receptors accompanied with no alterations of receptor expression. However, the induction of iNOS by cocaine may result in the increase in cyclic GMP production.
Keywords: Cocaine, methylecgonidine, carbachol, cyclic AMP, cyclic GMP, muscarinic receptor
Introduction
It is well known that acetylcholine (ACh) decreases heart rate, slows atrial-ventricular conduction, and attenuates catecholamine-stimulated effects. Several recent studies show that ACh had a direct chronotropic and negative inotropic effect on ferret and rat ventricular myocytes (Boyett et al., 1988; McMorn et al., 1993). In addition, ventricular myocytes isolated from cat, guinea-pig, and human hearts (Koumi & Wasserstrom, 1994), as well as from ferret hearts (Xiao & Morgan, 1998) expressed ACh-activated muscarinic K+ channels. Muscarinic cholinoceptor stimulation triggers its signal transduction system that leads to activation of guanylyl cyclase and an inhibition of adenylyl cyclase (Richardson et al., 1991). The cardiac negative inotropic effect of ACh was accompanied by a decrease in intracellular cyclic AMP content (Boyett et al., 1988; Fleming et al., 1987; Fleming & Watanabe, 1988), and at the same time by an increase in intracellular cyclic GMP production (George et al., 1970; Goldberg et al., 1975). Muscarinic receptor agonists reduced isoproterenol-stimulated contractility either by preventing the isoproterenol-enhanced formation of cyclic AMP via inhibition of adenylyl cyclase (Murad et al., 1962; Watson et al., 1988), or by stimulating the catabolism of cyclic AMP via activation of the cyclic GMP-dependent phosphodiesterase (Fischmeister & Hartzell, 1987). In contrast, MacDonell et al. (1995) reported that change in intracellular cyclic GMP levels was not responsible for the inhibitory effects of muscarinic receptor agonists on the contractility of rat ventricular cardiomyocytes during catecholamine stimulation. The mechanism underlying the muscarinic receptor-mediated inhibition of cyclic AMP-dependent physiological responses remains to be delineated (Ramesh et al., 1998).
Cocaine use has climbed rapidly and become a serious social and medical problem in many countries. A large variety of cardiovascular diseases have been associated with cocaine abuse, including acute myocardial ischaemia and infarction (Cregler & Mark, 1985; Pasternack et al., 1985; Isner et al., 1986), arrhythmias and sudden death (Nanji & Filipenko 1984; Benchimol et al., 1978; Isner et al., 1986), myocarditis (Virmani et al., 1988), cardiomyopathy (Weiner et al., 1986), hypertension (Resnick et al., 1977), accelerated atherosclerosis (Dressler et al., 1990), and endocarditis (Chambers et al., 1987). However, the mechanism about how cocaine affects the heart has not yet been fully elucidated. Our recent studies indicate that cocaine and MEG at relatively low concentrations produced a negative inotropic effect in ferret myocardium or isolated ventricular myocytes (Miao et al., 1996; Huang et al, 1997; Woolf et al., 1997). This negative inotropic effect could be abolished by addition of the muscarinic antagonist atropine and methoctramine, a selective M2 receptor blocker, but not by M1 (pirenzepine) and M3 (4-diphenylacetoxy-N-methylpiperidine methiodide) blocking agents (Huang et al., 1997). In addition, large doses of MEG caused structural damage to myocytes. These results suggest that the negative inotropic effects of cocaine and MEG may result from, at least in part, their muscarinic receptor stimulation.
Multiple muscarinic receptor subtypes have been identified in cardiac tissue of different species, but the M2 receptor subtype predominates in the heart (Goyal, 1989; Pappano & Mubagwa, 1992). In our previous study pharmacological methods were used to test muscarinic stimulation and the negative inotropic effects of cocaine and MEG in isolated ferret cardiomyocytes (Huang et al., 1997). The negative inotropic effects of cocaine and MEG were blocked by the M2 receptor blocker methoctramine, but not by M1 or M3 blocking agents. Selectivity and potency are always concerned when antagonists are used to distinguish receptor subtype functions. To provide conclusive evidence for our hypothesis of cocaine and MEG stimulation of M2 muscarinic receptors we used HEL299 cells, which specifically express the M2 receptor subtype, in the present experiments. The effects of cocaine and MEG on cyclic GMP and cyclic AMP production were investigated in cultured HEL299 cells. We also tested the effects of the cholinergic agonist carbachol and the muscarinic antagonists, atropine and methoctramine (a selective M2 blocker), on cyclic GMP and cyclic AMP levels in control and cocaine- or MEG-treated HEL299 cells. Western blot analysis was performed to determine whether the effects of cocaine and MEG on cyclic GMP and cyclic AMP production resulted from a stimulation of M2 protein expression. Other muscarinic receptor subtypes, such as M1 and M3 were also determined in cultured HEL299 cells.
Methods
Cell culture
All tissue culture media and supplements were obtained from Gibco BRL (Grand Island, NY, U.S.A.). Culturing flasks and dishes were from Becton Dickinson Company (Franklin Lakes, NJ, U.S.A.). Human embryonic lung (HEL299) cells, which specifically express M2 muscarinic receptors, were obtained from American Type Culture Collection (Manassas, VA, U.S.A.) and cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing non-essential amino acids, 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, at 37°C, in 5% CO2/humidified air. After incubation of cultured HEL299 cells with different concentrations of cocaine, MEG, carbachol and atropine for 24 h confluent cultures were harvested for Western Blotting analysis and cyclic AMP and cyclic GMP assay. Atropine and methoctramine (Sigma, St. Louis, MO, U.S.A.) was added to culture dishes 30 min ahead of the treatment of cocaine, MEG or carbachol to block muscarinic receptors. Pertussis toxin (PTX) obtained from Sigma was applied 3 h ahead of other treatments.
Measurement of cyclic AMP and cyclic GMP
Both cyclic AMP and cyclic GMP were measured by using commercially available enzyme immunoassay kits (Cayman Chemical Company, Ann Arbor, MI, U.S.A.) and by following the manufacturer's procedures. In brief, harvested cells were sonicated and centrifuged at 14,000×g for 10 min at 4°C. A small amount (50 μl) of supernatant was added to each well of a 96-well microtilter plate coated with anti-rabbit IgG monoclonal antibody. The plate was incubated for 18 h at room temperature. After rinsing five times with wash buffer (TBS containing 0.05% Tween-20), optimum development was obtained by incubating with 200 μl of Ellman's Reagent (provided by kit) in the dark for 90 min. The plate was read at 405 nm by using a Thermo max Microplate Reader (Molecular Devices Co., Menlo Park, CA, U.S.A.).
Immunoprecipitation and Western blot analysis
Dishes with 100 mm diameter containing confluent cells were washed three times with cold PBS. Cells were lysed in 10 mM Tris pH 7.4, 1 mM sodium orthovanadate, 1% SDS, 0.1 mM leupeptin and sonicated. Samples were centrifuged at 14,000×g for 5 min at 4°C. Protein concentrations of the supernatant were determined with a Bio-Rad Protein Assay Kit (Life Science, Hercules, CA, U.S.A.). One hundred microlitres total lysate was incubated with 0.5 – 5 μg of anti-goat muscarinic 1, 2, and 3 receptor polyclonal Ig G (Santa Cruz Biotechnology Inc., Santa Cruz, CA, U.S.A.) at 4°C for 1 h. After addition of 10 μl of Protein G Plus-Agarose (Calbiochem, Cambridge, MA, U.S.A.), samples were incubated with agitation overnight at 4°C. Agarose beads were resuspended in 30 μl of sample buffer (250 mM Tris pH 6.8, 4% SDS, 10% glycerol, 0.006% bromophenol blue, 2% β-mercaptoethanol) and boiled for 5 min before electrophoresis. After separation on 10% SDS – PAGE, proteins were transferred to nitrocellulose. The membrane was blocked in 5% nonfat dry milk in TBS with 0.1% Tween-20 (TTBS) for 2 h. Blots were incubated with specific antibodies to M1, M2, and M3 muscarinic receptors at 37°C for 1 h. After three TTBS washes, the membrane was incubated with an anti-goat peroxidase-conjugated antibody (Sigma Chemical Co., St. Louis, MO, U.S.A.) at 37°C for 30 min. Blots were detected with Western Blotting Luminol Reagent (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) and exposed to autoradiographic X-ray film and bands were quantified with ImageQuant (Molecular Dynamic, Sunnyvale, CA, U.S.A.) software.
Data analysis
To correct for variation in the number of cells, the data for cyclic GMP and cyclic AMP were normalized to total protein content. The expression levels of M2 receptor protein after HEK299 cells treated with different agents were normalized to cyclophilin A internal standard during the blotting measurement. Data were presented as mean±s.e.mean unless otherwise noted. Statistical differences between two groups were determined by the unpaired Student's _t_-test. Data derived from more than two experimental groups were tested by one-way analysis of variance (ANOVA). When a P value was less than 0.05, statistical significance of the difference was considered.
Results
Effect of carbachol on cyclic GMP and cyclic AMP production in HEL299 cells
Carbachol is a potent cholinergic stimulator. To determine whether HEL299 cells express muscarinic receptors and what was the effect of carbachol on cyclic GMP and cyclic AMP production, cultured HEL299 cells were incubated with 1 μM carbachol with or without 10 μM atropine for 24 h. Figure 1A shows that 1 μM carbachol significantly enhanced cyclic GMP production and reduced cyclic AMP content. The value of cyclic GMP content was increased from 1.28±0.08 pmol (mg protein)−1 for the control to 4.54±0.25 pmol (mg protein)−1 for carbachol (_n_=4, P<0.01). In contrast, cyclic AMP content was decreased from 612.7±30.3 pmol (mg protein)−1 for the control to 380.3±25.3 pmol (mg protein)−1 for carbachol (Figure 1B, _n_=4, P<0.05).
Figure 1.
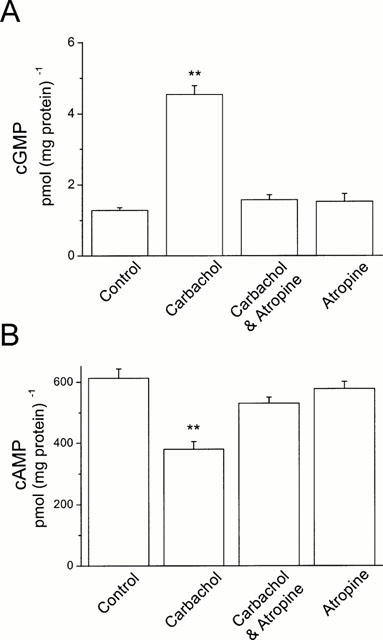
Effects of carbachol on cyclic GMP and cyclic AMP production in cultured HEL299 cells. (A) Content of cyclic GMP was measured in the absence (Control, _n_=4) and presence of 1 μM carbachol (Carbachol, _n_=4; P<0.01 vs control), 1 μM carbachol plus 10 μM atropine (Carbachol and Atropine, _n_=4; _P_>0.05 vs control), 10 μM atropine alone (Atropine, _n_=4; _P_>0.05 vs control). (B) Content of cyclic AMP was detected in the absence (Control, _n_=4) and presence of 1 μM carbachol (Carbachol, _n_=4; P<0.01 vs control), 1 μM carbachol plus 10 μM atropine (Carbachol and Atropine, _n_=4; _P_>0.05 vs control), 10 μM atropine alone (Atropine, _n_=4; _P_>0.05 vs control). Statistical significance was tested with unpaired Student's _t_-test. **P<0.01 vs control.
To test whether these effects of carbachol on cyclic GMP and cyclic AMP production were via stimulation of muscarinic receptors, the nonselective muscarinic antagonist atropine (10 μM) was applied to block muscarinic receptors in cultured HEL299 cells. While atropine alone had no significant effect on cyclic GMP and cyclic AMP levels in HEL299 cells, pre-application of atropine abolished the increase in cyclic GMP content and the decrease in cyclic AMP production in carbachol-treated HEL299 cells (Figure 1). These results demonstrate that HEL299 cells express ACh muscarinic receptors and carbachol stimulation increases cyclic GMP content and reduces cyclic AMP production.
Cocaine-induced alteration of cyclic GMP and cyclic AMP levels in HEL299 cells
Recently, we found that a negative inotropic effect of cocaine in ferret myocytes might result from a stimulation of muscarinic receptors, probably M2, because atropine and methoctramine (M2 blocker) abolished cocaine's effect (Huang et al., 1997). To test the hypothesis of cocaine stimulation of muscarinic receptors, specifically M2, we incubated HEL299 cells, which specifically express M2 muscarinic receptors, with cocaine for 24 h. The effects of cocaine on cyclic GMP and cyclic AMP production were evaluated in cocaine-treated cells. Figure 2A shows that cocaine at 1 μM significantly enhanced cyclic GMP production. The increase in cyclic GMP content was from 1.28±0.08 pmol (mg protein)−1 for the control to 4.01±0.35 pmol (mg protein)−1 for 1 μM cocaine (P<0.01, _n_=4). Pre-treatment of HEL299 cells with 10 μM atropine or 1 μM methoctramine prevented the cocaine-induced increase in cyclic GMP production (Figure 2A). The effect of cocaine on cyclic GMP production was concentration-dependent (Figure 2B). For example, cocaine at the highest concentration (100 μM) tested in our experiments increased cyclic GMP content approximately 4 fold, 5.8±0.71 pmol (mg protein)−1. The cocaine-induced increase in cyclic GMP production was almost abolished in HEL299 cells pre-treated with 10 μM atropine, especially at the points of 1 and 10 μM cocaine (Figure 2B).
Figure 2.
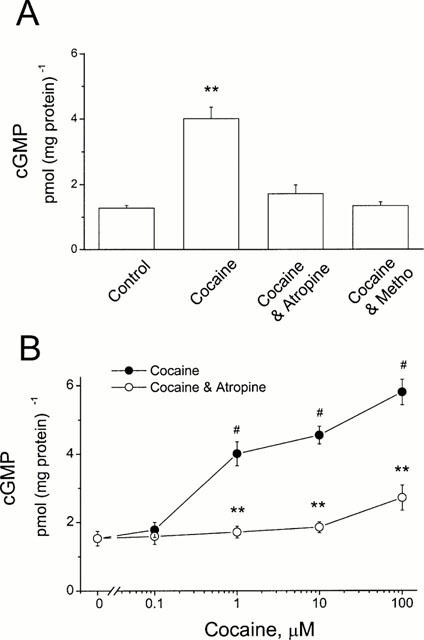
Enhancement of cyclic GMP production in HEL299 cells incubated with cocaine. (A) Compared to the control, content of cyclic GMP was significantly increased in the presence of 1 μM cocaine (Cocaine, _n_=4; P<0.01 vs control). Addition of 10 μM atropine (Cocaine and Atropine, _n_=4, _P_>0.05 vs control) or 1 μM methoctramine (Cocaine and Metho, _n_=5, _P_>0.05 vs control)) abolished the cocaine-induced increase in cyclic GMP production. (B) Concentration-dependent increases in cyclic GMP levels were observed in HEL299 cells incubated with various concentrations of cocaine. Addition of 10 μM atropine significantly attenuated the cocaine-induced enhancement of cyclic GMP production. Each point represents the mean±s.e.mean of at least four measurements. **P<0.01 (tested by unpaired Student's _t_-test between cocaine and cocaine plus atropine). #P<0.01 (tested by ANOVA among control and treated groups with various concentrations of cocaine alone or plus atropine).
In another series of experiments the effects of cocaine on cyclic AMP levels were assessed in cultured HEL299 cells. Figure 3A shows that a 30% reduction of cyclic AMP was observed in HEL299 cells incubated with 1 μM cocaine. Content of cyclic AMP was decreased from 614±22 for the control to 431±19 pmol (mg protein)−1 for 1 μM cocaine (P<0.01, _n_=6). Pre-incubation of HEL299 cells with atropine at 10 μM or the M2 blocker methoctramine at 1 μM abolished the cocaine-induced decrease in cyclic AMP production (Figure 3A). The cocaine-induced decrease in cyclic AMP production was concentration-dependent (Figure 3B). Cocaine at 100 μM decreased cyclic AMP production by 245±14 pmol (mg protein)−1 (P<0.01, _n_=4). Atropine (10 μM) markedly attenuated the cocaine-induced changes in cyclic AMP content (Figure 3B). In addition, a lower concentration (1 μM) of atropine had a smaller effect on the cocaine-induced decrease in cyclic AMP production (data not shown). For example, atropine blocked the effects of 1 μM cocaine on cyclic AMP production by 50% (_n_=6) and 76% (_n_=8) for 1 and 10 μM of the muscarinic antagonist, respectively.
Figure 3.
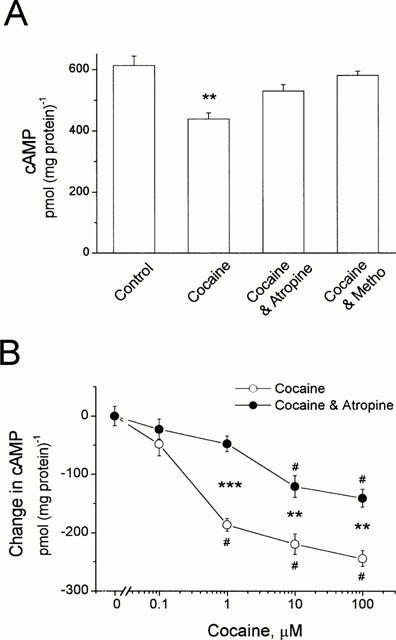
Inhibition of cyclic AMP production in HEL299 cells incubated with cocaine. (A) Incubation of HEL299 cells with 1 μM cocaine (Cocaine) significantly reduced cyclic AMP content (_n_=4; P<0.01 vs control). Addition of 10 μM atropine (Cocaine and Atropine, _n_=8, _P_>0.05) or 1 μM methoctramine (Cocaine and Metho, _n_=4, _P_>0.05) abolished the cocaine-induced decrease in cyclic AMP production. (B) Concentration-dependent changes in cyclic AMP content were observed in HEL299 cells incubated with various concentrations of cocaine. The differences of cyclic AMP levels between control and cocaine alone, and between control and cocaine plus atropine are shown. Addition of 10 μM atropine significantly attenuated the cocaine-induced decrease in cyclic AMP production. Each point represents the mean±s.e.mean of at least four measurements. **P<0.01 and ***P<0.001 (tested by unpaired Student's _t_-test between cocaine and cocaine plus atropine). #P<0.01 (tested by ANOVA among control and treated groups with various concentrations of cocaine alone or plus atropine).
To test whether co-application of cocaine and carbachol had a synergic effect on cyclic GMP and cyclic AMP production, HEL299 cells were treated with these compounds for 24 h in the absence or presence of atropine. Figure 4A shows that 1 μM cocaine plus 1 μM carbachol significantly increased cyclic GMP production from 1.28±0.08 pmol (mg protein)−1 for the control to 5.61±0.35 pmol (mg protein)−1 for the treatment (P<0.01, _n_=4). In contrast, cyclic AMP levels were decreased in the presence of 1 μM cocaine plus 1 μM carbachol, from 612.8±30.3 pmol (mg protein)−1 for the control to 358.8±37.3 pmol (mg protein)−1 for the treatment (Figure 4B, _P_<0.01, _n_=4). The effects of cocaine plus carbachol on either cyclic GMP or cyclic AMP production were abolished by addition of 10 μM atropine (Figure 4). It is interesting to note that the effects of 1 μM cocaine plus 1 μM carbachol on cyclic GMP and cyclic AMP production are greater than those of 1 μM cocaine alone (_P_<0.05, _n_=4), but similar to those of 1 μM carbachol alone (_P_>0.05, _n_=4). These results suggest that carbachol is a more potent agent than cocaine in terms of muscarinic receptor stimulation, because carbachol alone at 1 μM produced almost maximal stimulation of muscarinic receptors in cultured HEL299 cells, but cocaine alone at 1 μM did not.
Figure 4.
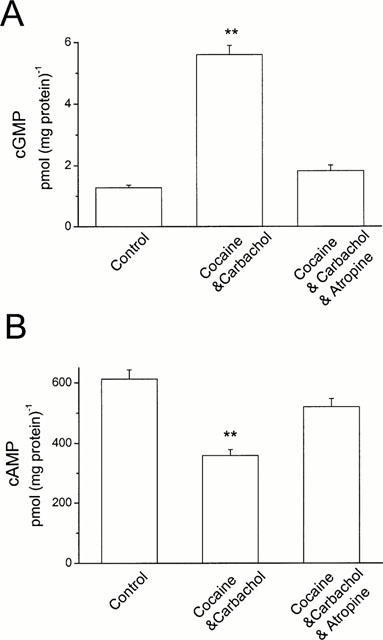
Synergic effects of cocaine and carbachol on cyclic GMP and cyclic AMP production in cultured HEL299 cells. (A) Content of cyclic GMP was measured in HEK299 cells for control (Control, _n_=4), 1 μM cocaine plus 1 μM carbachol (Cocaine and Carbachol, _n_=4; P<0.01 vs control), and 1 μM cocaine plus 1 μM carbachol plus 10 μM atropine (Cocaine and Carbachol and Atropine, _n_=4; _P_>0.05 vs control). (B) Compared to control (Control, _n_=4), cyclic AMP production was significantly decreased in HEK299 cells treated with 1 μM cocaine plus 1 μM carbachol (Cocaine and Carbachol, _n_=4; P<0.01 vs control). This reduction was abolished by addition of 10 μM atropine (Cocaine and Carbachol and Atropine, _n_=4; _P_>0.05 vs control). Statistical significance was tested with unpaired Student's _t_-test. **P<0.01 vs control.
Effect of MEG on cyclic GMP and cyclic AMP content in HEL299 cells
MEG is a major pyrolysis product formed from heating cocaine base (Meisels & Loke, 1993). MEG has been reported to have a cholinergic stimulatory effect on airway smooth muscle in some species (El-Fawal & Wood, 1995). This cholinergic action of MEG is probably due to its structural similarity to the muscarinic agonists arecoline and anatoxin A (Ettinger & Albin, 1989). To test the effects of MEG on cyclic GMP and cyclic AMP production and to compare the MEG effects with cocaine, HEL299 cells were incubated with MEG at various concentrations for 24 h in the absence and presence of 10 μM atropine. Figure 5A shows that MEG caused a concentration-dependent increase in cyclic GMP production in HEL299 cells. Pre-incubation of HEL299 cells with 10 μM atropine significantly attenuated the MEG-induced increase in cyclic GMP production at the concentrations of 0.1, 1 and 10 μM MEG. The levels of cyclic GMP were 1.77±0.36 pmol (mg protein)−1 for the control (_n_=6), 13.74±1.70 pmol (mg protein)−1 for 1 μM MEG (_n_=8), and 4.06±0.99 pmol (mg protein)−1 for 1 μM MEG plus 10 μM atropine (_n_=4), respectively. The MEG-induced increase in cyclic GMP production (7.8 fold) is much greater than those induced by 1 μM carbachol (3.1 fold, Figure 1A) and 1 μM cocaine (3.6 fold, Figure 2A). This result indicates that MEG is more potent in terms of stimulation of M2 muscarinic receptors.
Figure 5.
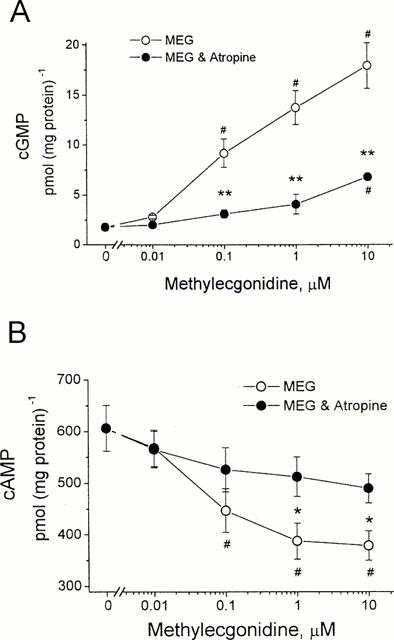
Effects of methylecgonidine on cyclic GMP and cyclic AMP production in cultured HEL299 cells. (A) Concentration-dependent increases in cyclic GMP production were observed in HEL299 cells incubated with various concentrations of MEG. Addition of 10 μM atropine significantly attenuated the MEG-induced increase in cyclic GMP production. (B) Concentration-dependent decreases in cyclic AMP were detected in HEL299 cells incubated with various concentrations of MEG. Addition of 10 μM atropine significantly attenuated the MEG-induced inhibition of cyclic AMP production. Each point represents the mean±s.e.mean of 4 – 8 measurements. **P<0.01 (tested by unpaired Student's _t_-test between MEG and MEG plus atropine). #P<0.01 (tested by ANOVA among control and treated groups with various concentrations of MEG alone or plus atropine).
To test whether MEG also produced an inhibition of cyclic AMP production as cocaine did, cultured HEL299 cells were incubated with various concentrations of MEG for 24 h. Figure 5B shows that a concentration-dependent decrease in cyclic AMP levels was observed in HEL299 cells incubated with MEG. It is interesting that the MEG-induced decrease in cyclic AMP production reached almost the maximum at 1 μM. This is similar to the result found in cocaine-treated HEL299 cells, but cocaine required 10 μM. Again, in terms of inhibition of cyclic AMP production, MEG is almost 10 fold more potent than cocaine. Figure 5B also shows that atropine at 10 μM significantly attenuated the MEG-induced decrease in cyclic AMP production in HEL299 cells treated with MEG. The differences at 1 and 10 μM MEG are significant between MEG and MEG plus atropine (P<0.001).
Prevention of the cocaine-induced decrease in cyclic AMP production by pertussis toxin
Stimulation of muscarinic receptors (M2) activates the pertussis toxin-sensitive inhibitory GTP-binding protein, Gi, which inhibits adenylate cyclase activity and reduces cyclic AMP production. To test whether the cocaine- or MEG-induced decrease in cyclic AMP content involved the G-protein, HEL299 cells were preincubated with 10 ng ml−1 pertussis toxin (PTX) for 3 h and then treated with cocaine or carbachol for 24 h. Figure 6 shows that the effects of cocaine and carbachol on cyclic AMP production were prevented in PTX-preincubated HEL299 cells. Compared to the control value, 597±28 pmol (mg protein)−1, of cyclic AMP production (_n_=6), the cyclic AMP levels were significantly decreased in HEK299 cells incubated with either 1 μM cocaine, 452±33 pmol (mg protein)−1 (_n_=6, P<0.05), or 1 μM carbachol, 402±34 pmol (mg protein)−1 (_n_=6, P<0.01). This reduction was abolished by addition of 10 ng ml−1 pertussis toxin. The values of cyclic AMP were 540±25 pmol (mg protein)−1 and 549±14 pmol (mg protein)−1 for HEL299 cells treated with cocaine plus PTX and carbachol plus PTX, respectively. However, cyclic AMP production was not significantly affected in HEL299 cells incubated with the toxin alone. This result indicates that the cocaine-induced reduction of cyclic AMP levels in HEL299 cells was mediated by the PTX-sensitive G-protein.
Figure 6.
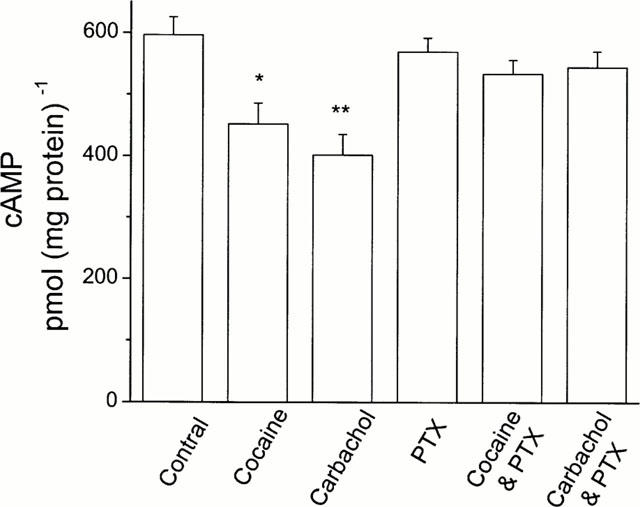
Abolition of the effects of cocaine and carbachol on cyclic AMP production by pertussis toxin. Content of cyclic AMP was measured in cultured HEK299 cells for control (Control), 1 μM cocaine (Cocaine), 1 μM carbachol (Carbachol), 10 ng ml−1 pertussis toxin (PTX), and 1 μM cocaine plus 10 ng ml−1 pertussis toxin (Cocaine and PTX), and 1 μM carbachol plus 10 ng ml−1 pertussis toxin (Carbachol and PTX). Compared to control (Control), cyclic AMP production was significantly decreased in HEK299 cells incubated with 1 μM cocaine (Cocaine) or 1 μM carbachol (Carbachol). This reduction was abolished by addition of 10 ng ml−1 pertussis toxin (Cocaine and PTX, and Carbachol and PTX). Each bar represents the mean±s.e.mean of six measurements. *P<0.05; **P<0.01; ***P<0.001; vs control.
Effect of cocaine and carbachol on muscarinic receptor expression
HEL299 cells are claimed to specifically express the ACh muscarinic receptor subtype, M2. To determine whether HEL299 cells solely express the M2 muscarinic receptor without expression of other muscarinic subtypes, the specific antibodies of M1, M2 and M3 protein isoforms were used in Western blot analysis. Figure 7a (control) shows that in the presence of the specific antibody to M2 receptor a band with about 50 kDs of molecular weight was blotted. In contrast, Western blot analysis of M1 and M3 in the presence of their specific antibodies did not detect any specific bands for these two receptor subtypes (data not shown). These results indicate that M2 muscarinic receptors are specifically expressed in HEL299 cells.
Figure 7.
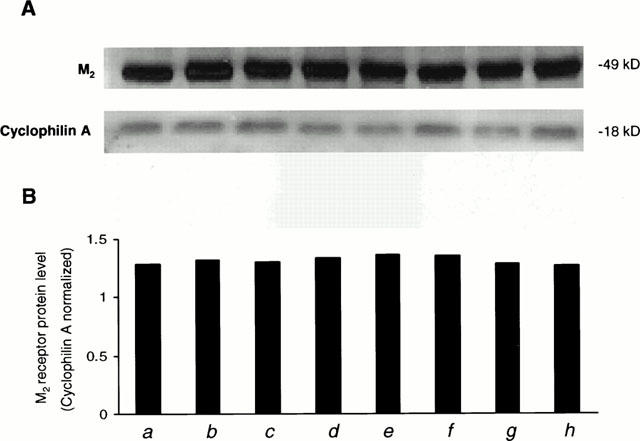
Effects of carbachol and cocaine on the expression of M2 muscarinic receptors in HEL299 cells. Western blot analysis was carried out in cultured HEL299 cells incubated with different compounds for 24 h. The experimental groups were divided as control (a), 1 μM cocaine (b), 1 μM carbachol (c), 10 μM atropine (d), 1 μM cocaine plus 1 μM carbachol (e), 1 μM cocaine plus 10 μM atropine (f), 1 μM cocaine plus 1 μM carbachol plus 10 μM atropine (g), 1 μM carbachol plus 10 μM atropine (h).
To examine whether the effects of cocaine and carbachol on cyclic GMP and cyclic AMP production were related to a change in the expression of M2 muscarinic receptor proteins, we analysed M2 protein levels in HEL299 cells after different treatments. HEL299 cells incubated with cocaine and/or carbachol at 1 μM did not significantly alter the expression of M2 protein levels (Figure 7b,c and e). In addition, atropine at 10 μM (Figure 7d) either with or without cocaine or carbachol (1 μM, Figure 7f – h) did not affect the expression of M2 protein levels. These results demonstrate that the effects of cocaine and carbachol on cyclic GMP and cyclic AMP production result from the stimulation of M2 muscarinic receptors, not from changes in the protein expression of M2 receptors.
Induction of iNOS by cocaine and carbachol
Stimulation of cardiac muscarinic receptors results in an increase in intracellular cyclic GMP content (Pappano et al., 1988; Eschenhagen, 1993; Stein et al., 1993). The primary activator of guanylate cyclase activity during stimulation of muscarinic receptors is nitric oxide (NO) produced by activation of NO synthase (Balligand et al., 1993; Schmidt et al., 1993). On the other hand, excessive NO production mediated by iNOS may contribute to the myocardial depression and beta-adrenergic hyporesponsiveness (Hare & Colucci, 1995). To understand whether the increase in cyclic GMP production by cocaine and carbachol was due to an induction of iNOS, we incubated HEL299 cells with cocaine or carbachol for 24 h. Western blot analysis shows that there was no detectable iNOS in the absence of cocaine or carbachol (Figure 8). However, a significant induction of iNOS was observed in cultured HEL299 cells incubated with 10 μM cocaine or 1 μM carbachol. The induction of iNOS by cocaine and carbachol was markedly attenuated after addition of 10 μM atropine or 1 μM methoctramine. These results indicate that the cocaine- or carbachol-induced increase in cyclic GMP production in cultured HEL299 cells may result from an increase in NO production by the induction of iNOS during the muscarinic stimulation.
Figure 8.
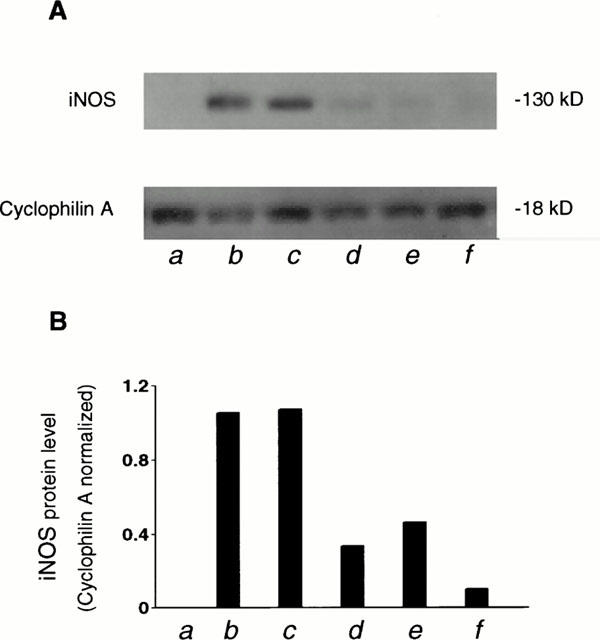
Induction of iNOS by cocaine in HEL299 cells. Western blot analysis was carried out in cultured HEL299 cells incubated with different compounds for 24 h. The experimental groups were divided as control (a), 10 μM cocaine (b), 1 μM carbachol (c), 10 μM cocaine plus 10 μM atropine (d), 1 μM carbachol plus 10 μM atropine (e), 10 μM cocaine plus 1 μM methoctramine (f). Note the induction of iNOS by cocaine and carbachol and the abolishment by addition of atropine or methoctramine.
Discussion
The present study demonstrates that cocaine, MEG, and the ACh receptor agonist carbachol enhanced cyclic GMP production in HEL299 cells, which specifically express M2 muscarinic receptors. The effects of cocaine and MEG on cyclic GMP production were concentration-dependent. The increase in cyclic GMP production in cocaine-, MEG-, and carbachol-treated cells was blocked by atropine, an antagonist of muscarinic receptors. In contrast, cyclic AMP levels of cultured HEL299 cells were decreased in the presence of cocaine, MEG or carbachol. The inhibitory effects of cocaine, MEG or carbachol on cyclic AMP content were abolished or significantly attenuated by atropine and methoctramine, a selective M2 blocker. These results suggest that the effects of cocaine, MEG, and carbachol on cyclic GMP and cyclic AMP production in cultured HEL299 cells result from stimulation of ACh M2 muscarinic receptors. In addition, atropine alone had no effects on cyclic GMP and cyclic AMP production in cultured HEL299 cells. Cocaine, an alkaloid extracted from the leaves of the Erythroxylon coca plant is one of the most widely abused drugs in many countries. Cocaine abuse causes a growing number of drug-related myocardial infarction, cardiac arrhythmia, rupture of the ascending aorta, pulmonary and obstetrical complications, and intestinal ischemia (Cregler & Mark, 1986). Our findings may provide new information for the understanding of drug-induced medical complications and deaths at cellular and molecular levels.
ACh muscarinic receptors regulate many important physiologic activities, such as heart rate, cardiac and smooth muscle contractility, exocrine and endocrine secretion, and neuronal functions. Therefore, our finding of cocaine-induced stimulation of ACh muscarinic receptors has clinical significance for explanation of medical complications caused by cocaine abuse. Most intracellular responses to stimulation of muscarinic receptors are mediated through guanine-binding proteins (G-proteins, Goyal, 1989; Eschenhagen, 1993). Activation of the inhibitory G-protein (Gi, a PTX-sensitive protein) inhibits adenylate cyclase activity, which causes a decrease in intracellular cyclic AMP levels. In our experiments, preincubation of HEL299 cells with PTX prevented the decrease in cyclic AMP production caused by cocaine. This result indicates that the cocaine-induced decrease in cyclic AMP production is mediated through the involvement of the PTX-sensitive GTP-binding protein.
One possible explanation for the increase in cyclic GMP production in HEL299 cells incubated with cocaine, MEG, or carbachol is via stimulation of muscarinic receptors, because atropine or methoctramine blocked the effects. It is well documented that muscarinic stimulation results in an increase in cardiomyocyte cyclic GMP content (Pappano et al., 1988; Eschenhagen, 1993; Stein et al., 1993). In many tissues, the production of nitric oxide (NO) by activation of NO synthase is the primary activator of guanylate cyclase activity during stimulation of muscarinic receptors (Balligand et al., 1993; Schmidt et al., 1993). Excessive NO production mediated by iNOS may cause the myocardial depression and beta-adrenergic hyporesponsiveness (Hare & Colucci, 1995). In the present study, we found that incubation of culture HEL299 cells with cocaine or carbachol induced an expression of iNOS and this induction was abolished by addition of atropine or methoctramine. Therefore, the increase in intracellular cyclic GMP levels in cocaine- or carbachol-incubated HEL299 cells may result from an increase in NO production due to the expression of iNOS. In addition, our recent experiments showed that incubation of cultured neonatal rat cardiomyocytes with cocaine or MEG for 2 days also induced an expression of iNOS and increased NO production by 3 fold of control (Yang & Xiao, unpublished data). The cocaine-induced increase in NO production was significantly attenuated by addition of atropine. It is interesting that the effects of cocaine on iNOS and NO were even greater (>2 fold) in cultured cardiomyocytes transfected with the M2 cDNA than in myocytes without transfection. The greater effects were not observed in cardiomyocytes with transfection of M1 or M3 (Yang & Xiao, unpublished data). Furthermore, Fischmeister & Hartzell (1987) found that stimulation of ACh muscarinic receptors caused an increase in intracellular cyclic GMP content and a decrease in cyclic AMP production in cardiac myocytes. The increase in cyclic GMP stimulated the cyclic-nucleotide-dependent phosphodiesterase to enhance breakdown of cyclic AMP. Our present data provide direct evidence that cocaine and MEG are able to modify intracellular cyclic GMP and cyclic AMP production via stimulation of ACh muscarinic receptors in cultured HEL299 cells expressing ACh M2 receptors. The cocaine-induced cyclic AMP reduction may result from both an inhibition of adenylate cyclase via activation of Gi and a stimulation of cyclic GMP-dependent phosphodiesterase when the drug activates muscarinic receptors. This modification leads to an inhibition of cyclic AMP-dependent kinase activity and a decrease in Ca2+ channel phosphorylation in cardiac myocytes. Therefore, cocaine and MEG reduced cardiac Ca2+ influx and contractility (Huang et al., 1997).
Cholinergic modulation of the cardiovascular system by cocaine has been demonstrated in several animal studies. For example, a study in conscious dogs showed that the significant modulation of an adrenergic response to cocaine was via the cholinergic mechanism (Stambler et al., 1993). This effect was eliminated by combined ganglionic and post-synaptic muscarinic receptor blockers. Cocaine at lower concentrations has a muscarinic agonist effect on isolated ferret myocytes (Boyett et al., 1988; Qiu & Morgan, 1993; Miao et al., 1996) and human ventricular trabeculae (Woolf et al., 1997). Diversity of cholinergic receptors in structure, signaling, and regulation was recognized in the last decade. Three pharmacologically identifiable types and at least five different molecular forms (M1 to M5) have been claimed (Goyal, 1989; Pappano & Mubagwa, 1992; Sharma et al., 1997). The heart is predominated by the M2 receptor subtype (Sharkey et al., 1988; Goyal, 1989; Pappano & Mubagwa 1992). Western blotting analysis in our present experiments confirms that HEL299 cells highly express M2 receptors. However, these cells have no detectable proteins of M1 and M3 receptors. Application of cocaine, MEG, and carbachol did not alter the level of protein expression of M2 receptors. Therefore, the increase in cyclic GMP production and the decrease in cyclic AMP content caused by cocaine and MEG are due to their functional stimulation of M2 receptors. As atropine alone had no effects on M2 protein expression, the antagonistic action of atropine on the drug-induced effects further supports the hypothesis of cocaine or MEG muscarinic stimulation.
On the other hand, cocaine as a competitive antagonist of muscarinic receptors has been reported in cardiac and brain tissues (Sharkey et al., 1988; Billman & Loppi, 1993). Billman & Loppi (1993) found that cocaine decreased the cardiac vagal tone index in conscious dogs. In addition, a binding study (Sharkey et al., 1988) presented evidence for cocaine as an antimuscarinic agent, particularly at higher concentrations, in heart and brain. Whereas cocaine interacts nonselectively with brain and heart muscarinic receptors (Flynn et al., 1992), the binding affinity of cocaine is markedly higher for M2 than for M1 with _K_i values of 19 and 40 μM, respectively (Sharkey et al., 1988). The mechanism of the antimuscarinic actions of cocaine has not been delineated. A possible explanation for cocaine as an antimuscarinic agent in the binding study and as an agonistic compound in our present experiments is that cocaine may have the ability of binding to different sites, primary and secondary recognition sites, of muscarinic receptors (Flynn et al., 1992). Therefore, cocaine may act as a partial agonist which produces different functional effects, especially at higher concentrations. Another explanation for functional studies is that cocaine may act behind muscarinic receptor levels and cause a suppression of cardiac vagal tone. The second hypothesis is supported by our recent finding that cocaine suppressed the ACh-activated K+ current (_I_K(ACh)) in ferret cardiomyocytes (Xiao & Morgan, 1998). The cocaine-induced suppression of _I_K(ACh)) is mainly by blocking the channel pore or by inhibiting the activity of the receptor-coupled G-protein, GK, not by blocking ACh muscarinic receptors. Animal species, tissue differences, and different concentrations used in different studies should also be counted for the disputed effects of cocaine on the cholinergic system.
In summary, our experiments demonstrate that cocaine, MEG, and carbachol caused an increase in cyclic GMP production in cultured HEL299 cells. In contrast, these compounds reduced intracellular cyclic AMP content. The muscarinic receptor antagonist atropine or methoctramine, a selective M2 blocker, abolished or significantly attenuated these effects. Since HEL299 cells specifically express the M2 receptor subtype, and since M2 receptor protein levels were not altered in the presence of cocaine and MEG, changes in cyclic GMP and cyclic AMP production after drug treatments mainly result from functional stimulation of M2 receptors. In the present study we provide new evidence that cocaine and MEG are able to stimulate M2 muscarinic receptors which affect many physiological functions.
Acknowledgments
This work was supported in part by research grants DA11762 and DA12774 (J.P. Morgan) from the National Institute of Drug Abuse, and 9930254N (Y.-F. Xiao) from the American Heart Association.
Abbreviations
ACh
acetylcholine
HEL299 cell
human embryonic lung cell
MEG
methylecgonidine
PTX
pertussis toxin
iNOS
the inducible isoform of nitric oxide synthase
References
- BALLIGAND J., KELLY R.A., MARSDEN P.A., SMITH T.W., MICHEL T. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc. Natl. Acad. Sci. U.S.A. 1993;90:347–351. doi: 10.1073/pnas.90.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENCHIMOL A., BARTALL H., DESSER K.B. Accelerated ventricular rhythm and cocaine abuse. Ann. Intern. Med. 1978;88:519–520. doi: 10.7326/0003-4819-88-4-519. [DOI] [PubMed] [Google Scholar]
- BILLMAN G.E., LOPPI M.D. Effects of cocaine on cardiac vagal tone before and during coronary artery occlusion: cocaine exacerbate the autonomic response to myocardial ischemia. J. Cardiovasc. Pharmacol. 1993;22:869–876. doi: 10.1097/00005344-199312000-00015. [DOI] [PubMed] [Google Scholar]
- BOYETT M.R., KIRBY M.S., ORCHARD C.H., ROBETTS A. The negative inotropic effect of acetylcholine on ferret ventricular myocardium. J. Physiol. (Lond.) 1988;404:613–635. doi: 10.1113/jphysiol.1988.sp017309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAMBERS H.F., MORRIS D.L., TAUBER M.G., MODIN G. Cocaine use and the risk for endocarditis in intravenous drug users. Ann. Intern. Med. 1987;106:833–836. doi: 10.7326/0003-4819-106-6-833. [DOI] [PubMed] [Google Scholar]
- CREGLER L.L., MARK H. Relation of acute myocardial infarction to cocaine abuse. Am. J. Cardiol. 1985;56:794. doi: 10.1016/0002-9149(85)91140-3. [DOI] [PubMed] [Google Scholar]
- CREGLER L.L., MARK H. Medical complications of cocaine abuse. N. Engl. J. Med. 1986;315:1495–1500. doi: 10.1056/NEJM198612043152327. [DOI] [PubMed] [Google Scholar]
- DRESSLER F.A., MALEKZADEH S., ROBERTS W.C. Quantitative analysis of amounts of coronary arterial narrowing in cocaine addicts. Am. J. Cardiol. 1990;65:303–308. doi: 10.1016/0002-9149(90)90292-9. [DOI] [PubMed] [Google Scholar]
- EL-FAWAL H.A., WOOD R.W. Airway smooth muscle relaxant effects of the cocaine pyrolysis product, Methylecgonidine. J. Pharmacol. Exp. Ther. 1995;272:991–996. [PubMed] [Google Scholar]
- ESCHENHAGEN T. G protein and the heart. Cell Biol. Int. 1993;17:723–749. doi: 10.1006/cbir.1993.1135. [DOI] [PubMed] [Google Scholar]
- ETTINGER N.A., ALBIN R.J. A review of the respiratory effects of smoking cocaine. Am. J. Med. 1989;87:664–668. doi: 10.1016/s0002-9343(89)80401-2. [DOI] [PubMed] [Google Scholar]
- FISCHMEISTER R., HARTZELL H.C. Cyclic guanosine 3′,5′-monophosphate regulates the calcium current in single cells from frog ventricle. J. Physiol. (Lond.) 1987;387:453–472. doi: 10.1113/jphysiol.1987.sp016584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLEMING J.W., STRAWBRIDGE R.A., WATANABE A.M. Muscarinic receptor regulation of cardiac adenylate cyclase activity. J. Mol. Cell. Cardiol. 1987;19:47–61. doi: 10.1016/s0022-2828(87)80544-8. [DOI] [PubMed] [Google Scholar]
- FLEMING J.W., WATANABE A.M. Muscarinic cholinergic-receptor stimulation of specific GTP hydrolysis related to adenylate cyclase activity in canine cardiac sarcolemma. Circ. Res. 1988;63:340–350. doi: 10.1161/01.res.63.2.340. [DOI] [PubMed] [Google Scholar]
- FLYNN D.A., VAISHNAV A.A., MASH D.C. Interactions of cocaine with primary and secondary recognition sites on muscarinic receptors. Mol. Pharmacol. 1992;41:736–742. [PubMed] [Google Scholar]
- GEORGE W.J., POISON J.B., O'TOOLE A.G., GOLDBERG N.D. Elevation of guanosine 3′,5′-cyclic phosphate in rat heart after perfusion with acetylcholine. Proc. Natl. Acad. Sci. U.S.A. 1970;66:398–402. doi: 10.1073/pnas.66.2.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLDBERG N.D., HADDOX M.K., NICHOL S.E., GLASS D.B., SANFORD C.H., KUEHL F.A., ESTENSEN E. Biologic regulation through opposing influences of cyclic GMP and cyclic AMP: the yin-yang hypothesis. Adv. Cyclic Nucleotide Res. 1975;5:307–330. [PubMed] [Google Scholar]
- GOYAL R.K. Muscarinic receptor subtypes. N. Engl. J. Med. 1989;321:1022–1028. doi: 10.1056/NEJM198910123211506. [DOI] [PubMed] [Google Scholar]
- HARE J.M., COLUCCI W.S. Role of nitric oxide in the regulation of myocardial function. Prog. Cardiovasc. Dis. 1995;38:155–166. doi: 10.1016/s0033-0620(05)80004-0. [DOI] [PubMed] [Google Scholar]
- HUANG L., WOOLF J.H., ISHIGURO Y., MORGAN J.P. Effect of cocaine and methylecgonidine on intracellular Ca2+ and myocardial contraction in cardiac myocytes. Am. J. Physiol. 1997;273:H893–H901. doi: 10.1152/ajpheart.1997.273.2.H893. [DOI] [PubMed] [Google Scholar]
- ISNER J.M., ESTES M., THOMPSON P.D., COSTANZO-NORDIN M.R., SUBRAMANIAN R., MILLER G., KATSAS G., SWEENEY K., STURNER W.Q. Acute cardiac events temporally related to cocaine abuse. N. Engl. J. Med. 1986;315:1438–1443. doi: 10.1056/NEJM198612043152302. [DOI] [PubMed] [Google Scholar]
- KOUMI S.-I., WASSERSTROM J.A. Acetylcholine-sensitive K+ channels in mammalian ventricular myocytes. Am. J. Physiol. 1994;266:H1812–H1821. doi: 10.1152/ajpheart.1994.266.5.H1812. [DOI] [PubMed] [Google Scholar]
- MACDONELL K.L., GLEN F.T., JACK D. cGMP elevation does not mediate muscarinic agonist-induced negative inotropy in rat ventricular cardiomyocytes. Am. J. Physiol. 1995;269:H1905–H1912. doi: 10.1152/ajpheart.1995.269.6.H1905. [DOI] [PubMed] [Google Scholar]
- MCMORN S.O., HARRISON S.M., ZANG W.-J., YU X.-J., BOYETT M.R. A direct negative effect of actylcholine on rat ventricular myocytes. Am. J. Physiol. 1993;265:H1393–H1400. doi: 10.1152/ajpheart.1993.265.4.H1393. [DOI] [PubMed] [Google Scholar]
- MEISELS I., LOKE J. The pulmonary effects of free-base cocaine A review. Cleveland Clin. J. Med. 1993;60:325–329. doi: 10.3949/ccjm.60.4.325. [DOI] [PubMed] [Google Scholar]
- MIAO L., QIU Z., MORGAN J.P. Cholinergic stimulation modulates the negative inotropic effect of cocaine on ferret ventricular myocardium. Am. J. Physiol. 1996;270:H678–H684. doi: 10.1152/ajpheart.1996.270.2.H678. [DOI] [PubMed] [Google Scholar]
- MURAD F., CHI Y.-M., RALL T.W., SUTHERLAND E.W. Adenyl cyclase III. The effect of catecholamines and choline esters on the formation of adenosine 3′,5′-phosphate by preparations from cardiac muscle and liver. J. Biol. Chem. 1962;237:1233–1238. [PubMed] [Google Scholar]
- NANJI A.A., FILIPENKO J.D. Asystole and ventricular fibrillation associated with cocaine intoxication. Chest. 1984;85:132–133. doi: 10.1378/chest.85.1.132. [DOI] [PubMed] [Google Scholar]
- PAPPANO A.J., MATSUMOTO K., TAJIMA T., AGNARSSON V., WEBB W. Pertussis toxin-insensitive mechanisms for carbachol-induced depolarization and positive inotropic effects in heart muscles. Trends Pharmacol. Sci. 1988. pp. 35–39. [PubMed]
- PAPPANO A.J., MUBAGWA K.Action of muscarinic agents and adenosine on the heart The Heart and Cardiovascular System 1992New York: Raven Press; 1765–1776.ed. Fozzard, H.A, et al [Google Scholar]
- PASTERNACK P.F., COLVIN S.B., BAUMANN F.G. Cocaine-induced angina pectoris and acute myocardial infarction in patients younger than 40 years. Am. J. Cardiol. 1985;55:847–848. doi: 10.1016/0002-9149(85)90172-9. [DOI] [PubMed] [Google Scholar]
- QIU Z., MORGAN J.P. Differential effects of cocaine and cocaethylene on intracellular Ca2+ and myocardial contraction in cardiac myocytes. Br. J. Pharmacol. 1993;109:293–298. doi: 10.1111/j.1476-5381.1993.tb13569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMESH C.G., JOACHIM N., AUGUST M.W., HANI N.S. Muscarinic-cholinoceptor mediated attenuation of phospholamban phosphorylation induced by inhibition of phosphodiesterase in ventricular cardiomyocytes: Evidence against a cAMP-dependent effect. Mol. Cell. Bioc. 1998;187:155–161. doi: 10.1023/a:1006899931151. [DOI] [PubMed] [Google Scholar]
- RESNICK R.B., KESTERNBAUM R.S., SCHWARTZ L.K. Acute systemic effects of cocaine in man: A controlled study by intranasal and intravenous routes. Science. 1977;195:696–698. doi: 10.1126/science.841307. [DOI] [PubMed] [Google Scholar]
- RICHARDSON R., MAYANIL C., HOSEY M. Subtype-specific antibodies for muscarinic cholinergic receptors. II Studies with reconstituted chick heart receptors and the GTP-binding protein G(0) Mol. Pharmacol. 1991;40:908–914. [PubMed] [Google Scholar]
- SCHMIDT H.H.H.W., LOHMANN S.M., WALTER U. The nitric oxide and cGMP signal transduction system: regulation and mechanism of action. Biochim. Biophys. Acta. 1993;1178:153–175. doi: 10.1016/0167-4889(93)90006-b. [DOI] [PubMed] [Google Scholar]
- SHARKEY J., RITZ M.C., SCHENDEN J.A., HANSON R.C., KUHAR M.J. Cocaine inhibits muscarinic cholinergic receptors in heart and brain. J. Pharmacol. Exp. Ther. 1988;246:1048–1052. [PubMed] [Google Scholar]
- SHARMA V.K., COLECRAFT H.M., RUBIN L.E., SHEU S.S. Does mammalian heart contain only the M2 muscarinic receptor subtype. Life Sci. 1997;60:1023–1029. doi: 10.1016/s0024-3205(97)00043-x. [DOI] [PubMed] [Google Scholar]
- STAMBLER B.S., KOMAMURA K., IHARA T., SHANNON R.P. Acute intravenous cocaine causes transient depression followed by enhanced left ventricular function in conscious dogs. Circulation. 1993;87:1687–1697. doi: 10.1161/01.cir.87.5.1687. [DOI] [PubMed] [Google Scholar]
- STEIN B., DROGEMULLER A., MULSCH A., SCHMITZ W., SCHOLZ H. Ca++-dependent constitutive nitric oxide synthase is not involved in the cyclic GMP-increasing effects of carbachol in ventricular cardiomyocytes. J. Pharmacol. Exp. Ther. 1993;266:919–925. [PubMed] [Google Scholar]
- VIRMANI R., ROBINOWITZ M., SMIALEK J.E., SMYTH D.F. Cardiovascular effects of cocaine: An autopsy study of 40 patients. Am. Heart J. 1988;115:1068–1075. doi: 10.1016/0002-8703(88)90078-6. [DOI] [PubMed] [Google Scholar]
- WATSON J.M., VOGEL S.M., COTTERELL D.J., DUBOCOVICH M.L. Cholinergic antagonism of -adrenergic stimulated action potentials and adenylate cyclase activity in rabbit ventricular cardiomyocytes. Eur. J. Pharmacol. 1988;155:101–108. doi: 10.1016/0014-2999(88)90407-4. [DOI] [PubMed] [Google Scholar]
- WEINER R.S., LOCKHART J.T., SCHWARTZ R.G. Dilated cardiomyopathy and cocaine abuse: Report of two cases. Am. J. Med. 1986;81:699–701. doi: 10.1016/0002-9343(86)90559-0. [DOI] [PubMed] [Google Scholar]
- WOOLF J.H., HUANG L., ISHIGURO Y., MORGAN J.P. Negative inotropic effect of methylecgonidine, a major product of cocaine base pyrolysis, on ferret and human myocardium. J. Cardiovasc. Pharmacol. 1997;30:352–359. doi: 10.1097/00005344-199709000-00013. [DOI] [PubMed] [Google Scholar]
- XIAO Y.-F., MORGAN J.P. Cocaine blockade of the acetylcholine-activated muscarinic K+ channel in ferret cardiac myocytes. J. Pharmacol. Exp. Thre. 1998;284:10–18. [PubMed] [Google Scholar]