Ambiguous genitalia: two decades of experience (original) (raw)
Abstract
BACKGROUND AND OBJECTIVES:
Ambiguous genitalia is a complex, medical and social emergency. The aim of this study is to present our experience over two decades, focusing on the pattern and clinical presentation.
DESIGN AND SETTING:
A retrospective study conducted in the pediatric endocrine clinic at a university hospital Saudi Arabia during the period 1989-2008.
PATIENTS AND METHODS:
Medical records of children with ambiguous genitalia were reviewed and the genitalia described.
RESULTS:
Of the 81 children with ambiguous genitalia, 53 (65.4%) patients were genetically females (46XX), with congenital adrenal hyperplasia being the common cause in 51 (96.5%) patients. Hyperpigmentation, variable degrees of salt wasting and a family history of a similar problem helped in diagnosis. Male genetic sex (46XY) was present in only 28 (34.6%) patients with a diversity of causes; multiple congenital anomalies in 9 (32.1%), local anorectal anomalies in 2 (7.1%), congenital adrenal hyperplasia (3-β-hydroxysteroid dehydrogenase deficiency) in 2 (7.14%), 5-α-reductase deficiency in 4 (14.28%), partial androgen insensitivity in 3 (10.7%), complete androgen insensitivity in 4 (14.28%), and hypogonadotrophin deficiency in 4 (14.3%).Twenty-five (47.2%) of females were wrongly assigned as males, where only two (7.1%) males were wrongly assigned as females.
CONCLUSION:
Ambiguous genitalia, currently termed disorders of sex development (DSD), is not uncommon in our community. Increased awareness, a detailed history, and a careful physical examination, coupled with appropriate laboratory and radiological investigations aid in early diagnosis and avoid serious sequelae.
Ambiguous genitalia are a complex and often confusing medical problem. In newborns it is a matter of an emergency to decide the appropriate sex for rearing and eventually to prevent associated metabolic disturbances. Male and female embryos both have bipotential gonads that will develop into ovaries unless testes-organizing factors encoded by the Y-chromosome induce the gonads to differentiate into a testis. Somatic sexual differentiation will constitutionally develop according to a female phenotype in the absence of testicular activity. The diagnostic procedures require a number of laboratory and radiological investigations, based on a schematic and simple view of normal sexual differentiation. Sexual ambiguity may result in a male fetus either from insufficient secretion of androgens and their metabolites or insensitivity to its effects, or a female fetus with excessive secretion or exposure to androgens.1–9 In this report we present our experience over two decades with genital ambiguity, focusing on the pattern and clinical presentation of this disorder at a pediatric clinic in Riyadh, Saudi Arabia.
PATIENTS AND METHODS
The medical records of 81 patients with ambiguous genitalia who were seen and evaluated at the Pediatric Endocrine Clinic, King Khalid University Hospital (KKUH), King Saud University, Riyadh, Saudi Arabia over the period 1989-2008 were reviewed. KKUH is the main teaching hospital of the King Saud University and is considered one of the major referral hospitals in the region. The hospital provides primary, secondary, and tertiary health care services for the local population and also receives patients referred from all over the country. The data collected included age, sex at presentation, relevant family and social history, pregnancy, clinical manifestations and results of all the laboratory, radiological and ancillary investigations. The degree of severity of virilization of the female external genitalia was determined by applying the Prader classification (Table 1).10
Table 1.
Degree of virilization of the external genitalia (Prader classification)

Genetic sex was based on chromosomal studies done on lymphocytes. Additional tests included genitography and pelvic ultrasonography.11 Definitive etiological diagnosis was based on detailed and specific hormonal investigations as recommended. Elevated 17-β-hydroxyprogesterone in 21-hydroxylase deficiency congenital adrenal hyperplasia (CAH), elevated 11-deoxycortisone in 11-β-hydroxylase deficiency while the presence of normal 17-β hydroxyprogesterone with increased dehydroepiandrosterone (DHEA) levels associated with low concentrations of androstenediones and testosterone suggests 3-β-hydroxysteroid dehydrogenase deficiency CAH. Elevated baseline and human chorionic gonadotrophin (HCG) stimulated testosterone (T) to dihydrotestosterone (DHT) ratio in 5-α-reductase deficiency, while a satisfactory penile length response to testosterone with a normal T/DHT ratio suggests partial androgen insensitivity. The available Islamic guidelines for sex assignment were applied.8
RESULTS
During the period under review, 81 children aged 1 day to 8 years were seen and evaluated by the author for ambiguous genitalia. Fifty-three (65.4%) were genetic females (46 XX), whose detailed clinical data is shown in Table 2. The majority (96.1%) were proven to have congenital adrenal hyperplasia with 25 (47.2%) patients wrongly assigned as a male due to severe virilization (Figure 1). Family history of a similar disorder, hyperpigmentation, and variable degrees of salt wasting suggested the diagnosis. Twenty-eight (34.6%) patients were genetic male (46XY) (Table 3), with an associated multiple congenital and local ano-rectal anomalies being the commonest, present in 9 (32.14%) and 2 (7.14%) patients, respectively. Others were due to a diversity of causes; congenital adrenal hyperplasia due to 3-β-hydroxysteroid dehydrogenase deficiency in 2 (7.14%), (Figure 2), 5-α-reductase deficiency in 4 (14.28%) patients; 2 of them were siblings who were wrongly assigned a female sex, which was corrected at 7 and 12 years of age. Partial androgen insensitivity (Figure 3) in 3 (10.7%) patients, while complete androgen insensitivity and hypopogonadotrophin hormone deficiency, in 4 (14.28%) patients each.
Table 2.
Clinical data of 53 patients with 46 XX genetic sex with ambiguous genitalia
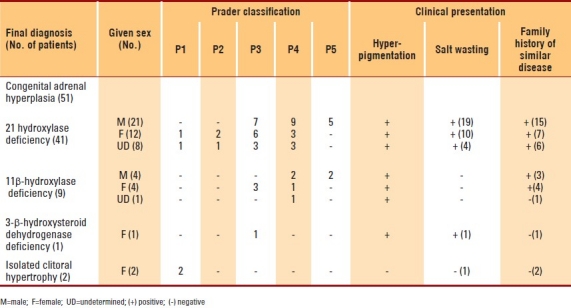
Figure 1.
Ambiguous genitalia in a 46 XX patient who was proved to have congenital adrenal hyperplasia, due to 21-b-hydroxylase deficiency. Note complete masculinization, with normal looking hyperpigmented male genitalia but no palpable testes.
Table 3.
Clinical data of 28 patients with 46 XY genetic sex with ambiguous genitalia
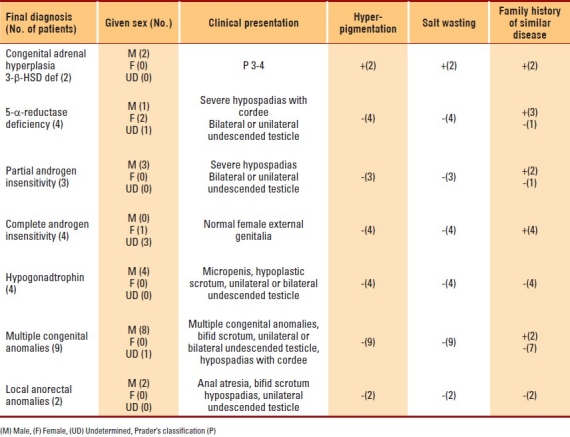
Figure 2.
Ambiguous genitalia in a 46 XY patient, who was proved to have congenital adrenal hyperplasia, due to 3-b-hydroxysteroid dehydrogenase deficiency. Note pigmented short curved phallus, central urogenital slit and separated labioscrotal testis.
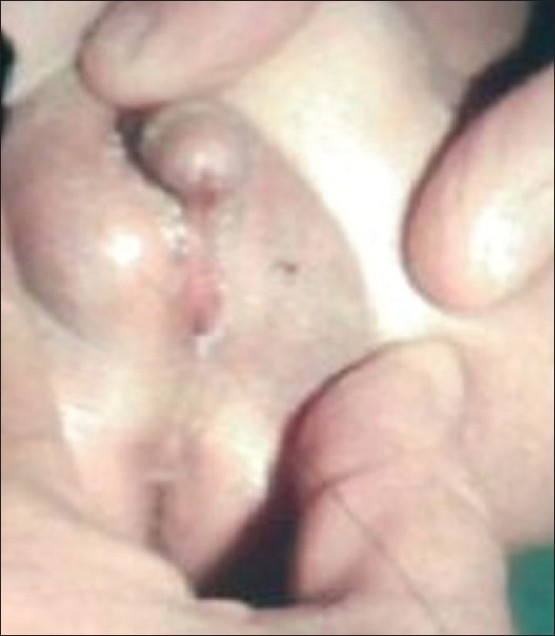
Figure 3.
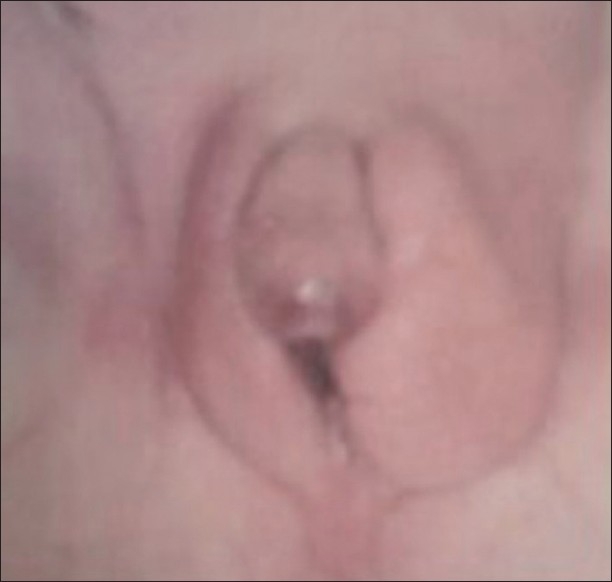
Ambiguous genitalia in a 46XY patient, who was proved to have partial androgen insensitivity. Note the micropenis, urogenital sinus, and labioscrotal folds with left palpable gonad.
DISCUSSION
Sexual differentiation is an orderly sequential process that begins with fertilization. A defect at any level can lead to sexual ambiguity. The newborn with ambiguous genitalia constitutes a major social and medical emergency, as several conditions produce significant salt loss which, if unrecognized, may lead to shock and death. A successful outcome in all cases depends on early recognition, correct assignment of sex within a short period of time and effectively dealing with the family's concerns and anxiety.1–9
In a genetic female-sex 46XX, with normal internal female organs and ovaries, exposure to elevated levels of testosterone in the first trimester will lead to variable degrees of virilization of the external genitalia, while, exposure to high androgen concentrations after 12 weeks of gestation will cause only growth of the clitoris, without fusion of the labia minora or majora or formation of a male-type urethra.1–9 Male external genitalia, in a genetic male-sex 46 XY, differentiate in response to dihydrotestosterone (DHT), which is formed in genital skin and other sensitive structures by the metabolism of testosterone by 5-α-reductase. DHT causes elongation of the genital tubercle and fusion of the genital folds to form the penis, while fusion of the labioscrotal folds form the scrotum. Thus, the lack of testosterone or its receptors or metabolites will lead to under-masculinization, the so-called male pseudohermaphrodite.1–9 In a true hermaphrodite, both testicular tubules and ovarian follicles are present, which is very rare.12
More than half of our patients (65.4%) had a genetic female-sex (46XX), the majority (96.5%) of which were due to various virilizing forms of congenital adrenal hyperplasia. This high occurrence is a reflection of multiple sibling involvement of a common autosomal recessive disorder in this community.13,14 Saedi-Wong et al15 showed a high rate of parity and consanguineous mating among the Saudi population. The diagnosis should be suspected after accurate physical examination, detailed history, including family history, hyperpigmentation and salt loss. The specific etiological diagnosis can be achieved by the appropriate hormonal investigations.13–14
The investigation and management of a child with genetic male sex 46XY and ambiguous genitalia is rather more difficult. In addition to radiological studies, specific diagnostic and therapeutic trials should be obtained. Hyperpigmentation with or without evidence of salt loss raises the suspicion of congenital adrenal hyperplasia due to 3-β-hydroxysteroid dehydrogenase deficiency which can be confirmed by increased dehydroepiandrosterone (DHEA) levels associated with low concentrations of androstenedione and testosterone.14 A defect in 5-α-reductase enzyme impairs conversion of testosterone (T) to dihydrotestosterone (DHT), and hence affects masculinization. The diagnosis is confirmed by the finding of an abnormally high T/DHT ratio (basal and post-human chorionic gonadotropin (HCG) stimulation.16,17 In unrecognized individuals raised as females, further virilization occurs at puberty, along with development of a male habitus. Two of our patients were siblings and wrongly assigned female sex at birth, both were re-assigned a male sex at 7 and 12 years of age.
Disorders affecting the end-organ androgen receptor are referred to as the androgen insensitivity syndrome. The defect may be complete or partial, resulting in a normal female external genitalia or ambiguous genitalia, respectively. These conditions are X-linked. Since anti-Mόllerian hormone (AMH) action is unaffected, fallopian tubes, uterus, and upper vagina are absent. In the complete form, the diagnosis may be suspected in a girl with bilateral inguinal hernia with palpable gonads. Confirmation of the diagnosis in individuals with partial defects is difficult. However, it should be suspected once other causes have been ruled out with normal T/DHT ratio. Diagnosis is further confirmed in the presence of favorable penile response to testosterone. A definitive diagnosis is also based on tissue receptor studies, though this was not feasible in our centre.18,19 Hypogonadotropin deficiency should be suspected in a newborn with micropenis, and cryptorchidism. It could be isolated or associated with other pituitary hormones deficiencies. The presence of other anomalies of morphogenesis usually indicates a non-endocrine cause for the appearance of the genitalia.20 This is clearly evident in our series.
Acknowledgments
The author would like to thank Loida M. Sese for her secretarial assistance.
REFERENCES
- 1.Bronx NY, Saenger P. Abnormal sex differentiation. J Pediatr. 1984;104:1–17. doi: 10.1016/s0022-3476(84)80581-8. [DOI] [PubMed] [Google Scholar]
- 2.Hughes IA. Disorders of sex development: a new definition and classification. Best Practice and Research Clinical Endocrinology and Metabolism. 2008;22:119–134. doi: 10.1016/j.beem.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Couch RM. Disorders of sexual differentiation. Medicine North America. 1987;14:2632–47. [Google Scholar]
- 4.Jini M, Sen S, Chacko J, Zachariah N, Raghurpathy P. Gender assignment in male-pseudohermaphroditism: An Indian Prospective. Pediatr Surg Int. 1993;8:500–1. [Google Scholar]
- 5.Rappaport R, Forest MG. Disorders of sexual differentiation. In: Bertrand J, Rappaport R, Sizomenko PC, editors. Pediatric Endocrinology Physiology, Pathophysiology and clinical aspects. 2nd ed. London: Williams and Wilkins; 1993. pp. 447–470. [Google Scholar]
- 6.Lee PA. Should we change our approach to ambiguous genitalia. The Endocrinologist. 2001;11:118–123. [Google Scholar]
- 7.Saenger PH. Physiology of sexual determination and differentiation. In: Brook CGD, Hindmarsh PC, editors. Clinical Pediatric Endocrinology. 4th edn. Oxford (UK): Blackwell Scientific Publisher; 2001. pp. 60–76. [Google Scholar]
- 8.Al Herbish AS, Al Jurayyan NA, Abo Bakr AM, Abdullah MA, Al Husain M, Al Rabeah AA, et al. Sex reassignment: a challenging problem – current medical and Islamic guidelines. Ann Saudi Med. 1996;16:12–15. doi: 10.5144/0256-4947.1996.12. [DOI] [PubMed] [Google Scholar]
- 9.Abdullah MA, Katugampola M, al-Habib S, al-Jurayyan N, al-Samarrai A, Al-Nuaim A, et al. Ambiguous genitalia: medical, socio-cultural and religious factors affecting management in Saudi Arabia. Ann Trop Paediatr. 1991;11:343–8. doi: 10.1080/02724936.1991.11747526. [DOI] [PubMed] [Google Scholar]
- 10.Prader A. Der Genitalbefund beim pseudohermaphroditism femininus des kongenitalen adrenogenitalen syndromes. Helven Paediatr Acta. 1954;9:231–248. [PubMed] [Google Scholar]
- 11.Al Jurayyan NA, Patel PJ, al Herbish AS, Abdullah MA, Abo-Bakr AM, al Rabeeah AA, et al. Ambiguous genitalia: comparative role of pelvic ultasonography and genitography. Ann Trop Paediatr. 1995;15:203–7. doi: 10.1080/02724936.1995.11747773. [DOI] [PubMed] [Google Scholar]
- 12.Van Niekerk WA. True hermaphroditism. In: Josso N, editor. The intersex child. (Pediatric and Adolescent Endocrinology) Basel: S Karger; 1981. pp. 80–99. [Google Scholar]
- 13.Salman H, Abanamy A, Ghassan B, Khalil M. Congenital adrenal hyperplasia. Ann Saudi Med. 1991;11:9–14. doi: 10.5144/0256-4947.1991.9. [DOI] [PubMed] [Google Scholar]
- 14.Al-Jurayyan NA, Al-Herbish AS, Abo Bakr AM, Al-Rabeeah AA, Al-Samarrai AI, Jawad AJ, et al. Congenital adrenal hyperplasia in a referral hospital in Saudi Arabia. Epidemiology, pattern and clinical presentation Ann Saudi Med. 1995;15:447–50. doi: 10.5144/0256-4947.1995.447. [DOI] [PubMed] [Google Scholar]
- 15.Saedi-Wong S, Al-Frayh AR, Wong HYM. Socio-economic epidemiology of consanguineous matings in the Saudi Arabian population. J Asian Afr Stu. 1989;24:247–52. [Google Scholar]
- 16.Griffin JE, Wilson JD. The androgen resistance syndromes: alpha-reductase deficiency, testicular feminization, and related syndromes. In: Scriver CR, Beaudet Al, Sly WS, Valle D, editors. The metabolic basis of inherited disease. 6th ed. New York: McGraw-Hill; 1989. pp. 1919–1944. [Google Scholar]
- 17.Imperato-McGinley J, Gautier T, Pichardo M. Daignosis of 5?-reductase deficiency in infancy. J Clin Endocrinol Metab. 1986;63:1313–8. doi: 10.1210/jcem-63-6-1313. [DOI] [PubMed] [Google Scholar]
- 18.Schweiket H. The androgen resistance syndrome: clinical and biochemical aspects. Eur J Pediatr. 1993;(Supp 2):550–7. doi: 10.1007/BF02125440. [DOI] [PubMed] [Google Scholar]
- 19.Brown TR, Scherer PA, Chang YT, Migeon CJ, Ghirri P, Murono K, et al. Molecular genetics of human androgen insensitivity. Eur J Pediatr. 1993;(Suppl 2):562–9. doi: 10.1007/BF02125442. [DOI] [PubMed] [Google Scholar]
- 20.Dincsoy MY, Salih MAM, Al Jurayyan N, Al Saadi M, Patel PJ. Multiple congenital malformations in two sibs reminiscent of hydrolethalus and pseudotrisomy 13 syndromes: new syndrome. Am J Med Genetics. 1995;56:317–21. doi: 10.1002/ajmg.1320560321. [DOI] [PubMed] [Google Scholar]