A QUANTITATIVE ASSESSMENT OF GLUTAMATE UPTAKE INTO HIPPOCAMPAL SYNAPTIC TERMINALS AND ASTROCYTES: NEW INSIGHTS INTO A NEURONAL ROLE FOR EXCITATORY AMINO ACID TRANSPORTER 2 (EAAT2) (original) (raw)
. Author manuscript; available in PMC: 2009 Nov 11.
Abstract
The relative distribution of the excitatory amino acid transporter 2 (EAAT2) between synaptic terminals and astroglia, and the importance of EAAT2 for the uptake into terminals is still unresolved. Here we have used antibodies to glutaraldehyde-fixed D-aspartate to identify electron microscopically the sites of D-aspartate accumulation in hippocampal slices. About 3/4 of all terminals in the stratum radiatum CA1 accumulated D-aspartate-immunoreactivity by an active dihydrokainate-sensitive mechanism which was absent in EAAT2 glutamate transporter knockout mice. These terminals were responsible for more than half of all D-aspartate uptake of external substrate in the slices. This is unexpected as EAAT2-immunoreactivity observed in intact brain tissue is mainly associated with astroglia. However, when examining synaptosomes and slice preparations where the extracellular space is larger than in perfusion fixed tissue, it was confirmed that most EAAT2 is in astroglia (about 80%). Neither D-aspartate uptake nor EAAT2 protein was detected in dendritic spines. About 6% of the EAAT2-immunoreactivity was detected in the plasma membrane of synaptic terminals (both within and outside of the synaptic cleft). Most of the remaining immunoreactivity (8%) was found in axons where it was distributed in a plasma membrane surface area several times larger than that of astroglia. This explains why the densities of neuronal EAAT2 are low despite high levels of mRNA in CA3 pyramidal cell bodies, but not why EAAT2 in terminals account for more than half of the uptake of exogenous substrate by hippocampal slice preparations. This and the relative amount of terminal versus glial uptake in the intact brain remain to be discovered.
Keywords: glutamate uptake, neurotransmitter transport, immunocytochemistry, synaptic transmission
Glutamate uptake into glia and neurons is essential for controlling the excitatory action of glutamate (for reviews see: Danbolt, 2001; Beart and O’Shea, 2007). It has been much debated whether the uptake activity of glutamatergic nerve terminals represents a major proportion of the total brain tissue uptake activity. The prevailing view is that most brain glutamate uptake is performed by astroglia, because (a) most of the glutamate uptake in forebrain tissue slices and synaptosome preparations is both dihydrokainate sensitive and dependent on the excitatory amino acid transporter (EAAT) 2 (GLT1; slc1a2) gene and protein, and (b) the highest numbers of dihydrokainate sensitive EAAT2 glutamate transporters are found in glial cells (for review see: Danbolt, 2001). This view, however, does not take account of a substantial amount of data showing a significant uptake into glutamatergic nerve terminals (e.g. Beart, 1976; Storm-Mathisen, 1977; Gundersen et al., 1993, 1996; Suchak et al., 2003; Xu et al., 2003; Waagepetersen et al., 2005; for a detailed discussion about the existence of nerve terminal glutamate up-take, see section 4.2 in Danbolt, 2001).
A carboxy-terminal splice variant of EAAT2 (the “b-variant”: GLT1b or GLT1v; for nomenclature see: Grewer and Rauen, 2005), as opposed to the original variant, now referred to as the “a-variant”: Pines et al., 1992) was recently cloned (Utsunomiya-Tate et al., 1997; Chen et al., 2002; Schmitt et al., 2002). Three groups (Chen et al., 2002; Schmitt et al., 2002; Reye et al., 2002) reported glial expression of the b-variant, while two of them reported additional neuronal expression at the light microscopic (Schmitt et al., 2002) or electron microscopic level (Chen et al., 2002). The latter investigators, however concluded in a subsequent report (Chen et al., 2004) that their antibodies were not sufficiently specific. Instead they found that antibodies to the a-variant labeled (in hippocampus CA1) 20–30% of axon terminals and about 5% of spines in addition to astroglia. They counted numbers of labeled structures, and not EAAT2 protein densities. Nevertheless, these results differ from previous observations, although it is well known that EAAT2 is expressed in neurons of the brain during early stages of development and in disease (for review see: Danbolt, 2001), including morphine withdrawal (Xu et al., 2003). Consequently, it is both important to validate these findings and to provide quantitative information on both distribution of EAAT2 protein and uptake activity.
Here, we provide four categories of new data: (a) quantitative visualization of EAAT2-protein amounts in terminals and confirmation of the observation that terminals in the stratum radiatum of hippocampus CA1 express the a-variant (Chen et al., 2004), (b) measurements of the plasma membrane surface area density of axon terminals to assess the transporter density, and (c) measurement of uptake activity and substrate selectivity using quantitative electron microscopy. The data show that D-aspartate up-take by terminals depends on the EAAT2 gene, and that terminals in slice preparations accumulate more externally added substrate than could be predicted based on EAAT2 protein levels alone. Possible causes for this discrepancy are discussed.
EXPERIMENTAL PROCEDURES
Materials
Glutaraldehyde, EM grade, was from TAAB (Reading, UK). D-[3H]Aspartic acid (30–40 Ci/mmol), L-[3H]glutamic acid (20–50 Ci/mmol), Sephadex G-25 and G-50, biotinylated anti-rabbit, anti-sheep and anti-mouse immunoglobulins, streptavidin-biotinylated horseradish peroxidase complex, and colloidal gold—labeled anti-rabbit and anti-mouse immunoglobulins were from Amersham (Buckinghamshire, UK). Paraformaldehyde and glutaraldehyde EM grade were from TAAB. Lowicryl HM20 was from Electron Microscopy Sciences (Fort Washington, PA, USA) or Agar Scientific (Stansted, UK). Nitrocellulose filters (HAWP; 0.45 _μ_m pores) were from Millipore (Carrigtwohill, C. Cork, Ireland). MK 801 (a non-competitive _N_-methyl-D-aspartate (NMDA) receptor antagonist), dihydrokainic acid, 6,7-dinitroquinoxaline-2,3-dione (DNQX; a selective non-NMDA receptor antagonist) were from Tocris (Bristol, UK). DL-_Threo-beta_-benzyloxyaspartate (TBOA: Shimamoto et al., 2000) was a generous gift from Dr. Keiko Shimamoto (Suntory Institute for Bioorganic Research, Mishima-gun, Osaka, Japan). All other reagents were either obtained from Sigma (St. Louis, MO, USA) or from Fluka (Buchs, Switzerland).
Antibodies
Antiserum to glutaraldehyde fixed D-aspartate (482 D-aspartate antiserum) and to L-glutamate (anti-L-Glu03) were kindly provided by J. Storm-Mathisen. The antiserum to D-aspartate was the same as that described previously (Zhang et al., 1993; Gundersen et al., 1993). The anti-D-aspartate antiserum recognizes D-aspartate and reacts weakly or not at all with other relevant amino acids when used as described. Testing freeze-substituted glutaraldehydefixed mixtures of albumin and D-aspartate in various concentrations showed that D-aspartate has to be present at about 1 mM in order to be detected with the immunocytochemical procedure used in this study (Ottersen, 1989). This implies that D-aspartate in the incubation medium must be concentrated almost two orders of magnitude in order to be detected. Consequently, the accumulation of D-aspartate in nerve terminals is caused by an active process or heteroexchange. The relatively low sensitivity of the assay may also explain why D-aspartate cross-linked to the surface of the plasma membranes (as a result of the glutaraldehyde fixation) is not detected to any significant degree.
The “anti-73 kDa antibodies” (AB#171; rabbit 85302: Danbolt et al., 1992) were raised against the purified rat EAAT2 protein (Danbolt et al., 1990), absorbed against aldehyde-treated proteins and affinity purified against pure deglycosylated EAAT2-protein (Fig. 1, lane 6). They recognize several EAAT2-epitopes (the 493–508 region in particular; Levy et al., 1993) and have been used in a number of studies (e.g. Chaudhry et al., 1995).
Fig. 1.
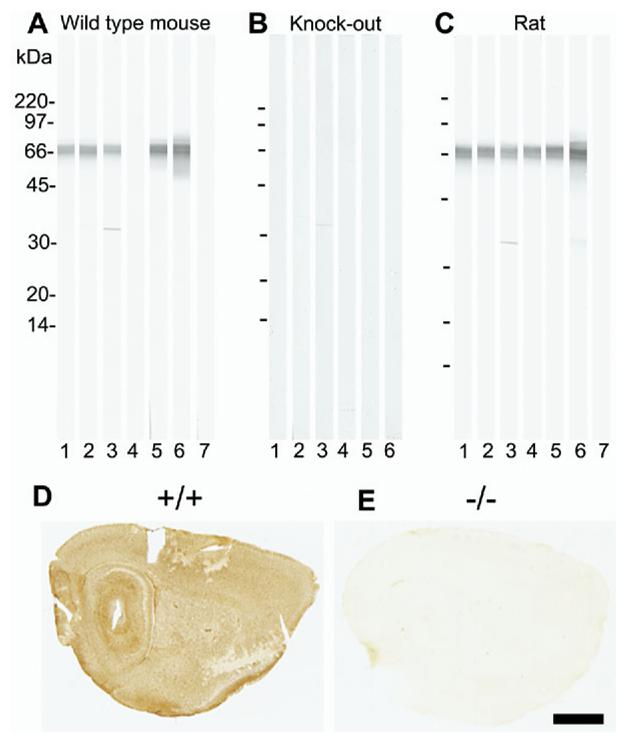
Specificity testing of GLT/EAAT2 antibodies by immunoblotting (A—C) and by immunocytochemistry (D—E). Whole brain tissue from wild-type (+/+) mouse (A), EAAT2 knockout (−/−) mouse (B) or adult Wistar rat (C) was solubilized in SDS, run on SDS-PAGE (10–20% gradient gels; 250 _μ_g protein per gel) and blotted onto nitrocellulose. The blots were cut into strips (25 strips per blot) and labeled with the following antibodies: 1: 0.2 _μ_g/ml anti-B12 (AB#360); 2: 0.2 _μ_g/ml anti-B12 (AB#150), 3: 0.1 _μ_g/ml anti-B493 (AB#95), 4: 0.5 _μ_g/ml anti-B518 (AB#94), 5: 0.2 _μ_g/ml anti-B563 (AB#355), 6: 0.1 _μ_g/ml anti-73 kDa (AB#171) and 7: no primary antibody (negative control). Mice brains (D: +/+;E: −/−) were immersion fixed in 4% formaldehyde. The sections were incubated with anti-EAAT2 antibodies in the presence of Triton X-100, followed by appropriate biotinylated secondary antibodies, then streptavidin peroxidase, and finally developed with diaminobenzidine. The figure only shows the test results for one of the antibodies: anti-B563 (2 _μ_g/ml; AB#355). Scale bar=1.5 mm.
Anti-peptide antibodies (Fig. 1, lanes 1–5) against glutamate transporters were prepared and tested as described previously (Lehre et al., 1995; Danbolt et al., 1998; Holmseth et al., 2006). All antibodies selected for this study recognize the transporter protein in its native state, after fixation with formaldehyde alone and after fixation with glutaraldehyde and formaldehyde. The peptides representing parts of rat EAAT1 (Storck et al., 1992; 543 amino acid residues) and rat EAAT2 (Pines et al., 1992; 573 amino acid residues) are referred to by capital letters “A” and “B,” respectively, followed by numbers indicating the corresponding amino acid residues in the sequences (given in parentheses): A522-541 (PYQLIAQDNEPEKPVADSET-amide), B12-26 (KQVEVRMHDSHLSSE-amide), B493-508 (YHLSKSELDTIDSQHR-amide), B518-536 (TQSVYDDTKNHRESNSNQC-amide), and B563-573 (SVEEEPWKREK-free acid).
The corresponding anti-peptide antibodies are named by adding “anti-” in front of the peptide names to which they are directed. As these names do not contain sufficient information to identify them unequivocally in our records, we have also included (in parentheses) the unique identification numbers they have in our internal database as well as the reference if these antibodies have previously been published: anti-A522 (AB#141; Lehre et al., 1995), anti-B12 (AB#150; Lehre et al., 1995), anti-B493 (AB#95;Haugeto et al., 1996), anti-B518 (AB#94; Lehre et al., 1995), and anti-B563 (AB#355). The anti-B563 antibodies are specific for the a-variant (the one first cloned: Pines et al., 1992). The rest of the anti-EAAT2 antibodies are directed to epitopes common to both variants.
Animals
Adult (200–250 g) male Wistar rats from Møllegaard Hansen (Denmark) were anesthetized with halothane and killed by decapitation. Mice lacking EAAT2 (generated by disrupting the part of the gene encoding the third transmembrane region of the EAAT2 protein), and their genotyping, have been described in detail previously and were kindly provided by Professor K. Tanaka (Tanaka et al., 1997). Knockout (−/−) and wild type (+/+) mice were produced by breeding heterozygote (+/−) mice which had been backcrossed six to nine times with C57Black/6 mice. Experiments were carried out on 2-week-old (P14) mice killed by cervical dislocation, and were always performed in pairs of +/+ and −/− mice from the same litter to reduce any variability that could occur between litters. The studies on mice were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986 and associated guidelines, and the studies on rats were carried out in the accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC). Formal approval to conduct the experiments was obtained from the animal subjects review boards of our institutions. All efforts were made to minimize the number of animals used and their suffering.
Preparation of hippocampal slices from adult and juvenile rat brains
Hippocampal slices were prepared and incubated with D-aspartate (supplemented with D-[3H]aspartate as stated) essentially as described previously (Gundersen et al., 1993, 1996). Briefly, rats were decapitated, the brains were quickly (1 min) taken out and chilled in ice cold Krebs’ phosphate solution (140 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 10 mM glucose and 15 mM sodium phosphate buffer, pH 7.4). The hippocampi were dissected free and 300 _μ_m transverse slices cut at +4 °C. The preparation procedure was completed in about 10 min. After cutting, the slices were placed on a nylon mesh, submerged in a glass cylinder containing 50 ml Krebs’ solution. Unless stated otherwise, the hippocampal slices were preincubated (45 min at 30 °C) and incubated (for 3–20 min at 30 °C or 20 °C as stated) in Krebs’ solution with 1–50 _μ_M D-aspartate. During pre-incubation and incubation in Krebs’ solution, the medium was continuously bubbled with O2. Glutamate receptor antagonists (1 mM kynurenic acid or 10 _μ_M MK-801 and 100 _μ_M DNQX) were added as stated. Negative controls were treated as above, but incubated either in the absence of D-aspartate or with the addition of 1 mM L-gluta-mate.
Ten other compounds were tested for their ability to inhibit D-[3H]aspartate uptake into slices, including dihydrokainate (0.2–1 mM), TBOA (0.01–1 mM), L-aspartate (0.01–1 mM), DL-_threo_hydroxyaspartate (0.01–0.5 mM), L-_trans_-pyrrolidine-2,4-dicarboxylate (0.01–0.5 mM), succinate (1 mM), D-glutamate (1 mM), GABA (1 mM), cystine (1 mM) and NMDA (1 mM). In agreement with previous studies (for review see Bridges et al., 1999; Danbolt, 2001) only the first five of these gave any significant inhibition (data not shown) as judged by accumulated radioactivity. The effect of dihydrokainate mediated inhibition was studied immunocytochemically. (Samples destined for measurement of radioactivity were treated in exactly the same way as samples destined for immunocytochemistry; thus all samples were aldehyde fixed as described below).
A number of other incubation solutions and conditions were also tested. These included direct incubation (without preincubation) in phosphate-buffered saline (PBS: 135 mM NaCl and 10 mM sodium phosphate buffer pH 7.4) with 50 _μ_M D-aspartate and other compounds as stated. The incubation in PBS was designed to mimic biochemical uptake assays in that the work was done without specific oxygenation, but solutions and samples were exposed to air. In order to mimic ischemia, Krebs’ solution without oxygenation or glucose, and with the addition of 2 mM cyanide and 1 mM iodoacetate to inhibit ATP production by mitochondria and by glycolysis, was also tested (Allen et al., 2005).
Following incubation, slices were fixed using 1% formalde-hyde and 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer. The slices were stored in the same fixative at 4 °C for up to 4 weeks. This is the fixative of choice for demonstrating amino acids, but it results in too harsh fixation for optimal detection of transporter proteins. Because of this, some of the slices were fixed for 2 h at room temperature in 4% formaldehyde and 0.1% glutaraldehyde in 0.1 M sodium phosphate buffer. These slices were then stored in the latter fixative diluted 1+9 with buffer for up to 4 weeks at 4 °C.
Some of the fixed slices were embedded by freeze substitution and processed for immunogold transmission electron microscopy (TEM) (see “Post-embedding electron microscopic immunocytochemistry” below) while the rest were dissolved and mixed with scintillation fluid for measurement of radioactivity.
Preparation of hippocampal slices from wild type and EAAT2 knockout mouse
The brains from 2-week-old +/+ and −/− EAAT2 mice (Tanaka et al., 1997) were dissected in ice cold Krebs’ solution containing 1 mM kynurenic acid, cut into 225 _μ_m slices on a vibrating slicer, washed and preincubated for 70 or 95 min in Krebs’ solution (in some cases 1 mM kynurenic acid was retained throughout in an attempt to reduce deterioration of slices through excitotoxic effects). Slices were then incubated with 50 _μ_M D-aspartate for 20 min with or without 1 mM kynurenic acid. Controls were prepared simultaneously, with the omission of D-aspartate as before. All slices were subsequently fixed in 2.5% glutaraldehyde and 1% formaldehyde in PBS. Some brains from EAAT2 deficient mice were also fixed (by immersion in 4% formaldehyde in NaPi, sodium phosphate buffer with pH 7.4) for light microscope immunocytochemistry.
Redundant brain tissue was immediately frozen (−80 °C) after the hippocampi were removed. Later, the tissue was dissolved in sodium dodecyl sulfate (SDS) for immunoblotting (as described by Lehre et al., 1995). It was confirmed that EAAT2 was absent in tissue from EAAT2 −/− mice (Fig. 1) while levels of EAAT1, EAAT3 and EAAT4 were comparable to those in tissue from wild type animals (data not shown).
Synaptosomes
Synaptosomes were made as described previously (Robinson, 1998) from adult rat hippocampus. The final synaptosome pellet was re-suspended in 5 to 50 volumes of sucrose. This suspension was kept on ice and was used within 2 h. One volume (350 _μ_l) of the synaptosome suspension was added to nine volumes of Krebs’ solution containing 0–50 _μ_M D-[3H]aspartate. When stated other compounds were added: dihydrokainate (300–500 _μ_M), L-glutamate (1 mM), succinate (1 mM), TBOA (0.01–0.5 mM), L-glutamate (0.01–1 mM), D-glutamate (1 mM), GABA (1 mM), L-α-aminoadipic acid (1 mM); cystine (1 mM), NMDA (1 mM), DL-_threo_-hydroxyaspartate (0.01–1 mM) or L-_trans_-pyrrolidine-2,4-dicarboxylate (0.01–1 mM). The synaptosomes were incubated (at 37 °C) for 10 s, 60 s, 3 min, 5 min and 20 min. The uptake was stopped by dilution into 10 volumes of ice-cold fixative (see above). 3 ml of the suspension was filtered through Millipore nitrocellulose—cellulose acetate filters (0.45 μ_m pores; HAWP) while the remainder was immediately pelleted by centrifugation (12,500 rpm, 20,000×_g, 4 °C for 20 min).
The radioactivity on the filters was determined by liquid scintillation counting after dissolving them in FilterCount (Packard, PerkinElmer, Downers Grove, IL, USA), while the pellets were suspended in 1.5 ml fresh fixative, transferred to 1.7 ml conical polyethylene tubes, centrifuged (17,000 rpm, 20 min, 4 °C) and stored overnight in the fixative. The substrate selectivity of the accumulation of radioactive D-aspartate (measured after fixation) corresponded (data not shown) to that reported previously using un-fixed synaptosomes (for review see: Danbolt, 2001). The lack of effect of succinate and α-aminoadipate suggests that neither dicarboxylate transporters nor the cystine—glutamate exchanger are interfering. Synaptosomes incubated (as indicated) without D-aspartate, with D-aspartate, or with D-aspartate and either L-glutamate or dihydrokainate were selected for electron microscopic analysis.
Post-embedding electron microscopic immunocytochemistry
The fixed synaptosome pellets and the fixed hippocampal slices, which were cut to include the stratum radiatum to the stratum moleculare of the central CA1, were rapidly frozen and embedded by freeze substitution in Lowicryl HM-20 resin according to methods described previously (Chaudhry et al., 1995; Dehnes et al., 1998). Ultrathin sections (50–100 nm) were cut orthogonal to the surface of the slices so that both the interior and the exposed surface could be compared. When two treatments were to be quantitatively compared (e.g. where the effect of dihydrokainate was being compared with a control), sections from both treatments were either placed on the same grid or incubated in the same drops of antibody to minimize experimental variation.
Sections were immunolabeled essentially as described before (Dehnes et al., 1998). Antibodies were diluted in a solution (TBS) consisting of 5–50 mM Tris—HCl (pH 7.4) and 50 mM NaCl with 2–3% human serum albumin (HSA). Goat—antirabbit secondary antibodies were conjugated to 10 or 15 nm gold particles diluted 1:20 in the above solution. Sections were counterstained in 2% aqueous uranyl acetate and (in some cases) 2% lead citrate prior to examination in a Phillips CM10, Phillips Tecnai 12, JEOL1230 or JEOL 100CX TEM. In some experiments, double labeling with Anti-D-Asp and transporter antibodies was also used in order to aid the identification of glial elements and was performed in two steps. The first step was as described above, and this was followed by a 1–2 h treatment with formaldehyde vapor at 80 °C to denature free anti-IgG binding sites (Wang and Larsson, 1985), and the labeling was repeated using the other antibodies.
Quantitative analysis of gold labeling
D-Aspartate immunoreactivity was quantified by counting the number of gold-particles associated with the various cellular compartments. Identification of terminals in slices or synaptosomes was made according to the presence of vesicle clusters and/or a distinct pre-synaptic specialization. To estimate the relative proportion of terminal versus non-terminal uptake, all slices incubated with D-aspartate were analyzed by means of a series of randomly taken photographs (usually 10) in which the labeling for D-aspartate in nerve terminals was compared with that in glia by counting the number of particles over one or the other compartment. The relative amounts of each compartment were estimated by means of a stereological technique (Baddeley et al., 1986) in which a grid was laid over each micrograph with evenly spaced points, and the number of points lying over terminals or glia were counted. In combination with the number of particles, an uptake ratio could then be calculated.
To estimate the relative density of glutamate transporter molecules in membranes of different structures, a different form of the above mentioned stereological technique was used. Up to 10 randomly selected images of hippocampal slices or synaptosome preparations labeled with antibodies to EAAT2 were analyzed by overlying a grid of evenly spaced lines. The number of times the lines crossed each type of membrane and the number of particles along each membrane category were determined. Particles were also counted over non-tissue (embedding resin) and unidentifiable areas. In addition, in the same images the lengths of selected membranes clearly definable as orthogonal to the section plane were also measured and the density of labeling determined, to account for potential overestimation of labeling density because of tangential membranes exposed to the section surface. Particles less than 40 nm from either side of the membrane were counted as membrane bound. When closer than 40 nm to two different types of membranes, particles were classified as “unattributable.”
For statistical analysis, data (numbers of particles or particle densities) were compared between different conditions and/or different tissue compartments using two-way analysis of variance (general linear model, Minitab14, Minitab Ltd., Coventry, UK) and Tukey’s all pairwise comparisons. A probability of 0.05 or less was considered to be significant. All data are given as “mean±S.E.M.”
Estimation of nerve terminal surface area
This was done in the stratum radiatum of hippocampus (subfield CA1) about 4 mm from the temporal pole. Electron micrographs of serial ultrathin sections cut at two different angles from stratum radiatum of hippocampus (subfield CA1) were from the same material as that used in Lehre and Danbolt (1998). The lengths of all identified dendritic (including spines) and astroglial cell membranes, and the area of the analyzed images (104 _μ_m2), were measured by a computer program (Image Analysis Tool in the electronic notebook database system version 1.0 from Science Linker AS, Oslo, Norway). Surface areas of dendrites and astroglia were calculated by multiplying the membrane lengths per image area by 4/π (Weibel, 1979).
Electrophoresis and immunoblotting
SDS-PAGE (Lehre et al., 1995), silver staining (Danbolt et al., 1990), electroblotting (Danbolt et al., 1992) and immunostaining (Lehre et al., 1995; Beckstrøm et al., 1999) were performed as described before.
RESULTS
D-Aspartate uptake in nerve terminals in rat hippocampal slices
Hippocampal slices were incubated with D-aspartate, fixed, embedded, cut, immunolabeled and studied electron microscopically (Fig. 2) mainly in the stratum radiatum of area CA1. Labeling above background was obtained after 3 min exposure to 10 _μ_M D-aspartate in the presence of sodium, with a mean proportion of 47% (of all gold particles) in terminals (in two slices from different animals). Higher substrate concentrations and longer incubation times gave stronger labeling (Fig. 3 shows labeling obtained using 50 _μ_M after 3 and 20 min). After 20 min most terminals (around 3/4) were immunopositive for D-aspartate. Slices incubated either without sodium (data not shown) or without D-aspartate had very little immunoreactivity (Fig. 3), while slices incubated with D-aspartate in combination with an uptake inhibitor (L-glutamate or dihydrokainate) were weakly positive (Fig. 3). This shows that the antibodies recognize D-aspartate and display little reactivity toward L-glutamate and compounds present endogenously in the tissue in agreement with previous studies using the same antibody (Gundersen et al., 1993).
Fig. 2.

Low magnification electron micrograph of D-aspartate uptake in a rat hippocampal slice. Hippocampal slices were incubated in D-aspartate (50 _μ_M, 3 min), fixed and labeled with antibodies to glutaraldehyde-fixed D-aspartate. Nerve terminals (t) and glia (g) were heavily labeled (note high number of black uniformly sized dots, three of which are indicated by arrows). Dendrites (d) and dendritic spines (s) were virtually unlabeled. Arrowheads indicate the location of a synaptic cleft. Note the large white areas which are not bounded by any continuous membranes and therefore probably represent extracellular space (es). Scale bar=300 nm.
Fig. 3.

Accumulation of D-aspartate immunoreactivity in rat slices. (A) Relative distribution of D-aspartate labeling in nerve terminals (solid bars) and astrocytes (open bars) in CA1 adult rat hippocampal slices from one representative experiment. The bars indicate mean±S.E.M. of number of gold particles per picture, and each bar represents eight pictures. Slices were incubated in Krebs’ solution with 50 _μ_M D-aspartate or no D-aspartate (negative control) for 3 or 20 min as indicated. Excess L-glutamate (1 mM) or dihydrokainate (DHK; 0.5 mM) was added to the incubation medium as indicated. Note that the anti-D-aspartate antibodies do not significantly label tissue which has been incubated in the absence of D-aspartate (negative control), and that the accumulation of D-aspartate immunoreactivity in nerve terminals can be substantially reduced by dihydrokainate or L-glutamate. This attests to the fact that the antibodies are highly specific. A predominance of nerve terminal labeling is already observed at 3 min arguing against a saturation phenomenon. Two-way ANOVA showed that the difference between nerve terminal labeling at 20 min and 3 min was not significant. However, the presence of L-Glu and DHK significantly reduced terminal labeling (P<0.001). No differences between glial labeling in any condition were significant, although the trend was for reduction by L-Glu and DHK. (B, C) TEM of nerve terminals in adult rat hippocampal slices double labeled for D-aspartate (large particles) and EAAT1/EAAT2 transporters (small particles) after incubation in 50 _μ_M D-aspartate. (B) Heavy labeling for D-aspartate occurs over terminals (t). The labeling for glutamate transporters is very low in the terminals. Some D-aspartate labeling is also detected over glial elements (g; the narrow process extending vertically between two other processes) which are heavily labeled for the transporters. (C) Micrograph taken from a slice treated as that in B except that 500 _μ_M dihydrokainate was present during the incubation. There is little labeling for D-aspartate over the terminals (t). D-aspartate labeling is, however, still present over the glial elements (g). Antibodies: 482 D-Asp, anti-73 kDa (AB#171) and anti-A522 (AB#141). Scale bars=250 nm.
From the time point when labeling could first be detected, D-aspartate immunoreactivity was observed in both astroglia and synaptic terminals. The majority of the identifiable nerve terminals making asymmetric synapses on spines were D-aspartate immunopositive when incubated with 50 _μ_M D-aspartate for 3 min or longer (Figs. 2 and 3A). Unambiguously identifiable spines were less labeled than any of the other structure types. Identifiable astroglia were often as strongly labeled as terminals (per area of section-surface). However, although both glial cell bodies (data not shown) and processes were labeled, synaptic terminals represented the compartment type that accumulated the largest fraction of the total labeling (Fig. 3A). This was a robust finding in spite of variation in incubation times and in other experimental conditions: in six slices from four animals after 50 _μ_M D-Asp incubation for 20 min, the proportion of all gold particles in terminals was 65±2.8% even though the overall labeling density varied dramatically. Terminals were also the strongest labeled structure observed in the stratum radiatum of hippocampi CA1 from juvenile rats (not shown) and juvenile mice (Fig. 4A, C). Furthermore, when electron micrographs were taken along the edges of the slices and compared with pictures taken in the middle of the slices, higher nerve terminal and glial labeling was observed as well as more efficient inhibition by L-glutamate (data not shown), but the ratio of nerve terminal to glial labeling was virtually unchanged (data not shown).
Fig. 4.

Accumulation of D-aspartate immunoreactivity in terminals depends on the EAAT2-gene. Hippocampal slices were incubated with D-aspartate (50 _μ_M, 20 min) and were protected with glutamate receptor blockers both during preincubation and incubation. (A) TEM of nerve terminals from juvenile wild type (+/+) mouse hippocampal slice preparations double labeled for D-aspartate (large particles) and EAAT1/EAAT2 transporters (small particles). As with the adult rat slices, terminals (t) containing vesicles can be seen to be heavily labeled for D-aspartate, but with low labeling for transporters. Glial profiles (g) are heavily labeled for the transporters and contain some labeling for D-Asp. (B) Hippocampal slice from a juvenile EAAT2-knockout (−/−) mouse. Note that vesicle containing terminals (t) are largely devoid of D-aspartate labeling, while glial elements express EAAT1 (small particles) and continue to show evidence of D-aspartate uptake by the presence of large particles over them. Antibodies: 482 D-Asp, anti-73 kDa (AB#171) and anti-A522 (AB#141). (C) Histogram showing relative distribution of D-aspartate labeling (gold particles) between nerve terminals (t) and astrocytes (g). Ten random TEMs were taken of the stratum radiatum in one slice from each of three +/+ and three −/− animals. Note that almost half of all the gold particles seen on the TEMs from the +/+ were located over nerve terminals. There was no significant difference between the proportions of labeling in terminals vs. glia (two-way ANOVA, _P_=0.94). It should be noted that no morphologically unambiguous terminals were labeled in the −/−, but all structures remotely resembling terminals were counted as such, implying that the number of particles in terminals in the −/− is likely to be overestimated. The difference in proportion of labeling between terminals and glia in the −/− was highly significant (P<0.001). NB: It was not possible to compare the uptake into glia directly between the +/+ and the −/− because of possible variations in labeling density between animals. Thus, the data show that glia account for most of the uptake still present after deletion of the EAAT2 gene, but not whether there is more or less uptake in +/+ or −/−. (D) TEM of L-glutamate labeling in the −/− mouse hippocampus. The picture shows heavy labeling for glutamate in terminals (t) and little labeling of dendrites (d). Thus, there are high concentrations of glutamate both in nerve terminals from −/− mice and in terminals from +/+ mice. Antibody: anti-L-Glu03. Scale bars=200 nm (A); 400 nm (B, D).
Addition of glutamate receptor blockers (1 mM kynurenic acid or 100 _μ_M DNQX and 10 _μ_M MK801) improved tissue morphology (thereby improving identification of cellular structures), but did not appear to affect the relative contribution of terminals to total labeling (data not shown). Poorer tissue morphology and lower overall labeling were observed in slices incubated directly (with no preincubation) in PBS with D-aspartate but the percentage of terminal labeling was still high (72% of all particles were found in terminals; mean of two experiments). Addition of cyanide and iodoacetate reduced overall labeling even further (data not shown).
For all of the experiments described above, most of the labeled unidentifiable structures are likely to be axons, implying that the uptake by neuronal compartments is even higher.
D-Aspartate uptake into terminals and glia is mediated by EAAT2
We used dihydrokainate, a selective competitive inhibitor of EAAT2 (Arriza et al., 1994; Bridges et al., 1999), to test the role of EAAT2 in D-aspartate accumulation (Fig. 3). Dihydrokainate reduced D-aspartate labeling in both glia and terminals. The presence of DNQX and MK801 in these experiments should rule out the possibility that dihydrokainate acted indirectly by activating glutamate receptors and thus weakening the ion and voltage gradients driving uptake.
To further test the role of EAAT2 we compared D-aspartate uptake into hippocampal slices from three pairs of wild-type (+/+) and gene modified mice (−/−) lacking EAAT2 (14 days old, P14). Slices were preincubated with oxygenated Krebs’ solution containing glucose as for the rat slices and prepared for immunogold labeling (Fig. 4A, B). Slices from EAAT2-deficient animals (−/−) incubated with D-aspartate in the absence of receptor blockers had too poor morphology to allow identification of the various cellular elements. Because of this, only slices incubated in the presence of receptor blockers were used for quantitative analyses. In these slices, virtually no labeling of terminals was detected in the −/− mouse (Fig. 4B). In fact, none of the morphologically unambiguous terminals were labeled. When counting as terminals all structures with the slightest resemblance to terminals, only 4±2% (_n_=3 animals) of the total number of gold particles could be attributed to terminals compared with 93±2% in glia-like processes (Fig. 4C). (NB: This does not imply increased glial uptake in the knockout. See legend to Fig. 4 for methodological comments). In contrast, the +/+ mice showed nerve terminal and glial labeling for D-aspartate similar to that seen in the rat (Fig. 4A). The proportions of total label in the terminal and glial compartments were 44±5 and 49±6%, respectively (_n_=3 animals; Fig. 4C). The almost complete absence of D-aspartate uptake in terminals from the −/− mice suggests that the nerve terminal uptake is dependent on the presence of an intact gene for EAAT2.
Although terminals in the −/− mice practically lacked ability to take up D-aspartate, they still contained high levels of endogenous glutamate (Fig. 4D). This indicates a normal ability to synthesize glutamate and also signifies that the antibodies to D-aspartate do not label L-glutamate as there was almost no terminal labeling in the D-aspartate treated −/− mouse slices.
Quantification of EAAT2 protein in synaptic terminals in hippocampal slices
The various cellular components making up brain tissue are not only small (often below the light microscopic resolution limit of around 0.25 _μ_m), but they also intermingle in close proximity to each other. This closeness makes it difficult to assess weak labeling of the plasma membrane of one structure neighboring a strongly labeled one, even at the electron microscopic level. The hippocampal slice preparations used in the present study, however, have been incubated in vitro and this treatment leads to an increase in the extracellular volume fraction compared with perfusion fixed brain tissue. It is therefore easier to distinguish neuronal EAAT2 labeling from glial EAAT2 labeling because the various cellular components are better separated. Fixed and embedded synaptosome preparations have been studied for the same reason.
As much as 80±6% (n=3 animals) of all gold-particles resulting from labeling with antibodies to EAAT2 was found in association with glial plasma membrane in rat slices (Fig. 5, and Table 1). There was variation in labeling intensity between glial membranes probably implying that there is more than one subtype of astroglial membranes (in agreement with: Matthias et al., 2003). Although most of the data shown are based on the anti-73 kDa antibodies (Ab#171), the other antibodies used alone or in combination gave similar labeling patterns attesting the specificity of the detection.
Fig. 5.

Electron microscopic visualization of EAAT2-protein in rat hippocampal slices. (A) Labeling was observed in glial (g) membrane (large arrowheads point to two of the gold particles) and also over nerve terminal (t) membranes (e.g. arrows). No significant labeling was found on the membranes of dendritic spines (s). Particles (e.g. small arrowheads) are counted as unattributable as they are closer than 40 nm to two different types of membranes. About 10% of the gold particles were classified as unattributable (Table 1). (B) Gold particles (arrows) were observed in axonal (a) membranes just prior to the axon terminal (t). (C) Glial membranes (g) were heavily labeled, while nerve terminals were weakly, but indisputably labeled. Some of the labeling of terminals was intracellular (asterisk), but this is likely to mostly represent unwanted reactivity of the antibodies as some labeling of terminal cytoplasm was also observed in −/− mice (see text). The arrow points to one of the only six gold particles (out of a total of 1532+741, Table 1) found in unambiguous association with identifiable spine (s) membranes. Although there are some unattributable particles, we believe that most of the attributable labeling is due to glia and to axons because the vast majority of identifiable spines were unlabeled. The one shown in A is typical (see also Results). (D) Labeling of the terminal membranes was also obtained with antibodies specific to the A-variant (the predominant form) of EAAT2. Antibodies: A—C, anti-73 kDa (AB#171) at 5 _μ_g/ml; D anti-B563 (AB#355) at 1 _μ_g/ml. Scale bars=200 nm.
Table 1.
Analysis of the relative density of EAAT2 labeling in terminals and glia in rat hippocampal slices
| Preparations (animals) | Mean±S.E.M. | |||
|---|---|---|---|---|
| A | B | C | ||
| Relative membrane length (number of line crossings on overlay screen) | ||||
| Glial membrane | 120 | 88 | 116 | |
| Terminal membrane | 39 | 98 | 121 | |
| Gold particle number | ||||
| Glial membrane | 2187 | 1113 | 569 | 79±4% |
| Terminal membrane | 63 | 91 | 68 | 5.9±2% |
| Terminal cytoplasm | 53 | 123 | 56 | 5.9±2% |
| Spine cytoplasm and membrane including post-synaptic density | ND | 6 | 0 | |
| Unattributable | 213 | 199 | 48 | 10±1.4% |
| Total | 2516 | 1532 | 741 | 100% |
| Relative membrane labeling density | ||||
| Glial gold/membrane length (crossings) | 18.2 | 12.6 | 4.91 | |
| Terminal gold/membrane length (crossings) | 1.62 | 0.93 | 0.56 | |
| Ratio of density (terminal/glia) | 0.089 | 0.073 | 0.11 |
The remaining 20% of the particles appear at first sight to be scattered all over the pictures, including parts only containing embedding medium and no tissue. Some of these particles therefore represent background (e.g. cross-reactivity, unspecific attachment or specific labeling which has detached during the drying of the sections after the incubation and washing steps). However, as is shown in Table 1 and Fig. 5, about 6% of the total number of gold-particles was associated with nerve terminal membranes. About half of the terminals were labeled (60±12% if only clearly labeled and clearly unlabeled are included in the calculation), but the number of gold particles per labeled terminal was low, usually one. Particles appeared qualitatively to be evenly distributed in nerve terminal membranes and were found both extra- and intra-synaptically, but no attempts were made to quantify the distribution because of the relatively low levels of labeling. Another 6% of the particles were found in terminal cytoplasm (Table 1), but this may represent background (see below).
About 8% of the EAAT2-labeling of rat tissue could not be attributed to any particular identifiable structure. Some of this unattributable category represented labeling of mitochondria (and is therefore due to cross-reactivity with other molecules), but most of it represents labeling of small axons implying that the combined labeling of axons and terminals is between 10 and 20% of the total labeling. The reasons for this are that the number of axons is high, and that the axons are hard to distinguish from dendritic spine necks when cut transversely. Thus, small axons are overrepresented in the group of “unidentifiable” structures. In agreement with the fact that no D-aspartate uptake into spines was detected (Fig. 2), we did not observe statistically significant EAAT2-immunoreactivity in clearly identifiable dendrites with any of the anti-EAAT2 antibodies used. Only six gold particles, out of 2273, were closer than 40 nm to a dendritic spine membrane and more than 40 nm from another type of membrane. One of these six was in a postsynaptic density (not shown). Another of these six is shown in Fig. 5C (arrow). Fig. 5C also shows that there are a number of particles located less than 40 nm from either membrane. These are classified as “unattributable.” Consequently, the present material from the stratum radiatum (CA1) does not provide any evidence for expression of EAAT2 in spines, but does not rigorously rule out the possibility that a small subset of spines could be different (Chen et al., 2004). For the overwhelming majority of spines in this region, however, it seems clear that if there is any EAAT2 there at all, then the concentration must be even lower than that in terminals and axons. The total number of gold particles in myelin and dendritic shafts also appears insignificant in the present material.
Tissue from EAAT2 (+/+) and (−/−) mice (Tanaka et al., 1997) was used to confirm EAAT2 antibody specificity in labeling of the terminal membrane. For animal welfare reasons mice deficient in EAAT2 were killed at P14. The levels of EAAT2 protein are lower at this age than in the adult (Ullensvang et al., 1997) so antigen detectability is likely to be lower than in tissue from adult rats. Nevertheless, clear labeling of glial membranes was observed in the P14 +/+ mice, and there was a weaker but consistent labeling of terminal membranes. Approximately 22% and 15%, respectively, of terminals had apparently specific membrane labeling in slices from two animals (labeled with the anti-73 kDa antibody).
In the EAAT2 −/− mice, no labeling was detected at the light microscopic level with EAAT2 antibodies (Fig. 1). At the EM level, there was very little labeling of any type of plasma membrane in tissue from the EAAT2-deficient mice with either anti-73 kDa antibody or anti-B563 antibodies (data not shown). In slices from two −/− animals, gold particles were found in about 3% and 0.9% of terminals respectively with the anti-73 kDa antibody, or about one tenth overall the number found in the +/+. Occasional particles were observed in the terminal cytoplasm, with about the same frequency in the −/− (15% and 11%, respectively) and in the +/+ (9% and 9%, respectively), implying that the cytosolic labeling represents unwanted reactivity.
It might be argued that only a part of the gene has been deleted in the EAAT2 −/− (Tanaka et al., 1997) and that the possibility exists that the remaining labeling in terminals represents truncated EAAT2 protein. This seems unlikely, however, as there is no sign of such a protein in glial cells in tissue sections and also not on Western blots based (Fig. 1) on 10–20% gradient gels (which are able to separate proteins down to about 2 kDa).
Rat synaptic terminals and axons express the a-variant of EAAT2
Most of the antibodies used in these studies recognize both a- and b-variants of EAAT2. The only exception is the anti-B563 (AB#355) antibodies, which are specific for the a-variant. The anti-B563 antibodies (Ab#355) labeled astroglia (not shown) and terminals and axons (Fig. 5D) just like the antibodies recognizing all variants of EAAT2.
The plasma membrane surface densities of axons and terminals (boutons)
A low EAAT2-concentration in axons and terminals could still represent a significant amount of EAAT2 protein if it is distributed in a large surface area. To address this question, it was important to estimate the total surface areas of the various cellular structures.
As reported previously (Lehre and Danbolt, 1998) the astroglial plasma membrane surface density (surface area per unit volume) in the hippocampus stratum radiatum CA1 is 1.4 _μ_m2/_μ_m3 (or 10% of the total). Here we estimated the surface density of dendrites (including spines) to be 1.5 _μ_m2/_μ_m3. This is similar to the surface density of spines in the molecular layer of the cerebellum which is 1.1 _μ_m2/_μ_m3 (Dehnes et al., 1998). Based on the numbers above, the surface density of nerve terminals and axons was calculated by subtracting the glial and dendritic area from the total area [14−(1.4+1.5)=11.1]. Some glial and dendritic structures may have been misidentified as axons, so the real surface density of axons and terminals is probably less than 11.1 _μ_m2/_μ_m3, but closer to 11 _μ_m2/_μ_m3 than to 10 _μ_m2/_μ_m3. Because about 80% of nerve terminals in stratum radiatum are glutamatergic and synapse onto spines (for review see: Ottersen and Storm-Mathisen, 1984), it follows that the combined surface density of the glutamatergic axons and terminals is about 9 _μ_m2/_μ_m3.
In the stratum radiatum of the adult rat, each pyramidal cell spine receives only one synaptic contact (Megías et al., 2001). The ratio of membrane surface area of the spines to that of the terminals synapsing onto them is of the order of 1:1 (considering terminals as cylinders with diameter 500 nm and about 800 nm long, and spines as cylinders with a mean diameter of 300 nm and 1 _μ_m long (Sorra and Harris, 2000)). Thus, the terminals make up about one of the 9 _μ_m2/_μ_m3 membrane belonging to excitatory axons and terminals, and so contribute almost as large a membrane area as astrocytes (1.4 _μ_m2/_μ_m3).
D-Aspartate uptake and EAAT2 immunoreactivity in synaptosomes
To ensure that the results described above are not due to some unexpected diffusion barrier that hampers access of D-aspartate to parts of the tissue, or due to D-aspartate transfer between compartments, we studied D-aspartate uptake in synaptosome preparations which should not have any diffusion barriers or transfer between compartments. As shown in Fig. 6, D-aspartate immunoreactivity accumulates preferentially in nerve terminals in the synaptosome preparation. Gold labeling was noted on clearly identifiable synaptic terminals in all of the D-aspartate incubated preparations. A range of D-aspartate concentrations and incubation times was tested (Table 2). At 10 s incubation with 50 _μ_M D-aspartate, very weak labeling was obtained. The majority of terminals were unlabeled and most of the labeled ones had only one gold particle. However, although the total number of gold particles was low, about two thirds of them were associated with terminals. Longer incubation with D-aspartate increased labeling density per terminal dramatically (Table 2). After 20 min of 50 _μ_M D-aspartate, 85.5% of the gold particles were associated with terminals (mean of two experiments). In agreement with earlier reports uptake into glial-like elements was noted (e.g. Henn et al., 1976; Nakamura et al., 1993), but at a relatively low frequency. At 10 _μ_M D-aspartate for 3 min, 500 _μ_M dihydrokainate inhibited accumulation of radioactive D-[3H]aspartate and D-aspartate immunoreactivity to a similar degree, around two thirds (data not shown), suggesting that EAAT2 mediates much of the uptake.
Fig. 6.
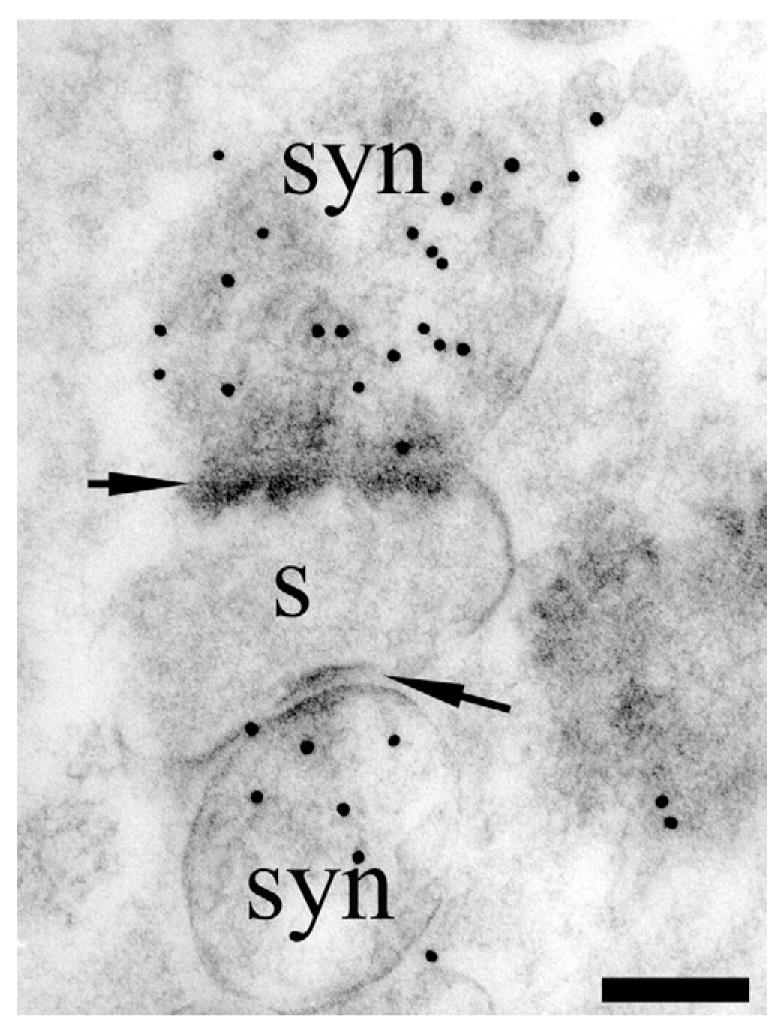
TEM visualization of D-aspartate uptake in synaptosome preparations. Rat synaptosomes (syn) were incubated for 20 min in 50 _μ_M D-aspartate. Note heavy labeling over synaptosomes (syn) that are attached to a remnant of a dendritic spine (s). The synaptosomes are clearly identifiable by the presence of synaptic densities in the opposing membranes (arrows) and vesicles. Antibody: 482 D-Asp. Scale bar=100 nm.
Table 2.
Synaptosomes: density of D-aspartate labeling in terminals compared to glia
| Incubation time with 50 _μ_M D-Asp | ||||
|---|---|---|---|---|
| 0 s | 10 s | 60 s | 20 min | |
| Gold particles over terminals | 0.3 | 1.8 | 10.5 | 42.4 |
| Gold particles over non-terminals | 0.7 | 0.8 | 4.9 | 9.1 |
| Labeled terminals as percent of total number of terminals | Not counted | Not counted | 76% | 68% |
| Relative density of gold particles over terminals per unit area | 1 | 6.7 | 32 | 160 |
The synaptosome preparations also provide a good way to confirm EAAT2 presence in the terminal membranes as the synaptosomes are well separated from glial profiles in these preparations. Labeling with anti-73 kDa antibodies was found on terminal membranes and cytosol, as in the slice preparations (Fig. 7), with slightly higher relative densities compared with glia (Table 3). The latter may be due to greater ambiguity and a higher level of inappropriate assignment in the slices, where glial membranes are not as well separated from terminals as in the synaptosome samples.
Fig. 7.
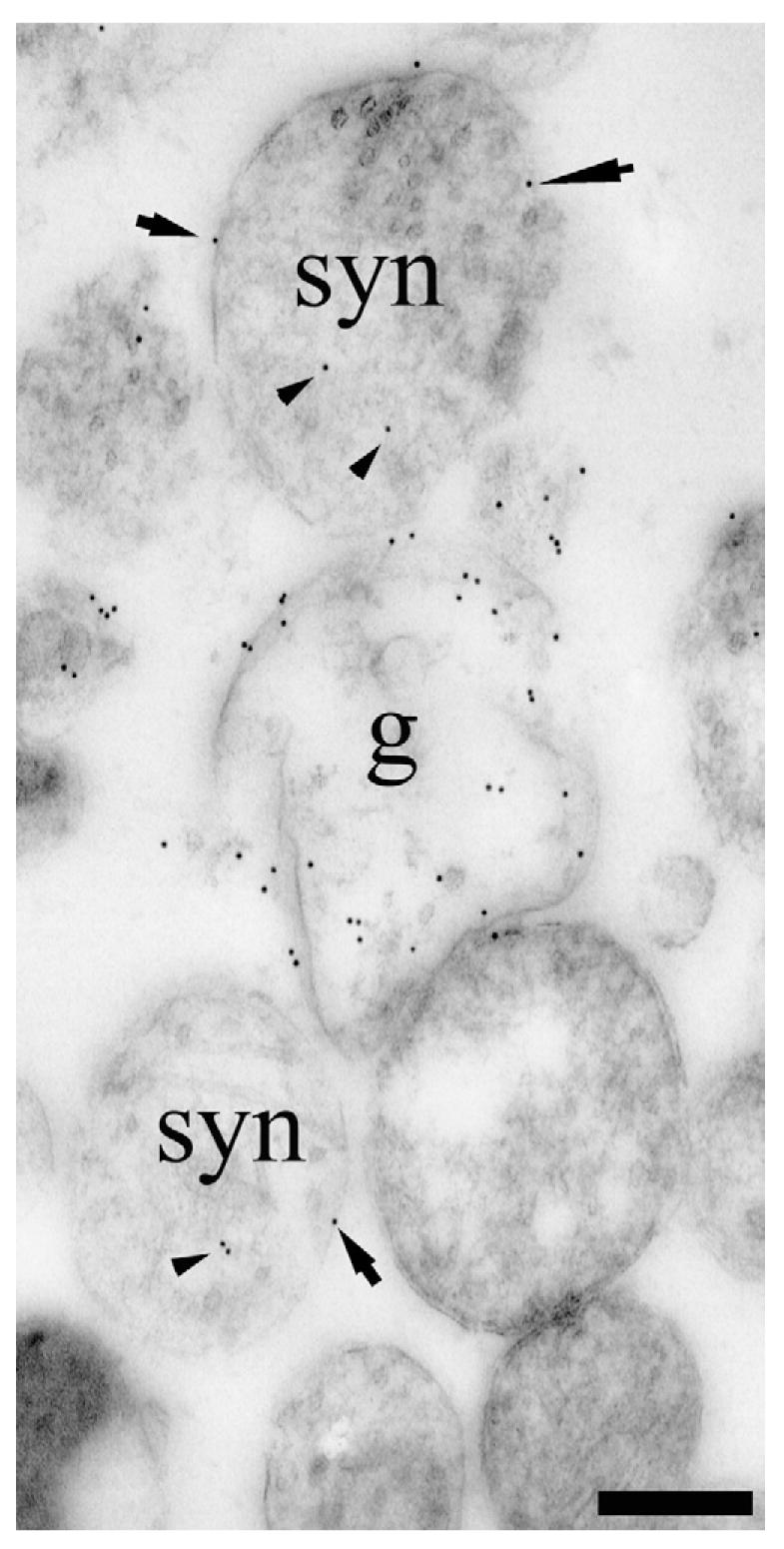
TEM visualization of EAAT2-protein in a synaptosome preparation containing nerve terminals (syn) and astrocytic processes (g). Note that some terminal membrane labeling is detectable (arrows point to three clear examples) which cannot be attributed to glial membranes because the synaptosome is isolated from surrounding tissue. Also note that there was some labeling of terminal cytoplasm (arrowheads), but this is of unclear significance (see text). Antibodies: a mixture of the anti-B12 (AB#150), anti-B493 (AB#95) and anti-B518 (AB#94) antibodies to EAAT2. Scale bar=300 nm.
Table 3.
Density of EAAT2 in terminals compared to glia in synaptosomal preparations
| Mix | Anti-73 kDa | |
|---|---|---|
| Terminals (synaptosomes) | ||
| Total number of terminals assessed | 70 | 41 |
| Length of labeled membrane | 19.7 | 7.47 |
| Number of gold particles in membrane | 21 | 8 |
| Mean density | 0.99 | 1.5 |
| Number of gold particles in cytosol | 14 | 3 |
| Glia | ||
| Total number of profiles assessed | 8 | 7 |
| Length of labeled membrane | 5.7 | 6.6 |
| Number of gold particles in membrane | 50 | 33 |
| Mean density | 6.3 | 5.0 |
| Ratio of density (terminals/glia) | 0.16 | 0.3 |
DISCUSSION
Hippocampal terminals accumulate D-aspartate by means of EAAT2
The data presented here show that most small CA1 nerve terminals with asymmetric synaptic specializations (putative excitatory synapses) accumulate D-aspartate by a sodium-dependent mechanism that concentrates it in relation to the external medium. This mechanism is likely to be based on EAAT2 because the build-up of D-aspartate immunoreactivity is inhibited by compounds known to inhibit EAAT2, including dihydrokainate (Arriza et al., 1994; Bridges et al., 1999), and because the uptake into terminals is absent in EAAT2-deficient mice.
The accumulation is likely to be via direct uptake of D-aspartate, rather than the by-product of a metabolic process, such as the glutamine-glutamate cycle. The antibodies detect D-aspartate itself which is unlikely to be metabolized in astrocytes, transferred into neurons and then reconverted into something recognized by the antibodies, especially in view of the slow metabolism of D-aspartate (see below). In addition, the large volume of the incubation medium should dilute any released intermediates to insignificant levels thereby interrupting the cycle, particularly in the synaptosome suspensions and at the surface of the slices where D-aspartate immunoreactivity tended to be highest.
The anti-D-Asp antibodies do not appear to recognize L-glutamate because there was no labeling in slices that have not been incubated in D-aspartate, nor was there labeling of the slices from EAAT2 −/− mice incubated with D-aspartate, whereas there was labeling in the latter for glutamate. This indicates that despite the lack of direct uptake in the EAAT2 −/−, L-glutamate is still present in the terminals presumably because of normal synthetic processes taking place.
D-Aspartate preferentially accumulates in synaptic terminals
One reason for the apparent predominance of uptake into terminals compared with glia could be that the latter metabolize D-aspartate faster than nerve terminals, converting it to a substance that is not recognized by the antibody. But if glial uptake in these preparations is considerably larger than that of nerve terminals, then it follows that most of the D-aspartate that has been taken up has been metabolized. A rapid metabolism of D-aspartate, however, does not agree with published experimental data. In fact, D-aspartate was chosen for this study precisely because its overall metabolism is very slow in adult brain tissue (Davies and Johnston, 1976; Takagaki, 1978). Further, although D-aspartate oxidase has been detected in cultured astrocytes (Urai et al., 2002), it seems to be predominantly expressed in neurons in the intact brain (Schell et al., 1997; Zaar et al., 2002). Finally, we find a predominance of terminal uptake even at incubation times as short as 3 min. Thus, metabolism of D-aspartate can be ruled out as a cause of the predominance of nerve terminal labeling. The low levels of D-aspartate in spines could be a sign of metabolism as D-aspartate oxidase is expressed there (Schell et al., 1997) or, more likely, it could simply be due to a low level of glutamate transporters. We failed to detect EAAT2 in spines, and although EAAT3 is expressed in them, it is at low levels compared with EAAT2 in glia. The total tissue concentration of EAAT3 in young adult rat hippocampus is 100 times lower than that of EAAT2 (Dan-bolt et al., 2006) and the total plasma membrane surface density of cellular compartments expressing EAAT3 in hippocampus CA1 is about the same as that of astroglia (S. Holmseth and K. P. Lehre, unpublished observations). This implies that the density of EAAT3 in spine membranes is an order of magnitude lower than the density of EAAT2 in terminals and two orders of magnitude lower than the EAAT2 density in astroglia (see below).
Another possibility is that the damage to plasma membranes during cutting of the slices differentially affects nerve terminals and glia. Damage anywhere on glial cells may compromise the driving forces of the uptake in the entire cell with all its extensions, while similar damage to neuronal dendrites and cell bodies may not necessarily compromise the activities in the terminals. A difference between axons and glial cell processes in their ability to reseal after being cut would have the same effect. We think these possibilities are unlikely, however, because glial cells in slices maintain a large negative resting potential, and their extensive gap junctional coupling should help to maintain the membrane potential and intracellular ion concentrations near normal values even if one particular cell is somewhat damaged (Bergles and Jahr, 1997).
Consequently, the most likely interpretation is that the measured amounts of accumulated D-aspartate in glia and terminals reflects the relative uptake into these compartments.
The density of EAAT2 molecules in terminals is low
About 6% of the total number of gold-particles representing EAAT2 was observed in the terminal plasma membranes, while about 80% was found in astrocyte membranes. Because glial and terminal membranes are found at a density of 1.4 and 1 _μ_m2/_μ_m3, respectively, it follows that the average densities of EAAT2 protein molecules in the membranes differ by a factor of about 10. [Ten times 6% divided by 1 _μ_m2/_μ_m3 equals 80% divided by 1.4 _μ_m2/_μ_m3.] This number, however, is just an approximation considering (a) that the labeling of both terminals and glia is variable, (b) that the immunoreactivity found in the terminal cytoplasmis not included because it probably mostly represents background (another 6% of the total), and (c) that we assume all of the glial EAAT2 is in the membranes. Although the labeling is associated with the plasma membranes, the resolution of the method used is insufficient to tell if all of the EAAT2 protein is actually in the membrane (and thereby in a position where it can be actived) or if some of it is present in the cytoplasm close to the plasma membrane. It should also be noted that the immunocyto-chemical data are obtained from slices while the data on membrane surface densities are obtained from perfusion fixed intact brains. Further, since neurons have been reported to express increased levels of EAAT2 in disease, it is possible that neuronal expression may be highly variable (for references and review, see: Danbolt, 2001).
Most of the remaining 10% of EAAT2 immunoreactivity that is not accounted for in the preceding calculation is distributed in axons (membranes and axoplasm). The diameter of these axons is too small to allow membrane-inserted and cytoplasmic EAAT2 to be distinguished. The combined plasma membrane surface density of these axons is 8 _μ_m2/_μ_m3. This implies that the EAAT2 density in axonal membranes is less than 2% of that in glial membranes [(10/8)/(80/1.4)=0.022].
A key question is whether EAAT2 has to be expressed above a certain threshold to be detectable. If so, this could have a significant impact on the calculation of protein concentrations because of the large plasma membrane surface density of the small axons. A related question is whether the labeling intensity is a linear function of the protein concentration within the relevant concentration ranges. This is unknown, and it should be noted that membrane labeling per labeled terminal is usually one gold particle. About half of the terminals were labeled in rats while three quarters take up D-aspartate and therefore probably express EAAT2. This is not an argument against a linear relationship, but it clearly shows that the method is used at its limits and that some caution is warranted.
Nevertheless, the finding that 10–20% of all EAAT2 is neuronal in the stratum radiatum of hippocampus CA1 explains why there are the high levels of EAAT2 mRNA in CA3 pyramidal neurons (Torp et al., 1994, 1997; Chen et al., 2004; Berger et al., 2005).
The mismatch between EAAT2 distribution and sites of D-aspartate accumulation
In view of the dependence of the uptake on EAAT2 and that about 6% of the EAAT2 protein is present in the nerve terminal membranes, it is tempting to speculate how terminals appear to take up D-aspartate as well as glia with only a fraction of the total EAAT2 protein. It could be that terminal EAAT2 is more active than astroglial EAAT2 (i.e. different regulatory control or different lipidic microenvironments). However, because of the higher internal levels of glutamate in terminals (e.g. Bramham et al., 1990; Ji et al., 1991; Ottersen et al., 1992), it could also be that the disproportionately strong labeling of terminals is due to a greater capacity for heteroexchange, i.e. exchange of external and internal substrates at a 1:1 relationship (for review see: Kanner and Schuldiner, 1987; Danbolt, 2001; Grewer and Rauen, 2005).
CONCLUSIONS
(1) Our data confirm that the “old” studies on hippocampal synaptosomes and slices (see section 4.2 in Danbolt, 2001) predominantly measured nerve terminal uptake as originally believed. Although it remains to be discovered if and to what extent the present findings apply to other regions, it has been demonstrated electron microscopically that terminals in some other regions, e.g. the neocortex (Beart, 1976) and the striatum (Gundersen et al., 1996) also have glutamate uptake capabilities. (2) We confirm that antibodies recognizing all forms of EAAT2 and antibodies specific for the a-variant positively identify EAAT2 protein in both astroglia and terminals (Chen et al., 2004). While Chen and co-workers used a pre-embedding technique, we have used a post-embedding technique in combination with stereology and we show that the density of EAAT2 protein associated with astroglial membranes is on average 10 times higher than those of terminals. (3) About three quarters of small nerve terminals with asymmetric synaptic specializations (putative excitatory synapses) in the stratum radiatum (hippocampus CA1) are able to accumulate D-aspartate via a mechanism that depends on the EAAT2 gene. This is a higher number than that reported by Chen and co-workers (2004), and may be due in part to our technique being more sensitive, and that Chen and co-workers probably only detected the terminals with the highest levels. (4) Neither EAAT2 nor D-aspartate up-take was detected in spines, but the study was not aimed at ruling out the possibility (Chen et al., 2004) that a subset of spines could be different.
Acknowledgments
We thank Bjørg Riber and Jon Storm-Mathisen for antibodies to D-aspartate and L-glutamate, Kohichi Tanaka for knockout mice, and Helene Marie and Steve McGuinness for genotyping. We are grateful to Rolf Seljelid without whose encouragement and support the project would not have been possible. This work was supported by the Norwegian Top Research Program (Toppforskningsprogrammet), the Norwegian Research Council (grant to N.C.D., fellowships to Y.D., K.U., A.Q.A. and V.G.; Medisin og Helsegruppe), EU BIOMED (contract QLG3-CT-2001-02004), by the Wellcome Trust and a Wolfson-Royal Society Award (to D.A.) and by grant 1R01NS051561 (to D.J.R.) from the National Institute of Neurological Disorders and Stroke/National Institute of Mental Health.
Abbreviations
DNQX
6,7-dinitroquinoxaline-2,3-dione
EAAT
excita-tory amino acid transporter (=glutamate transporter)
MK801
dizocilpine
NMDA
_N_-methyl-D-aspartate
PBS
phosphate-buffered saline
SDS
sodium dodecyl sulfate
TBOA
DL-_threo-beta_-benzyloxyaspartate
TEM
transmission electron microscopy
REFERENCES
- Allen NJ, Karadottir R, Attwell D. A preferential role for glycolysis in preventing the anoxic depolarization of rat hippocampal area CA1 pyramidal cells. J Neurosci. 2005;25:848–859. doi: 10.1523/JNEUROSCI.4157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baddeley AJ, Gundersen HJG, Cruz-Olive LM. Estimation of surface area from vertical sections. J Microsc. 1986;142:259–276. doi: 10.1111/j.1365-2818.1986.tb04282.x. [DOI] [PubMed] [Google Scholar]
- Beart PM. The autoradiographic localization of L-[3H] glutamate in synaptosomal preparations. Brain Res. 1976;103:350–355. doi: 10.1016/0006-8993(76)90804-0. [DOI] [PubMed] [Google Scholar]
- Beart PM, O’Shea RD. Transporters for L-glutamate: An update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstrøm H, Julsrud L, Haugeto Ø, Dewar D, Graham DI, Lehre KP, Storm-Mathisen J, Danbolt NC. Interindividual differences in the levels of the glutamate transporters GLAST and GLT, but no clear correlation with Alzheimer’s disease. J Neurosci Res. 1999;55:218–229. doi: 10.1002/(SICI)1097-4547(19990115)55:2<218::AID-JNR9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Berger UV, Desilva TM, Chen WZ, Rosenberg PA. Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. J Comp Neurol. 2005;492:78–89. doi: 10.1002/cne.20737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Torp R, Zhang N, Storm-Mathisen J, Ottersen OP. Distribution of glutamate-like immunoreactivity in excitatory hippocampal pathways: a semiquantitative electron microscopic study in rats. Neuroscience. 1990;39:405–417. doi: 10.1016/0306-4522(90)90277-b. [DOI] [PubMed] [Google Scholar]
- Bridges RJ, Kavanaugh MP, Chamberlin AR. A pharmacological review of competitive inhibitors and substrates of high-affinity, sodium-dependent glutamate transport in the central nervous system. Curr Pharm Des. 1999;5:363–379. [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, Campagne MV, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Chen W, Aoki C, Mahadomrongkul V, Gruber CE, Wang GJ, Blitzblau R, Irwin N, Rosenberg PA. Expression of a variant form of the glutamate transporter GLT1 in neuronal cultures and in neurons and astrocytes in the rat brain. J Neurosci. 2002;22:2142–2152. doi: 10.1523/JNEUROSCI.22-06-02142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WZ, Mahadomrongkul V, Berger UV, Bassan M, Desilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci. 2004;24:1136–1148. doi: 10.1523/JNEUROSCI.1586-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Attwell D, Bergles D, Dehnes Y, Furness DN, Gundersen V, Hamann M, Holmseth S, Lehre KP, Qureshi A, Rossi D, Ullensvang K. Glutamate transporters around synapses. J Neurochem. 2006;96:87. [Google Scholar]
- Danbolt NC, Lehre KP, Dehnes Y, Chaudhry FA, Levy LM. Localization of transporters using transporter-specific antibodies. Methods Enzymol. 1998;296:388–407. doi: 10.1016/s0076-6879(98)96028-1. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Pines G, Kanner BI. Purification and reconstitution of the sodium- and potassium-coupled glutamate transport glyco-protein from rat brain. Biochemistry US. 1990;29:6734–6740. doi: 10.1021/bi00480a025. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Storm-Mathisen J, Kanner BI. An [Na+ + K+]coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience. 1992;51:295–310. doi: 10.1016/0306-4522(92)90316-t. [DOI] [PubMed] [Google Scholar]
- Davies LP, Johnston GAR. Uptake and release of D- and L-aspartate by rat brain slices. J Neurochem. 1976;26:1007–1014. doi: 10.1111/j.1471-4159.1976.tb06485.x. [DOI] [PubMed] [Google Scholar]
- Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci. 1998;18:3606–3619. doi: 10.1523/JNEUROSCI.18-10-03606.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewer C, Rauen T. Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J Membr Biol. 2005;203:1–20. doi: 10.1007/s00232-004-0731-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen V, Danbolt NC, Ottersen OP, Storm-Mathisen J. Demonstration of glutamate/aspartate uptake activity in nerve endings by use of antibodies recognizing exogenous D-aspartate. Neuroscience. 1993;57:97–111. doi: 10.1016/0306-4522(93)90114-u. [DOI] [PubMed] [Google Scholar]
- Gundersen V, Ottersen OP, Storm-Mathisen J. Selective excitatory amino acid uptake in glutamatergic nerve terminals and in glia in the rat striatum: quantitative electron microscopic immunocytochemistry of exogenous D-aspartate and endogenous gluta-mate and GABA. Eur J Neurosci. 1996;8:758–765. doi: 10.1111/j.1460-9568.1996.tb01261.x. [DOI] [PubMed] [Google Scholar]
- Haugeto Ø, Ullensvang K, Levy LM, Chaudhry FA, Honoré T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- Henn FA, Anderson DJ, Rustad DG. Glial contamination of synaptosomal fractions. Brain Res. 1976;101:341–344. doi: 10.1016/0006-8993(76)90274-2. [DOI] [PubMed] [Google Scholar]
- Holmseth S, Lehre KP, Danbolt NC. Specificity controls for immunocytochemistry. Anat Embryol (Berl) 2006;211:257–266. doi: 10.1007/s00429-005-0077-6. [DOI] [PubMed] [Google Scholar]
- Ji Z, Aas J-E, Laake J, Walberg F, Ottersen OP. An electron microscopic, immunogold analysis of glutamate and glutamine in terminals of rat spinocerebellar fibres. J Comp Neurol. 1991;307:296–310. doi: 10.1002/cne.903070210. [DOI] [PubMed] [Google Scholar]
- Kanner BI, Schuldiner S. Mechanism of transport and storage of neurotransmitters. CRC Crit Rev Biochem. 1987;22:1–38. doi: 10.3109/10409238709082546. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Lehre KP, Rolstad B, Danbolt NC. A monoclonal antibody raised against an [Na+-K+]coupled L-glutamate transporter purified from rat brain confirms glial cell localization. FEBS Lett. 1993;317:79–84. doi: 10.1016/0014-5793(93)81495-l. [DOI] [PubMed] [Google Scholar]
- Matthias K, Kirchhoff F, Seifert G, Huttmann K, Matyash M, Ketten-mann H, Steinhauser C. Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J Neurosci. 2003;23:1750–1758. doi: 10.1523/JNEUROSCI.23-05-01750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megías M, Emri Z, Freund TF, Gulyás AI. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience. 2001;102:527–540. doi: 10.1016/s0306-4522(00)00496-6. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Iga K, Shibata T, Shudo M, Kataoka K. Glial plasmalemmal vesicles: a subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia. 1993;9:48–56. doi: 10.1002/glia.440090107. [DOI] [PubMed] [Google Scholar]
- Ottersen OP. Postembedding immunogold labelling of fixed glutamate: an electron microscopic analysis of the relationship between gold particle density and antigen concentration. J Chem Neuroanat. 1989;2:57–66. [PubMed] [Google Scholar]
- Ottersen OP, Storm-Mathisen J. Neurons containing or accumulating transmitter amino acids. In: Bjorklund A, Hökfelt T, Kuhar MJ, editors. Handbook of chemical neuroanatomy: classical transmitters and transmitter receptors in the CNS. Elsevier Science Publishers B.V.; Amsterdam: 1984. pp. 141–246. [Google Scholar]
- Ottersen OP, Zhang N, Walberg F. Metabolic compartmentation of glutamate and glutamine: morphological evidence obtained by quantitative immunocytochemistry in rat cerebellum. Neuroscience. 1992;46:519–534. doi: 10.1016/0306-4522(92)90141-n. [DOI] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjørås M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. Cloning and expression of a rat brain L-glutamate transporter. Nature. 1992;360:464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- Reye P, Sullivan R, Scott H, Pow DV. Distribution of two splice variants of the glutamate transporter GLT-1 in rat brain and pituitary. Glia. 2002;38:246–255. doi: 10.1002/glia.10059. [DOI] [PubMed] [Google Scholar]
- Robinson MB. Examination of glutamate transporter heterogeneity using synaptosomal preparations. Methods Enzymol. 1998;296:189–202. doi: 10.1016/s0076-6879(98)96015-3. [DOI] [PubMed] [Google Scholar]
- Schell MJ, Cooper OB, Snyder SH. D-aspartate localizations imply neuronal and neuroendocrine roles. Proc Natl Acad Sci U S A. 1997;94:2013–2018. doi: 10.1073/pnas.94.5.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Lesch KP, Kugler P. A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: cloning and localization in rat nervous system. Neuroscience. 2002;109:45–61. doi: 10.1016/s0306-4522(01)00451-1. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Shigeri Y, Yasuda-Kamatani Y, Lebrun B, Yumoto N, Nakajima T. Syntheses of optically pure beta-hydroxyaspartate derivatives as glutamate transporter blockers. Bioorg Med Chem Lett. 2000;10:2407–2410. doi: 10.1016/s0960-894x(00)00487-x. [DOI] [PubMed] [Google Scholar]
- Sorra KE, Harris KM. Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus. 2000;10:501–511. doi: 10.1002/1098-1063(2000)10:5<501::AID-HIPO1>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci U S A. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm-Mathisen J. Localization of transmitter candidates in the brain: the hippocampal formation as a model. Prog Neurobiol. 1977;8:119–181. doi: 10.1016/0301-0082(77)90013-2. [DOI] [PubMed] [Google Scholar]
- Suchak SK, Baloyianni NV, Perkinton MS, Williams RJ, Meldrum BS, Rattray M. The “glial” glutamate transporter, EAAT2 (Glt-1) accounts for high affinity glutamate uptake into adult rodent nerve endings. J Neurochem. 2003;84:522–532. doi: 10.1046/j.1471-4159.2003.01553.x. [DOI] [PubMed] [Google Scholar]
- Takagaki G. Sodium and potassium ions and accumulation of labelled D-aspartate and GABA in crude synaptosomal fraction from rat cerebral cortex. J Neurochem. 1978;30:47–56. doi: 10.1111/j.1471-4159.1978.tb07033.x. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Torp R, Danbolt NC, Babaie E, Bjørås M, Seeberg E, Storm-Mathisen J, Ottersen OP. Differential expression of two glial glutamate transporters in the rat brain: an in situ hybridization study. Eur J Neurosci. 1994;6:936–942. doi: 10.1111/j.1460-9568.1994.tb00587.x. [DOI] [PubMed] [Google Scholar]
- Torp R, Hoover F, Danbolt NC, Storm-Mathisen J, Ottersen OP. Differential distribution of the glutamate transporters GLT1 and rEAAC1 in rat cerebral cortex and thalamus: an in situ hybridization analysis. Anat Embryol (Berl) 1997;195:317–326. doi: 10.1007/s004290050051. [DOI] [PubMed] [Google Scholar]
- Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur J Neurosci. 1997;9:1646–1655. doi: 10.1111/j.1460-9568.1997.tb01522.x. [DOI] [PubMed] [Google Scholar]
- Urai Y, Jinnouchi O, Kwak KT, Suzue A, Nagahiro S, Fukui K. Gene expression of D-amino acid oxidase in cultured rat astrocytes: regional and cell type specific expression. Neurosci Lett. 2002;324:101–104. doi: 10.1016/s0304-3940(02)00184-2. [DOI] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Endou H, Kanai Y. Tissue specific variants of glutamate transporter GLT-1. FEBS Lett. 1997;416:312–316. doi: 10.1016/s0014-5793(97)01232-5. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Qu H, Sonnewald U, Shimamoto K, Schousboe A. Role of glutamine and neuronal glutamate uptake in gluta-mate homeostasis and synthesis during vesicular release in cultured glutamatergic neurons. Neurochem Int. 2005;47:92–102. doi: 10.1016/j.neuint.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Wang BL, Larsson LI. Simultaneous demonstration of multiple antigens by indirect immunofluorescence or immunogold staining. Novel light and electron microscopical double and triple staining method employing primary antibodies from the same species. Histochemistry. 1985;83:47–56. doi: 10.1007/BF00495299. [DOI] [PubMed] [Google Scholar]
- Weibel ER. Practical methods for biological morphometry. Academic Press; London: 1979. Stereological methods Vol. 1. [Google Scholar]
- Xu NJ, Bao L, Fan HP, Bao GB, Pu L, Lu YJ, Wu CF, Zhang X, Pei G. Morphine withdrawal increases glutamate uptake and surface expression of glutamate transporter GLT1 at hippocampal synapses. J Neurosci. 2003;23:4775–4784. doi: 10.1523/JNEUROSCI.23-11-04775.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaar K, Kost HP, Schad A, Volkl A, Baumgart E, Fahimi HD. Cellular and subcellular distribution of D-aspartate oxidase in human and rat brain. J Comp Neurol. 2002;450:272–282. doi: 10.1002/cne.10320. [DOI] [PubMed] [Google Scholar]
- Zhang N, Storm-Mathisen J, Ottersen OP. A model system for quantitation in single and double labelling postembedding electron microscopic immunocytochemistry. Elsevier Neurosci Protoc. 1993;93-050-13:1–20. [Google Scholar]