Role of Endothelial-Mesenchymal Transition (EndoMT) in the Pathogenesis of Fibrotic Disorders (original) (raw)
Abstract
The accumulation of a large number of myofibroblasts is responsible for exaggerated and uncontrolled production of extracellular matrix during the development and progression of pathological fibrosis. Myofibroblasts in fibrotic tissues are derived from at least three sources: expansion and activation of resident tissue fibroblasts, transition of epithelial cells into mesenchymal cells (epithelial-mesenchymal transition, EMT), and tissue migration of bone marrow–derived circulating fibrocytes. Recently, endothelial to mesenchymal transition (EndoMT), a newly recognized type of cellular transdifferentiation, has emerged as another possible source of tissue myofibroblasts. EndoMT is a complex biological process in which endothelial cells lose their specific markers and acquire a mesenchymal or myofibroblastic phenotype and express mesenchymal cell products such as α smooth muscle actin (α-SMA) and type I collagen. Similar to EMT, EndoMT can be induced by transforming growth factor (TGF-β). Recent studies using cell-lineage analysis have demonstrated that EndoMT may be an important mechanism in the pathogenesis of pulmonary, cardiac, and kidney fibrosis, and may represent a novel therapeutic target for fibrotic disorders.
The fibrotic diseases encompass a wide spectrum of clinical entities including multisystemic diseases such as systemic sclerosis,1,2 multifocal fibrosclerosis,3,4 sclerodermatous graft versus host disease in bone marrow transplant recipients,5 and the recently recognized nephrogenic systemic fibrosis,6,7 as well as organ-specific disorders such as pulmonary, liver, and kidney fibrosis.8–10 Although their etiology and causative mechanisms are vastly different, they share the common feature of disordered and exaggerated deposition of extracellular matrix in affected tissues. Elevated expression of genes encoding matrix proteins is a common and characteristic feature of these conditions, and the resulting fibrosis disrupts the normal architecture of the affected organs, ultimately leading to their dysfunction and failure.11–13 Indeed, it is the persistent activation of fibroblastic cells regarding the production of extracellular matrix macromolecules that distinguishes controlled repair such as that occurring during normal wound healing from the uncontrolled fibrosis that is the hallmark of fibrotic diseases. Because fibrotic diseases affect a wide spectrum of organs and a large number of individuals, their devastating effects cause an enormous burden on health resources, with severe economic consequences. The usually progressive nature of these diseases and the current absence of effective treatment compound the seriousness of the problem.
Although the etiology of the fibrotic disorders is quite diverse, and their pathogenesis is variable and dependent on the causative agent or initiating event, a common feature is the presence in affected tissues of large numbers of activated fibroblasts or myofibroblasts. These cells play a prominent role in the fibrotic process and display unique biological functions, including increased production of fibrillar type I and type III collagens, initiation of expression of α-smooth muscle actin (α-SMA), a molecular marker of activated myofibroblasts, and reduction in the expression of genes encoding extracellular matrix–degradative enzymes.14–17 Regardless of the etiological event, the accumulation of activated fibroblasts (myofibroblasts) in affected tissues and the persistence of their elevated biosynthetic functions are crucial determinants of the extent and rate of progression of the fibrotic diseases, and of their clinical course, response to therapy, prognosis, and mortality.
The origins of the mesenchymal cells responsible for the exaggerated and uncontrolled production of collagen and other extracellular matrix proteins in the fibrotic disorders have not been completely elucidated. Extensive research studies have shown that these cells originate from several sources, including expansion of resident tissue fibroblasts, migration and tissue accumulation of bone marrow–derived circulating fibrocytes, or from epithelial cells that have undergone epithelial to mesenchymal transition (EMT). More recent studies, however, have demonstrated that endothelial cells are also capable of undergoing endothelial to mesenchymal transition (EndoMT) and that this transition might be an important source of the mesenchymal cells participating in the fibrotic process. Here, the experimental evidence supporting the role of EndoMT in the pathogenesis of fibrotic disorders will be reviewed.
Origin of Activated Fibroblasts/Myofibroblasts in Fibrotic Tissues
The origins of activated fibroblasts in tissues affected by a fibrotic process are diverse and may differ depending on the affected organ and the initiating event. There are three general mechanisms responsible for the tissue accumulation of activated fibroblasts. i) Proliferation and activation of tissue resident fibroblasts or perivascular and vascular adventitial fibroblasts in response to specific profibrotic cues originating from infiltrating inflammatory cells leading to a progressive evolution from quiescent fibroblasts to cells expressing a myofibroblast phenotype.18 ii) Recruitment of fibroblast precursor cells from bone marrow as a result of the local release of activated chemokines. These bone marrow precursor cells are fibrocytes, a unique cell population expressing bone marrow cellular surface markers (CD34 protein) and capable of production of fibroblastic proteins (type I procollagen). These cells possess the ability to migrate from the bloodstream in response to specific chemokine gradients and localize in tissues undergoing pathological fibrogenesis.19,20 iii) Transformation of epithelial cells to myofibroblasts, a process known as EMT, which is induced by transforming growth factor β (TGF-β) and perhaps other polypeptides such as endothelin-1 (ET-1) or insulin growth factor.21–23 Such EMT has been shown to occur in the course of renal fibrosis, pulmonary fibrosis, and liver fibrosis.24–27
Endothelial-Mesenchymal Transition: A Recently Identified Source of Activated Mesenchymal Cells
Recently, EndoMT, another type of cellular transition, has emerged as a possible mechanism in pathological fibrosis. EndoMT is a complex biological process in which endothelial cells lose their specific endothelial cell markers, such as vascular endothelial cadherin (VE cadherin), and acquire a mesenchymal or myofibroblastic phenotype initiating expression of mesenchymal cell products such as α-SMA, vimentin, and type I collagen. Besides acquisition of an activated pro-fibrogenic phenotype, these cells also become motile and are capable of migrating into surrounding tissues. Similar to EMT, EndoMT can be induced by TGF-β as illustrated in Figure 1.
Figure 1.
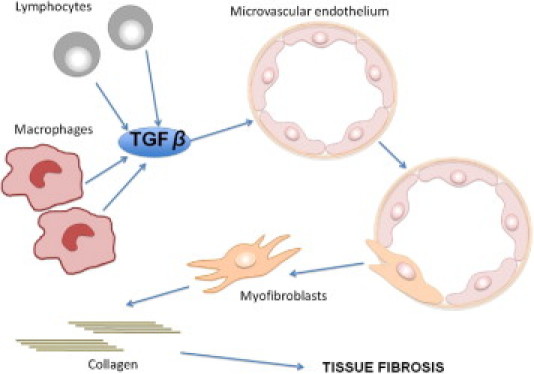
Schematic illustration of the process of EndoMT in tissue fibrosis. TGF-β secreted by tissue-infiltrating chronic inflammatory cells such as macrophages and lymphocytes initiates the transition of endothelial cells into myofibroblasts. These mesenchymal cells of endothelial origin migrate into the interstitium and participate in tissue fibrosis.
Although in the past EndoMT was believed to be a rare phenomenon confined to embryonic development, its occurrence in the adult vasculature was demonstrated in mature bovine vascular endothelium.28 These studies showed that adult bovine endothelial cells with an endothelial-specific phenotype could differentiate into smooth muscle cells. Initiation of transdifferentiation correlated with the disruption of cell–cell contacts and was induced by TGF-β1. Thus, these pioneering studies showed that mature bovine large vessel endothelial cells were able to acquire a smooth muscle or myofibroblastic phenotype via a transdifferentiation process initiated and driven by TGF-β1. Following these early studies, extensive experimental efforts were devoted to examine whether, in similarity with the participation of EMT in the pathogenesis of tissue fibrosis, EndoMT may also play a role in pathological organ fibrosis. These studies took advantage of the development of transgenic mice strains that allow endothelial cell lineage analysis based on the expression of microscopically detectable cell markers under the control of endothelial cell–specific gene promoters using the Cre-LoxP recombination system as shown in Figure 2. As described in greater detail below, endothelial cell lineage analyses during the development of experimentally induced tissue fibrosis have demonstrated the participation of EndoMT in the pathogenesis of interstitial fibrotic processes in various organs.
Figure 2.
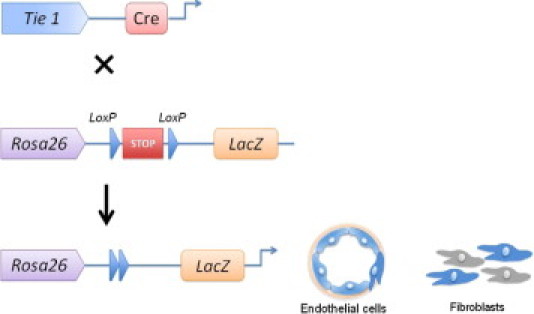
Endothelial cell lineage analysis in transgenic mice. The Tie 1 promoter is specific for endothelial cells and drives expression of the Cre recombinase exclusively in endothelial cells and any endothelial lineage cells. The Rosa 26 reporter construct drives expression of a marker (in this illustration, LacZ) and contains two LoxP sites bracketing a stop codon. When Tie 1-Cre mice are crossed with Rosa 26 mice, the Cre recombinase exclusively expressed in endothelial cells and all cells of endothelial lineage will cause excision and deletion of the stop codon bracketed by the LoxP sites in the Rosa26 mice. As a result, all cells from endothelial origin will become labeled with the LacZ marker (blue).
EndoMT in Cardiac Fibrosis
In a landmark study published in 2007, Zeisberg and collaborators29 examined the role of EndoMT in the development of experimentally induced cardiac fibrosis. For this purpose, they used two strains of transgenic mice. One of these strains consisted of mice in which the endothelial cell–specific promoter Tie 1, encoding a tyrosine kinase receptor that is exclusively expressed in endothelial cells, is used to mark irrevocably endothelial cells with the lacZ marker. The second strain consisted of FSP1-GFP transgenic mice in which green fluorescent protein is expressed under control of the promoter for the fibroblast-specific protein FSP1. Cardiac fibrosis was induced in these mice either by aortic banding or by heterotopic cardiac allograft rejection. In addition, in vitro studies were performed in human coronary artery endothelial cells. In these studies, the authors conclusively demonstrated that cardiac fibrosis is associated with the emergence of fibroblasts originating from endothelial cells. Furthermore, they showed that TGF-β1 induced endothelial cells to undergo EndoMT since there was a significant reduction in EndoMT-derived mesenchymal cells in aortic banded Smad3 +/− transgenic mice in which the TGF-β response is blunted due to the deficiency of Smad3. They also showed that recombinant human bone morphogenic protein 7, a TGF-β antagonistic protein, allowed preservation of the endothelial cell phenotype, and that its systemic administration significantly inhibited EndoMT and the progression of cardiac fibrosis in mouse models of pressure overload and chronic allograft rejection. These studies showed that from 27% to 35% of fibroblasts present in the fibrotic myocardium of mice with aortic banding originated from endothelial cells.
Goumans et al30 recently reviewed the role that TGF-β plays in cardiac fibrosis and emphasized the important role of the growth factor in mediating EndoMT. The authors noted that the generation of mesenchymal cells from endothelial cells by this process appears to recapitulate the transdifferentiation of endothelial cells that leads to the formation of cardiac valves during embryonic development. They also pointed out that in cardiac fibrosis, EndoMT represents the most important contributor to the generation of fibrotic tissue, and they suggested that EndoMT may act as a profibrotic switch in cardiac fibrosis as well as in other fibrotic disorders and that this process may represent a novel target for the prevention of tissue fibrosis.
In another study, Widyantoro et al31 showed that endothelial cell–derived ET-1 promotes cardiac fibrosis in diabetic hearts through stimulation of EndoMT. Streptozotocin-induced diabetes mellitus resulted in increased cardiac ET-1 expression, and it was shown that the elevated ET-1 levels promoted severe cardiac fibrosis and heart failure through the accumulation of fibroblasts via EndoMT. All of these features were abolished in hearts from transgenic mice with endothelial cell–specific ET-1 deletion. Other studies showed that small-interfering RNA–targeted ET-1 gene silencing in cultured human endothelial cells ameliorated the high glucose–induced phenotypic transition of endothelial cells and their subsequent acquisition of a fibroblast phenotype through the inhibition of TGF-β signaling. These results provide strong evidence that diabetes mellitus–induced cardiac fibrosis is associated with the emergence of fibroblasts originating from EndoMT and that this process is initiated and/or stimulated by ET-1. These results further suggested that targeting EndoMT induced by endothelial cell–derived ET-1 might be beneficial in the prevention of diabetic cardiomyopathy.
EndoMT in Kidney Fibrosis
The transition of renal epithelial cells into myofibroblasts in renal fibrosis has been intensively investigated, and numerous studies have demonstrated the contribution of EMT to the pool of activated fibroblasts responsible for interstitial kidney fibrosis in several experimental models as well as in some human diseases15,25,27 although some recent studies have raised doubts about the occurrence of this process in vivo.32 Recently, EndoMT has emerged as another potentially important mechanism in the development and progression of pathological kidney fibrosis. In 2008, Zeisberg et al33 demonstrated that EndoMT contributes to the accumulation of activated fibroblasts and myofibroblasts in fibrotic kidneys. These authors examined the contribution of EndoMT in three mouse models of chronic kidney disease: unilateral ureteral obstructive nephropathy, streptozotocin-induced diabetic nephropathy, and a model of Alport renal disease. The transition of endothelial cells to activated fibroblasts or myofibroblasts was assessed by the expression of FSP-1 and α-SMA in cells carrying the endothelial lineage–specific cell surface marker CD-31. Approximately 30% to 50% of fibroblasts in the fibrotic kidneys were found to co-express the endothelial marker CD31 along with FSP-1–specific and α-SMA–specific markers of fibroblasts and myofibroblasts, respectively. Endothelial lineage tracing further demonstrated that EndoMT was an important mechanism contributing to the accumulation of activated fibroblasts and myofibroblasts in kidney fibrosis.
Several other studies have confirmed the occurrence of EndoMT in various animal models of kidney fibrosis. For example, a recent study by Li et al34 demonstrated that EndoMT contributes to the early development of renal interstitial fibrosis in streptozotocin-induced diabetic mice independently of microalbuminuria. Subsequently, the same investigators35 examined whether blockade of EndoMT would reduce the early development of streptozotocin-induced diabetic nephropathy. In this latter study, EndoMT was induced in a mouse pancreatic microvascular endothelial cell line in the presence of advanced glycation end-products and in an endothelial lineage–traceable mouse line by administration of advanced glycation end-products. Activation of the TGF-β pathway was assessed by study of phosphorylated Smad3 and TGF-β blocking studies used a specific inhibitor of Smad3 (SIS3). These results demonstrated that advanced glycation end-products induced EndoMT in the microvascular endothelial cells in vitro and in the transgenic mice in vivo and that this process was mediated by Smad3 activation. TGF-β pathway inhibition with SIS3 abrogated EndoMT, reduced renal fibrosis, and retarded progression of nephropathy. These results confirmed that EndoMT is a novel pathway leading to fibrotic development in diabetic nephropathy and suggested that blockade of EndoMT using inhibitors of the TGF-β pathway, such as, for example, the specific Smad3 inhibitor SIS3, may provide a new strategy to retard the progression of diabetic nephropathy and other fibrotic processes.
EndoMT in Experimentally Induced Pulmonary Fibrosis
Hashimoto et al36 performed studies to evaluate whether endothelial cells could represent a source of interstitial fibroblasts involved in the development of pulmonary fibrosis. They examined bleomycin-induced lung fibrosis in double-transgenic mice in which LacZ was stably expressed in endothelial cells and in any cells originated from an endothelial cell lineage. Morphological evaluation of lungs from the transgenic mice following endotracheal injection of bleomycin showed severe pulmonary fibrosis with the areas of fibrotic involvement containing large numbers of LacZ-positive fibroblasts indicative of their endothelial origin. To directly demonstrate the presence of endothelial cell–derived fibroblasts, lung fibroblasts from either saline-injected control mice or from mice that received bleomycin injections were isolated and cultured. LacZ detection revealed that approximately 16% of lung fibroblasts in the cultures from bleomycin-treated mice were endothelial cell derived. Immunocytochemical staining for type I collagen and α-SMA showed that some cells from the bleomycin-treated mice expressed LacZ, type I collagen, and α-SMA, conclusively demonstrating that they originated from endothelial cells (Figure 3). To explore the underlying mechanisms, the effects of activated Ras and TGF-β on a microvascular endothelial cell line were analyzed. Combined treatment with activated Ras and TGF-β caused a significant loss of endothelial-specific markers while inducing strong de novo mesenchymal phenotypes with a 20-fold increase in fibronectin expression and a 10-fold increase in collagen I expression. The altered expression of these markers in the microvascular endothelial cells with activated Ras persisted after withdrawal of TGF-β both in vitro and in vivo, indicative that the phenotypic change was a permanently acquired trait and was not reversible. These findings conclusively showed that lung endothelial cells could give rise to significant numbers of activated fibroblasts through EndoMT in the bleomycin-induced lung fibrosis model.
Figure 3.
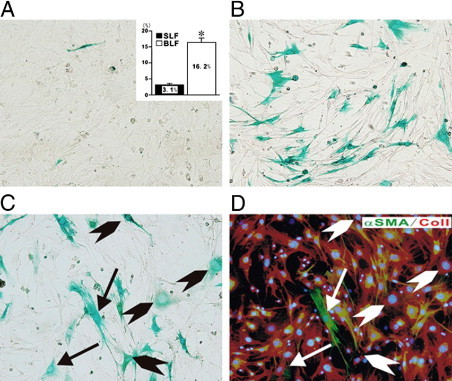
Demonstration of endothelial cell–derived fibroblasts in fibroblast cultures established from lung parenchyma of mice with bleomycin-induced pulmonary fibrosis. Fibroblast cultures were established from lungs from mice injected intratrachealy with either normal saline or bleomycin. When the cultures reached confluency, they were stained with X-Gal to identify the cells from endothelial lineage. Note the absence of X-Gal–staining cells in the cultures from saline-injected control mice (A) in contrast with the marked abundance of X-Gal–staining fibroblasts in the cultures from bleomycin-injected mice (B). The inset in A shows the percentage of X-Gal–positive cells in four separate samples of cultured fibroblasts from saline-injected mice (SLF) compared to eight separate samples of fibroblasts cultured from bleomycin-injected mice (BLF). *P < 0.05. C and D show sequential staining of a fibroblast culture from bleomycin-injected mice with X-Gal (C) followed by immunocytochemistry for the mesenchymal cell markers type I collagen (red) and α- SMA (green). The arrows indicate cells positive for X-Gal, type I collagen, and α-SMA, whereas the arrowheads indicate cells positive for X-Gal and type I collagen.
Reproduced from Hashimoto et al.36 with permission.
EndoMT in Other Fibrotic Diseases
Idiopathic portal hypertension is a noncirrhotic presinuosidal portal hypertension disorder of unknown etiology. Histopathologically, idiopathic portal hypertension is characterized by fibrotic narrowing of smaller portal veins with prominent portal fibrosis. In a recent study, the possibility that EndoMT of portal vein endothelium via TGF-β/Smad activation may also be involved in portal venopathy was examined.37 The results showed enhanced expression of phosphorylated Smad2 (pSmad2) in venous endothelium of smaller portal veins in idiopathic portal hypertension, which was associated with colocalization of either the endothelial cell marker CD34 and the fibroblast protein product, S100A4 or of CD34 and type I collagen. The authors concluded that such conversion of portal vein endothelial cells into cells expressing a myofibroblastic phenotype may be responsible for exaggerated periportal-venous deposition of collagen and other fibrous tissue proteins, and may cause portal venous obliteration in idiopathic portal hypertension.
Molecular Mechanisms of EndoMT
In contrast to the extensive studies focused on the molecular mechanisms of EMT, there is a paucity of information regarding EndoMT, and few studies have examined the intimate mechanisms involved in the EndoMT process. A recent study examined the in vitro conversion of endothelial cells derived from mouse embryonic stem cells into mural mesenchymal cells under the influence of TGF-β2.38 These studies demonstrated that TGF-β2 treatment of the endothelial cells induced their expression of mesenchymal cell markers, including α-SMA, transgrelin, and calponin, and reduced the expression of the endothelial cell marker claudin 5. The crucial role of TGF-β2 in this process was confirmed using a TGF-β receptor kinase inhibitor that inhibits activated TGF-β. Subsequent studies concluded that Snail1, a zinc finger–containing transcription factor that has been implicated in EMT, also mediates the actions of endogenous TGF-β signals that induce EndoMT.38 In a related study, it was shown that human dermal microvascular endothelial cells treated with TGF-β2 were capable of undergoing EndoMT with the appearance of mesenchymal cells also coexpressing endothelial cell–specific markers. Subsequent characterization of these mesenchymal cells indicated that they displayed markers of multipotential stem cells including STRO-1 and were capable of undergoing subsequent differentiation into osteoblasts, chondrocytes, and adipocytes under appropriate in vitro culture conditions.39
The source or sources of TGF-β and related proteins capable of initiating the EndoMT process have not been studied. Several possible sources could be proposed, including circulating inflammatory cells attaching to the endothelium or penetrating into the subendothelial space, other inflammatory cells present in the perivascular milieu, or latent TGF-β molecules produced by other mesenchymal cells that are sequestered in the extracellular matrix of the subendothelial or perivascular connective tissue.
We recently examined the mechanisms involved in the induction of EndoMT by TGF-β in primary cultures of murine pulmonary endothelial cells. Our studies also examined the intracellular transduction pathways involved in this process.40 We confirmed the occurrence of EndoMT following TGF-β1 treatment and demonstrated that the downstream signaling pathway initiated by TGF-β1 resulted in strong up-regulation of the transcriptional repressor Snail1. This effect appeared to be quite specific since transcript and protein levels of Snail2, the other member of this family of transcription factors, were not affected. Snail1 causes potent inhibition of E-cadherin gene transcription in cultured cells41 and plays an important role in the process of EMT.42,43 Our observations that TGF-β1 also induced Snail1 expression in pulmonary endothelial cells confirm previous EMT studies44 and indicate that EndoMT and EMT share similar molecular mechanisms.
The intracellular signaling studies we performed showed that TGF-β1–induced EndoMT occurred independently of Smad2/3 activation and represented, therefore, a non-Smad pathway of TGF-β signaling. Furthermore, we demonstrated that this non-Smad pathway involves the participation of important kinases, including the c-Abl protein kinase (c-Abl), protein kinase Cδ (PKC-δ), and glycogen synthase kinase 3β (GSK-3β). Our observations of the crucial role of GSK-3β are in agreement with recent studies on the regulatory mechanisms of EMT demonstrating that phosphorylation of specific serine residues in GSK-3β can result in inactivation of the kinase, which in turn induces the nuclear accumulation of Snail1. The nuclear accumulation of Snail1 results in a profound increase in the expression of its corresponding gene and, at the same time, in stabilization of the protein.45,46 These events result in a marked increase in the transcriptional effects of Snail1 and eventually in the expression of mesenchymal cell–specific proteins such as α-SMA. In contrast, in the absence of GSK-3β phosphorylation, the GSK-3β kinase is active and induces the proteosomal degradation of Snail1, thus abrogating endothelial cell transition into a mesenchymal phenotype. Figure 4 illustrates the TGF-β–activated pathways that are involved in EndoMT in murine pulmonary endothelial cells, emphasizing the pathway that involves the phosphorylation of GSK-3β mediated by PKCδ and cAbl. Of substantial importance is our observation that the inhibition of GSK-3β phosphorylation by specific inhibition of PKC-δ with rottlerin or the specific inhibition of c-Abl activity with imatinib effectively abolished the acquisition of the myofibroblastic phenotype, and therefore, it is expected that these interventions may cause a potent abrogation of the fibrotic response initiated by the EndoMT process. Although rottlerin is not a specific inhibitor of PKC-δ, the level of specificity for PKC-δ inhibition at the concentrations used in this study is very high. Furthermore, the role of PKC-δ was confirmed using specific PKC-δ small-interfering RNA.40
Figure 4.
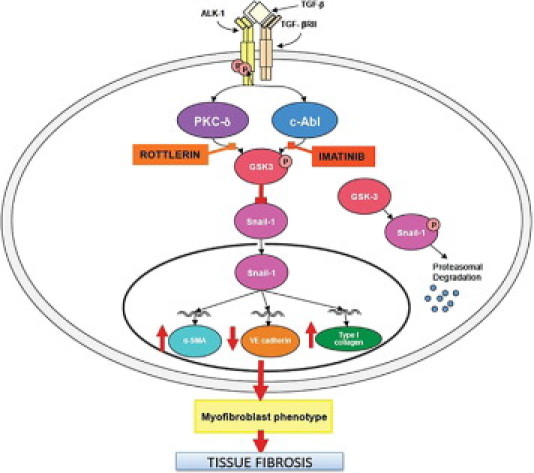
Schematic diagram showing the putative TGF-β signaling pathways involved in EndoMT in pulmonary endothelial cells. Following ligand-initiated activation of the Smad-independent TGF-β pathway, there is phosphorylation of GSK-3β mediated by PKC-δ and the cAbl nonreceptor kinase. Phosphorylation of GSK-3β at serine 9 (ser9) causes its inhibition, which then allows Snail1 to enter the nucleus. Nuclear accumulation of Snail1 results in marked stimulation of Snail1 expression, which then leads to acquisition of the myofibroblast phenotype with stimulation of α-SMA. The inhibition of GSK-3 ser9 phosphorylation by specific inhibition of PKC-δ with rottlerin, or the specific inhibition of c-Abl activity with imatinib, allows GSK-3β to phosphorylate Snail1, targeting it for proteosomal degradation and thus, effectively abolishes the acquisition of the myofibroblastic phenotype and the fibrotic response.
Modified from Li and Jimenez.40
Concluding Remarks
The studies reviewed here provide strong evidence for the prominent participation of EndoMT in the generation of activated myofibroblasts during the development of tissue fibrosis in a wide spectrum of fibrotic diseases. However, it is essential to point out that all these studies except for the study on idiopathic portal hypertension37 describe results from animal models. Thus, the results discussed in this review will need to be further validated in other human clinical pathological conditions. Furthermore, the studies discussed in the review used cell lineage analysis that was based on the assumption that gene expression directed by the Tie-2 promoter is highly specific for endothelial cells and should not occur in any other cell type. Thus, some caution is necessary for interpretation of the results in the unlikely circumstance that Tie-2 expression proves to occur in other cell types besides endothelial cells. The studies reviewed here further suggest that a greater understanding of the molecular mechanisms involved in the EndoMT process and the subsequent pharmacological blockade of this pathway in fibrotic disorders40 may represent a novel therapeutic approach for these devastating diseases. The feasibility of this approach has recently been supported by the demonstration that pharmacological inhibition of c-Abl kinase and protein kinase C delta, two kinases intimately involved in the EndoMT process, resulted in abrogation of EndoMT in vitro.40
Future Directions
The participation of EndoMT in the pathogenesis of various fibrotic disorders requires confirmation and validation from further studies of human clinical pathological conditions. Such confirmation should lead to a change in the paradigm of the origin of cells involved in the fibrotic process. Furthermore, identification of the source or sources of TGF-β and other active ligands that are capable of initiating the EndoMT process should also be of great value to provide cellular targets amenable of therapeutic interventions. Future efforts should also be devoted to further understanding of the molecular mechanisms and the regulatory controls involved in this process. These efforts would eventually lead to the development of novel therapeutic approaches for these incurable and often devastating disorders.
Acknowledgments
The expert assistance of Melissa Bateman in the preparation of the manuscript is gratefully acknowledged.
Footnotes
Supported by National Institutes of Health grant 5 R01 AR19616 to S.A.J.
CME Disclosure: None of the authors disclosed any relevant financial relationships.
A guest editor acted as Editor-in-Chief for this manuscript. No person at Thomas Jefferson University was involved in the peer review process or final disposition for this article.
References
- 1.Varga J., Abraham D. Systemic sclerosis: a prototypic multi-system fibrotic disorder. J Clin Invest. 2007;117:557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denton C.P., Black C.M. Scleroderma: clinical and pathological advances. Best Pract Res Clin Rheumatol. 2004;18:271–290. doi: 10.1016/j.berh.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Gleeson M.H., Taylor S., Dowling R.H. Multifocal fibrosclerosis. Proc Roy Soc Med. 1970;63:1309–1311. [PMC free article] [PubMed] [Google Scholar]
- 4.Neild G.H., Rodriguez-Justo M., Wall C., Connolly J.O. Hyper-IgG4 disease: report and characterization of a new disease. BMC Med. 2006;4:23. doi: 10.1186/1741-7015-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White J.M., Creamer D., du Vivier A.W., Pagliuca A., Ho A.Y., Devereux S., Salisbury J.R., Mufti G.J. Scleroderma graft-versus-host disease: clinical spectrum and therapeutic challenges. Br J Dermatol. 2007;156:1032–1038. doi: 10.1111/j.1365-2133.2007.07827.x. [DOI] [PubMed] [Google Scholar]
- 6.Cowper S.E. Nephrogenic systemic fibrosis: an overview. J AM Coll Radiol. 2008;5:23–28. doi: 10.1016/j.jacr.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Mendoza F.A., Artlett C.M., Sandorfi N., Latinis K., Piera-Velazquez S., Jimenez S.A. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35:238–249. doi: 10.1016/j.semarthrit.2005.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noble P.W. Idiopathic pulmonary fibrosis: natural history and prognosis. Clin Chest Med. 2006;27(Suppl 1):S11–S16. doi: 10.1016/j.ccm.2005.08.003. v. [DOI] [PubMed] [Google Scholar]
- 9.Bataller R., Brenner D.A. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schnaper H.W., Kopp J.B. Renal fibrosis. Front Biosci. 2003;8:e68–e86. doi: 10.2741/925. [DOI] [PubMed] [Google Scholar]
- 11.Rosenbloom J., Castro S.V., Jimenez S.A. Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Ann Intern Med. 2010;152:159–166. doi: 10.7326/0003-4819-152-3-201002020-00007. [DOI] [PubMed] [Google Scholar]
- 12.Wei J., Bhattacharyya S., Tourtellotte W.G., Varga J. Fibrosis in systemic sclerosis: emerging concepts and implications for targeted therapy. Autoimmun Rev. 2011;10:267–275. doi: 10.1016/j.autrev.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wynn T.A. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krieg T., Abraham D., Lafyatis R. Fibrosis in connective tissue disease: the role of the myofibroblast and fibroblast-epithelial cell interactions. Arthritis Res Ther. 2007;9(Suppl 2):S4. doi: 10.1186/ar2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neilson E.G. Mechanisms of disease: fibroblasts-a new look at an old problem. Nat Clin Pract Nephrol. 2006;2:101–108. doi: 10.1038/ncpneph0093. [DOI] [PubMed] [Google Scholar]
- 16.Abraham D.J., Eckes B., Rajkumar V., Krieg T. New developments in fibroblast and myofibroblast biology: implications for fibrosis and scleroderma. Curr Rheumatol Rep. 2007;9:136–143. doi: 10.1007/s11926-007-0008-z. [DOI] [PubMed] [Google Scholar]
- 17.McAnulty R.J. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39:666–671. doi: 10.1016/j.biocel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Postlethwaite A.E., Shigemitsu H., Kanangat S. Cellular origins of fibroblasts: possible implications for organ fibrosis in systemic sclerosis. Curr Opin Rheumatol. 2004;16:733–738. doi: 10.1097/01.bor.0000139310.77347.9c. [DOI] [PubMed] [Google Scholar]
- 19.Herzog E.L., Bucala R. Fibrocytes in health and disease. Exp Hematol. 2010;38:548–556. doi: 10.1016/j.exphem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bellini A., Mattoli S. The role of the fibrocytes, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest. 2007;87:858–870. doi: 10.1038/labinvest.3700654. [DOI] [PubMed] [Google Scholar]
- 21.Thiery J.P., Acloque H., Huang R.Y., Nieto M.A. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Thiery J.P., Sleeman J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 23.Kalluri R., Neilson E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willis B.C., Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 25.Rastaldi M.P. Epithelial-mesenchymal transition and its implications for the development of renal tubulointerstitial fibrosis. J Nephrol. 2006;19:407–412. [PubMed] [Google Scholar]
- 26.Willis B.C., duBois R.M., Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3:377–382. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeisberg M., Kalluri R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front Biosci. 2008;13:6991–6998. doi: 10.2741/3204. [DOI] [PubMed] [Google Scholar]
- 28.Arciniegas E., Sutton A.B., Allen T.D., Schor A.M. Transforming growth factor beta 1 promotes the differentiation of endothelial cells into smooth muscle-like cells in vitro. J Cell Sci. 1992;103:521–529. doi: 10.1242/jcs.103.2.521. [DOI] [PubMed] [Google Scholar]
- 29.Zeisberg E.M., Tarnavski O., Zeisberg M., Dorfman A.L., McMullen J.R., Gustafsson E., Chandraker A., Yuan X., Pu W.T., Roberts A.B., Neilson E.G., Sayegh M.H., Izumo S., Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 30.Goumans M., van Zonneveld A.J., ten Dijke P. Transforming growth factor β-induced endothelial-to-mesenchymal transition: a switch to cardiac fibrosis. Trends Cardiovasc Med. 2008;18:293–298. doi: 10.1016/j.tcm.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Widyantoro B., Emoto N., Nakayama K., Anggrahini D.W., Adiarto S., Iwasa N., Yagi K., Miyagawa K., Rikitake Y., Suzuki T., Kisanuki Y.Y., Yanagisawa M., Hirata K. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121:2407–2418. doi: 10.1161/CIRCULATIONAHA.110.938217. [DOI] [PubMed] [Google Scholar]
- 32.Wilhelm K., Kaissling B., Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrois: fact or fantasy? J Clin Invest. 2011;121:468–474. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeisberg E.M., Potenta S.E., Sugimoto H., Zeisberg M., Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J., Qu X., Bertram J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol. 2009;175:1380–1388. doi: 10.2353/ajpath.2009.090096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J., Qu X., Yao J., Caruana G., Ricardo S.D., Yamamoto Y., Yamamoto H., Bertram J.F. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:2612–2624. doi: 10.2337/db09-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashimoto N., Phan S.H., Imaizumi K., Matsuo M., Nakashima H., Kawabe T., Shimokata K., Hasegawa Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010;43:161–172. doi: 10.1165/rcmb.2009-0031OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakanuma Y., Sato Y., Kiktao A. Pathology and pathogenesis of portal venopathy in idiopathic portal hypertension: hints from systemic sclerosis. Hepatol Res. 2009;39:1023–1031. doi: 10.1111/j.1872-034X.2009.00555.x. [DOI] [PubMed] [Google Scholar]
- 38.Kokudo T., Suzuki Y., Yoshimatsu Y., Yamazaki T., Watabe T., Miyazono K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci. 2008;121:3317–3324. doi: 10.1242/jcs.028282. [DOI] [PubMed] [Google Scholar]
- 39.Medici D., Shore E.M., Lounev V.Y., Kaplan F.S., Kalluri R., Olsen B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Z., Jimenez S.A. Protein kinase C delta and the c-abl kinase are required for transforming growth factor-beta induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. 2011 Mar 18 doi: 10.1002/art.30317. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Batlle E., Sancho E., Franci C., Dominguez D., Monfar M., Baulida J., Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 42.Cano A., Perez-Moreno M.A., Rodrigo I., Locascio A., Blanco M.J., del Barrio M.G., Portillo F., Nieto M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 43.Barrallo-Gimeno A., Nieto M.A. The snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 44.Nieto M.A. Epithelial-mesenchymal transitions in development and disease: old views and new perspectives. Int J Dev Biol. 2009;53:1541–1547. doi: 10.1387/ijdb.072410mn. [DOI] [PubMed] [Google Scholar]
- 45.Zhou B.P., Deng J., Xia W., Xu J., Li Y.M., Gunduz M., Hung M.C. Dual regulation of snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 46.Doble B.W., Woodgett J.R. Role of glycogen synthase kinase-3 in cell fate and epithelial-mesenchymal transitions. Cells Tissues Organs. 2007;185:73–84. doi: 10.1159/000101306. [DOI] [PubMed] [Google Scholar]