Daily Acyclovir Delays HIV-1 Disease Progression Among HIV-1/HSV-2 Dually-Infected Persons: A Randomized Trial (original) (raw)
. Author manuscript; available in PMC: 2010 May 27.
Abstract
Background
Well-tolerated medications that slow HIV-1 disease progression and delay initiation of antiretroviral therapy (ART) are needed. Most HIV-1-infected persons are dually-infected with herpes simplex virus type 2 (HSV-2). Daily HSV-2 suppression reduces plasma HIV-1 levels, but whether HSV-2 suppression delays HIV-1 disease progression is unknown.
Methods
Within a randomized, placebo-controlled trial of HSV-2 suppressive therapy (acyclovir 400 mg orally bid) to decrease HIV-1 transmission, 3381 HSV-2/HIV-1 dually-infected heterosexual Africans who at enrollment had CD4 counts ≥250 cells/mm3 and were not taking ART were followed for up to 24 months. We evaluated the effect of acyclovir on HIV-1 disease progression, defined by a primary composite endpoint of first occurrence of CD4 count <200 cells/mm3, ART initiation, or non-trauma related death. As an exploratory analysis, we evaluated the endpoint of CD4 decline to <350 cells/mm3.
Findings
At enrollment, median CD4 was 462 cells/mm3 and median HIV-1 plasma RNA was 4.1 log10 copies/mL. Acyclovir reduced risk of HIV-1 disease progression: 284 participants on acyclovir versus 324 on placebo reached the primary endpoint (hazard ratio [HR] 0.84, 95% confidence interval [CI] 0.71–0.98, p=0.03). Among participants with CD4 counts ≥350 cells/mm3, acyclovir delayed risk of CD4 decline to <350 cells/mm3 (HR 0.81, 95% CI 0.71–0.93, p=0.002).
Interpretation
HSV-2 suppression with acyclovir reduced the risk of HIV-1 disease progression by 16% (95% CI 2–29%). The role of HSV-2 suppression in reducing HIV-1 disease progression prior to ART initiation warrants consideration (ClinicalTrials.gov #NCT00194519).
Keywords: HIV-1 disease progression, HIV-1 discordant couples, HSV-2, genital herpes, herpes suppression, acyclovir, randomized clinical trial
Introduction
Recent expansion in antiretroviral therapy (ART) access has had a significant impact on HIV-1 disease progression and mortality among HIV-1 infected persons in resource-limited countries. Still, only one-third of HIV-1 infected persons who meet international ART initiation guidelines are estimated to currently be receiving medications.(1) The number of persons requiring ART will continue to grow despite ART programmatic and resource constraints, particularly if higher CD4 thresholds are adopted for ART initiation (e.g., 350 cells/mm3). Moreover, the majority of HIV-1 infected persons worldwide currently have CD4 counts above the 200 or 350 cells/mm3 CD4 thresholds for ART initiation. Thus, low-cost interventions to slow HIV-1 disease progression are needed for persons not meeting ART initiation guidelines.
Herpes simplex virus type 2 (HSV-2) is the most common cause of genital ulcer disease (GUD) globally. HSV-2 seroprevalence among HIV-1 infected persons ranges from 70% to >90%.(2) HSV-2 reactivation is common and often asymptomatic among HIV-1 infected persons, occurring on approximately 30% of days.(3) Plasma and genital HIV-1 levels increase during HSV-2 reactivations,(4–8) suggesting that HSV-2 reactivation enhances HIV-1 replication, possibly through binding of HSV proteins to the HIV-1 long terminal repeat, elevation of pro-inflammatory cytokines, or infiltration of HIV-1 target cells in the genital tract.(9–11)
Given the strong relationship between higher plasma HIV-1 concentrations and faster HIV-1 disease progression,(12, 13) HSV-2 suppression has been considered as a potential strategy to reduce HIV-1 levels and slow HIV-1 disease progression. Five recent randomized trials among HIV-1/HSV-2 dually-infected persons not taking ART found daily HSV-2 suppressive therapy with acyclovir or valacyclovir for 8–12 weeks reduced plasma HIV-1 levels by 0.25–0.5 log10 copies/mL.(14–18)
We conducted a multicenter trial of daily HSV-2 suppression among HIV-1/HSV-2 dually-infected Africans with CD4 counts ≥250 cells/mm3 and not meeting national guidelines for ART initiation at study entry. Here, we present the efficacy of suppressive acyclovir on measures of HIV-1 disease progression.
Methods
Study Design
The Partners in Prevention HSV/HIV Transmission Study was a randomized, double-blind, placebo-controlled trial of twice daily acyclovir 400 mg for HSV-2 suppressive therapy provided to the HIV-1/HSV-2 dually-infected partner within heterosexual HIV-1 serodiscordant couples (i.e., one partner HIV-1 infected and the other HIV-1 uninfected). The primary aim of the trial was to measure the efficacy of acyclovir on HIV-1 transmission. As reported elsewhere, acyclovir did not reduce HIV-1 transmission within the couples, despite reducing HSV-2-positive GUD by 73% and HIV-1 plasma levels by 0.25 log10 copies/mL.(19) Study procedures have been described elsewhere.(19–21)
After the trial was underway, the investigators observed that the number of clinical events related to HIV-1 disease (e.g., decline of CD4 count to <200 cells/mm3 and ART initiation) was sufficient to warrant an analysis of HIV-1 disease progression by study arm. The Data and Safety Monitoring Board (DSMB) accepted an addendum to the statistical analysis plan describing this analysis.
Study Population
HIV-1 serodiscordant couples were recruited at sites in southern Africa (Gaborone, Botswana; Cape Town, Orange Farm, and Soweto, South Africa; Kitwe, Lusaka, and Ndola, Zambia) and East Africa (Eldoret, Kisumu, Nairobi, and Thika, Kenya; Kigali, Rwanda; Moshi, Tanzania; and Kampala, Uganda) between November 2004 and April 2007. HIV-1 infected partners were required to be ≥18 years of age, seropositive for HIV-1 and HSV-2, and have a CD4 count ≥250 cells/mm3. Those who had an AIDS-defining diagnosis, reported taking ART, had prior adverse reactions to acyclovir or planned use of antivirals, or were pregnant were excluded.(19)
Randomisation
The randomisation method was developed and implemented by the statistician, JPH, and used block sizes of 4, 6, 8, and 10, stratified by site. The randomisation list was used to assemble sequentially numbered, identical sealed kits containing, in a 1:1 ratio, sufficient acyclovir (400 mg, orally, twice daily) or matched placebo (Ranbaxy Laboratories, Haryana, India) for the entire study period. At enrollment, HIV-1 infected partners were assigned the next sequentially numbered kit. Participants were instructed to take one tablet in the morning and one in the evening, and to double the next dose if a dose was missed. Investigators (except for an unblinded statistician and two coordinating center data managers) remained blinded to randomization assignments throughout the study follow-up.
Follow-up of HIV-1 Infected Partners
Participants were followed monthly for up to 24 months after enrollment. At each monthly visit, a one-month supply of study drug was provided and adherence counseling was performed. Study drug adherence was assessed by pill count and self-report of 100% or <100%. Women were tested for pregnancy quarterly and when they reported missed menses; those who became pregnant had study drug interrupted for the duration of pregnancy and were referred to local antenatal clinics for prevention of mother-to-child transmission (PMTCT) services. CD4 counts were measured semi-annually and clinical assessment was performed quarterly. Participants meeting national CD4 and clinical criteria for ART initiation during follow-up were offered ART through referral to local clinics or at the study site. For participants who died during follow-up, cause of death was obtained from family members and medical records, where available. At enrollment and follow-up visits, participants received intensive risk reduction counseling (both individual and as a couple), free condoms, and treatment of sexually transmitted infections. Participants who reached an HIV-1 disease progression endpoint were continued in follow-up.
Ethical Review
The University of Washington Human Subjects Review Committee and ethical review committees at each local institution, collaborating organization, and national regulatory board approved the study protocol. All participants provided written informed consent. The trial was registered through ClinicalTrials.gov (NCT00194519).
Laboratory Analyses
As detailed previously (19, 20), HIV-1 serology was by dual rapid tests with confirmatory EIA; HSV-2 serology was by HerpeSelect-2 EIA (Focus Technologies, Cypress CA) using an index value ≥3.5 to improve test specificity.(22–24). HIV-1 and HSV-2 serostatus were confirmed in batch at the University of Washington by Western blot using enrollment sera with those not confirmed by Western blot excluded from analysis.(20) CD4 testing was performed at study sites using standard flow cytometry (BD Biosciences, San Jose, USA).
Definition of HIV-1 Disease Progression Endpoints
Three measures were identified prior to study unblinding to evaluate the effect of acyclovir on HIV-1 disease progression: 1) decline of CD4 count to <200 cells/mm3, 2) first reported use of ART (excluding ART used for PMTCT), and 3) death from non-trauma causes. The primary analysis was a composite endpoint defined as the first occurrence of any of these three outcomes; if a participant experienced multiple HIV-1 disease progression endpoints (e.g., CD4 decline to <200 followed by ART initiation), only the first was included in the primary composite endpoint. Similar composite measures have been used as outcomes in previous studies of ART and have been proposed as outcomes for trials of preventive HIV-1 vaccines that may alter viral load and disease progression.(25–27) Secondary analyses considered each outcome measure separately. After unblinding of study randomization, an exploratory analysis also evaluated CD4 count decline to <350 cells/mm3 among those with CD4 counts ≥350 cells/mm3 at study entry, to reflect recent changes in ART initiation guidelines.(28) Due to the impact of ART on CD4 decline and mortality, participants initiating ART (for any reason) were censored thereafter from the risk pool for any analyses using the death or CD4 endpoints.
Statistical Analysis
Statistical analyses were performed using SAS 9.2 (SAS Institute, Inc., Cary, NC, USA). All analyses were intent-to-treat. We used Cox proportional hazards regression models, stratified by site, to compare time to occurrence of HIV-1 disease progression outcomes between the two intervention arms and applied the Efron method for handling ties.(29) The Kaplan-Meier method was used to estimate and plot the cumulative probability of reaching the study endpoint by intervention arm.
Cox proportional hazards analyses were also performed for the composite disease progression endpoint for pre-specified subgroups defined by baseline characteristics: gender, HIV-1 plasma viral load, and CD4 count. Tests for differential treatment effects across subgroups were based on likelihood ratio comparisons between models with and without appropriate interaction terms.
Study drug adherence was calculated as the product of the proportion of dispensed drug taken (assessed by monthly pill count of returned study drug bottles for 99.2% of visits or self-report of 100% adherence for 0.8% visits) and the proportion of visits at which drug was dispensed. This measure indicates study drug coverage during follow-up and accounts for drug not dispensed (primarily for missed visits and pregnancy). Participants contributed to the summaries of adherence through the time of the composite endpoint.
A post-randomization subgroup analysis assessed the impact of study drug coverage over time on the risk of developing the composite primary endpoint. For this analysis, we analyzed drug coverage averaged over each quarter of study follow-up as a time-varying covariate and categorized as <75%, 75–89%, ≥90%.
We calculated the number of participants one would have to treat with acyclovir to prevent one event in one year (the number needed to treat, NNT) using survival over a year in the placebo arm (calculated from the average hazard over all follow-up) and the hazard ratio comparing the acyclovir to the placebo arm.(30) Since the median time to each outcome was not observed during study follow-up, we projected the median times assuming a constant hazard in each arm.
A sensitivity analysis was conducted to evaluate the possible effect of missing follow-up data on our primary analysis. A “sensitivity-adjusted” RR (sRR) was calculated as
sRR=(pyrs(A)+myrs(A)∗α(A))/(pyrs(A)+myrs(A))(pyrs(P)+myrs(P)∗α(P))/(pyrs(P)+myrs(P))RR
where RR is the observed relative risk, pyrs is the number of observed person-years in the indicated arm (A for acyclovir and P for placebo), myrs is the number of missing person-years in the indicated arm and α is the relative incidence during the missing person-years compared to the observed person-years. We allowed α to vary from 0.75 to 1.5 in each arm. We also computed a “sensitivity-adjusted” p-value by dividing log (sRR) by the standard error of the estimated log hazard ratio from the primary analysis and comparing to a standard normal table.
Role of the Funding Source
The Bill and Melinda Gates Foundation funded the study but did not assume responsibility for review and approval of the protocol and protocol revisions. The authors designed the study and wrote the protocol, had full access to the raw data, performed all analyses, wrote the manuscript, and had final responsibility for the decision to submit for publication.
Results
Among 3408 HIV-1/HSV-2 dually-infected persons enrolled, 27 were excluded based on confirmatory HIV-1 and HSV-2 serologic testing (Figure 1). Of the remaining 3381, 1693 were randomized to acyclovir and 1688 to placebo. Baseline demographic and clinical characteristics were similar between the two study arms (Table 1). Two-thirds (68%) of participants were female. The median baseline CD4 count and HIV-1 plasma RNA were 462 cells/mm3 (inter-quartile range [IQR] 347–631) and 4.1 log10 copies/mL (IQR 3.4–4.7), respectively. Most participants had asymptomatic HIV-1 disease, with few (≤5%) participants reporting pneumonia, tuberculosis or herpes zoster in the prior year.
Figure 1.

Trial Profile
*HSV-2 seropositivity at enrollment determined by Focus HerpeSelect-2 EIA at site laboratory; inclusion in primary analysis based on HSV-2 Western blot at University of Washington, as described in the Methods.
** Numerator includes attended visits only. Denominator includes all expected visits and reflects staged site close-out. During follow-up, 3 subjects were dispensed a drug kit for the incorrect randomization arm; follow-up time has been censored at the visit when that occurred.
Table 1.
Enrollment characteristics, by study arm
| Characteristic | Female (N= 2284) | Male (N= 1097) | ||
|---|---|---|---|---|
| Acyclovir (n=1132) | Placebo (n=1152) | Acyclovir (n=561) | Placebo (n=536) | |
| Age, median (IQR) | 29 (25–34) | 29 (25–34) | 37 (31–44) | 37 (32–44) |
| CD4, median, cells/mm3 (IQR) | 484 (363–674) | 480 (349–655) | 435 (340–580) | 414 (331–559) |
| HIV-1 plasma RNA, median, log10 copies/mL (IQR) | 4.0 (3.2–4.6) | 3.9 (3.2–4.5) | 4.3 (3.7–4.9) | 4.4 (3.6–4.9) |
| HIV-1-associated symptoms | ||||
| Weight loss >10%, prior year | 57 (5%) | 47 (4%) | 26 (5%) | 21 (4%) |
| Fever >1 mo, prior year | 34 (3%) | 38 (3%) | 22 (4%) | 33 (6%) |
| Diarrhea >1 mo, prior year | 7 (1%) | 11 (1%) | 3 (1%) | 8 (1%) |
| Cough >1 mo, prior year | 48 (4%) | 59 (5%) | 39 (7%) | 51 (10%) |
| Genital ulcers, prior 3 months | 242 (21%) | 257 (22%) | 145 (26%) | 119 (22%) |
| Clinical diagnoses, by self-report | ||||
| Pneumonia, prior year | 54 (5%) | 50 (4%) | 24 (4%) | 17 (3%) |
| Tuberculosis, prior year | 31 (3%) | 39 (3%) | 22 (4%) | 29 (5%) |
| Herpes zoster, prior year | 37 (3%) | 39 (3%) | 29 (5%) | 25 (5%) |
| Physical examination findings | ||||
| Lymphadenopathy | 148 (13%) | 162 (14%) | 92 (16%) | 94 (18%) |
| Oral candidiasis | 5 (0%) | 2 (0%) | 3 (1%) | 4 (1%) |
| Herpes zoster | 11 (1%) | 13 (1%) | 10 (2%) | 5 (1%) |
| GUD | 38 (3%) | 35 (3%) | 12 (2%) | 12 (2%) |
Retention and Study Drug Adherence
Retention of participants at 24 months of follow-up was 92% overall (91% in the acyclovir arm and 93% in the placebo arm) (Figure 1). Participants contributed 4826 person-years of follow-up for analysis of the primary composite endpoint. A total of 96.3% of dispensed doses were taken and 93.7% of monthly study drug dispensed, resulting in overall drug coverage of 90.2% (Table 2). During each quarter of study follow-up, 80.8% to 85.3% of participants achieved ≥90% drug coverage.
Table 2.
Study drug adherence during follow-up*, by randomization arm
| Month of Study Follow-Up | 1–3 | 4–6 | 7–9 | 10–12 | 13–15 | 16–18 | 19–21 | 22–24 |
|---|---|---|---|---|---|---|---|---|
| Mean adherence | ||||||||
| Acyclovir | 94.2% | 92.4% | 90.0% | 88.4% | 87.9% | 88.8% | 88.2% | 88.1% |
| Placebo | 93.6% | 91.9% | 89.7% | 88.2% | 88.0% | 89.3% | 89.7% | 89.0% |
| Proportion of participants with drug coverage ≥90% | ||||||||
| Acyclovir | 84.1% | 85.0% | 85.3% | 84.0% | 82.1% | 84.7% | 82.9% | 81.9% |
| Placebo | 82.8% | 84.5% | 85.0% | 82.2% | 80.8% | 83.3% | 84.3% | 85.2% |
Effect of Acyclovir on Measures of HIV-1 Disease Progression
During follow-up, a total of 430 participants had CD4 decline to <200 cells/mm3 (incidence 9.1 per 100 person-years), 331 initiated ART, excluding ART initiated for PMTCT (incidence 6.7 per 100 person-years), and 61 died from non-trauma causes (incidence 1.2 per 100 person-years). The 61 deaths were attributed to pneumonia (13), tuberculosis (10), gastrointestinal infections (7), other infectious processes (6), malaria (5), and other causes (20). Four participants died from trauma, 2 in the acyclovir arm and 2 in the placebo arm, and 11 participants died after initiating ART (6 on acyclovir and 5 on placebo); these deaths were not included in the analyses. Among participants initiating ART, the median CD4 count prior to ART initiation was 195 cells/mm3 (IQR 159–246), with 34% initiating ART at CD4 counts between 200 and 350 cells/mm3 and 11% at CD4 counts >350 cells/mm3.
Table 3 and Figure 2 show the comparison of disease progression outcomes by study arm. A total of 608 participants reached the primary composite endpoint (first occurrence of either CD4 count <200 cells/mm3, ART initiation, or non-trauma death): 284 in the acyclovir arm and 324 in the placebo arm (hazard ratio [HR] 0.84, 95% confidence interval [CI] 0.71–0.98, p=0.03), a 16% reduction. Of these 608 composite endpoints, 425 (70%) were CD4 declines to <200 cells/mm3, 129 (21%) were ART initiations (five of whom also had a first CD4<200 at the same visit), and 54 (9%) were non-trauma deaths. When each component of the composite endpoint was analyzed separately, acyclovir reduced the risk of HIV-1 disease progression by 17–24% (corresponding p values 0.05 to 0.29 for the components of the primary outcome). Among 2431 participants with CD4 counts ≥350 cells/mm3 at enrollment, acyclovir reduced risk of progression to CD4 <350 cells/mm3 by 19% (395 versus 441 events for acyclovir versus placebo; HR 0.81, 95% CI 0.71–0.93, p=0.002).
Table 3.
Effect of acyclovir on measures of HIV-1 disease progression during the study follow-up period
| Measure of HIV-1 disease progression | Acyclovir | Placebo | Total events | HR (95% CI) | P | NNT†† | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N* | Events | Person-years at risk | Incidence per 100 person-years | N* | Events | Person-years at risk | Incidence per 100 person-years | |||||
| Primary composite endpoint | ||||||||||||
| First occurrence of CD4 decline to <200 cells/mm3, ART initiation†, or non-trauma death | 1686 | 284 | 2446 | 11.6 | 1677 | 324 | 2380 | 13.6 | 608 | 0.84 (0.71–0.98) | 0.03 | 43 |
| Components of the primary endpoint | ||||||||||||
| CD4 decline to <200 cells/mm3 ‡ | 1642 | 200 | 2401 | 8.3 | 1635 | 230 | 2333 | 9.9 | 430 | 0.83 (0.69–1.01) | 0.06 | 53 |
| ART initiation† | 1665 | 151 | 2500 | 6.0 | 1658 | 180 | 2441 | 7.4 | 331 | 0.81 (0.65–1.00) | 0.05 | 65 |
| Non-trauma related death‡ | 1686 | 27 | 2519 | 1.1 | 1677 | 34 | 2462 | 1.4 | 61 | 0.76 (0.46–1.26) | 0.29 | 324 |
| Exploratory endpoint | ||||||||||||
| CD4 decline to <350 cells/mm3 ‡, ** | 1236 | 395 | 1646 | 24.0 | 1195 | 441 | 1505 | 29.3 | 836 | 0.81 (0.71–0.93) | 0.002 | 20 |
Figure 2.

Cumulative probability of select HIV-1 disease progression endpoints (Kaplan-Meier estimates) by treatment arm.
a) Primary composite endpoint, defined as first occurrence of CD4 decline to <200 cells/mm3, non-PMTCT ART initiation, or non-trauma death. Hazard ratio 0.83, 95% confidence interval 0.71–0.96, p=0.03.
b) First occurrence of CD4 decline to <350 cells/mm3 among participants with CD4 counts ≥350 cells/mm3 at study enrollment. Hazard ratio 0.81, 95% confidence interval 0.71–0.93, p=0.002.
The effect of acyclovir on the composite measure of HIV-1 disease progression was assessed within pre-specified subgroups defined by gender, baseline HIV-1 plasma RNA concentration, and baseline CD4 count (Table 4). No statistically significant differences were observed. Among the subgroup of participants in our study with CD4 counts ≥ 500 cells/mm3 at enrollment, we found no suggestion of delayed HIV-1 disease progression as a result of acyclovir (HR 1.34), although there was no statistical evidence that the effect of acyclovir differed across the CD4 categories defined in Table 4 (p=0.19). Although participants with study drug coverage >90% showed increased effectiveness of the intervention against HIV-1 disease progression compared to those with <90% drug coverage, this difference was not significant (p=0.17).
Table 4.
Subgroup analyses for the effect of acyclovir on the primary composite endpoint (first occurrence of CD4 decline to <200 cells/mm3, non-PMTCT ART initiation, or non-trauma death)

Overall, 3.3% of expected follow-up time was missing (3.2% and 3.5% in the acyclovir and placebo arms, respectively). In sensitivity analyses, the RR for the composite primary endpoint varied between 0.81 and 0.86. Assuming that the incidence of infection during missing follow-up periods was identical in both groups and equal to the observed incidence in the placebo arm, the sensitivity-adjusted RR was 0.84 (p = 0.03).
Assuming that the incidence of HIV-1 disease progression endpoints remained constant beyond the 24 months of our study follow-up, we estimated that acyclovir would delay median time to the composite endpoint by 10.7 months (62.0 months in the placebo arm vs. 72.7 months in the acyclovir arm) and median time to CD4 <350 cells/mm3 by 6.3 months (28.8 months in the placebo arm vs. 35.1 months in the acyclovir arm).
Discussion
In this international multi-center trial, standard doses of acyclovir for HSV-2 suppression in HIV-1/HSV-2 dually-infected African women and men with CD4 counts ≥250 cells/mm3 reduced the risk of HIV-1 disease progression by 16% (95% CI 2–29%). We have previously reported that acyclovir reduced HIV-1 plasma RNA by 0.25 log10 copies/mL in this trial population.(19) This result was similar to that seen in prior trials of short-term HSV-2 suppression (1 to 3 months) showing a 0.25–0.5 log10 copies/mL reduction in HIV-1 levels (summarized in Table 5).(14–18) We infer that the reduction in HIV-1 levels during acyclovir suppression mediated a reduction in risk of HIV-1 disease progression. Consistent with this hypothesis, a recent systematic review of U.S. and African observational studies found a 0.3 log10 copies/mL reduction in plasma HIV-1 levels would be predicted to reduce risk of HIV-1 progression by 25%.(31) Our results demonstrate that a non-ART based strategy (i.e., HSV-2 suppression) that reduces plasma HIV-1 levels by a lesser amount than current combination ART regimens can modestly delay HIV-1 disease progression.
Table 5.
Comparison of clinical trials of HSV-2 suppression on HIV-1 plasma RNA in HSV-2/HIV-1 dually-infected persons
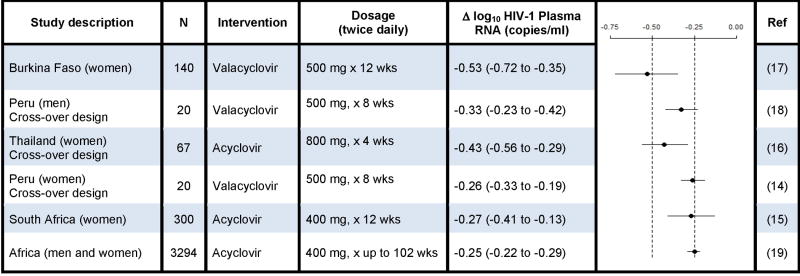
Early studies of zidovudine monotherapy showed similar reductions in HIV-1 plasma RNA(32) and decreased risk of disease progression and mortality(33) as were demonstrated in the present study. Zidovudine effects waned over 3–6 months as resistant HIV-1 variants were selected. Acyclovir is a highly HSV-specific chain terminator requiring HSV thymidine kinase for initial phosphorylation, and is preferentially incorporated by HSV DNA polymerase. This, in conjunction with the 73% reduction in the incidence of HSV-2 positive GUD among those randomized to acyclovir in our study,(19) leads us to hypothesize that acyclovir’s effect in reducing HIV-1 levels is mediated through HSV-2 suppression. Notably, recent in vitro studies suggest that acyclovir may directly inhibit HIV-1 replication, possibly utilizing kinases from other ubiquitous herpesviruses (e.g., human herpesvirus 6);(34, 35) and one in vitro study using high-dose acyclovir demonstrated selection of an uncommon HIV-1 mutation, V75I.(35) However, the 0.25 log10 average decreased plasma HIV-1 levels observed in our study persisted through 24 months of follow-up(19) without an HIV-1 plasma RNA ‘rebound’ that might be expected from selection of resistant variants. Future investigations will assess incidence of HIV-1 mutations in the acyclovir versus placebo arms during follow-up to evaluate specific mechanisms underlying HIV-1 plasma RNA reductions.
Acyclovir has a much lower frequency of adverse effects than many ART regimens currently used in resource-poor settings; as previously reported, we found no serious adverse events associated with acyclovir in the present trial.(19) High tolerability of acyclovir likely contributed to the high adherence in this study. In addition, the lack of need for specific laboratory monitoring for acyclovir toxicity during HSV-2 suppression is particularly important where laboratory infrastructure for monitoring and access to care are limited.
Our selection of a standard dose of acyclovir (comparable to valacyclovir 500 mg twice daily (36)) was based on demonstrated efficacy in reducing frequency of symptomatic GUD and asymptomatic genital HSV-2 reactivation in HIV-1/HSV-2 dually-infected persons,(37, 38) well-documented safety, generic availability, and relative low cost. A meta-analysis of several small studies of high-dose HSV-2 suppression ( 3200 mg/day) in conjunction with mono- or dual-nucleoside ART identified a similar magnitude of effect on HIV-1 associated mortality (HR 0.78, 95% CI 0.65–0.93) to what we observed.(39) Additional studies are needed to directly assess whether higher doses of herpes suppressive therapy have greater impact on HIV-1 plasma levels and disease progression.
Further study is needed to determine if HSV-2 suppression could be implemented to slow HIV-1 disease progression until HIV-1/HSV-2 dually-infected persons reach guidelines for ART initiation. Table 6 presents our summary results in the context of other non-ART biomedical interventions evaluated for their effect on measures of HIV-1 disease progression. For example, trimethoprim-sulfamethoxazole (TMP-SMX) prophylaxis and multivitamins in HIV-1 infected persons have become standard practice in many resource-poor settings, based on trials showing a reduction in HIV-1 associated mortality of ~45%(40–42) and 27%(43), respectively. However, those non-ART interventions to reduce HIV-1 disease progression were conducted in the era before combination ART was widely available and thus included considerable follow-up of persons with advanced HIV-1 disease. Furthermore, subgroup analyses found that TMP-SMX had greatest efficacy among individuals with CD4 <200 cells/mm3 or symptoms of advanced immunosuppression.(40) In contrast, we found that HSV-2 suppression delayed HIV-1 disease progression in a low-resource setting among men and women with a wide range of ages and CD4 counts ≥250 cells/mm3 at enrollment. The International AIDS Society-USA Panel recently revised recommendations to initiate ART at CD4 <350 cells/mm3 in some settings.(28) Earlier ART initiation will likely have a greater impact on disease progression than we found with acyclovir in this study and may have an ancillary benefit of reducing HIV-1 transmission. However, currently there are insufficient resources in many settings to provide ART even to those with CD4 counts <200/mm3.(44) Furthermore, given the interest in identifying interventions for persons with higher CD4 counts, more detailed evaluation of HSV-2 suppression among persons with CD4 >500 is needed. A recent cost-effectiveness analysis found that HSV-2 suppression meets the World Development Report cost-effectiveness threshold ($1000 per life-year gained) at the lowest available pricing for generic acyclovir ($25 per year for twice daily acyclovir 400 mg tablets).(45, 46) However, the local pricing of acyclovir varies widely and can exceed the international reference price by 6 to 10-fold in subSaharan Africa.(47) Clearly, efforts are needed to improve drug procurement, distribution and access across sub-Saharan Africa in order to maximize the impact of acyclovir on the HIV-1 epidemic. Mathematical modeling may be useful to define how to best use HSV-2 suppression to impact the HIV-1 epidemic by quantifying the benefits, costs, and potential impact of implementing HSV-2 suppression or other non-ART strategies compared to earlier ART initiation to delay HIV-1 disease progression.
Table 6.
Comparison of biomedical clinical trials of non-ART interventions to reduce HIV-1 disease progression
| Intervention | Study Population | N | Endpoint | Estimate of Effect (95% CI) | Ref. |
|---|---|---|---|---|---|
| Co-trimoxazole for prophylaxis of bacterial infections | Cote d’Ivoire (adults) | 771 | Death | HR=0.54 (0.38 to 0.77) | (42) |
| Cote d’Ivoire (adults) | 545 | Death or hospitalization | HR=0.57 (0.43 to 0.75) | (49) | |
| South Africa (adults) | 562 | Death | HR=0.40 (0.22 to 0.75) | (41) | |
| Uganda (adults) | 509 | Death | HR=0.54 (0.35 to 0.84) | (40) | |
| Multivitamins | Tanzania (pregnant women) | 1078 | WHO Stage 4 or death | HR=0.71 (0.51 to 0.98) | (43) |
| Albendazole for treatment of helminth infection | Kenya (adults) | 208 | HIV-1 plasma RNA | Δ HIV-1 plasma RNA: −0.54 (−1.17 to 0.09) | (50) |
| HSV-2 suppression (acyclovir) | |||||
| High dose oral (>3200 mg/day) | Meta-analysis of 8 randomized trials conducted in the United States and Europe | 1792 | Death | HR=0.28, (0.21 to 0.37) | (39) |
| Standard dose | East and southern African adults | 3363 | First of CD4<200, ART initiation, or death | HR=0.84, (0.71 to 0.98) | Present study |
One limitation to this study was the low frequency of diagnostic testing and autopsies to inform the etiology of deaths. Furthermore, although most participants initiated ART at CD4 counts ≥200 cells/mm3, reasons for ART initiation at higher CD4 counts were not captured since ART care was commonly provided outside the study clinics. TMP-SMX prophylaxis data was also not collected at all sites; however, at five sites where this information was recorded, participants reported TMP-SMX use at 73% of follow-up visits and this did not differ by treatment arm (data not shown). Finally, although studies suggest HIV-1 disease progression may differ by HIV-1 subtype,(48) subtype data are not currently available for our cohort and will be evaluated in future analyses.
In summary, we have demonstrated that acyclovir for HSV-2 suppression among HIV-1/HSV-2 dually-infected persons with CD4 >250 cells/mm3 who are not taking ART can modestly reduce the risk of HIV-1 disease progression. Further study is needed to determine if HSV-2 suppression has a role in HIV-1 treatment for persons not eligible for ART.
Acknowledgments
We are grateful to the study participants for their dedication. We thank the referral partners, Community Advisory Boards, institutions, and communities that partnered with the study sites; the members of the Data and Safety Monitoring Board (Drs. Richard Whitley [chair], Ann Arvin, Francis Kasolo, Alejandros Llanos, Jonathan Matenga, James Neaton, and David Serwadda) for their oversight and wisdom; Toni Maddox for manuscript preparation; and Dr. Deborah Donnell for insightful comments on the analysis and interpretation of the data.
Funding: The Partners in Prevention HSV/HIV Transmission Study was funded by the Bill and Melinda Gates Foundation (grant ID #26469). Development of HIV testing and recruitment strategies for HIV serodiscordant couples conducted in part with support from the US National Institutes of Health (National Institute of Mental Health R01 66767) and the Emory-Rwanda-Zambia HIV Research Group.
Appendix
The members of the Partners in Prevention HSV/HIV Transmission Study Team are: University of Washington Coordinating Center, Seattle, USA: Connie Celum (principal investigator), Anna Wald (protocol co-chair), Jairam Lingappa (medical director), Amalia Magaret, James P. Hughes (protocol statisticians), Lawrence Corey, Jared Baeten, M. Juliana McElrath (co-investigators).
Footnotes
Role of Contributors
The core protocol team (CC, AW, JRL, JMB, LC, AM, JPH) designed the study and JPH and KT performed the primary data analysis. All investigators were involved in data collection, reviewed manuscript drafts and approved the final manuscript; JRL, JMB and CC wrote the initial manuscript draft and vouch for the data, analysis, and manuscript submission.
Study site principal investigators and study coordinators: Cape Town, South Africa (University of Cape Town): David Coetzee, Mercy Kamupira; Eldoret, Kenya (Moi University, Indiana University): Kenneth Fife, Edwin Were, Cosmas Apaka; Gaborone, Botswana (Botswana Harvard Partnership): Max Essex, Joseph Makhema, Patrick Ndase; Kampala, Uganda (Infectious Disease Institute, Makerere University): Elly Katabira, Allan Ronald, Linda Kavuma; Kigali, Rwanda (Rwanda Zambia HIV Research Group, and Emory University): Susan Allen, Kayitesi Kayitenkore, Etienne Karita, Brigitte Bekan; Kisumu, Kenya (Kenya Medical Research Institute, University of California San Francisco): Elizabeth Bukusi, Craig Cohen, Josephine Odoyo; Kitwe, Zambia (Rwanda Zambia HIV Research Group, and Emory University): Susan Allen, William Kanweka, Rachel Blacher; Lusaka, Zambia (Rwanda Zambia HIV Research Group, and Emory University): Susan Allen, Bellington Vwalika; Moshi, Tanzania (Kilimanjaro Christian Medical College, Harvard University): Saidi Kapiga, Rachel Manongi, Paul Magao; Nairobi, Kenya (University of Nairobi, University of Washington): Carey Farquhar, Grace John-Stewart, James Kiarie, Harrison Tamooh; Ndola, Zambia (Rwanda Zambia HIV Research Group, and Emory University): Susan Allen, Mubiana Inambao, Frank Wong; Orange Farm, South Africa (Reproductive Health Research Unit, University of the Witwatersrand): Sinead Delany-Moretlwe, Helen Rees, Nonkululeko Mlaba; Soweto, South Africa (Perinatal HIV Research Unit, University of the Witwatersrand): Guy de Bruyn, Glenda Gray, James McIntyre, Puleng Dhlamini; Thika, Kenya (University of Nairobi, University of Washington): Nelly Rwamba Mugo, Kenneth Ngure.
DF/Net Research, Inc. (Seattle, USA) provided data management. Contract Lab Services (University of the Witwatersrand, Johannesburg, South Africa) provided study site laboratory oversight.
Potential Conflicts of Interest: CC has received research grant support from GlaxoSmithKline (GSK), which did not include salary support, and has served on an advisory board for GSK. AW has received grant support from Astellas, GSK, and Antigenics; she has been a consultant for Astellas and Aicuris. KF has received research grant funding from Astellas Pharma USA and GSK. RWC is on an advisory board for Merck. RM has served on an advisory board for Abbott Molecular, Biokit USA and Roche Diagnostics. MJM has served on an advisory board for the AIDS Vaccine Research Subcommittee. LC is a consultant for AiCuris and GenPhar. He is the head of the Scientific Advisory Board of Immune Design and receives financial remuneration for this, including equity shares that are <1% ownership. The University of Washington Virology Division Laboratories have received grant funding from GSK and Novartis to perform herpes simplex virus serologic assays and polymerase chain reaction assays for studies funded by these companies; LC directs these laboratories, but receives no salary support from these grants.
References
- 1.UN AIDS/World Health Organization. Report on the global AIDS epidemic. Geneva: UNAIDS/World Health Organization; 2008. Dec, [Google Scholar]
- 2.Weiss H. Epidemiology of herpes simplex virus type 2 infection in the developing world. Herpes. 2004 Apr;11( Suppl 1):24A–35A. [PubMed] [Google Scholar]
- 3.Posavad CM, Wald A, Kuntz S, Huang ML, Selke S, Krantz E, et al. Frequent reactivation of herpes simplex virus among HIV-1-infected patients treated with highly active antiretroviral therapy. J Infect Dis. 2004 Aug 15;190(4):693–6. doi: 10.1086/422755. [DOI] [PubMed] [Google Scholar]
- 4.Baeten JM, McClelland RS, Corey L, Overbaugh J, Lavreys L, Richardson BA, et al. Vitamin A supplementation and genital shedding of herpes simplex virus among HIV-1-infected women: a randomized clinical trial. J Infect Dis. 2004 Apr 15;189(8):1466–71. doi: 10.1086/383049. [DOI] [PubMed] [Google Scholar]
- 5.Mbopi-Keou FX, Legoff J, Gresenguet G, Si-Mohamed A, Matta M, Mayaud P, et al. Genital shedding of herpes simplex virus-2 DNA and HIV-1 RNA and proviral DNA in HIV-1- and herpes simplex virus-2-coinfected African women. J Acquir Immune Defic Syndr. 2003 Jun 1;33(2):121–4. doi: 10.1097/00126334-200306010-00001. [DOI] [PubMed] [Google Scholar]
- 6.Mole L, Ripich S, Margolis D, Holodniy M. The impact of active herpes simplex virus infection on human immunodeficiency virus load. J Infect Dis. 1997 Sep;176(3):766–70. doi: 10.1086/517297. [DOI] [PubMed] [Google Scholar]
- 7.Schacker T, Ryncarz AJ, Goddard J, Diem K, Shaughnessy M, Corey L. Frequent recovery of HIV-1 from genital herpes simplex virus lesions in HIV-1-infected men. JAMA. 1998 Jul 1;280(1):61–6. doi: 10.1001/jama.280.1.61. [DOI] [PubMed] [Google Scholar]
- 8.Schacker T, Zeh J, Hu H, Shaughnessy M, Corey L. Changes in plasma human immunodeficiency virus type 1 RNA associated with herpes simplex virus reactivation and suppression. J Infect Dis. 2002 Dec 15;186(12):1718–25. doi: 10.1086/345771. [DOI] [PubMed] [Google Scholar]
- 9.Moriuchi M, Moriuchi H, Williams R, Straus SE. Herpes simplex virus infection induces replication of human immunodeficiency virus type 1. Virology. 2000 Dec 20;278(2):534–40. doi: 10.1006/viro.2000.0667. [DOI] [PubMed] [Google Scholar]
- 10.Mosca JD, Bednarik DP, Raj NB, Rosen CA, Sodroski JG, Haseltine WA, et al. Herpes simplex virus type-1 can reactivate transcription of latent human immunodeficiency virus. Nature. 1987 Jan 1–7;325(6099):67–70. doi: 10.1038/325067a0. [DOI] [PubMed] [Google Scholar]
- 11.Rebbapragada A, Wachihi C, Pettengell C, Sunderji S, Huibner S, Jaoko W, et al. Negative mucosal synergy between Herpes simplex type 2 and HIV in the female genital tract. AIDS. 2007 Mar 12;21(5):589–98. doi: 10.1097/QAD.0b013e328012b896. [DOI] [PubMed] [Google Scholar]
- 12.Lavreys L, Baeten JM, Chohan V, McClelland RS, Hassan WM, Richardson BA, et al. Higher set point plasma viral load and more-severe acute HIV type 1 (HIV-1) illness predict mortality among high-risk HIV-1-infected African women. Clin Infect Dis. 2006 May 1;42(9):1333–9. doi: 10.1086/503258. [DOI] [PubMed] [Google Scholar]
- 13.Mellors JW, Rinaldo CR, Jr, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996 May 24;272(5265):1167–70. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 14.Baeten JM, Strick LB, Lucchetti A, Whittington WL, Sanchez J, Coombs RW, et al. Herpes Simplex Virus (HSV)-Suppressive Therapy Decreases Plasma and Genital HIV-1 Levels in HSV-2/HIV-1 Coinfected Women: A Randomized, Placebo-Controlled, Cross-Over Trial. J Infect Dis. 2008 Dec 15;198(12):1804–8. doi: 10.1086/593214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delany S, Mlaba N, Clayton T, Akpomiemie G, Capovilla A, Legoff J, et al. Impact of aciclovir on genital and plasma HIV-1 RNA in HSV-2/HIV-1 co-infected women: a randomized placebo-controlled trial in South Africa. AIDS. 2009 Feb 20;23(4):461–9. doi: 10.1097/QAD.0b013e32831db217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunne EF, Whitehead S, Sternberg M, Thepamnuay S, Leelawiwat W, McNicholl JM, et al. Suppressive acyclovir therapy reduces HIV cervicovaginal shedding in HIV- and HSV-2-infected women, Chiang Rai, Thailand. J Acquir Immune Defic Syndr. 2008 Sep 1;49(1):77–83. doi: 10.1097/QAI.0b013e3181831832. [DOI] [PubMed] [Google Scholar]
- 17.Nagot N, Ouedraogo A, Foulongne V, Konate I, Weiss HA, Vergne L, et al. Reduction of HIV-1 RNA levels with therapy to suppress herpes simplex virus. N Engl J Med. 2007 Feb 22;356(8):790–9. doi: 10.1056/NEJMoa062607. [DOI] [PubMed] [Google Scholar]
- 18.Zuckerman RA, Lucchetti A, Whittington WL, Sanchez J, Coombs RW, Zuniga R, et al. Herpes simplex virus (HSV) suppression with valacyclovir reduces rectal and blood plasma HIV-1 levels in HIV-1/HSV-2-seropositive men: a randomized, double-blind, placebo-controlled crossover trial. J Infect Dis. 2007 Nov 15;196(10):1500–8. doi: 10.1086/522523. [DOI] [PubMed] [Google Scholar]
- 19.Celum C, Wald A, Lingappa JR, et al. Acyclovir and transmission of HIV-1 from persons infected with HIV-1 and HSV-2. N Engl J Med. 2010;362:427–39. doi: 10.1056/NEJMoa0904849. for the Partners in Prevention HSV/HIV Transmission Study Team. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lingappa JR, Kahle E, Mugo N, Mujugira A, Magaret A, Baeten J, et al. Characteristics of HIV-1 discordant couples enrolled in a trial of HSV-2 suppression to reduce HIV-1 transmission: the partners study. PLoS ONE. 2009;4(4):e5272. doi: 10.1371/journal.pone.0005272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lingappa JR, Lambdin B, Bukusi EA, Ngure K, Kavuma L, Inambao M, et al. Regional Differences in Prevalence of HIV-1 Discordance in Africa and Enrollment of HIV-1 Discordant Couples into an HIV-1 Prevention Trial. PLoS ONE. 2008;3(1):e1411. doi: 10.1371/journal.pone.0001411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gamiel JL, Tobian AA, Laeyendecker OB, Reynolds SJ, Morrow RA, Serwadda D, et al. Improved performance of enzyme-linked immunosorbent assays and the effect of human immunodeficiency virus coinfection on the serologic detection of herpes simplex virus type 2 in Rakai, Uganda. Clin Vaccine Immunol. 2008 May;15(5):888–90. doi: 10.1128/CVI.00453-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golden MR, Ashley-Morrow R, Swenson P, Hogrefe WR, Handsfield HH, Wald A. Herpes simplex virus type 2 (HSV-2) Western blot confirmatory testing among men testing positive for HSV-2 using the focus enzyme-linked immunosorbent assay in a sexually transmitted disease clinic. Sex Transm Dis. 2005 Dec;32(12):771–7. doi: 10.1097/01.olq.0000175377.88358.f3. [DOI] [PubMed] [Google Scholar]
- 24.Laeyendecker O, Henson C, Gray RH, Nguyen RH, Horne BJ, Wawer MJ, et al. Performance of a commercial, type-specific enzyme-linked immunosorbent assay for detection of herpes simplex virus type 2-specific antibodies in Ugandans. J Clin Microbiol. 2004 Apr;42(4):1794–6. doi: 10.1128/JCM.42.4.1794-1796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hammer SM, Katzenstein DA, Hughes MD, Gundacker H, Schooley RT, Haubrich RH, et al. A trial comparing nucleoside monotherapy with combination therapy in HIV-infected adults with CD4 cell counts from 200 to 500 per cubic millimeter. AIDS Clinical Trials Group Study 175 Study Team. N Engl J Med. 1996 Oct 10;335(15):1081–90. doi: 10.1056/NEJM199610103351501. [DOI] [PubMed] [Google Scholar]
- 26.MacArthur RD, Novak RM, Peng G, Chen L, Xiang Y, Hullsiek KH, et al. A comparison of three highly active antiretroviral treatment strategies consisting of non-nucleoside reverse transcriptase inhibitors, protease inhibitors, or both in the presence of nucleoside reverse transcriptase inhibitors as initial therapy (CPCRA 058 FIRST Study): a long-term randomised trial. Lancet. 2006 Dec 16;368(9553):2125–35. doi: 10.1016/S0140-6736(06)69861-9. [DOI] [PubMed] [Google Scholar]
- 27.Gilbert PB, Sun Y. Failure time analysis of HIV vaccine effects on viral load and antiretroviral therapy initiation. Biostatistics. 2005 Jul;6(3):374–94. doi: 10.1093/biostatistics/kxi014. [DOI] [PubMed] [Google Scholar]
- 28.Hammer SM, Eron JJ, Jr, Reiss P, Schooley RT, Thompson MA, Walmsley S, et al. Antiretroviral treatment of adult HIV infection: 2008 recommendations of the International AIDS Society-USA panel. JAMA. 2008 Aug 6;300(5):555–70. doi: 10.1001/jama.300.5.555. [DOI] [PubMed] [Google Scholar]
- 29.Efron B. Efficiency of Cox’s likelihood function for censored data. J Amer Statist Assoc. 1977;72:557–65. [Google Scholar]
- 30.Altman DG, Andersen PK. Calculating the number needed to treat for trials where the outcome is time to an event. BMJ. 1999 Dec 4;319(7223):1492–5. doi: 10.1136/bmj.319.7223.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Modjarrad K, Chamot E, Vermund SH. Impact of small reductions in plasma HIV RNA levels on the risk of heterosexual transmission and disease progression. AIDS. 2008 Oct 18;22(16):2179–85. doi: 10.1097/QAD.0b013e328312c756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katzenstein DA, Hammer SM, Hughes MD, Gundacker H, Jackson JB, Fiscus S, et al. The relation of virologic and immunologic markers to clinical outcomes after nucleoside therapy in HIV-infected adults with 200 to 500 CD4 cells per cubic millimeter. AIDS Clinical Trials Group Study 175 Virology Study Team. N Engl J Med. 1996 Oct 10;335(15):1091–8. doi: 10.1056/NEJM199610103351502. [DOI] [PubMed] [Google Scholar]
- 33.Volberding PA, Lagakos SW, Grimes JM, Stein DS, Balfour HH, Jr, Reichman RC, et al. The duration of zidovudine benefit in persons with asymptomatic HIV infection. Prolonged evaluation of protocol 019 of the AIDS Clinical Trials Group. JAMA. 1994 Aug 10;272(6):437–42. [PubMed] [Google Scholar]
- 34.Lisco A, Vanpouille C, Tchesnokov EP, Grivel JC, Biancotto A, Brichacek B, et al. Acyclovir is activated into a HIV-1 reverse transcriptase inhibitor in herpesvirus-infected human tissues. Cell Host Microbe. 2008 Sep 11;4(3):260–70. doi: 10.1016/j.chom.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMahon MA, Siliciano JD, Lai J, Liu JO, Stivers JT, Siliciano RF, et al. The antiherpetic drug acyclovir inhibits HIV replication and selects the V75I reverse transcriptase multidrug resistance mutation. J Biol Chem. 2008 Nov 14;283(46):31289–93. doi: 10.1074/jbc.C800188200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta R, Wald A, Krantz E, Selke S, Warren T, Vargas-Cortes M, et al. Valacyclovir and acyclovir for suppression of shedding of herpes simplex virus in the genital tract. J Infect Dis. 2004 Oct 15;190(8):1374–81. doi: 10.1086/424519. [DOI] [PubMed] [Google Scholar]
- 37.Gupta R, Wald A. Genital herpes: antiviral therapy for symptom relief and prevention of transmission. Expert Opin Pharmacother. 2006 Apr;7(6):665–75. doi: 10.1517/14656566.7.6.665. [DOI] [PubMed] [Google Scholar]
- 38.Lingappa JR, Celum C. Clinical and therapeutic issues for herpes simplex virus-2 and HIV co-infection. Drugs. 2007;67(2):155–74. doi: 10.2165/00003495-200767020-00001. [DOI] [PubMed] [Google Scholar]
- 39.Ioannidis JP, Collier AC, Cooper DA, Corey L, Fiddian AP, Gazzard BG, et al. Clinical efficacy of high-dose acyclovir in patients with human immunodeficiency virus infection: a meta-analysis of randomized individual patient data. J Infect Dis. 1998 Aug;178(2):349–59. doi: 10.1086/515621. [DOI] [PubMed] [Google Scholar]
- 40.Mermin J, Lule J, Ekwaru JP, Malamba S, Downing R, Ransom R, et al. Effect of co-trimoxazole prophylaxis on morbidity, mortality, CD4-cell count, and viral load in HIV infection in rural Uganda. Lancet. 2004 Oct 16–22;364(9443):1428–34. doi: 10.1016/S0140-6736(04)17225-5. [DOI] [PubMed] [Google Scholar]
- 41.Badri M, Ehrlich R, Wood R, Maartens G. Initiating co-trimoxazole prophylaxis in HIV-infected patients in Africa: an evaluation of the provisional WHO/UNAIDS recommendations. AIDS. 2001 Jun 15;15(9):1143–8. doi: 10.1097/00002030-200106150-00009. [DOI] [PubMed] [Google Scholar]
- 42.Wiktor SZ, Sassan-Morokro M, Grant AD, Abouya L, Karon JM, Maurice C, et al. Efficacy of trimethoprim-sulphamethoxazole prophylaxis to decrease morbidity and mortality in HIV-1-infected patients with tuberculosis in Abidjan, Cote d’Ivoire: a randomised controlled trial. Lancet. 1999 May 1;353(9163):1469–75. doi: 10.1016/s0140-6736(99)03465-0. [DOI] [PubMed] [Google Scholar]
- 43.Fawzi WW, Msamanga GI, Spiegelman D, Wei R, Kapiga S, Villamor E, et al. A randomized trial of multivitamin supplements and HIV disease progression and mortality. N Engl J Med. 2004 Jul 1;351(1):23–32. doi: 10.1056/NEJMoa040541. [DOI] [PubMed] [Google Scholar]
- 44.Baggaley RF, Griffin JT, Chapman R, Hollingsworth TD, Nagot N, Delany S, et al. Estimating the public health impact of the effect of herpes simplex virus suppressive therapy on plasma HIV-1 viral load. AIDS. 2009 May 15;23(8):1005–13. doi: 10.1097/QAD.0b013e32832aadf2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vickerman P, Devine A, Meyer-Rath G, Foss A, Delany-Moretlwe S, Mayaud P. Modelling the cost-effectiveness of (HSV-2) suppressive therapy among dually HIV and HSV-2 infected women in Johannesburg, South Africa (Poster P4.148). 18th International Society for Sexually Transmitted Diseases Research; 2009 28 June - 1 July; London, England. 2009. [Google Scholar]
- 46.Devine A, Meyer-Rath G, Foss A, Vickerman P, Edwards M, Bachmann M, et al. Cost and cost effectiveness of herpes simplex virus-type 2 (HSV-2) and suppressive therapy in HIV-1 and HSV-2 infected women in Johannesburg, South Africa (Poster P4.152). 18th International Society for Sexually Transmitted Diseases Research; London, England. 2009. [Google Scholar]
- 47.Corbell C, Stergachis A, Ndowa F, Ndase P, Barnes L, Celum C. Genital ulcer disease treatment policies and access to acyclovir in eight sub-Saharan African Countries. Sex Transm Dis. doi: 10.1097/OLQ.0b013e3181e212e5. in press. [DOI] [PubMed] [Google Scholar]
- 48.Baeten JM, Chohan B, Lavreys L, Chohan V, McClelland RS, Certain L, et al. HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. J Infect Dis. 2007 Apr 15;195(8):1177–80. doi: 10.1086/512682. [DOI] [PubMed] [Google Scholar]
- 49.Anglaret X, Chene G, Attia A, Toure S, Lafont S, Combe P, et al. Early chemoprophylaxis with trimethoprim-sulphamethoxazole for HIV-1-infected adults in Abidjan, Cote d’Ivoire: a randomised trial. Cotrimo-CI Study Group. Lancet. 1999 May 1;353(9163):1463–8. doi: 10.1016/s0140-6736(98)07399-1. [DOI] [PubMed] [Google Scholar]
- 50.Walson JL, Otieno PA, Mbuchi M, Richardson BA, Lohman-Payne B, Macharia SW, et al. Albendazole treatment of HIV-1 and helminth co-infection: a randomized, double-blind, placebo-controlled trial. AIDS. 2008 Aug 20;22(13):1601–9. doi: 10.1097/QAD.0b013e32830a502e. [DOI] [PMC free article] [PubMed] [Google Scholar]