Genomic and Genic Deletions of the FOX Gene Cluster on 16q24.1 and Inactivating Mutations of FOXF1 Cause Alveolar Capillary Dysplasia and Other Malformations (original) (raw)
Abstract
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV) is a rare, neonatally lethal developmental disorder of the lung with defining histologic abnormalities typically associated with multiple congenital anomalies (MCA). Using array CGH analysis, we have identified six overlapping microdeletions encompassing the FOX transcription factor gene cluster in chromosome 16q24.1q24.2 in patients with ACD/MPV and MCA. Subsequently, we have identified four different heterozygous mutations (frameshift, nonsense, and no-stop) in the candidate FOXF1 gene in unrelated patients with sporadic ACD/MPV and MCA. Custom-designed, high-resolution microarray analysis of additional ACD/MPV samples revealed one microdeletion harboring FOXF1 and two distinct microdeletions upstream of FOXF1, implicating a position effect. DNA sequence analysis revealed that in six of nine deletions, both breakpoints occurred in the portions of Alu elements showing eight to 43 base pairs of perfect microhomology, suggesting replication error Microhomology-Mediated Break-Induced Replication (MMBIR)/Fork Stalling and Template Switching (FoSTeS) as a mechanism of their formation. In contrast to the association of point mutations in FOXF1 with bowel malrotation, microdeletions of FOXF1 were associated with hypoplastic left heart syndrome and gastrointestinal atresias, probably due to haploinsufficiency for the neighboring FOXC2 and FOXL1 genes. These differences reveal the phenotypic consequences of gene alterations in cis.
Introduction
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV) (Alveolar Capillary Dysplasia, Congenital; CACD [MIM 265380]) is a rare and lethal developmental disorder of the lung that affects both acinar structure and the intrinsic pulmonary vasculature, producing a constellation of histologic changes that when present together define the entity.1 Infants affected with ACD/MPV develop respiratory distress and severe pulmonary hypertension within the first two days of life, have no sustained response to supportive measures, and die of respiratory failure within the first month of life, although longer survivals and later presentations have been reported.1, 2, 3, 4, 5, 6, 7, 8, 9, 10 More than 80% of infants with ACD/MPV have additional malformations affecting the cardiac, gastrointestinal, and genitourinary systems. Intestinal malrotation is the most commonly observed of these anomalies, and hypoplastic left heart together with hypoplasia or coarctation of the aortic arch are the most common associated cardiovascular abnormalities.10, 11
Almost 200 ACD/MPV cases have been reported in the literature, approximately 10% having a familial association.4, 5, 6, 10, 12, 13, 14 The genetic etiology of ACD/MPV has remained elusive.10 Recently, STRA6 (MIM 610745) has been implicated in a malformation syndrome that includes anophthalmia, additional malformations, and developmental lung abnormalities said to be ACD.15 However, the reported lung changes in children with STRA6 mutations do not include misalignment of pulmonary veins, the defining histologic feature, and the clinical course is vastly different than that of ACD/MPV.
We initiated a study of the genetic basis of esophageal atresia and tracheo-esophageal fistula, ascertaining patients with malformations of this type, especially those that were associated with other congenital anomalies. We carried out an array comparative genomic hybridization (array CGH) screen to detect copy number variation (CNV) at critical genomic loci, particularly those harboring genes that were good candidates on the basis of their already established roles in foregut development in model organisms.
In the present study, we demonstrate a crucial role for FOXF1 (MIM 601089) in human lung and intrinsic pulmonary vascular development by identifying inactivating mutations in patients with ACD/MPV. These patients have additional congenital malformations that together define a syndrome of ACD/MPV, intestinal malrotation, and urinary tract malformations. We show that patients with deletions harboring FOXF1 and the neighboring FOXC2 (MIM 602402) and FOXL1 (MIM 603252) genes at 16q24.1 have not only ACD/MPV, as expected, but also distinct malformations comprising congenital heart defect, in particular hypoplastic left heart syndrome, and gastrointestinal atresias, including esophageal atresia, as well as urinary tract malformations and other malformations.
Subjects and Methods
Subject Recruitment
We obtained DNA samples from probands with ACD/MPV and their family members after obtaining their informed consent, using protocols approved by the Institutional Review Board for Human Subject Research at Baylor College of Medicine (H8712). The Cambridgeshire 4 Research Ethics Committee, UK, approved the study of a cohort of patients with esophageal atresia and associated malformations (reference 04/5/022). Patient D2 was ascertained via a study of prenatal malformations conducted at Addenbrooke's Hospital. This study was approved by Addenbrooke's Hospital Local Research Ethics Committee and by Cambridgshire 1 Research Ethics Committee, UK (reference 08/H0304/).
Histopathology
Histologic slides of lung tissue obtained at autopsy (deletion cases D1, D3, D4, D8, and all four mutation cases M1–M4) or biopsy (deletion cases D9 and D10) were reviewed by C.L. for the diagnostic histologic features of ACD/MPV. All cases had slides stained with hematoxylin and eosin, and patient D1 also had elastic tissue stains.
DNA Isolation
Patients' genomic DNA was extracted from peripheral blood via the Puregene DNA isolation kit (Gentra System, Minneapolis, MN, USA). DNA was extracted from frozen tissue and peripheral leukocytes via the Puregene DNA Extraction Kit, as well. Alternatively, DNA from paraffin blocks was isolated via the QIAGEN Kit in accordance with the vendor's instruction or as described previously.10
Array CGH Analysis
Initial array CGH analysis was performed with the use of a 244K commercial array (Agilent Technologies, Santa Clara, CA, USA) in patient D1 and with an Affymetrix GeneChip 6.0 array in patient D2, in accordance with the manufacturers' instructions; no additional pathogenic CNVs were identified. Chromosomal microarray analysis was performed with the use of the V6.1 BAC-based array (patient D4), V6.3 OLIGO (patients D5 and D7), and V7.2 OLIGO (patient D3), designed by Baylor Medical Genetics laboratories and manufactured by Agilent Technology as previously described.16, 17 Array CGH in patient D5 was performed with the use of the BAC clone SignatureChipWG whole-genome microarray, in patient D6 with the use of the SignatureChipOS, a 105K-feature whole-genome microarray (made for Signature Genomic Laboratories by Agilent Technologies), in accordance with the manufacturer's instructions. Patient D5 was also analyzed with the use of the Affymetrix 250K SNP array.
Whole-genome high-resolution oligonucleotide microarray CGH analyses in patients D1–D9, for fine mapping of the sizes and extents of the deletions, and in subject D10, for confirmation of the deletion, were performed with the NimbleGen array HG18_WG_CGH_v1 with 385,000 or 2.1M oligonucleotides (NimbleGen Systems, Madison, WI, USA), in accordance with the manufacturer's instructions.
Custom 16q24 region-specific high-resolution 44K microarrays were designed with the use of eArray (Agilent Technologies) and used for CNV screening in 14 mutation-negative patients with ACD/MPV.
Fluorescence In Situ Hybridization Analysis
Confirmatory fluorescence in situ hybridization (FISH) analysis in patients D2–D5 was performed via standard procedures.
Long-Range Polymerase Chain Reaction and DNA Sequencing
Long-range polymerase chain reaction (LR-PCR), performed in accordance with the manufacturer's instructions (Takara Bio, Japan), amplified the predicted junction fragments from the breakpoint regions in patients D1–D9. PCR products were purified with the PCR Purification Kit (QIAGEN, Valencia, CA, USA) and bidirectionally sequenced by Sanger di-deoxynucleotide sequencing (Lone Star Labs, Houston, TX, USA).
Mutation Analysis of the FOXF1 Gene
Overlapping amplicons covering the entire coding region of exons 1 and 2 of FOXF1 were amplified and sequenced by conventional Sanger di-deoxynucleotide sequencing (Lone Star Labs, Houston, TX; Molecular Core Lab, BCM, Houston, TX). DNA sequences were analyzed by comparison with reference sequence (NM_001451.2) with the use of Sequencher v4.2 (GeneCodes, Ann Arbor, MI, USA). None of the identified mutations was found in 150 ethnically matched control chromosomes. Individual primer sequences and PCR conditions are available on request.
Mutation Cloning
A 1482 bp region covering the mutation and an informative heterozygous (C/G) SNP, rs2078304, in patient M4 was amplified by LR-PCR and cloned with the Copy Control Cloning Kit (Epicenter Biotechnologies, Madison, WI, USA). Clone DNA was isolated with the Qiprep Miniprep Kit (QIAGEN, Valencia, CA, USA) and sequenced by conventional Sanger di-deoxynucleotide sequencing (Lone Star Labs, Houston, TX, USA).
Polymorphic SNP and Microsatellite Marker Analysis
SNP markers rs12596341, rs10660430, rs11398689, rs1364225, rs1424019, rs9941308, rs58016760, rs1364224, rs1424016, rs58557724, and rs1064259 and the microsatellite marker between chr16:84,723,490–84,723,542 were amplified for parental studies via routine methods with PCR primers obtained from Sigma (Woodlands, TX, USA). The SNP PCR products were bidirectionally sequenced (Lone Star Labs, Houston, TX, USA). The microsatellite amplicons were separated on 8% polyacrylamide gels and visualized by ethidium bromide staining. The loci order, locations, and sequences were obtained from the UCSC Genome Browser database.
Bioinformatics and In Silico Sequence Analysis
Genomic sequence based on the oligonucleotide coordinates from the array CGH experiment was downloaded from the UCSC genome browser (build 36, March 2006) and assembled with the Sequencher v4.2 software. Interspersed repeat sequences were analyzed by RepeatMasker.
Results
Initial Array CGH Screening
Using BAC clone and oligonucleotide array CGH, we identified overlapping microdeletions in 16q24.1q24.2, ranging in size from ∼100 kb to ∼3.5 Mb, in seven patients referred for high-resolution genome analysis (Figures 1A and 1C, Table 1). The first of these patients (D1) was ascertained through our study of the genetic basis of esophageal atresia, for which, to date, 80 syndromic cases have been studied. The remainder were ascertained by queries of databases of patients referred for high-resolution genome analysis for a variety of reasons. Parental samples were available in six out of seven patients (not D2). One deletion (D7) was inherited from a phenotypically abnormal parent; the remaining five were de novo, supporting their likely pathogenic effect. No deletion CNVs in this genomic region were found, either in the Database of Genomic Variants or in the BCM and Signature Genomics databases of over 30,000 patients studied by array CGH. Five of the seven deletions (D2–D5 and D7) were independently verified by FISH analysis (Figure 1D). High-resolution oligonucleotide array CGH (385K or 2.1M NimbleGen) confirmed the deletions and enabled breakpoint characterization in all seven cases (Figures 1E and 1F).
Figure 1.
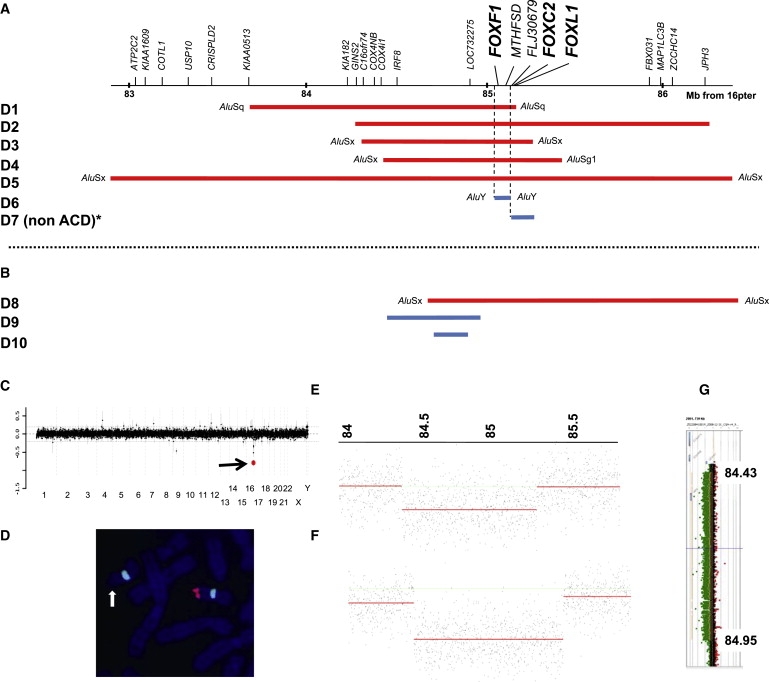
Summary of the Results in Patients with Microdeletions in 16q24.1q24.2
(A) Schematic representation of the genomic region harboring the FOX transcription gene cluster, FOXF1, FOXC2, and FOXL1, showing the extent and gene content of the regions deleted in seven patients analyzed by array CGH (D1–D7). A non-ACD/MPV patient, D7, with a deletion of FOXC2 and FOXL1 and an intact FOXF1, is also shown.
(B) Microdeletions identified via a custom designed 16q24-specific high-resolution 4×44K Agilent array in three patients with ACD/MPV (D8–D10). Note that the microdeletion in subject D8 contains FOXF1, whereas those in patients D9 and D10 are located upstream of FOXF1, indicating a position effect on this gene. Group 1 patients are indicated in red, group 2 in blue (see main text for explanation). In six out of nine deletions analyzed, breakpoints map within Alu repetitive elements, suggesting MMBIR/FoSTeS or NAHR mechanisms of formation.
(C) Targeted array CGH plot obtained with V7.2 OLIGO (Agilent 105K) in patient D3, showing a deletion in 16q24.1 (red mark designated by arrow).
(D) Metaphase chromosomes of patient D4 after FISH with a 16q24-specific BAC clone, RP11-542M13 (red), and a control chromosome 16 alfa satellite (Vysis) probe (green), showing absence of signal (arrow) on one chromosome 16, consistent with a heterozygous deletion.
(E and F) NimbleGen whole-genome oligonucleotide array CGH profiles for subjects D3 (E) and D4 (F).
(G) The ∼524 kb deletion in patient D9, located ∼52 kb upstream of FOXF1, was detected via a custom designed 16q24-specific high-resolution 4×44K Agilent array. Green dots shifted to the left between 84.43–84.95 represent the deleted segment.
Table 1.
Summary of the Clinical Findings in Patients with FOX Gene Cluster Microdeletions in 16q24.1q24.2
| Patient | Deletion | M/F | Gest. (wks) | LS | Birth Wt. (g) | Respiratory Findings | Cardiac Findings | Gastrointestinal Findings | Genitourinary Findings | Vertebral/Axial Findings | Other Findings |
|---|---|---|---|---|---|---|---|---|---|---|---|
| D1 | ∼1.5 Mb FOXF1, FOXC2, FOXL1 | F | 28 | 1 day | 1091 | ACD/MPV, pulmonary lymphangiectasia | PDA | Esophageal atresia, tracheo-esophageal fistula; abnormal placement of anus | - | - | Single umbilical artery |
| D2 | ∼2.0 Mb FOXF1, FOXC2, FOXL1 | M | 22; TOP | N/A | NK | - | HLHS | - | Dilated renal pelvices | - | - |
| D3 | ∼0.9 Mb FOXF1, FOXC2, FOXL1 | M | 38; elective C-section | 40 days | 2900 | ACD/MPV; persistent pulmonary hypertension; ECMO dependent (veno-veno) | Tetralogy of Fallot; PDA | Duodenal atresia with annular pancreas; imperforate anus | Bilateral hydronephrosis, mod right, severe left | - | Single umbilical artery |
| D4 | ∼1.0 Mb FOXF1, FOXC2, FOXL1 | F | 37; elective C-section | 15 days | - | ACD/MPV; left pneumothorax; hypoxemia; Surfactant therapy; ECMO | HLHS | - | Mild uretero-pelvic caliectasis | T11 butterfly vertebra | Cleft lip, cleft palate, brachycephaly; single umbilical artery |
| D5 | ∼3.5 Mb FOXF1, FOXC2, FOXL1 | M | - | 18 days | - | ECMO dependent | IAA, dilated PA, large PDA, decreased size left ventricle, pulmonary hypertension | - | Bilateral renal pelviectasis | Posterior rib fusions: 10/11 (right side); 9/10 and 11/12 (left side) | - |
| D6 | ∼103 Kb FOXF1, MTHFSD | M | 35 | 5 days | 1700 | Respiratory distress requiring inhaled nitrous oxide, hydrocortisone, prostaglandin and FiO2 100%, attributed to RDS | AVSD, ostium primum ASD, small VSD, PDA, mildly hypoplastic LV, bicuspid AV, | Possible duodenal atresia, possible intestinal malrotation | Moderate bilateral pelvicaliectasis, mild to moderate proximal urethral dilatation; probable mild to moderate distal ureteral dilatation | - | Absent spleen; transverse orientation of the liver, compatible with abdominal heterotaxy |
| D7 | 131 Kb, FOXC2 and FOXL1 | F | - | Alive at 3 yrs | - | - | Spontaneous ASD closing | Diastasis recti (6 cm at 14 months of age) | Dilated and tortuous ureters bilaterally with left hydronephrosis | - | CNS, ventriculomegaly, Chiari malformation and syrinx, meningeal cysts at T2-T3, developmental delay, o/e hypertelorism and broad forehead similar to mother, low-set ears, CAL R buttock and L lower scapula, healing capillary hemangioma L buttock |
| D8a | ∼1.8 Mb FOXF1, FOXC2, FOXL1 | F | term | 25 days | 3676 | ACD/MPV; persistent pulmonary hypertension; lung biopsy done | PDA, ligated; structurally normal heart by echocardiogram | Adhesions between bowel loops, second part of duodenum and gallbladder; no malrotation seen at laparotomy | - | - | - |
| D9 | 524 Kb, Upstream of FOXF1 | F | term | 20 days | 3742 | ACD/MPV; persistent pulmonary hypertension, 2 courses of ECMO days 2-7 and again beginning on day 10. Lung biopsy at 17 days | Multiple normal echocardiograms | Suspected intestinal malrotation; imperforate anus | Bicornuate uterus with cervical duplication | Multiple butterfly vertebrae | - |
| D10 | 145 Kb, Upstream of FOXF1 | F | term | 13 days | 3600 | ACD/MPV respiratory distress with pulmonary hypertension, requiring mechanical ventilation, HFOV, inhaled nitric oxide, ECMO. Lung biopsy at 12 days | Echocardiogram on day 2 showed changes related to severe pulmonary hypertension and small PDA with no other abnormality | - | - | - | - |
The deletions centered around the FOX transcription factor gene cluster at 16q24.1. All but one harbored FOXF1, a gene with a role in lung and foregut development ascertained on the basis of previous studies of mice.18, 19, 20 Patient D7 had an ∼131 kb deletion encompassing FOXC2 and FOXL1 but not FOXF1.
Clinical Characterization
Of the six patients with chromosomal deletions harboring FOXF1 (D1–D6), five (D1 and D3–D6) died from pulmonary insufficiency in the first two months of life, and the mother of a sixth (D2) underwent elective termination of pregnancy at 22 weeks. Three patients (D1, D3, and D4) had ACD/MPV documented by histopathological examination (Figure 2A). Two other patients (D5 and D6) had severe respiratory distress, raising the possibility of ACD/MPV, but no postmortem examination was performed. Cardiac malformations were present in five of the six patients; two patients (D2 and D4) had a hypoplastic left heart, and a third (D5) had a small left ventricle. One patient (D1) manifested esophageal atresia with tracheo-esophageal fistula, whereas duodenal and anal atresias were present in another patient (D3). Five patients (D2–D6) had abnormalities consistent with urinary tract obstructive lesions, ranging from uretero-pelvocaliectasis to severe hydronephrosis. The phenotype in patient D7 was clearly distinct from that in the other six patients. This patient did not have respiratory insufficiency in the neonatal period and was still alive at the age of 3 years, having presented with atrial septal defect, bilaterally dilated and tortuous ureters with left hydronephrosis, diastasis recti, and developmental delay (Figure 2A and Table 1). The deletion was inherited from the patient's mother, whose sole recognized phenotypic abnormality was the presence of an umbilical hernia.
Figure 2.
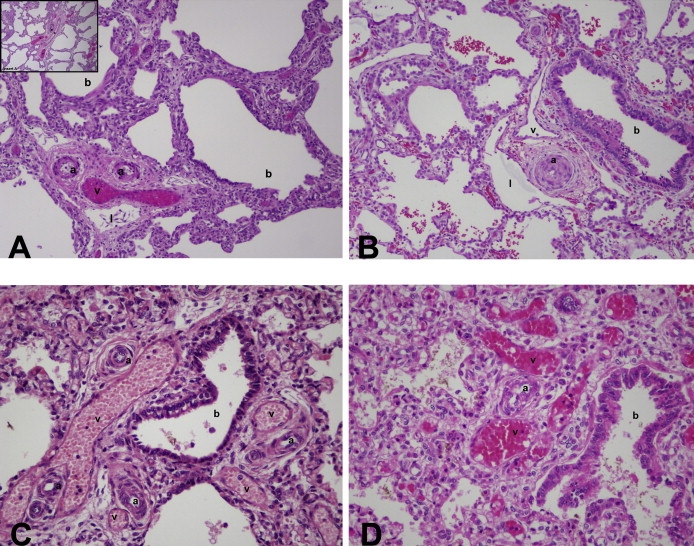
Histopathology Studies
Histopathology studies show that all deletion and mutation patients have characteristic changes of ACD/MPV, with medial hyperplasia of small pulmonary arteries, abnormally positioned pulmonary veins adjacent to membranous and terminal bronchioles and coursing with small pulmonary arteries (misalignment), lobular underdevelopment, and deficient numbers of normally positioned airspace wall capillaries with abnormal enlarged and centrally placed thin-walled vascular channels in airspace walls. The deletion cases often show marked airspace enlargement, thinner airspace walls, and pulmonary lymphangiectasis; these findings are uncommon in mutation cases. Hematoxylin and eosin, initial magnification 25x. (A) Patient D3 with FOXF1 deletion. Note two small pulmonary artery branches with moderately thickened smooth muscle (a), an adjacent malpositioned congested pulmonary vein (v), and a dilated lymphatic channel (l) all located adjacent to a dilated terminal bronchiole (b). In the inset, this same abnormal vascular configuration is seen adjacent to a membranous bronchiole and the lobular parenchyma is formed of markedly enlarged and simplified airspaces with thinner walls, compared to mutation cases. (B) Patient D9 with deletion upstream of FOXF1. Small pulmonary arteries (a) have strikingly thickened medial smooth muscle and only pin-point lumens; they share a common connective tissue sheath with pulmonary vein branches (v) that are neither dilated nor congested and both are adjacent to a membranous bronchiole (b). Normally positioned capillaries are not seen in airspace walls, although there are more centrally placed congested thin-walled vascular channels. A prominent lymphatic (l) is adjacent to the artery/vein combination. Airspaces appear prominently enlarged. (C) Patient M2 with frameshift mutation in exon 1. Multiple thick-walled small pulmonary arteries (a) and dilated congested malpositioned pulmonary veins (v) often share the same connective tissue sheath adjacent to a small bronchiole. The lobular parenchyma is formed of enlarged, simplified and poorly subdivided airspaces; however airspace enlargement is less dramatic than in the deletion cases. (D) Patient M3 with frameshift mutation in exon 2. Thick-walled small pulmonary arteries (a) and adjacent dilated and congested veins (v) are located next to a terminal bronchiole (b). The lobular parenchyma is underdeveloped with simplified and poorly subdivided airspaces separated by thickened septa in which there are dilated and congested central vascular channels, but only rare capillaries.
Mutation Analysis of the FOXF1 Gene
Two lines of evidence suggested that FOXF1 haploinsufficiency might be responsible for ACD/MPV in our initial cohort. First, patients with deletions harboring FOXF1 (D1–D6) had a severe respiratory phenotype (confirmed histologically as ACD/MPV in three cases), whereas a patient with a deletion harboring FOXC2 and FOXL1 but not FOXF1, patient D7, did not. Second, mice haploinsufficient for Foxf1 have abnormal alveolar development, notwithstanding the fact that the histological changes are not identical to those of ACD/MPV.18, 19 We therefore sequenced FOXF1 in a cohort of 18 patients with ACD/MPV and other malformations.10 We identified four de novo heterozygous mutations in the coding sequence of FOXF1 in four unrelated patients with sporadic ACD/MPV. Patient M1 had a nonsense mutation (c.150C→A; p.Y50X) in exon 1; patient M2 had a frameshift mutation (c.775dupT; p.Y259Lfs11X) in exon 1; patient M3 had a frameshift mutation (c.956_957delTT; p.F319CfsX66) in exon 2 adding 29 amino acids to the protein, as predicted by conceptual translation; and patient M4 had a T→C substitution in the first base of the stop codon, a no-stop mutation (c.1063T→C; p.X355RextX74) adding 73 amino acids to the protein, as predicted by conceptual translation (Figures 3A–3D). All four patients had associated malformations, including a partial atrioventricular canal defect (1/4 cases), patent ductus arteriosus (1/4), bowel malrotation (3/4), a congenital short bowel (1/4), an annular pancreas (1/4), and urinary tract malformations (3/4) (Table 2).
Figure 3.
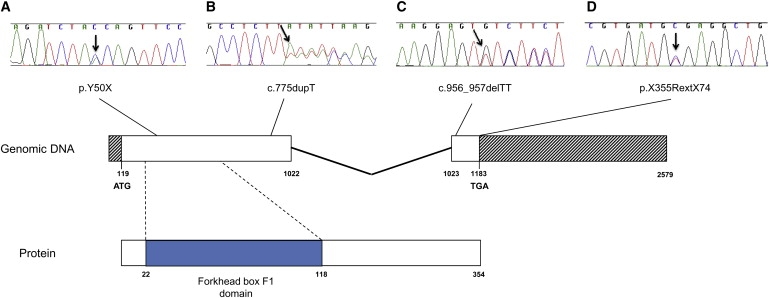
Genomic and Protein Structure of FOXF1 and DNA Sequencing Chromatograms
Genomic and protein structure of FOXF1 and DNA sequencing chromatograms showing four mutations in the coding sequence of FOXF1 identified in patients with ACD/MPV.
(A) A nonsense mutation c.150C > A; p.Y50X in exon 1 in patient M1.
(B) A frameshift mutation c.775 dupT; p.Y259Lfs11X in exon 1 in subject M2.
(C) A frameshift mutation c.956_957 delTT; p.F319CfsX66 in exon 2 in patient M3.
(D) A no-stop mutation c.1063T > C; p.X355RextX74 in exon 2 in individual M4. The base numbering for the FOXF1 gene refers to A of the second ATG start codon as position 1 (nucleotide 119 of GenBank accession number NM_001451.2).34
Table 2.
Summary of the clinical findings in patients with FOXF1 mutations
| Patient | Mutation | M/F | Gest. (wks) | LS | Birth Wt. (g) | Respiratory Findings | Cardiac Findings | Gastrointestinal Findings | Genitourinary Findings | Vertebral/Axial Findings | Other Findings |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M1a | Exon 1, Nonsense | M | 32 | 10 days | 2470 | ACD/MPV; bilobed right lung | Partial atrio-ventricular canal defect, PDA | Intestinal malrotation; Annular pancreas and duodenal stenosis | Bilateral hydronephrosis, hydroureter, dilatation urinary bladder | - | - |
| M2 | Exon 1, Frameshift | M | 32 | 12 hrs | 2460 | ACD/MPV | - | - | Obstructive renal dysplasia | - | - |
| M3 | Exon 2, Frameshift | F | 35 | 1 day | 2490 | ACD/MPV; partial anomalous pulmonary venous connection with absent left upper lobe pulmonary vein | - | Malrotation bowel; congenital short bowel; small omphalocele | Bilateral severe hydronephrosis, hydroureter, and bladder dilatation | - | Micrognathia, low set ears |
| M4 | Exon, 2, No-stop | M | 38 | 47 days | 5057 | ACD/MPV | - | Intestinal malrotation, Meckel's diverticulum | - | - | - |
Custom-Designed Array CGH
We hypothesized that some of the cases of ACD/MPV in the cohort might be due to whole-gene deletions, as was seen for patients D1–D6. We therefore designed a 16q24 region-specific high-resolution 44K oligonucleotide microarray and used it to screen the 14 mutation-negative patients from our ACD cohort. This analysis revealed an ∼1.8 Mb microdeletion harboring FOXF1 in patient D8. In addition, two microdeletions, one ∼524 kb (patient D9) and one ∼145 kb (patient D10) in size, located ∼52 kb and ∼259 kb upstream of FOXF1, respectively, were detected (Figures 1B and 1G, Table 1). All three rearrangements arose de novo.
Histopathology
Of 14 patients, ten had lung tissue available for histologic review. All ten showed the constellation of changes required for the histologic diagnosis of ACD/MPV (Figure 2). Many also showed other superimposed changes; consequences of respiratory failure and therapeutic support, terminal events, postmortem changes, and focal inflammation (rarely). However, these did not obscure the diagnostic hallmarks of ACD/MPV. Pulmonary lymphangiectasis was evident in all analyzed deletion cases (D1, D3, D4, and D8–D10), as well as in one (M2) of four examined patients with mutations. In the patients with lung biopsy but without autopsy (D9 and D10), information related to other malformations was based on clinical and/or imaging observations without pathologic confirmation; for those with neither lung biopsy nor autopsy (D2, D5, and D6), lung histology remains unknown and information related to other malformations is based on clinical and/or imaging observations only. No pulmonary tissue is available from patient D7, and that patient remains alive at three years of age, without respiratory symptomatology.
DNA Sequence Analysis
DNA sequence analysis enabled breakpoint sequencing in patients D1–D9. This confirmed the deletions and revealed that in patients D1, D3–D6, and D8, both breakpoints occurred in the portions of Alu elements showing eight to 43 base pairs (bp) of perfect microhomology. In patients D2 and D7, microhomologies of 4 and 3 bp, respectively, were found, and in patient D9, a 3 bp AGC insertion was identified at the junction fragments (Table 3).
Table 3.
Array CGH-Detected Microdeletions in 16q24
| Patient | Initial Array CGH Detection | FISH Verification | Parental Origin of Deletion | High-Resolution Array CGH Verification and Characterization | Coordinates | Size | Micro-homology (bp) | Mechanism of Formation |
|---|---|---|---|---|---|---|---|---|
| D1 | 244 K (Agilent Technologies) | ND | de novo maternal | 385 K (NimbleGen) | 83,705,765–85,204,004 | 1,498,239 bp | 17 | Alu-Alu MMBIR/ FoSTeSx1 or NAHR |
| D2 | Affymetrix SNP 6.0 | RP11-565I1 | ND | 385 K (NimbleGen) | 84,275,154–86,275,754 | 2,000,600 bp | 4 | NHEJ or MMBIR/FoSTeSx1 |
| D3 | CMA V7.2 OLIGO | RP11-542M13 | de novo maternal | 2.1 M (NimbleGen) | 84,374,208–85,277,007 | 902,799 bp | 18 | Alu-Alu MMBIR/FoSTeSx1 or NAHR |
| D4 | CMA V6.1 BAC | RP11-542M13 | de novo maternal | 2.1 M (NimbleGen) | 84,402,571–85,435,712 | 1,033,141 bp | 8 | Alu-Alu MMBIR/FoSTeSx1 or NAHR |
| D5 | SignatureChipWG | RP11-463O9 | de novo ND | 385 K (NimbleGen) Affymetrix 250K | 82,908,199–86,405,076 | 3,496,877 bp | 12 | Alu-Alu MMBIR/FoSTeSx1 or NAHR |
| D6 | SignatureChipOS | ND | de novo ND | 2.1 M (NimbleGen) | 85,051,032–85,153,914 | 102,882 bp | 19 | Alu-Alu MMBIR/FoSTeSx1 or NAHR |
| D7 | CMA V6.3 OLIGO | RP11-58A18 | inherited maternal | 2.1 M (NimbleGen) | 85,157,843–85,288,903 | 131,060 bp | 3 | NHEJ or MMBIR/FoSTeSx1 |
| D8 | 4×44 K (Agilent Technologies) | ND | de novo maternal | 385 K (NimbleGen) | 84,648,160–86,478,255 | 1,830,095 bp | 43 | Alu-Alu MMBIR/FoSTeSx1 or NAHR |
| D9 | 4×44 K (Agilent Technologies) | ND | de novo maternal | 385 K (NimbleGen) | 84,425,269–84,949,662 | 524,393 bp | 1 | NHEJ AGC insertion |
| D10 | 4×44 K (Agilent Technologies) | ND | de novo maternal | 385 K (NimbleGen) | ∼ 84,698–84,843 kb | ∼ 145 kb | ND | ND |
Parental Origin of Deletions and Mutation
We investigated, when possible, the parent of origin, and we showed by microsatellite polymorphism and SNP studies that the deletions in patients D1, D3, D4, D8, D9, and D10 are maternal in origin. In patient M4, DNA sequencing of the cloned PCR product revealed that the no-stop mutation c.1063T→C was associated with the C allele inherited from the patient's father, whereas the G allele was associated with the wild allele, demonstrating that the mutation occurred on the paternal chromosome (Table S1).
Discussion
Our report of deletions and point mutations within the FOX transcription factor gene cluster at 16q24.1 highlights the differing phenotypic consequences of gene alterations in cis. The patients described in this report can be classified into three groups on the basis of genotype and phenotype. Patients in group 1 have deletions that encompass all three FOX genes in the 16q24.1 cluster—FOXF1, FOXC2, and _FOXL1_—as well as MTHFSD (genomic deletions). Group 2 patients have deletions spanning either none (patients D9 and D10) or some (patients D6 and D7; genic deletions) but not all of the genes in the FOX cluster. Group 3 patients comprise those with inactivating point mutations in FOXF1. All group 1 and group 3 patients with histologic evaluation had ACD/MPV, but there were consistent differences in the spectrum of associated malformations between the two groups. All six group 1 patients had cardiovascular malformations. These ranged in severity from patent ductus arteriosus (PDA) (D1, D3, D5, and D8) through interrupted aortic arch (D5) and tetralogy of Fallot (D3) to reduced left ventricular size (D5) and hypoplastic left heart (D2 and D4). Just one of the four patients in group 3 (M1) had a cardiac malformation, a partial AV canal defect, together with PDA. Group 1 patients had gastrointestinal atresias (esophageal in D1, duodenal and anal in D3), whereas group 3 patients had intestinal malrotation or congenital short bowel. As expected, patients in group 2 were less readily classifiable. Patients with deletions, either upstream of (D9 and D10) or encompassing (D6) FOXF1, had ACD/MPV; one patient (D7) with a downstream deletion encompassing just FOXC2 and FOXL1 did not. Likewise, malformations occurring in this group were of group 1 type, such as hypoplastic left heart and imperforate anus, and group 3 type, such as intestinal malrotation. Additional malformations not seen in either group 1 or group 3 occurred in this group: possible abdominal heterotaxy in patient D6, Chiari malformation and meningeal cysts in patient D7.
We propose that ACD/MPV results from haploinsufficiency of FOXF1 and that the differences in the associated malformations between groups 1 and 3 relate to haploinsufficiency for FOXC2 and FOXL1. Point mutations in FOXC2, probably leading to its haploinsufficiency, are responsible for lymphoedema-distichiasis syndrome;21, 22 a small proportion of these patients also have congenital heart defects (6.8% in one series,21 including tetralogy of Fallot and ventricular septal defect but not hypoplastic left heart). Mice with targeted inactivation of the orthologous Foxc2 have interrupted aortic arch, as well as defects of the skull and vertebral column.23, 24 Haploinsufficiency of FOXL1 in humans has not previously been described. Targeted disruption of Foxl1 in mice is associated with aberrant epithelial cell positioning in the small intestine, hyperplasia of the gastric mucosa and retarded growth.25, 26, 27 MTHFSD is a methylene tetrahydrofolate synthase domain-containing gene of unknown function.
The lack of intestinal atresia in group 3 suggests that either FOXC2 or FOXL1 may also contribute to this phenotype in humans, despite the fact that, in mice, haploinsufficiency for Foxf1 alone is associated with esophageal atresia. It will be fascinating to determine whether deletions or inactivating mutations contribute to malformations reported here when they occur in isolation, in particular for hypoplastic left heart syndrome and intestinal malrotation, for which no gene has yet been causally implicated. The presence of overlapping deletions upstream of the FOX cluster in two patients (D9 and D10) with ACD/MPV strongly suggests that critical regulatory sequences are harbored within the minimal interval, and future work will focus on the identification of any evolutionarily conserved elements within this interval and the elucidation of their functional role.
Although the abnormal lung phenotype of Foxf1+/− mice has some overlap with that of ACD/MPV in humans with alveolarization abnormalities, paucity of alveolar capillaries and pulmonary lymphangiectasia being common to both, there is no evidence of MPV, the defining histologic feature of ACD/MPV, in the mutant mice. Two different mouse models showed that _Foxf1_−/− embryos die by 8.5 days postcoitum (dpc) from defects in extraembryonic mesoderm development.18, 19, 28 Interestingly, in one model, half of the newborn Foxf1+/− mice had reduced (20% of wild-type) levels of pulmonary Foxf1, died from pulmonary hemorrhage, and showed severe defects in alveolarization and vasculogenesis (lung immaturity, hypoplasia) with fusion of the right lung lobes and lobar arteries, as well as an abnormal gall bladder, narrowing of the esophagus and trachea, esophageal atresia with tracheo-esophageal fistula, and defects in liver and lung repair. The other half of the Foxf1+/− mice had normal Foxf1 levels and normal lung morphology, interpreted as a “compensation” for the alveolarization defect.28 We suggest that the occurrence of these two distinct populations of mice is consistent with imprinting of Foxf1, in which the expression level of Foxf1 in Foxf1+/− mice is dependent upon the parent of origin of the normal Foxf1 allele. Supporting this notion, the expression from the Foxf1 promoter has been shown to be regulated by tissue-specific methylation29 and FOXF1 was computationally predicted to be paternally imprinted in humans.30 However, the facts that all six de novo deletions that we studied for the parental origin arose on a maternal chromosome and one mutation of FOXF1 (in patient M4) occurred on a paternal chromosome, as well as the observation of a relatively normal phenotype associated with paternal uniparental disomy 16,31 argue against FOXF1 imprinting in humans; nevertheless, a complex tissue-specific genomic imprinting cannot be excluded.
Foxf1 has been shown to be downregulated in Shh−/− mice, probably through Gli proteins,32 and activated by bone morphogenetic protein 4 (BMP4).32, 33 In _Foxf1_−/+ mice, a diminished expression of Vegfa, Vegfr2, Bmp-4, Tbx, Lklf, Fgf-10, Gli3, and Notch-2 has been found.19, 28, 34, 35, 36 Some of these genes, critical for lung development, may be mutated in patients with ACD/MPV without detectable abnormalities in FOXF1.
DNA sequence analysis of the deletion breakpoints in nine patients (D1–D9) revealed that in six patients (D1, D3–D6, and D8), both breakpoints map in the portions of Alu elements showing perfect microhomology (eight to 43 base pairs) (Table 3), suggesting replication error MMBIR/FoSTeS as a mechanism of their formation.37 Alternatively, these deletions might have occurred via nonallelic homologous recombination (NAHR). In patients D2, D7, and D9, the presence of short microhomologies (D2 and D7) and AGC insertion (D9) at the junction fragments indicate that these deletions probably arose via nonhomologous end joining or the MMBIR/FoSTeS mechanism.37
Our data may contribute to a better understanding of the pathogenesis of malformations seen in patients with the VACTERL (vertebral, anal, cardiac, tracheo-esophageal, renal and limb malformations) association (MIM 192350).38 Cardiac malformations, esophageal and anal atresia, renal and vertebral/axial malformations, and a single umbilical artery appear to be common to both VACTERL association and 16q24.1 microdeletion. However, although lung malformations have been reported occasionally in cases of VACTERL association, ACD/MPV has not been described in this context. We suggest that microdeletions at 16q24.1 should be considered in cases of apparent VACTERL association for which there is a severe respiratory distress and/or neonatal lethality. We propose that patients with intestinal atresia, isolated diastasis recti, or umbilical hernia should be screened for mutations and deletions in FOXL1. Given that FOXF1 maps only ∼3.7 Mb from 16qter, we also suggest that a balanced rearrangement involving 16qter (e.g., submicroscopic subtelomeric reciprocal translocation) should be excluded in parents with an ACD/MPV-affected child from whom no material is available for testing.
Acknowledgments
The authors thank all of the families who participated in this study and the Alveolar Capillary Dysplasia Association, the parent organization, for help in recruitment of patients. We thank A.L. Beaudet, J.W. Belmont, M. Jamrich, M. Justice, J.R. Lupski, F.J. Probst, D. Scott, and M. Shinawi for helpful discussion. P. Sen would like to thank M. Haymond for support and encouragement. We acknowledge J. Rosenfeld for collecting clinical information on the patients and helping with data collection. P.St. was supported in part by grant R13-0005-04/2008 from the Polish Ministry of Science and Higher Education. Part of this study was supported by a National Association of Rare Disorders (NORD) grant to P. Sen. We thank T.F. Beck for technical assistance. M.S., V.M., D.R., T.F., S.G., E.P., N.P.C., and C.S.-S. are funded by the Wellcome Trust; C.S.-S. is the recipient of a Wellcome Trust Intermediate Clinical Fellowship. M.T., I.S., L.W., J.P., and S.M. gratefully acknowledge funding from the Biomedical Research Centre Grant from the National Institute for Health Research (NIHR), UK. C.S.-S., V.M., and M.S. gratefully acknowledge additional funding from the Addenbrooke's Charitable Trust and Tracheo-Oesophageal Fistula Support, the UK patient support group for patients with esophageal atresia and tracheo-esophageal fistula. C.S.-S. thanks N. Conte, A. Bradley, R. Hennekam, and P. Scambler for helpful discussion; and C. Hall, H. Martin, and B. Treacy for technical assistance.
Some authors are based in the Department of Molecular and Human Genetics at Baylor College of Medicine (BCM), which offers extensive genetic laboratory testing, including use of arrays for genomic copy number analysis, and derives revenue from this activity. B.A.B. and L.G.S. have ownership and sit on the Members' Board of Signature Genomic Laboratories, LLC. The UK authors declare no competing interest.
Published online: June 4, 2009
Footnotes
Supplemental Data include two tables and can be found with this article online at http://www.ajhg.org/.
Web Resources
The URLs for data presented herein are as follows:
- Database of Genomic Variants, http://projects.tcag.ca/variation/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
- RepeatMasker, http://www.repeatmasker.org
- University of California, Santa Clara (UCSC) Genome Browser, http://genome.ucsc.edu/
Supplemental Data
Document S1. Two Tables
References
- 1.Langston C. Misalignment of pulmonary veins and alveolar capillary dysplasia. Pediatr. Pathol. 1991;11:163–170. doi: 10.3109/15513819109064753. [DOI] [PubMed] [Google Scholar]
- 2.Wagenvoort C.A. Misalignment of lung vessels: a syndrome causing persistent neonatal pulmonary hypertension. Hum. Pathol. 1986;17:727–730. doi: 10.1016/s0046-8177(86)80182-4. [DOI] [PubMed] [Google Scholar]
- 3.Al-Hathlol K., Phillips S., Seshia M.K., Casiro O., Alvaro R.E., Rigatto H. Alveolar capillary dysplasia. Report of a case of prolonged life without extracorporeal membrane oxygenation (ECMO) and review of the literature. Early Hum. Dev. 2000;57:86–94. doi: 10.1016/s0378-3782(99)00065-1. [DOI] [PubMed] [Google Scholar]
- 4.Eulmesekian P., Cutz E., Parvez B., Bohn D., Adatia I. Alveolar capillary dysplasia: a six-year single center experience. J. Perinat. Med. 2005;33:347–352. doi: 10.1515/JPM.2005.067. [DOI] [PubMed] [Google Scholar]
- 5.Vassal H.B., Malone M., Petros A.J., Winter R.M. Familial persistent pulmonary hypertension of the newborn resulting from misalignment of the pulmonary vessels (congenital alveolar capillary dysplasia) J. Med. Genet. 1998;35:58–60. doi: 10.1136/jmg.35.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Licht C., Schickendantz S., Sreeram N., Arnold G., Vierzig A., Mennicken U., Roth B. Prolonged survival in alveolar capillary dysplasia syndrome. Eur. J. Pediatr. 2004;163:181–182. doi: 10.1007/s00431-003-1385-6. [DOI] [PubMed] [Google Scholar]
- 7.Ahmed S., Ackerman V., Faught P., Langston C. Profound hypoxemia and pulmonary hypertension in a 7 month old: Late presentation of alveolar capillary dysplasia. Pediatr. Crit. Care Med. 2008;9:e43–e46. doi: 10.1097/PCC.0b013e31818e383e. [DOI] [PubMed] [Google Scholar]
- 8.Steinhorn R.H., Cox P.N., Fineman J.R., Finer N.N., Rosenberg E.M., Silver M.M., Tyebkhan J., Zwass M.S., Morin F.C., 3rd Inhaled nitric oxide enhances oxygenation but not survival in infants with alveolar capillary dysplasia. J. Pediatr. 1997;130:417–422. doi: 10.1016/s0022-3476(97)70203-8. [DOI] [PubMed] [Google Scholar]
- 9.Michalsky M.P., Arca M.J., Groenman F., Hammond S., Caniano D.A. Alveolar capillary dysplasia: a logical approach to a fatal disease. J. Pediatr. Surg. 2005;40:1100–1195. doi: 10.1016/j.jpedsurg.2005.03.067. [DOI] [PubMed] [Google Scholar]
- 10.Sen P., Thakur N., Stockton D.W., Langston C., Bejjani B.A. Expanding the phenotype of alveolar capillary dysplasia (ACD) J. Pediatr. 2004;145:646–651. doi: 10.1016/j.jpeds.2004.06.081. [DOI] [PubMed] [Google Scholar]
- 11.Rabah R., Poulik J.M. Congenital alveolar capillary dysplasia with misalignment of pulmonary veins associated with hypoplastic left heart. Pediatr. Dev. Pathol. 2001;4:167–174. doi: 10.1007/s100240010125. [DOI] [PubMed] [Google Scholar]
- 12.Simonton S., Chrenka B. Familial persistent pulmonary hypertension in two siblings with phocomelia and alveolar capillary dysplasia (ACD) - a new syndrome? {abstract. Mod. Pathol. 1993;6:9P. [Google Scholar]
- 13.Boggs S., Harris M.C., Hoffman D.J., Goel R., McDonald-McGinn D., Langston C., Zackai E., Ruchelli E. Misalignment of pulmonary veins with alveolar capillary dysplasia: affected siblings and variable phenotypic expression. J. Pediatr. 1994;124:125–128. doi: 10.1016/s0022-3476(94)70267-5. [DOI] [PubMed] [Google Scholar]
- 14.Gutierrez C., Rodriguez A., Palenzuela S., Forteza C., Rossello J.L. Congenital misalignment of pulmonary veins with alveolar capillary dysplasia causing persistent neonatal pulmonary hypertension: report of two affected siblings. Pediatr. Dev. Pathol. 2000;3:271–276. doi: 10.1007/s100249910035. [DOI] [PubMed] [Google Scholar]
- 15.Pasutto F., Sticht H., Hammersen G., Gillessen-kaesbach G., Fitzpatrick D.R., Nürnberg G., Brasch F., Schirmer-Zimmermann H., Tolmie J.L., Chitayat D. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007;80:550–560. doi: 10.1086/512203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu X., Shaw C.A., Patel A., Li J., Cooper M.L., Wells W.R., Sullivan C.M., Sahoo T., Yatsenko S.A., Bacino C.A. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PLoS ONE. 2007;2:e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ou Z., Kang S.H., Shaw C.A., Carmack C.E., White L.D., Patel A., Beaudet A.L., Cheung S.W., Chinault A.C. Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet. Med. 2008;10:278–289. doi: 10.1097/GIM.0b013e31816b4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalinichenko V.V., Lim L., Shin B., Costa R.H. Differential expression of forkhead box transcription factors following butylated hydroxytoluene lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001;280:L695–L704. doi: 10.1152/ajplung.2001.280.4.L695. [DOI] [PubMed] [Google Scholar]
- 19.Mahlapuu M., Enerbäck S., Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development. 2001;128:2397–2406. doi: 10.1242/dev.128.12.2397. [DOI] [PubMed] [Google Scholar]
- 20.Maeda Y., Dave V., Whitsett J.A. Transcriptional control of lung morphogenesis. Physiol. Rev. 2007;87:219–244. doi: 10.1152/physrev.00028.2006. [DOI] [PubMed] [Google Scholar]
- 21.Brice G., Mansour S., Bell R., Collin J.R., Child A.H., Brady A.F., Sarfarazi M., Burnand K.G., Jeffery S., Mortimer P. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis in 74 patients with FOXC2 mutations or linkage to 16q24. J. Med. Genet. 2002;39:478–483. doi: 10.1136/jmg.39.7.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell R., Brice G., Child A.H., Murday V.A., Mansour S., Sandy C.J., Collin J.R., Brady A.F., Callen D.F., Burnand K. Analysis of lymphoedema-distichiasis families for FOXC2 mutations reveals small insertions and deletions throughout the gene. Hum. Genet. 2001;108:546–551. doi: 10.1007/s004390100528. [DOI] [PubMed] [Google Scholar]
- 23.Iida K., Koseki H., Kakinuma H., Kato N., Mizutani-Koseki Y., Ohuchi H., Yoshioka H., Noji S., Kawamura K., Kataoka Y. Essential roles of the winged helix transcription factor MFH-1 in aortic arch patterning and skeletogenesis. Development. 1997;124:4627–4638. doi: 10.1242/dev.124.22.4627. [DOI] [PubMed] [Google Scholar]
- 24.Kanzaki-Kato N., Tamakoshi T., Fu Y., Chandra A., Itakura T., Uezato T., Tanaka T., Clouthier D.E., Sugiyama T., Yanagisawa M. Roles of forkhead transcription factor Foxc2 (MFH-1) and endothelin receptor A in cardiovascular morphogenesis. Cardiovasc. Res. 2005;65:711–718. doi: 10.1016/j.cardiores.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 25.Fukamachi H., Kukuda K., Suzuki M., Furumoto T., Ichinose M., Shimizu S., Horie S., Suzuki Y., Saito Y., Watanabe K. Mesenchymal transcription factor Fkh6 is essential for development and differentiation of parietal cells. Biophys. Biochem. Res. Commun. 2001;280:1069–1076. doi: 10.1006/bbrc.2001.4247. [DOI] [PubMed] [Google Scholar]
- 26.Fukuda K., Yoshida H., Sato T., Furumoto T.A., Mizutani-Koseki Y., Suzuki Y., Saito Y., Takemori T., Kimura M., Sato H. Mesenchymal expression of Foxl1, a winged helix transcriptional factor, regulates generation and maintenance of gut-associated lymphoid organs. Dev. Biol. 2003;255:278–289. doi: 10.1016/s0012-1606(02)00088-x. [DOI] [PubMed] [Google Scholar]
- 27.Takano-Maruyama M., Hase K., Fukamachi H., Kato Y., Koseki H., Ohno H. Foxl1-deficient mice exhibit aberrant epithelial cell positioning resulting from dysregulated EphB/EphrinB expression in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;291:G163–G170. doi: 10.1152/ajpgi.00019.2006. [DOI] [PubMed] [Google Scholar]
- 28.Kalinichenko V.V., Lim L., Stolz D.B., Shin B., Rausa F.M., Clark J., Whitsett J.A., Watkins S.C., Costa R.H. Defects in pulmonary vasculature and perinatal lung hemorrhage in mice heterozygous for the Forkhead Box f1 transcription factor. Dev. Biol. 2001;235:489–506. doi: 10.1006/dbio.2001.0322. [DOI] [PubMed] [Google Scholar]
- 29.Chang V.W., Ho Y. Structural characterization of the mouse Foxf1a gene. Gene. 2001;267:201–211. doi: 10.1016/s0378-1119(01)00400-0. [DOI] [PubMed] [Google Scholar]
- 30.Luedi P.P., Dietrich F.S., Weidman J.R., Bosko J.M., Jirtle R.L., Hartemink A.J. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17:1723–1730. doi: 10.1101/gr.6584707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohlhase J., Janssen B., Weidenauer K., Harms K., Bartels I. First confirmed case with paternal uniparental disomy of chromosome 16. Am. J. Med. Genet. 2000;91:190–191. doi: 10.1002/(sici)1096-8628(20000320)91:3<190::aid-ajmg6>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 32.Astorga J., Carlsson P. Hedgehog induction of murine vasculogenesis is mediated by Foxf1 and Bmp4. Development. 2007;134:3753–3761. doi: 10.1242/dev.004432. [DOI] [PubMed] [Google Scholar]
- 33.Tseng H.-T., Shah R., Jamrich M. Function and regulation of FoxF1 during Xenopus gut development. Development. 2004;131:3637–3647. doi: 10.1242/dev.01234. [DOI] [PubMed] [Google Scholar]
- 34.Hellqvist M., Mahlapuu M., Samuelsson L., Enerbäck S., Carlsson P. Differential activation of lung-specific genes by two forkhead proteins, FREAC-1 and FREAC-2. J. Biol. Chem. 1996;271:4482–4490. doi: 10.1074/jbc.271.8.4482. [DOI] [PubMed] [Google Scholar]
- 35.Lim L., Kalinichenko V.V., Whitsett J.A., Costa R.H. Fusion of lung lobes and vessels in mouse embryos heterozygous for the forkhead box f1 targeted allele. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;282:L1012–L1022. doi: 10.1152/ajplung.00371.2001. [DOI] [PubMed] [Google Scholar]
- 36.Kalinichenko V.V., Gusarova G.A., Kim I.M., Shin B., Yoder H.M., Clark J., Sapozhnikov A.M., Whitsett J.A., Costa R.H. Foxf1 haploinsufficiency reduces Notch-2 signaling during mouse lung development. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L521–L530. doi: 10.1152/ajplung.00212.2003. [DOI] [PubMed] [Google Scholar]
- 37.Gu W., Zhang F., Lupski J.R. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw-Smith C. Oesophageal atresia, tracheo-oesophageal fistula and the VACTERL association: review of genetics and epidemiology. J. Med. Genet. 2006;43:545–554. doi: 10.1136/jmg.2005.038158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Two Tables