“Methanoplasmatales,” Thermoplasmatales-Related Archaea in Termite Guts and Other Environments, Are the Seventh Order of Methanogens (original) (raw)
Abstract
The Euryarchaeota comprise both methanogenic and nonmethanogenic orders and many lineages of uncultivated archaea with unknown properties. One of these deep-branching lineages, distantly related to the Thermoplasmatales, has been discovered in various environments, including marine habitats, soil, and also the intestinal tracts of termites and mammals. By comparative phylogenetic analysis, we connected this lineage of 16S rRNA genes to a large clade of unknown mcrA gene sequences, a functional marker for methanogenesis, obtained from the same habitats. The identical topologies of 16S rRNA and mcrA gene trees and the perfect congruence of all branches, including several novel groups that we obtained from the guts of termites and cockroaches, strongly suggested that they stem from the same microorganisms. This was further corroborated by two highly enriched cultures of closely related methanogens from the guts of a higher termite (Cubitermes ugandensis) and a millipede (Anadenobolus sp.), which represented one of the arthropod-specific clusters in the respective trees. Numerous other pairs of habitat-specific sequence clusters were obtained from the guts of other termites and cockroaches but were also found in previously published data sets from the intestinal tracts of mammals (e.g., rumen cluster C) and other environments. Together with the recently described Methanomassiliicoccus luminyensis isolated from human feces, which falls into rice cluster III, the results of our study strongly support the idea that the entire clade of “uncultured _Thermoplasmatales_” in fact represents the seventh order of methanogenic archaea, for which the provisional name “_Methanoplasmatales_” is proposed.
INTRODUCTION
Methanogenesis is an important process in the carbon cycle, with a significant impact on global warming. Methane is produced exclusively by methanogenic archaea—strictly anaerobic microorganisms that occur in almost all anoxic habitats on earth, from the marine environment to freshwater sediments to soils, including hot springs and the deep subsurface, in sewage sludge, and in the digestive tracts of animals and humans (33).
All methanogens belong to the phylum Euryarchaeota. They presently comprise members of six orders. The basal groups are Methanopyrales, Methanococcales, and Methanobacteriales (class I); Methanomicrobiales (class II) (3); and Methanosarcinales (class III) (2), with the recently recognized sister group Methanocellales (50). It has been hypothesized that the genes for hydrogenotrophic methanogenesis were already present in a common ancestor and were vertically inherited in a broader monophyletic unit encompassing all methanogens (3). Consequently, it has to be postulated that methanogenesis was lost in the Archaeoglobales (which fall among class I methanogens), the Thermoplasmatales, and the Halobacteriales (which fall between class I and class II) (3).
In addition, there are many deep-branching lineages of archaea that are exclusively represented by their 16S rRNA genes (19, 53, 60) and whose properties cannot be safely predicted for lack of any cultivated representatives. One of these lineages is a diverse clade of sequences distantly related to the Thermoplasmatales. Originally discovered in the marine environment (9, 17) and the deep subsurface (59), related clones were subsequently obtained from rice field soil (20), the water column and sediment of freshwater lakes (27, 44), and soil and leachate of landfills (24, 37). Other members of this clade were found in the guts of termites (16, 42, 54), wood-feeding cockroaches (22), and scarab beetle larvae (14). Also, studies of the mammalian digestive tract reported sequences of uncultured archaea distantly related to the Thermoplasmatales in cattle (10, 26, 58, 62), sheep (63), and wallabies (15) and in the guts and subgingival pockets of humans (32, 39, 40, 51). Although concrete evidence was lacking, several of these earlier reports had already suggested that such “uncultured _Thermoplasmatales_” may represent a novel lineage of methanogens.
The mcrA gene, which encodes the α-subunit of methyl coenzyme M (methyl-CoM) reductase, has been established as a molecular marker for methanogenic archaea (36). Studies of the diversity of methanogens in landfill soil yielded several novel mcrA gene sequences that formed a deep-branching cluster separate from those of the established orders of methanogens (37). Related sequences were soon discovered in a eutrophic lake (13) and in salt marsh sediments (7). Later studies of vertebrate guts also revealed the presence of novel mcrA genes in the cow rumen (10); feces of pigs, chickens, and horses (61); the guts of humans (39, 51); and the foregut of wallabies (15).
Kemnitz et al. (28) observed a correlation between the abundance of rice cluster III (RC-III) archaea and the rate of methanogenesis in enrichment cultures. Mihajlovski et al. (39) claimed that a new mcrA phylotype and a new 16S rRNA phylotype obtained from the same stool sample belonged to the same organism and subsequently postulated that they represent a putative new order of methanogens (40). Also, Evans et al. (15) had speculated that the unknown mcrA gene sequences in the foreguts of wallabies and ruminants belong to a lineage of uncultivated archaea encountered in these habitats. However, the final proof for this hypothesis is still lacking.
Previous studies have shown that 16S rRNA and mcrA genes in the established methanogenic lineages have the same phylogeny (36, 37). This allows the correlation of unknown mcrA sequences with the corresponding 16S rRNA gene sequences, a strategy that has been successfully employed to predict the methanogenic nature of the uncultivated archaea in rice cluster I (36), which eventually led to the enrichment and isolation of Methanocella paludicola (50).
In this study, we comprehensively analyzed the phylogeny of all _Thermoplasmatales_-related 16S rRNA genes available to date and the unknown mcrA genes from the respective habitats. To further corroborate the hypothetical congruence of the resulting trees, we obtained additional sequence sets of archaeal 16S rRNA and mcrA genes from the hindguts of various higher termites and wood-feeding cockroaches, which are known to harbor abundant and diverse populations of uncultured Thermoplasmatales (5). In addition, we initiated enrichment cultures from the hindguts of termites and millipedes to isolate a potentially methanogenic member of this novel lineage.
MATERIALS AND METHODS
Termites and cockroaches.
Cubitermes ugandensis and Ophiotermes sp. were collected in Kakamega Forest Reserve (Kenya), and Macrotermes michaelseni was collected near Kajiado (Kenya). Trinervitermes sp. and Alyscotermes trestus originated from the campus of the Jomo Kenyatta University of Agriculture and Technology, Gachororo, Kenya. Only worker caste termites were used for this work. The wood-feeding cockroaches Salganea esakii and Panesthia angustipennis were collected in the vicinity of the Keta Shrine in Ishikawa Prefecture, Japan, by Kiyoto Maekawa, Toyama University. The millipede Anadenobolus sp. was obtained from a commercial breeder (b.t.b.e. Insektenzucht, Schnürpflingen, Germany). All animals were kept in plastic containers at room temperature in the dark.
DNA extraction and purification.
The hindguts of 10 to 20 termites were dissected with sterile fine-tipped forceps, pooled in 2-ml tubes containing 750 μl sodium phosphate buffer (120 mM; pH 8.0), and homogenized. Homogenates of individual cockroach hindguts were prepared in a similar manner. DNA was prepared using a bead-beating protocol combined with phenol-chloroform extraction. The homogenate was transferred to a 2-ml bead-beating vial, and 250 μl sodium dodecyl sulfate (SDS) solution (10% SDS, 0.5 M Tris-HCl, pH 8.0, 0.1 M NaCl), and 0.7 g heat-sterilized zirconia-silica beads (0.1-mm diameter; Carl Roth, Karlsruhe, Germany) was added. Cells were lysed by shaking with a cell disruptor (FastPrep-24; MP Biomedicals, Ilkirch, Germany) for 45 s at a velocity of 6.5 m/s. Cell debris was sedimented by centrifugation at 20,000 × g for 4 min. The supernatant was extracted with 1 volume of phenol-chloroform-isoamyl alcohol (24:24:1 by volume; pH 8.0). After a second centrifugation step, the supernatant was extracted with 1 volume of chloroform-isoamyl alcohol (24:1 [vol/vol]) and centrifuged again in a 2-ml phase lock gel heavy tube (Eppendorf, Hamburg, Germany). The DNA was precipitated by mixing the aqueous phase with 2 volumes of polyethylene glycol (PEG) solution (30% PEG 6000 in 1.6 M NaCl). After centrifugation for 30 min, the pellet was washed with 500 μl ice-cold ethanol (70%) and dried under vacuum. DNA was dissolved in 50 μl elution buffer (MinElute PCR Purification Kit; Qiagen, Hilden, Germany), checked photometrically for purity (Nanodrop; PeqLab, Erlangen, Germany), quantified fluorimetrically (Qubit; Invitrogen, Eugene, OR), and stored at −20°C.
PCR amplification and cloning.
16S rRNA genes were amplified using either the archaeon-specific primer pair Ar109f (5′-AMDGCTCAGTAACACGT-3′) (25) and Ar912r (5′-CTCCCCCGCCAATTCCTTTA-3′) (35) or the archaeon-specific primer Ar109f and the prokaryote-specific primer 1490R with the modification of Hatamoto et al. (23) (5′-GGHTACCTTGTTACGACTT-3′), a combination that yields only archaeal 16S rRNA genes (43). Each PCR mixture (50 μl) contained reaction buffer, 2.5 mM MgCl2, 1 U Taq DNA polymerase (all Invitrogen, Carlsbad, CA), 50 μM deoxynucleoside triphosphate mixture, 0.3 μM each primer, 0.8 mg/ml bovine serum albumin, and 1 μl DNA extract. The PCR program consisted of an initial denaturation step (94°C for 3 min) followed by 32 cycles of denaturation (94°C for 20 s), annealing (52°C for 20 s), and extension (72°C for 50 s) and a final extension step (72°C for 7 min). For the amplification of the mcrA gene, the primer pair _mcrA_-f (5′-GGTGGTGTMGGATTCACACARTAYGCWACAGC-3′) and _mcrA_-r (5′-TTCATTGCRTAGTTWGGRTAGTT-3′; 37) was used; the reaction mixture and the PCR protocol were the same as described above, except for the annealing temperature (53.5°C) and the cycle number (35) and a decreased ramp temperature rate of 1°C/s. The PCR products were purified and cloned as described by Schauer et al. (52).
Sequence analysis.
The 16S rRNA gene sequences obtained in this study were imported into the current Silva database (version 106) (48; http://www.arb-silva.de) using the ARB software package (34). Sequences from other studies that were not included in Silva were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/). The sequences were automatically aligned, and the alignments were refined manually. A 30% consensus filter was used to exclude highly variable positions. Phylogenetic trees of almost full-length sequences (1,250 bp) were calculated using RAxML, a maximum-likelihood method (56). Tree topology and node support (100 bootstraps) were tested using the maximum-parsimony method (DNAPARS) implemented in ARB. The mcrA gene sequences were imported into a seed alignment complemented with sequences of unknown origin that were retrieved from the NCBI database. Trees were calculated at the amino acid level (140 amino acids) using PhyML, a maximum-likelihood method (21) implemented in ARB. Tree topology and node support (100 bootstraps) were tested using the maximum-parsimony method (PROTPARS) implemented in ARB.
Cultivation.
Enrichment cultures were set up in anoxic, bicarbonate-buffered AM5 medium under an atmosphere of N2-CO2 (80:20 [vol/vol]) (4), but dithiothreitol (DTT) was omitted. The basal medium was supplemented with Casamino Acids (2 g/liter), coenzyme M (10 mg/liter), cysteine (2 mM), and palladium on activated charcoal (10 ml/liter) and (optionally) with yeast extract (2 g/liter) or rumen fluid (10%). The medium (4.5 ml) was dispensed into 15-ml rubber-stoppered glass vials, and hydrogen gas (5 ml) was added to the headspace. Substrates were added from sterile stock solutions (final concentrations): formate (50 mM), methanol (50 mM), acetate (30 mM), or xylan (9 g/liter). Tubes were inoculated (0.5 ml) with gut homogenates of C. ugandensis or Anadenobolus sp. prepared in basal medium (1 gut per ml), and the tubes were incubated at 30°C in the dark. The methane content in the headspace was measured every week. The culture headspace (0.2 ml) was sampled with a gas-tight syringe, and the methane content was analyzed using a gas chromatograph with a flame ionization detector (38).
Quantitative PCR and pyrotag sequencing.
DNA was extracted from the enrichment culture (2 ml) (see above), and the copy numbers of archaeal 16S rRNA genes were determined by quantitative real-time PCR (qPCR) as described by Kemnitz et al. (28) using the primers A364aF (5′-CGGGGYGCASCAGGCGCGAA-3′) (6) and A934b (5′-GTGCTCCCCCGCCAATTCCT-3′) (20). Bacterial 16S rRNA genes were quantified as described by Stubner (57) using the primer pair 519fc (5′-CAGCMGCCGCGGTAANWC-3′) and 907r (5′-CCGTCAATTCMTTTRAGTT-3′) (31). In addition, the bacterial community structure of the sample was determined by 454 pyrotag sequencing as described elsewhere (30).
Nucleotide sequence accession numbers.
The sequences obtained in this study were submitted to GenBank. The accession numbers are JX266062 to JX266091 for 16S rRNA genes and JX266092 to JX266145 for mcrA genes from hindgut homogenates. The accession numbers for the corresponding genes of strains MpT1 and MpM2 are JX266068, JX266097, JX648297, and JX648298.
RESULTS
Comparison of 16S rRNA and mcrA clone frequencies.
Analysis of the archaeal 16S rRNA gene sequences from the hindguts of several higher termites revealed a diverse community of methanoarchaea, consisting of Methanobacteriales, Methanosarcinales, and Methanomicrobiales, although not all lineages were represented in each species (Table 1). In addition, each termite species yielded a substantial proportion of clones that clustered with a deep-branching lineage distantly related to Thermoplasmatales previously obtained from termite guts and other intestinal environments. A detailed analysis of the entire archaeal diversity in the different termite species will be published in a different context (J. O. Nonoh, K. Paul, D. K. Ngugi, and A. Brune, unpublished data).
Table 1.
Clone frequencies in libraries of archaeal 16S rRNA genes and mcrA genes obtained from the hindguts of higher termites, documenting the cooccurrence of a novel lineage of _Thermoplasmatales-_related archaea and a cluster of novel mcrA genes
| Termite species_a_ | 16S rRNA genes (%)b | No. of clones | mcrA genes (%)c | No. of clones | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Methanomicrobiales | Methanobacteriales | Methanosarcinales | Novel lineage | Methanomicrobiales | Methanobacteriales | Methanosarcinales | Novel cluster | |||
| C. ugandensis | 27 | 26 | 20 | 28 | 66 | 19 | 33 | 14 | 35 | 34 |
| Ophiotermes sp. | 0 | 7 | 65 | 28 | 80 | 0 | 49 | 28 | 23 | 19 |
| Trinervitermes sp. | 50 | 31 | 0 | 19 | 42 | 18 | 64 | 0 | 18 | 11 |
| M. michaelseni | 0 | 52 | 45 | 2 | 44 | 0 | 74 | 16 | 11 | 19 |
| A. trestus | 3 | 84 | 0 | 14 | 37 | 4 | 64 | 0 | 32 | 25 |
Clone libraries of the mcrA genes amplified from the same samples not only yielded the mcrA genes expected of the methanogens identified in the rRNA-based analysis, but each contained an additional cluster of mcrA genes of unknown origin (Table 1). In each termite, the distribution patterns of the different clone groups were in agreement with the assumption that the novel mcrA genes stemmed from the uncultured lineage of Thermoplasmatales.
Phylogenetic analysis of the 16S rRNA genes.
Since the 16S rRNA gene sequences obtained in the first data set were relatively short (800 bp), the phylogenetic resolution was not sufficient for the requirements of our study. Therefore, we also constructed smaller clone libraries with the primer pair Ar109f and Ar1490r for the termites C. ugandensis and Ophiotermes sp. and for the cockroaches S. esakii and P. angustipennis to obtain longer sequences (1,380 bp), together with those already present from previous studies, for all lineages of _Thermoplasmatales_-related archaea affiliated with termites and cockroaches. We included 16S rRNA gene sequences from all established lineages of Euryarchaeota from the Silva database and GenBank, including all sequences of uncultured Thermoplasmatales obtained in previous studies. The resulting phylogenetic trees showed the same major lineages of methanoarchaea previously documented by others, with the Thermoplasmatales and their uncultured relatives clearly falling within the radiation of methanogens, confirming the paraphyletic character of methanoarchaea as a taxonomic group (Fig. 1).
Fig 1.

Phylogenetic tree showing the relationships among uncultured archaea related to Thermoplasmatales and to representatives of all other orders of methanogenic archaea and the ANME-1 group. Clusters of clones from termite (TC) and cockroach (CC) guts are indicated. The tree is based on a maximum-likelihood analysis of an alignment of archaeal 16S rRNA genes (1,250 bp) of archaea in public databases; sequences obtained in this study are marked in boldface. Sequences of Trinervitermes sp., M. michaelseni, and A. trestus were shorter and were added to the tree using the ARB parsimony tool. The bullets indicate bootstrap support (●, >95%; ○, >70%). The scale bar indicates substitutions per site.
The sequences of _Thermoplasmatales_-related archaea obtained from termites and cockroaches fell into a distinct clade of clones obtained exclusively from intestinal environments that was clearly separated from previously published clades containing sequences from diverse marine and freshwater habitats. Next, relatives of this clade were clones previously obtained from an anaerobic digestor (Vadin Group) (18). Within the intestinal cluster, the sequences from insect guts formed two distinct lineages, each comprising both termite-specific and cockroach-specific lineages, with well-supported subclusters reflecting the phylogeny of their respective hosts. Other lineages in the intestinal cluster consisted of clones from vertebrate guts, which were previously obtained from the intestinal tracts of cattle, wallabies, chickens, and humans (see the introduction), and clones obtained from a manure pit.
Phylogenetic analysis of the mcrA genes.
To test the phylogenetic positions of the novel mcrA genes obtained in this study, we added the sequences to a comprehensive set of mcrA sequences from public databases, comprising all major lineages of methanogens and including all mcrA genes of uncertain origin from environmental studies. Phylogenetic analysis confirmed the presence of mcrA genes in insect guts belonging to representatives of the orders Methanosarcinales, Methanobacteriales, and Methanomicrobiales, which was in agreement with the results of the 16S rRNA analysis (Fig. 2). The clones of unknown origin obtained from termite guts (Table 1) and from the guts of the cockroaches S. esakii and P. angustipennis (this study) formed two distinct insect-specific lineages in a larger cluster of mcrA genes from intestinal habitats, including cows, wallabies, pigs, chickens, and humans. Also, the mcrA genes from the intestinal tracts of termites and cockroaches formed well-supported subclusters reflecting the phylogeny of their respective hosts.
Fig 2.

Phylogenetic tree showing the relationships among the novel mcrA genes and to representative mcrA genes of all other orders of methanogenic archaea and the ANME-1 group. Clusters of clones from termite (TC) and cockroach (CC) guts are indicated. The tree is based on a maximum-likelihood analysis of an alignment of the mcrA genes (140 amino acids) of archaea in public databases; sequences obtained in this study are marked in boldface. The bullets indicate bootstrap support (●, >95%; ○, >70%). The scale bar indicates substitutions per site.
As in case of the 16S rRNA gene sequences of _Thermoplasmatales_-related archaea, the novel mcrA genes from intestinal environments were most closely related to clones from an anaerobic digestor and clearly separated from other, previously published clades containing sequences from diverse marine and freshwater habitats, including additional sequences of intestinal origin.
Enrichment of novel methanogens from arthropod guts.
A hindgut homogenate of C. ugandensis was inoculated into basal medium with or without yeast extract with optional additions of methanol, formate, or xylan and incubated under a headspace containing H2 and CO2. After a lag phase of several weeks, the culture containing methanol and yeast extract started to form CH4. No methane formation was observed under any other conditions, even after 6 months of incubation, and also not if rumen fluid was added to the cultures. Subsequent transfers of the culture on the same medium led to robust CH4 formation (up to 17-kPa headspace partial pressure); rumen fluid was not required. Transfers of the enrichment culture to medium lacking methanol showed no methanogenesis; transfers to medium containing methanol in the absence of H2 produced much less methane than with H2. No methanogenesis occurred with acetate as the sole substrate (Fig. 3).
Fig 3.

Time course of methane partial pressure in the headspace of the enrichment culture MpT1 (N2-CO2; 80/20) inoculated from a methanol-starved preculture into basal medium supplemented with different substrates: H2 (50 kPa in headspace), methanol (50 mM), or acetate (30 mM). The values are means of two cultures; mean standard deviations are shown only if they are larger than the symbols.
After the initial transfers, the culture already consisted mostly of small, roundish cells (ca. 0.6 to 1 μm in diameter) (Fig. 4A). DNA was extracted from several subcultures, and the archaeal 16S rRNA genes were amplified using specific primers (Ar109f and 1490R). Each PCR product could be sequenced without cloning, and the sequencer traces indicated that in each case only a single phylotype of archaea was present. The sequences obtained from the different subcultures were identical. Phylogenetic analysis revealed that the archaeal 16S rRNA sequence (phylotype MpT1) fell into the apical cluster of putative methanogens consisting exclusively of clones from higher termites (Fig. 1, TC-1a). Also, the mcrA genes amplified from the same samples yielded identical sequences, which fell into the corresponding cluster of novel mcrA genes in the phylogenetic tree (Fig. 2, TC-1a).
Fig 4.
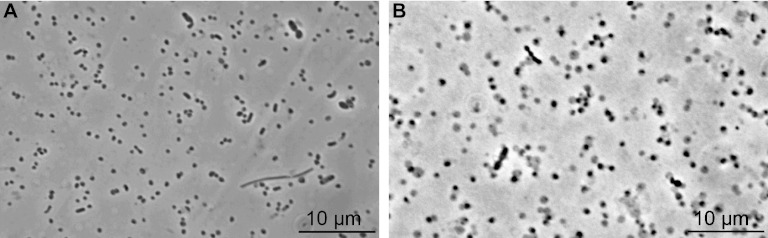
Phase-contrast photomicrographs of the methanogenic enrichment cultures MpT1 (A) and MpM2 (B) after several transfers in basal medium supplemented with H2 and methanol. Both cultures consisted mostly of small roundish cells (diameter, 0.6 to 1.0 μm).
When the abundance of archaeal 16S rRNA genes in the enrichment culture had increased to 64% (based on the total copy numbers of archaeal and bacterial 16S rRNA, determined by qPCR), the bacterial contaminants remaining in the enrichment culture were determined by 454 pyrotag sequencing. Classification of the bacterial sequences revealed that the bacteria remaining in the enrichment culture represented several lineages of so-far uncultivated Clostridiales (Fig. 5). All attempts to isolate strain MpT1 in pure culture have so far been unsuccessful.
Fig 5.

Composition of the enrichment culture of strain MpT1, determined by quantitative real-time PCR of bacterial and archaeal 16S rRNA genes. 454 pyrotag sequencing revealed that the bacterial contaminants belonged almost exclusively to families of the order Clostridiales.
Meanwhile, we also obtained a second methanogenic enrichment culture from the hindgut homogenate of a millipede (Anadenobolus sp.), using the same medium and enrichment strategy as for strain MpT1. The culture accumulated even larger amounts of methane (45 kPa in the headspace) than strain MpT1. Again, the PCR products obtained with specific primers for archaeal 16S rRNA genes and mcrA genes could be sequenced without cloning, which indicated that this enrichment culture was also dominated by a single strain of methanogens. Strain MpM2 had the same coccoid morphology as strain MpT1 but was slightly larger (Fig. 4B); both strains did not show the typical cofactor F420 autofluorescence of many methanogens. Phylogenetic analysis showed that the 16S rRNA sequence of strain MpM2 also fell into the intestinal cluster of the novel methanogens within the radiation of sequences from termites and cockroaches (Fig. 1, TC-1 and CC-1). The mcrA gene of strain MpM2 clustered with the corresponding mcrA genes of the TC-1 subcluster (Fig. 2).
DISCUSSION
The results of this study are the final proof that the deep-branching lineage of so far uncultured Euryarchaeota distantly related to the Thermoplasmatales represents the seventh order of methanogens. This is supported by the congruence of the phylogenies of 16S rRNA and mcrA genes, which indicates that the corresponding gene sets obtained from termite and cockroach guts (this study) and from mammalian guts and several other environments (previous studies) stem from the same organisms (Fig. 6). Further evidence for the methanogenic nature of the entire lineage comes from the highly enriched strains of methanogens from the hindguts of termites and millipedes and the isolate Methanomassiliicoccus luminyensis from human feces (12) (see below).
Fig 6.

Tanglegram illustrating the congruence of the phylogenies of _Thermoplasmatales_-related archaea (16S rRNA) and the mcrA genes of unknown origin (for details, see Fig. 1 and 2). Sequence pairs stemming from the same study are connected by dashed lines. Sequences obtained in this study are marked in boldface. The bullets indicate bootstrap support (●, >95%; ○, >70%). The scale bars indicate substitutions per site.
Novel archaea in the guts of termites and cockroaches.
Previous studies of archaeal diversity in the hindguts of C. orthognathus (subfamily Termitinae) and Nasutitermes takasagoensis (subfamily Nasutitermitinae) had revealed the presence of four major lineages of Euryarchaeota in higher termites: Methanosarcinales, Methanomicrobiales, Methanobacteriales (for references, see reference 5), and a deep-branching clade distantly related to Thermoplasmatales (16, 42). Clones from the same lineages were also recovered from C. ugandensis and Ophiotermes sp., Trinervitermes sp., M. michaelseni, and A. trestus (this study), which indicated that representatives of the clade are consistently present in all subfamilies of higher termites. In addition, clones of this lineage were also obtained from the wood-feeding cockroaches S. esakii and P. angustipennis (22; this study), which are distantly related to termites.
Interestingly, the novel archaea from insect guts form two distinct lineages, each comprising clones from higher termites and wood-feeding cockroaches that seem to be specific for their respective hosts. The general absence of the group from lower termites is in agreement with previous studies reporting that the insects are exclusively colonized by members of the genus Methanobrevibacter (45); the single clone of _Thermoplasmatales_-related archaea obtained from Reticulitermes speratus (54) is affiliated with cluster TC-1b (Fig. 1).
Methanogenic nature of novel archaea.
The tree topologies of the 16S rRNA genes of novel archaea (Fig. 1) and the mcrA genes of unknown origin (Fig. 2) strongly resemble each other. A simplified tanglegram of the two trees illustrates that the phylogenetic positions of the major clusters of 16S rRNA and mcrA genes match perfectly (Fig. 6). This is true for all studies that reported both 16S rRNA and mcrA clones from the same environments: termite and cockroach guts (this study), wallaby gut (15), human gut (39, 40), and salt marsh sediment (7). In addition, other opposing clusters in the tree contain clones that originated from the same (e.g., the rumen) or related (i.e., from the guts and the manure of farm animals) habitats. Also the internal topologies of the respective groups are highly coincident (Fig. 1 and 2), which provides strong support for the idea that the sequence pairs from different animals originated from the same archaeal lineages. This is corroborated further by the similar clone frequencies of 16S rRNA and mcrA genes in the corresponding libraries of different termite gut species (Table 1), although the results are probably affected by differences in copy numbers of the 16S rRNA gene in Methanosarcinales and Methanobacteriales (1).
Further proof of the methanogenic nature of the new lineage came from the successful enrichment of strains MpT1 and MpM2, the only archaea present in the highly methanogenic enrichment cultures from termite and millipede guts. The 16S rRNA and mcrA gene sequences of both strains cluster with corresponding clones obtained from the guts of termites and cockroaches (Fig. 1 and 2, TC-1 and CC-1). They are part of the “intestinal cluster” of putative methanogens that also comprises clones from the rumen (RCC) (58) and the human gut (40). More distant relatives are found in anaerobic digestors (VADIN), rice field soil (RC-III), sediments, and other terrestrial environments (TMEG-1 and -2). Since matching mcrA genes were obtained from most of these habitats, it is safe to assume that all these lineages are methanogenic.
The final piece of evidence for the methanogenic nature of the new lineage was provided by the study of Dridi et al. (12), which was published during the revision stage of the present study. They isolated and described a new genus and species of methanogens, M. luminyensis, from human feces and reported that its 16S rRNA gene sequence was most closely related to several clones of uncultured Thermoplasmatales previously obtained from the digestive tracts of various mammals. They claimed that these clones and their isolate represent a new order of methanogens, but their phylogenetic analysis was superficial and comprised only a limited set of taxa. Our detailed phylogenetic analysis of both 16S rRNA and mcrA genes revealed that M. luminyensis is not a member of the vertebrate clones in the intestinal cluster (Fig. 1 and 2), which comprises most of the clones previously obtained from the digestive tracts of mammals. Instead, the isolate falls within the radiation of RC-III, where it clusters with clones from rice field soil (20, 8) and a single clone previously obtained from the human gut (39).
The methanogenic character of euryarchaeota in RC-III had been suggested earlier by Kemnitz et al. (28), who observed that the abundance of RC-III clones in a methanogenic enrichment culture from rice field soil was reduced by the addition of bromoethanesulfonate (BES), a specific inhibitor of methanogenesis. Considering the methanogenic character of M. luminyensis and the fact that mcrA sequences corresponding to RC-III have been obtained from rice paddies and other soils (Fig. 6), it is likely that all members of RC-III are methanogens.
The tanglegram (Fig. 6) shows that the most basal cluster in the new lineage of mcrA genes belongs to marine benthic group D, based on the matching positions of two sets of 16S rRNA and mcrA genes (ARC-7 and MCR-2) obtained from the same salt marsh samples (7). Although there are no mcrA genes matching the deeper-branching lineages, this may be due to the general lack of _mcrA_-based studies of methanogenic diversity, particularly in marine habitats. Therefore, it is not possible to predict whether the deeper-branching lineages are also methanogenic.
Interestingly, we observed a consistent and moderately supported sister group position of the novel mcrA genes and those of the ANME-1 group, an uncultivated lineage of methane-oxidizing archaea that may involve a methyl-CoM reductase in anaerobic methane oxidation (29). However, in view of the methanogenic properties of M. luminyensis (12) and our enrichment cultures and the cumulative evidence for the absence of methane oxidation in termite guts (47), a methanotrophic character of this novel lineage of archaea can be excluded.
Physiological properties of the enrichment cultures.
In the highly enriched cultures of strains MpT1 and MpM2, methanogenesis was strongly stimulated by the simultaneous supply of both H2 and methanol. The small amount of methane formation in the enrichment culture containing only methanol is most likely due to hydrogen formation by the clostridial members of the enrichment culture during fermentation of substrates stemming from yeast extract. Although a final statement on the substrate requirements will have to wait until these strains have been brought into pure culture, it seems that the metabolism of strains MpT1 and MpM2 (intestinal cluster) resembles that of M. luminyensis (RC-III) and obligately H2-requiring methylotrophic methanogens from other lineages, like Methanosphaera stadtmanae (Methanobacteriales) and Methanomicrococcus blatticola (Methanosarcinales). Interestingly, such organisms have so far been isolated exclusively from the intestinal tracts of humans (12, 41) and cockroaches (55). It is likely that this mode of methanogenesis is an adaptation to the intestinal habitat.
In a study of the archaeal diversity in the hindgut of the termite N. takasagoensis, the relative abundance of uncultured Thermoplasmatales (Fig. 1, clusters TC-1a and TC-2) increased when the animals were fed with xylan (42), a substrate that contains substantial amounts of _O_-methylated glucuronic acid residues (49). It is possible that the apparent enrichment of these methanogens was an indirect effect caused by methanol formation during the fermentative breakdown of xylan.
The seventh order of methanogens.
Although it has been repeatedly proposed that environmental clones distantly affiliated with the Thermoplasmatales represent a separate order of methanogens (15, 39), none of these studies provided enough evidence to substantiate this claim. Analysis of the entire set of sequences available to date clearly documents the diversity of the new lineage, including numerous habitat-specific clades and its sister group relationship to the Thermoplasmatales, and provides robust evidence for the presence of mcrA genes in all members. The methanogenic nature of the lineage is further corroborated by the isolation of M. luminyensis (12) and the enrichment cultures of strains MpT1 and MpM2 (this study). Based on this evidence, we propose the provisional name “_Methanoplasmatales_” for the entire deep-branching lineage of euryarchaeota outlined in Fig. 6. Although a first representative of the lineage has been isolated and described (12), we suggest postponing a formal description of any higher taxa until further representatives have been obtained in culture, their cell envelopes have been characterized, and the presence of mcrA genes in the basal lineages (particularly the marine groups) has been assessed.
Considering the apparently obligate hydrogen dependence of methanol reduction both in M. luminyensis and in the enrichment cultures, it may be promising to also use such combinations of methanogenic substrates for enrichments from other habitats. There are several other deep-branching lineages of euryarchaeota that may also be methanogenic, and even more diversity may be present because of a bias of commonly used PCR primers against hitherto undetected lineages (60). This is underlined by two studies of archaeal diversity in termite guts that had failed to detect clones affiliated with “Methanoplasmatales.” In one case (46), this was most likely due to a mismatch in the reverse primer to the consensus sequence of “Methanoplasmatales,” whereas in the other case (11), the sequence of the forward primer differed slightly from that of the forward primer successfully used by Hara et al. (22).
It is striking that the majority of the mcrA genes of the “_Methanoplasmatales_” have so far been retrieved only from intestinal samples. The fact that there are only a few clones from other environments may simply be due to the lack of such studies, particularly in marine environments. More cultivation efforts are required to expand our knowledge about this novel group of methanogens, not least to investigate their metabolic relationship to Thermoplasmatales, a clade of Euryarchaeota that may have experienced a secondary loss of the capacity for methanogenesis (3).
ACKNOWLEDGMENTS
We are grateful to Michael Friedrich for providing a core database of mcrA genes, Kiyoto Maekawa for wood-feeding cockroaches, and Rudolf Scheffrahn for termite identification. We thank David Ngugi and Eugen Bauer for providing DNA samples and Katja Meuser and Aram Mikaelyan for help with the clone libraries and the 454 analysis.
Footnotes
Published ahead of print 21 September 2012
REFERENCES
- 1.Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF. 2004. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186:2629–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson I, et al. 2009. Genomic characterization of Methanomicrobiales reveals three classes of methanogens. PLoS One 4:e5797 doi:10.1371/journal.pone.0005797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bapteste E, Brochier C, Boucher Y. 2005. Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea 1:353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boga HI, Brune A. 2003. Hydrogen-dependent oxygen reduction by homoacetogenic bacteria isolated from termite guts. Appl. Environ. Microbiol. 69:779–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brune A. 2010. Methanogens in the digestive tract of termites, p 81–100_In_Hackstein JHP. (ed), (Endo)symbiotic methanogenic archaea. Springer, Heidelberg, Germany [Google Scholar]
- 6.Burggraf S, Huber H, Stetter KO. 1997. Reclassification of the crenarchaeal orders and families in accordance with 16S rRNA sequence data. Int. J. Syst. Bacteriol. 47:657–660 [DOI] [PubMed] [Google Scholar]
- 7.Castro H, Ogram A, Reddy KR. 2004. Phylogenetic characterization of methanogenic assemblages in eutrophic and oligotrophic areas of the Florida Everglades. Appl. Environ. Microbiol. 70:6559–6568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chin KJ, Lukow T, Conrad R. 1999. Effect of temperature on structure and function of the methanogenic archaeal community in an anoxic rice field soil. Appl. Environ. Microbiol. 65:2341–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeLong EF. 1992. Archaea in coastal marine environments. Proc. Natl. Acad. Sci. U. S. A. 89:5685–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denman SE, Tomkins NW, McSweeney CS. 2007. Quantitation and diversity analysis of ruminal methanogenic populations in response to the antimethanogenic compound bromochloromethane. FEMS Microbiol. Ecol. 62:313–322 [DOI] [PubMed] [Google Scholar]
- 11.Donovan SE, Purdy KJ, Kane MD, Eggleton P. 2004. Comparison of Euryarchaea strains in the guts and food-soil of the soil-feeding termite Cubitermes fungifaber across different soil types. Appl. Environ. Microbiol. 70:3884–3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dridi B, Fardeau M-L, Ollivier B, Raoult D, Drancourt M. 2012. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 62:1902–1907 [DOI] [PubMed] [Google Scholar]
- 13.Earl J, Hall G, Pickup RW, Ritchie DA, Edwards C. 2003. Analysis of methanogen diversity in a hypereutrophic lake using PCR-RFLP analysis of mcr sequences. Microb. Ecol. 46:270–278 [DOI] [PubMed] [Google Scholar]
- 14.Egert M, Wagner B, Lemke T, Brune A, Friedrich MW. 2003. Microbial community structure in midgut and hindgut of the humus-feeding larva of Pachnoda ephippiata (Coleoptera: Scarabaeidae). Appl. Environ. Microbiol. 69:6659–6668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans PN, et al. 2009. Community composition and density of methanogens in the foregut of the Tammar wallaby (Macropus eugenii). Appl. Environ. Microbiol. 75:2598–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedrich MW, Schmitt-Wagner D, Lueders T, Brune A. 2001. Axial differences in community structure of Crenarchaeota and Euryarchaeota in the highly compartmentalized gut of the soil-feeding termite Cubitermes orthognathus. Appl. Environ. Microbiol. 67:4880–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuhrman JA, McCallum NN, Davis AA. 1992. Novel archaebacterial group from marine plankton. Nature 356:148–149 [DOI] [PubMed] [Google Scholar]
- 18.Godon J-J, Zumstein E, Dabert P, Habouzit F, Moletta R. 1997. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 63:2802–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gribaldo S, Brochier-Armanet C. 2006. The origin and evolution of Archaea: a state of the art. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361:1007–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grosskopf R, Stubner S, Liesack W. 1998. Novel euryarchaeotal lineages detected on rice roots and in the anoxic bulk soil of flooded rice microcosms. Appl. Environ. Microbiol. 64:4983–4989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the perfomance of PhyML 3.0. Syst. Biol. 59:307–321 [DOI] [PubMed] [Google Scholar]
- 22.Hara K, Shinzato N, Seo M, Oshima T, Yamagishi A. 2002. Phylogenetic analysis of symbiotic archaea living in the gut of xylophagous cockroaches. Microb. Environ. 17:185–190 [Google Scholar]
- 23.Hatamoto M, Imachi H, Ohashi A, Hara H. 2007. Identification and cultivation of anaerobic, syntrophic long-chain fatty acid-degrading microbes from mesophilic and thermophilic methanogenic sludges. Appl. Environ. Microbiol. 73:1332–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang L-N, et al. 2002. Diversity and structure of the archaeal community in the leachate of a fullscale recirculating landfill as examined by direct 16S rRNA gene sequence retrieval. FEMS Microbiol. Lett. 214:235–240 [DOI] [PubMed] [Google Scholar]
- 25.Imachi H, et al. 2006. Non-sulfate-reducing, syntrophic bacteria affiliated with Desulfotomaculum cluster I are widely distributed in methanogenic environments. Appl. Environ. Microbiol. 72:2080–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janssen PH, Kirs M. 2008. Structure of the archaeal community of the rumen. Appl. Environ. Microbiol. 74:3619–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jurgens G, et al. 2000. Identification of novel Archaea in bacterioplankton of a boreal forest lake by phylogenetic analysis and fluorescent in situ hybridization. FEMS Microbiol. Ecol. 34:45–56 [DOI] [PubMed] [Google Scholar]
- 28.Kemnitz D, Kolb S, Conrad R. 2005. Phenotypic characterization of Rice Cluster III archaea without prior isolation by applying quantitative polymerase chain reaction to an enrichment culture. Environ. Microbiol. 7:553–565 [DOI] [PubMed] [Google Scholar]
- 29.Knittel K, Boetius A. 2009. Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63:311–334 [DOI] [PubMed] [Google Scholar]
- 30.Köhler T, Dietrich C, Scheffrahn RH, Brune A. 2012. High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Appl. Environ. Microbiol. 78:4691–4701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175_In_Stackebrandt E, Goodfellow M. (ed), Nucleic acids techniques in bacterial systematics. John Wiley & Sons, Chichester, United Kingdom [Google Scholar]
- 32.Li CL, et al. 2009. Prevalence and molecular diversity of Archaea in subgingival pockets of periodontitis patients. Oral Microbiol. Immunol. 24:343–346 [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Whitman WB. 2008. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N. Y. Acad. Sci. 1125:171–189 [DOI] [PubMed] [Google Scholar]
- 34.Ludwig W, et al. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lueders T, Friedrich M. 2000. Archaeal population dynamics during sequential reduction processes in rice field soil. Appl. Environ. Microbiol. 66:2732–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lueders T, Chin KJ, Conrad R, Friedrich M. 2001. Molecular analyses of methyl-coenzyme M reductase α-subunit (mcrA) genes in rice field soil and enrichment cultures reveal the methanogenic phenotype of a novel archaeal lineage. Environ. Microbiol. 3:194–204 [DOI] [PubMed] [Google Scholar]
- 37.Luton PE, Wayne JM, Sharp RJ, Riley PW. 2002. The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148:3521–3530 [DOI] [PubMed] [Google Scholar]
- 38.McWilliam IG, Dewar RA. 1958. Flame ionization detector for gas chromatography. Nature 181:760 [Google Scholar]
- 39.Mihajlovski A, Alric M, Brugère J-F. 2008. A putative new order of methanogenic Archaea inhabiting the human gut, as revealed by molecular analyses of the mcrA gene. Res. Microbiol. 159:516–521 [DOI] [PubMed] [Google Scholar]
- 40.Mihajlovski A, Dore J, Levenez F, Alric M, Brugère JF. 2010. Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ. Microbiol. Rep. 2:272–280 [DOI] [PubMed] [Google Scholar]
- 41.Miller TL, Meyer JW. 1985. Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch. Microbiol. 141:116–122 [DOI] [PubMed] [Google Scholar]
- 42.Miyata R, et al. 2007. Phylogenetic relationship of symbiotic archaea in the gut of the higher termite Nasutitermes takasagoensis fed with various carbon sources. Microb. Environ. 22:157–164 [Google Scholar]
- 43.Mochimaru H, et al. 2007. Methanogen diversity in deep subsurface gas-associated water at the Minami-Kanto gas field in Japan. Geomicrobiol. J. 24:93–100 [Google Scholar]
- 44.Nüsslein B, Chin KJ, Eckert W, Conrad R. 2001. Evidence for anaerobic syntrophic acetate oxidation during methane production in the profundal sediment of subtropical Lake Kinneret (Israel). Environ. Microbiol. 3:460–470 [DOI] [PubMed] [Google Scholar]
- 45.Ohkuma M, Noda S, Horikoshi K, Kudo T. 1995. Phylogeny of symbiotic methanogens in the gut of the termite Reticulitermes speratus. FEMS Microbiol. Lett. 134:45–50 [DOI] [PubMed] [Google Scholar]
- 46.Ohkuma M, Noda S, Kudo T. 1999. Phylogenetic relationships of symbiotic methanogens in diverse termites. FEMS Microbiol. Lett. 171:147–153 [DOI] [PubMed] [Google Scholar]
- 47.Pester M, Tholen A, Friedrich MW, Brune A. 2007. Methane oxidation in termite hindguts: absence of evidence and evidence of absence. Appl. Environ. Microbiol. 73:2024–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pruesse E, et al. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosell K-G, Svennson S. 1974. Studies of the distribution of the 4-_O_-methyl-d-glucuronic acid residues in birch xylan. Carbohydr. Res. 42:297–304 [Google Scholar]
- 50.Sakai S, et al. 2008. Methanocella paludicola gen. nov., sp. nov., a methane-producing archaeon, the first isolate of the lineage ‘Rice Cluster I’, and proposal of the new archaeal order Methanocellales ord. nov. Int. J. Syst. Evol. Microbiol. 58:929–936 [DOI] [PubMed] [Google Scholar]
- 51.Scanlan PD, Shanahan F, Marchesi JR. 2008. Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC Microbiol. 8:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schauer C, Thompson CL, Brune A. 2012. The bacterial community in the gut of the cockroach Shelfordella lateralis reflects the close evolutionary relatedness of cockroaches and termites. Appl. Environ. Microbiol. 78:2758–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schleper C, Jurgens G, Jonuscheit M. 2005. Genomic studies of uncultivated archaea. Nat. Rev. Microbiol. 3:479–488 [DOI] [PubMed] [Google Scholar]
- 54.Shinzato N, Matsumoto T, Yamaoka I, Oshima T, Yamagishi A. 1999. Phylogenetic diversity of symbiotic methanogens living in the hindgut of the lower termite Reticulitermes speratus analyzed by PCR and in situ hybridization. Appl. Environ. Microbiol. 65:837–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sprenger WW, van Belzen MC, Rosenberg J, Hackstein JHP, Keltjens JT. 2000. Methanomicrococcus blatticola gen. nov., sp. nov., a methanol- and methylamine-reducing methanogen from the hindgut of the cockroach Periplaneta americana. Int. J. Syst. Evol. Microbiol. 50:1989–1999 [DOI] [PubMed] [Google Scholar]
- 56.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690 [DOI] [PubMed] [Google Scholar]
- 57.Stubner S. 2002. Enumeration of 16S rDNA of Desulfotomaculum lineage 1 in rice field soil by real-time PCR with SybrGreen detection. J. Microbiol. Methods 50:155–164 [DOI] [PubMed] [Google Scholar]
- 58.Tajima K, Nagamine T, Matsui H, Nakamura M, Aminov RI. 2001. Phylogenetic analysis of archaeal 16S rRNA libraries from the rumen suggests the existence of a novel group of archaea not associated with known methanogens. FEMS Microbiol. Lett. 200:67. [DOI] [PubMed] [Google Scholar]
- 59.Takai K, Horikoshi K. 1999. Genetic diversity of archaea in deep-sea hydrothermal vent environments. Genetics 152:1285–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teske A, Sorensen K. 2008. Uncultured archaea in deep marine subsurface sediments: have we caught them all? ISME J. 2:3–18 [DOI] [PubMed] [Google Scholar]
- 61.Ufnar JA, Ufnar DF, Wang SY, Ellender RD. 2007. Development of a swine-specific fecal pollution marker based on host differences in methanogen mcrA genes. Appl. Environ. Microbiol. 73:5209–5217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wright A-DG, Auckland CH, Lynn DH. 2007. Molecular diversity of methanogens in feedlot cattle from Ontario and Prince Edward Island, Canada. Appl. Environ. Microbiol. 73:4206–4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wright A-DG, et al. 2004. Molecular diversity of rumen methanogens from sheep in Western Australia. Appl. Environ. Microbiol. 70:1263–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]