Endothelial 12(S)-HETE vasorelaxation is mediated by thromboxane receptor inhibition in mouse mesenteric arteries (original) (raw)
Abstract
Arachidonic acid (AA) metabolites mediate endothelium-dependent relaxation in many vascular beds. Previously, we identified the major AA 12/15-lipoxygenase (12/15-LO) metabolite of mouse arteries as 12-hydroxyeicosatetraenoic acid (12-HETE). The goal was to determine the stereospecific configuration of mouse vascular 12-HETE and characterize the role of 12-HETE stereoisomers in the regulation of vascular tone. Using normal, reverse phase, and chiral HPLC, the stereospecific configuration was identified as 12(S)-HETE. 12(S)-HETE relaxed U46619-, carbocyclic thromboxane A2-, PGF2α-, and 8-iso PGF2α-preconstricted mesenteric arteries, but not phenylephrine-preconstricted arteries. 12(R)-HETE was more potent than 12(S)-HETE in relaxing U46619-preconstricted mouse arteries (maximum relaxations = 91.4 ± 2.7% and 71.8 ± 5.9%, respectively). Neither 12-HETE isomer caused constriction. Pretreatment with 12(S)- or 12(R)-HETE (1 μM) inhibited constrictions to U46619 but not phenylephrine. To investigate the role of thromboxane A2 (TP) receptors in 12-HETE vascular actions, [3H]SQ29548 radioligand binding studies were performed in mouse platelets. U46619, 12(R)-HETE, and 12(S)-HETE displaced [3H]SQ29548 binding with IC50s of 0.07, 0.32, and 1.73 μM, respectively. Both 12(S)- and 12(R)-HETE inhibited intracellular calcium increases induced by U46619 (10 nM) in HEK293 cells overexpressing TPα receptor (65.5% and 45.1%, respectively) and coexpressing prostacyclin (IP) and TPα receptors (58.0% and 27.1%, respectively). The LO inhibitor NDGA (10 μM) reduced AA relaxations in arteries preconstricted with U46619 but not phenylephrine. These results indicate that exogenous and endogenous 12(S)-HETE relax mouse mesenteric arteries that are preconstricted with thromboxane agonists. These 12(S)-HETE relaxations are mediated by TP receptor competitive inhibition and inhibition of TP agonist-induced increases in intracellular calcium.
Keywords: 12-HETE, arachidonic acid, lipoxygenase, thromboxane receptors, vasorelaxation
the dynamic regulation of vascular relaxation and constriction is extremely important for the regulation of tissue blood flow. Disruption of this regulation occurs in many cardiovascular diseases. The vascular endothelium plays a central role in the regulation of vascular tone by producing a variety of dilator and constrictor substances. Three major dilator mediators are nitric oxide (NO), prostaglandin I2 (PGI2), and a group of compounds called endothelium-derived hyperpolarizing factors (EDHFs) (9, 13, 19). PGI2, an arachidonic acid (AA) cyclooxygenase (COX) metabolite, was the first endothelial relaxing factor discovered (19). In addition to PGI2, AA is also metabolized by cytochrome P450 (CYP450) to epoxyeicosatrienoic acids (EETs) that are EDHFs in numerous arteries. They cause relaxation by activating smooth muscle potassium channels to cause hyperpolarization [reviewed by Campbell and Falck (2)]. EETs as well as PGI2 mediate vasorelaxation in bovine coronary arteries (28). In addition to COX and CYP450, AA is metabolized by lipoxygenase (LO) to vasodilator metabolites in rabbit aorta (3) and rat small mesenteric arteries (18). Recently, Gauthier et al. reported that 12-hydroxyeicosatetraenoic acid (12-HETE) was the major AA metabolite in mouse abdominal aorta, carotid, femoral, and mesenteric arteries (10). In addition to 12-HETE, 15-HETE, hydroxyepoxyeicosatrienoic acids (HEETAs) and 11,12,15-trihydroxyeicosatrienoic acids (11,12,15-THETAs) were also found. Although 12-HETE is the major metabolite, its role in the regulation of vascular tone in mouse arteries has not been investigated.
Several receptors have been identified that mediate the actions of 12-HETE including the thromboxane A2 (TP) receptor (15), the low-affinity leukotriene B4 (BLT2) receptor (32), and the G protein coupled receptor 31 (GPR31) (11). In human platelets, both 12(S)- and 12(R)-HETE inhibited binding of the thromboxane mimetic, [125I]-I-BOP (15). In Chinese hamster ovary (CHO) cells stably expressing the BLT2 receptor, 12(S)-HETE competed for [3H]leukotriene B4 binding (32), and in CHO cells transiently expressing GPR31, [3H]-12(S)-HETE showed specific, saturable, high-affinity binding (11). Herein, we eliminate the possibility of BLT2 receptors and GPR31 in mediating the vascular actions of 12-HETE and show that inhibition of the TP receptor mediates 12-HETE relaxations in mouse mesenteric arteries.
MATERIALS AND METHODS
Animals
Eight- to fourteen-week-old male C57BL6 mice (24–28 g) were obtained from Jackson Laboratory (Bar Harbor, ME). Animal protocols were approved by the Animal Care Committee of the Medical College of Wisconsin, and procedures were carried out in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals (2011).
[14C]Arachidonic Acid Metabolism
Mouse aorta were dissected, placed in cold HEPES buffer (in mM: 10 HEPES, 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 6 glucose, pH 7.4) and cleaned of adhering connective tissues. The aorta were pretreated with indomethacin (10 μM) for 10 min at 37°C. [14C]AA (0.05 μCi, 50 μM) and unlabeled AA (0.1 μM) were added and further incubated for 5 min. Thereafter, calcium ionophore A23187 (20 μM) was added and the arteries were incubated for additional 15 min. The reaction was stopped by the addition of ice-cold ethanol to a final concentration of 15%. The arteries were removed and the incubation buffer acidified (pH < 3.5) with glacial acetic acid and extracted on Bond Elut C18 extraction columns as previously described (3). The extracts were dried under a stream of nitrogen gas and stored at −30°C until analysis by HPLC.
HPLC Separation of Arachidonic Acid Metabolites
Reverse phase (RP)-HPLC.
Extracts were resolved on a Nucleosil-C18 column (5 μm, 4.6 × 250 mm) using solvent system I. Solvent A was deionized water and solvent B was acetonitrile containing 0.1% glacial acetic acid. The program was a linear gradient from 50% solvent B in solvent A to 100% solvent B, for 40 min. The flow rate was 1 ml/min. Column elute was collected (5 fractions/min), and aliquots counted for radioactivity by a liquid scintillation counter. UV absorbance was detected at 205 nm. Fractions corresponding to the HETEs were collected and further extracted with 50:50 cyclohexane and ethyl acetate. The extracts were dried under a stream of nitrogen and stored at −30°C until further analysis.
Normal phase (NP)-HPLC.
The determination of HETE isomers and their stereoisomer conformations were performed as previously described (26) with some modifications. HETEs collected from RP-HPLC were resolved on NP-HPLC (Nucleosil silica column, 5 μm, 4.6 × 250 mm) using solvent system II. Solvent A was hexane containing 0.1% glacial acetic acid. Solvent B was hexane with 0.1% glacial acetic acid and 2% isopropanol. The program consisted of a 5-min isocratic phase with 25% B in A followed by a 40-min linear gradient to 75% B in A. The flow rate was 3 ml/min. UV absorbance was detected at 235 nm. Column elute was collected (5 fractions/min), and aliquots counted for radioactivity. Fractions corresponding to 12-HETE were pooled, dried under a stream of nitrogen and stored at −30°C. 12-HETE stereoisomer composition was established using a Chiracel OC chiral phase column (Baker). Solvent system III was hexane containing 0.1% glacial acetic acid and 2% isopropanol. The program was a 70-min isocratic separation with 50% B in A at a flow rate of 0.5 ml/min. UV absorbance was monitored at 235 nm. Column elute was collected (5 fractions/min) and radioactivity counted.
Vascular Activity
Isometric tension recording was performed as previously described (34). Mouse mesenteric arteries 150 to 300 μm in diameter were cut into small rings (1.5 to 1.8 mm in length), and mounted in a four-chamber wire myograph (model 610M, Danish Myo Technology A/S). The arteries were maintained in physiological saline solution (PSS, in mM: 119 NaCl, 4.7 KCl, 2.5 CaCl2, 1.17 MgSO4, 24 NaHCO3, 1.18 KH2PO4, 0.026 EDTA, and 5.5 glucose), at 37°C, supplied with 95% O2/5% CO2. Thereafter, the arteries were stretched to a tension of 0.25–0.80 mN, where optimum isometric length-tension was achieved. KCl (60 mM) and the thromboxane mimetic, U46619 (100 nM), were used to stimulate the arteries 3–4 times until they reached maximum active tension. Appropriate inhibitors as well as vehicle controls were added 10 min before preconstriction and nordihydroguaiaretic acid (NDGA, 10 μM) were added 30 min before preconstriction. Arteries were preconstricted to approximately 50–70% of maximum active tension. The arteries were preconstricted with phenylephrine (2–20 μM) or a TP receptor agonist (20–200 nM U46619, 5 μM–1 mM 8-iso PGF2α, 10–40 μM PGF2α, 20–300 nM CTA2 or 0.3–10 nM I-BOP) in the presence of indomethacin (10 μM) and l-NA (30 μM). After a stable constriction, increasing concentrations of test compounds were added and tension was recorded. Results are expressed as %relaxation with basal tension representing 100% relaxation. In some instances, test compounds were added to basal tension and constriction responses recorded. Constriction responses are expressed as %constriction with maximum active tension being 100%.
Western Immunoblotting
The preparation of protein lysates was carried out as previously described (10). Briefly, cleaned mouse mesenteric arteries were homogenized and lysed in lysis buffer [in mM: 50 HEPES, 150 NaCl, 1.5 MgCl2, and 1 EGTA and 10% glycerol, 1% Triton X-100, and protease inhibitor cocktail (Roche Molecular Biochemicals, Germany)]. The homogenates were centrifuged and the supernatant collected. Protein concentrations were determined by the Bio-Rad protein assay. Each lane was loaded with 30 μg of protein and was subjected to SDS-PAGE using a 10% resolving gel and 4% stacking gel. Proteins were transferred to nitrocellulose membranes and then blocked for nonspecific binding with 5% nonfat dry milk (Bio-Rad) in TBS-T buffer (20 mM Tris base, 50 mM NaCl, 0.1% Tween 20, pH 7.8) at room temperature for 1 h. The membranes were incubated at 4°C overnight with appropriate primary antibodies (rabbit GPR31 polyclonal antibody; 1:750 dilution; Abcam, and rabbit BLT2 receptor polyclonal antibody; 1:200 dilution; Cayman Chemical Co) in 0.5% milk TBS-T buffer. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:3,000 dilution; Invitrogen) and HRP-conjugated donkey anti-rabbit IgG (1:10,000 dilution; GE Healthcare) in 0.5% milk TBS-T buffer were used as secondary antibodies for GPR31 and BLT2 receptor, respectively. The secondary antibodies were incubated at room temperature for 90 min. After each step of antibody incubation, the blot was washed with TBS-T buffer. Immunoreactive bands were detected using the Pierce ECL Western blotting substrate (Thermo Scientific) and Kodak BioMax ML film. β-Actin was used as a loading control (1:10,000 dilution; mouse monoclonal anti-β-actin primary antibody; Santa Cruz Biotechnology, and 1:10,000 dilution; HRP goat anti-mouse secondary antibody; Invitrogen).
Mouse Platelet Preparation
Mouse blood was collected from the right ventricle using a 21-gauge needle filled with heparin solution (180 U/ml) in HEPES-Tyrode buffer (in mM: 12 NaHCO3, 138 NaCl, 5.5 glucose, 2.9 KCl, 10 HEPES, pH 7.4). Whole blood was centrifuged at 86 g for 8 min and platelet-rich plasma (PRP) was collected. Platelet-poor plasma (PPP) was obtained by washing platelet-poor blood with 200 μl of HEPES-Tyrode buffer and 100 μl of 100 mM EDTA and centrifugation at 86 g for 8 min. An additional wash was done with HEPES-Tyrode buffer. PRP and PPP were pooled and centrifuged at 718 g for 6 min. The supernatant was discarded and the platelet pellet was resuspended in the buffer and then centrifuged at 500 g for 30 s. Supernatant platelets were used.
[3H]SQ29548 Competitive Binding Assay
The reaction buffer (final volume of 200 μl) was prepared (in mM: 25 HEPES, 10 CaCl2, 20 MgCl2, 10 μM indomethacin). Mouse platelets (1.5 × 108 platelets/assay tube) were incubated with 2 nM [3H]SQ29548 (48 Ci/mmol, New England Nuclear), a TP antagonist, and increasing concentration of 12(S)-HETE, 12(R)-HETE, or U46619 at 30°C for 30 min. Nonspecific binding was determined in the presence of 10 μM unlabeled SQ29548. The reaction was stopped by addition of 4 ml ice-cold wash buffer (in mM: 50 Tris-HCl, 100 NaCl, pH 7.4). The reaction solution was filtered through GF/B Whatman filters on a Brandel apparatus. The filters were then washed 3 times with 4 ml wash buffer and filter radioactivity was counted using a liquid scintillation counter.
Cell Culture
HEK293 cells overexpressing the human TPα receptor (TPα-HEK) were produced as previously described (31). Since vascular smooth muscle cells express both the TP and IP receptors, studies were also performed in HEK293 cells coexpressing human IP and TPα receptor (IP/TPα-HEK). The cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 25 mM HEPES, 4.5 g/l d-glucose, 4 mM l-glutamine in the presence of 0.5 mg/ml G418 for TPα-HEK and 0.5 mg/ml G418 plus 75 μg/ml hygromycin B for IP/TPα-HEK. The cells were maintained in a humidified atmosphere of 5% CO2 and 95% O2 at 37°C.
Intracellular Calcium Measurement Using a Fluorometric Imaging Plate Reader (FLIPR)
The assay was performed in a black CellBIND surface 96-well plate with clear bottoms (Corning). TPα-HEK or IP/TPα-HEK cells were seeded (5 × 104 cells per well) in phenol red-free growth media, allowed to adhere overnight and kept until a confluent monolayer was formed. Thereafter, the media was carefully removed and replaced with 100 μl of assay buffer (Fluo-4 NW calcium assay kit, Molecular Probes) containing Hanks' balanced salt solution (HBSS, 1X), HEPES (20 mM), probenecid (2.5 mM), and Fluo-4 dye mix (1X). Probenecid was used to prevent an efflux of the dye from cells. Cells were incubated with dye for 15 min at 37°C followed by an additional 10 min at room temperature. Test compounds, i.e., U46619, 12(S)-HETE, and 12(R)-HETE, were prepared in assay buffer without probenecid and dye in a V-shape bottom microplate (Greiner Bio-one, Germany). The calcium assay was performed using a fluorometric imaging plate reader (FLIPR) instrument (Molecular Devices) in two sequences. First, vehicle control, 12(S)-HETE or 12(R)-HETE was added and the fluorescence signal measured. After 4 min, U46619 was added and the signal monitored.
Intracellular calcium was calculated by subtracting the average basal fluorescence from the maximum fluorescence and normalizing to total protein in each well. To perform protein concentration measurements, the dye solution was carefully removed after the FLIPR assay and cells were lysed with 30 μl of NaOH (2 N) for 2 h at 60°C. Next, 30 μl of HCl (2 N) was added to neutralize the NaOH. Samples were then used for protein concentration measurement using BCA assay (Pierce BCA protein assay kit, Thermo scientific).
Drugs and Chemicals
12(S)-HETE, 12(R)-HETE, U46619, I-BOP ([1_S_-[1α,2α(Z),3β(1_E_,3_S_),4α]]-7-[3-[3-hydroxy-4-(4-iodophenoxy)-1-butenyl]-7-oxabicyclo[2.2.1]hept-2-yl]-5-heptenoic acid), carbocyclic TxA2 (CTA2), PGF2α, 8-iso PGF2α, CAY10583, and leukotriene B4 were purchased from Cayman Chemical; DMEM, penicillin, streptomycin, HEPES, l-glutamine, hygromycin B, and G418 (Geneticin) were purchased from Invitrogen. Apamin was purchased from Tocris Bioscience. Hyclone fetal bovine serum was purchased from Thermo Scientific. Indomethacin, nitro-l-arginine (l-NA), AA, phenylephrine, nordihydroguaiaretic acid (NDGA), calcium ionophore A23187, iberiotoxin and other chemicals were purchased from Sigma. All 12(S)- and 12(R)-HETE standards were tested for the chiral composition by chiral HPLC (solvent system III) prior to use.
Data Analysis
Data are presented as means ± SE. Significant differences between mean values were evaluated by Student _t_-test or ANOVA followed by the Student-Newman-Keuls multiple comparison test. A value of P < 0.05 was considered statistically significant. The binding data were analyzed using the nonlinear regression analysis of PRISM (GraphPad Software). EC50 is the concentration of test compound that causes half maximum effect. IC50 represents the concentration that causes 50% inhibition.
RESULTS
In RP-HPLC analysis of AA metabolites from mouse aorta using solvent system I, radioactive metabolites comigrated with the THETAs, HEETAs, 15- and 12-HETE standards (Fig. 1, A and B), consistent with our previous study (10). The [14C]-labeled HETE fractions from RP-HPLC (18–22 min) were collected and rechromatographed on NP-HPLC using solvent system II. An aliquot of each fraction was measured for radioactivity (Fig. 1_C_). The retention time of major radioactive peak comigrated with the 12-HETE standard (Fig. 1_D_), confirming that 12-HETE is the major metabolite in mouse arteries. The peak comigrating with 15-HETE standard was only vaguely detected in the biological sample. The column elute that comigrated with 12-HETE (5.4–6.6 min) was collected and used for stereoisomer determination by chiral NP-HPLC solvent system III. The sample was spiked with 12(R)-HETE and 12(S)-HETE (1:2 ratio) and the UV absorbance was monitored at 235 nm. Retention time of [14C]-labeled 12-HETE of 48.6 min (Fig. 1_E_) corresponded with the retention time of the 12(S)-HETE standard (Fig. 1_F_). A metabolite peak co-migrating with 12(R)-HETE was not observed.
Fig. 1.
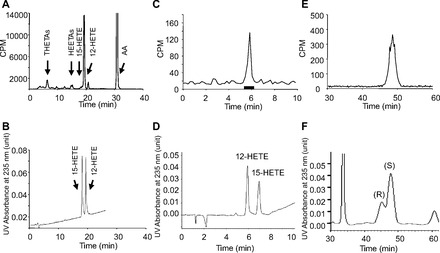
HPLC analysis of arachidonic acid (AA) lipoxygenase metabolites showing the radioactivity counts/min (CPM) (A, C, E) and UV absorbance at 235 nm (B, D, F). A: [14C]AA metabolites of mouse aorta in the presence of a cyclooxygenase inhibitor, indomethacin (10 μM). Aortic rings were incubated with [14C]AA, and metabolites were extracted and resolved on RP-HPLC using solvent system I. The hydroxyeicosatetraenoic acid (HETE) fraction was collected for further analysis. Migration times of known standards are indicated. The UV absorbance at 235 nm of 12- and 15-HETE standards is shown in B. C: normal phase (NP)-HPLC of the HETE fraction collected from reverse phase (RP)-HPLC (A) using solvent system II. The [14C]-labeled metabolite comigrated with 12-HETE standard (UV absorbance was monitored at 235 nm) (D). E and F: the biological 12-HETE peak from C was collected, unlabeled 12(S)- and 12(R)-HETE (2:1 ratio) added, and resolved on a chiral HPLC column using solvent system III. UV absorbance was monitored at 235 nm. Retention time of the radioactive biological 12-HETE (E) comigrated with the 12(S)-HETE standard (F).
Given that 12-HETE is the dominant LO product from AA in mouse vascular tissues, we next determined the vascular activity of 12-HETE stereoisomers in mouse mesenteric arteries. 12(S)-HETE relaxed U46619-preconstricted mouse mesenteric arteries in a concentration-dependent manner (Fig. 2, A and C) with maximum relaxations of 71.8 ± 5.9%. 12(R)-HETE also relaxed mouse mesenteric arteries preconstricted with U46619 (maximum relaxation = 91.4 ± 2.7%) (Fig. 2, B and C). The EC50, a concentration that causes 50% maximal effect, was 0.63 and 0.05 μM for 12(S)-HETE and 12(R)-HETE, respectively. Interestingly, 12(R)-HETE was more potent than the 12(S)-HETE.
Fig. 2.
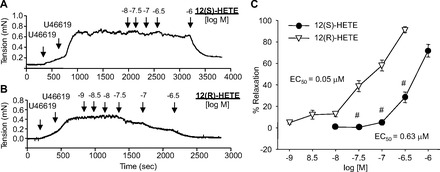
Effect of 12(S)-HETE and 12(R)-HETE on relaxations of mouse mesenteric arteries preconstricted with U46619. A and B: representative isometric tension recordings of mouse mesenteric rings. The arteries were preconstricted with U46619 until they reached 50–70% maximum tension. Upon a stable tension, increasing concentrations of 12(S)- or 12(R)-HETE were added. C: relaxation responses to 12(S)-(●) and 12(R)-HETE (▽) presented as %relaxation (as the basal tension representing 100%). EC50 refers to the concentration of test compound that causes 50% of maximum response. Each value represents the mean ± SE; n = 8–12. #P < 0.05 compared with 12(R)-HETE.
Our next goal was to investigate the mechanism of 12-HETE relaxations. We first examined the expression of the proposed 12-HETE receptors, GPR31 and BLT2 receptor. Western immunoblotting detected GPR31 receptor expression in mouse platelets (used as a positive control) but not in mouse aorta and mesenteric arteries (Fig. 3_A_). BLT2 receptor expression was detected in mouse aorta, mesenteric arteries, liver and spleen at molecular weight of 43 kDa (Fig. 3_B_). Similarly, immunohistochemistry staining confirmed mouse mesenteric artery smooth muscle cell BLT2 receptor expression (data not shown).
Fig. 3.
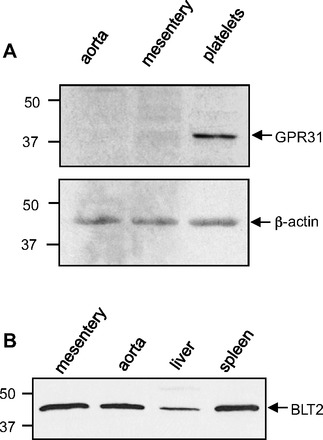
Western immunoblot analysis of GPR31 and BLT2 receptor in mouse tissues. A: GPR31 expression in mouse aorta, mesenteric arteries, and platelets. The platelets were used as a positive control. β-Actin was used as a loading control. B: BLT2 receptor expression in mouse mesenteric arteries, aorta, liver, and spleen. The liver and spleen were used as positive controls for BLT2 receptor expression.
To determine the role of BLT2 receptors in vascular relaxations of mouse mesenteric arteries, we looked at the relaxations to the BLT2 receptor agonist, CAY10583. CAY10583 had no effect on a basal tension (Fig. 4_B_). Additionally, it failed to relax either U46619- or phenylephrine-preconstricted arteries (Fig. 4_A_). Similar results were observed with leukotriene B4 (LTB4), a potent ligand of BLT2 receptor (Fig. 4_C_). This suggests that BLT2 receptor activation does not mediate relaxation of mouse mesenteric arteries and therefore the BLT2 receptor could not contribute to 12-HETE relaxations.
Fig. 4.
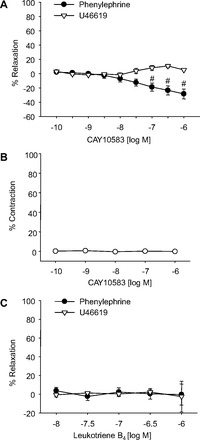
Effect of BLT2 receptor agonists on vascular tone in mouse mesenteric arteries. Relaxation responses to the BLT2 receptor agonists CAY10583 (A) and leukotriene B4 (C) in mouse mesenteric arteries preconstricted with either phenylephrine (●) or U46619 (▽). CAY10583 had no effect on the basal tension (B). Each value represents the mean ± SE; n = 4–15. #P < 0.05 compared with U46619-preconstricted arteries.
Next, we determined if 12(S)-HETE relaxations were mediated through K+ channels, an EDHF-type mechanism. In U46619-preconstricted arteries, 12(S)-HETE relaxation was not changed by treatment with K+ channel blockers (1 μM apamin, 100 nM iberiotoxin, and 1 μM apamin plus 1 μM TRAM-34) (Fig. 5, A–C). Apamin, TRAM-34, and iberiotoxin are selective inhibitors for small (SK), intermediate (IK), and large (BK) conductance Ca2+ activated K+ channels, respectively. These data indicate that 12(S)-HETE is not an EDHF in mouse mesenteric arteries.
Fig. 5.
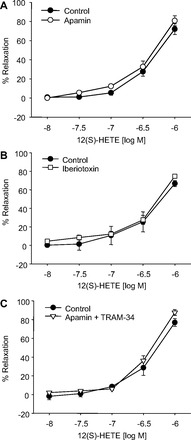
Effect of K+ channel inhibition on 12(S)-HETE relaxations in mouse mesenteric arteries preconstricted with U46619. The arteries were pretreated with apamin (SKCa blocker, 1 μM; A), iberiotoxin (BKCa blocker, 100 nM; B), and apamin plus TRAM-34 (IKCa blocker, 1 μM; C). Each value represents the mean ± SE; n = 4–12.
To examine the contribution of TP receptors in 12-HETE relaxations, we repeated the vascular relaxation responses but constricted the arteries with phenylephrine, a selective α1-adrenergic receptor agonist. In mesenteric arteries preconstricted with phenylephrine, 12(S)- and 12(R)-HETE failed to cause relaxation (Fig. 6_A_) although potent relaxations were observed in arteries preconstricted with U46619 (Fig. 2). In addition, in arteries pretreated with 12(S)- or 12(R)-HETE (1 μM), U46619-induced contractions were significantly inhibited (Fig. 6_D_). The right shift caused by 12(R)-HETE was greater than with 12(S)-HETE. Moreover, pretreatment of the arteries with 12(S)-HETE (1 μM) also right shifted the concentration-response curve of I-BOP, a potent TP receptor agonist (Fig. 6_E_). Phenylephrine contractions were not altered by either 12(S)- or 12(R)-HETE (Fig. 6_F_). Neither 12(S)- nor 12(R)-HETE caused constriction when added to arteries under basal tension (Fig. 6_C_). This suggests that 12(S)- and 12(R)-HETE relax mouse mesenteric arteries through a TP receptor-dependent mechanism and do not alter α1-adrenergic-induced contractions.
Fig. 6.
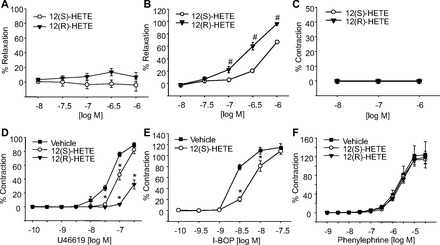
Effect of 12(S)-HETE and 12(R)-HETE on vascular tone of mouse mesenteric arteries. Concentration-dependent relaxations of 12(S)- (○) and 12(R)-HETE (▼) on arteries preconstricted with phenylephrine (A) or carbocyclic thromboxane A2 (CTA2; B). C: responses of 12(S)- (○) and 12(R)-HETE (▼) on basal tension of the arteries. Concentration-dependent contractions to U46619 (D), I-BOP (E), or phenylephrine (F) in the presence of 12(S)-HETE (1 μM, ○) or 12(R)-HETE (1 μM, ▼). Each value represents the mean ± SE; n = 6–12. *P < 0.05 compared with vehicle control (■). #P < 0.05 compared with 12(S)-HETE.
We next examined the ability of 12(S)-HETE to alter constrictions induced by a series of TP receptor agonists including U46619, CTA2, PGF2α, 8-iso-PGF2α, and I-BOP. These compounds constricted the mouse arteries with different potencies (Fig. 7_A_). While I-BOP, U46619, and CTA2 caused very potent constrictions, PGF2α and 8-iso PGF2α were weaker constrictor agonists. The EC50 rank orders of the constrictions were I-BO_P_ < CTA2 < U46619 < PGF2α ≤ 8-iso PGF2α. SQ29548, a TP receptor antagonist, inhibited U46619-induced contractions in a concentration-related manner (Fig. 7_C_). Similarly, the contractions to PGF2α were inhibited by SQ29548 (5 nM) (Fig. 7_D_); thus, PGF2α is acting as a TP receptor agonist in mouse arteries. When the mesenteric arteries were preconstricted with the various TP receptor agonists, 12(S)-HETE-induced relaxations were observed with each TP receptor agonist except with I-BOP (Fig. 7_B_). Moreover, 12(R)-HETE had a greater effect than 12(S)-HETE for relaxing arteries preconstricted with CTA2 (Fig. 6_B_). This finding with CTA2 was consistent with the results from U46619-preconstricted arteries. This suggests that 12(S)-HETE relaxes the mesenteric arteries by inhibiting or competing with the TP agonists for TP receptor binding. Since I-BOP was the most potent TP agonist in causing constriction, 12(S)-HETE, in the concentrations used, was not able to compete with I-BOP binding to the TP receptor when added after I-BOP. However, pretreatment with 12(S)-HETE inhibited I-BOP (Fig. 6_E_).
Fig. 7.
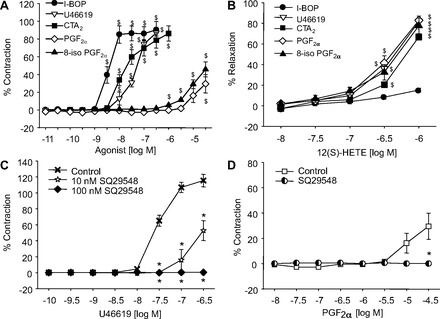
Effects of TP receptor agonists on vascular tone of mouse mesenteric arteries. A: constriction responses induced by different TP receptor agonists; I-BOP (●), U46619 (▽), CTA2 (■), PGF2α (◇), and 8-iso PGF2α (▲). B: relaxations induced by 12(S)-HETE in mouse mesenteric arteries preconstricted with I-BOP (●), U46619 (▽), CTA2 (■), PGF2α (◇), and 8-iso PGF2α (▲). C: U46619-induced contractions in the presence of SQ29548 (10 nM ☆ or 100 nM ◆) or vehicle control (X). D: PGF2α-induced contractions in the arteries pretreated with either vehicle (□) or SQ29548 (5 nM, half filled circle). Each value represents the mean ± SE; n = 4–13. $P < 0.05 compared with basal; *P < 0.05 compared with vehicle control.
To verify that 12(S)-HETE competed with TP receptor ligands at the TP receptor, radioligand competitive binding studies were performed. Mouse platelets, a rich source of TP receptors, were incubated with [3H]SQ29548, a TP receptor antagonist, and increasing concentrations of U46619, 12(S)-HETE, or 12(R)-HETE. 12(S)-HETE, 12(R)-HETE, and U46619 reduced [3H]SQ29548 binding in a concentration-dependent manner (Fig. 8). The IC50 values were 0.07, 0.32, and 1.73 μM for U46619, 12(R)-HETE, and 12(S)-HETE, respectively. 12(R)-HETE was more potent than 12(S)-HETE, consistent with the vascular relaxation studies.
Fig. 8.
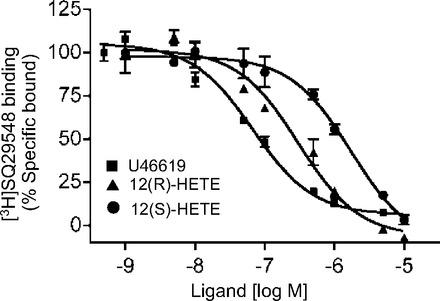
Inhibition of [3H]SQ29548 specific binding to mouse platelets by U46619 (■), 12(R)-HETE (▲), and 12(S)-HETE (●). Specific binding (total binding − nonspecific binding) was normalized to 100% of maximum binding. IC50 were 0.07, 0.32, and 1.73 μM for U46619, 12(R)-HETE, and 12(S)-HETE, respectively. Data were analyzed using the nonlinear regression analysis of PRISM (GraphPad). Each value represents the mean ± SE; n = 3.
Intracellular calcium measurements were performed using a FLIPR assay in HEK293 cells overexpressing the human TPα receptor (TPα-HEK, Fig. 9) or co-expressing the TPα and the IP receptor (IP/TPα-HEK, Fig. 10). In TPα-HEK cells, U46619 induced concentration-dependent increases in fluorescence with a maximum fluorescence of 172.3 ± 17.3 fluorescence units by 0.1 μM U46619 (Fig. 9_B_). In IP/TPα-HEK cells maximum fluorescence induced by U46619 was 268.8 ± 15.1 fluorescence units by 1 μM U46619 (Fig. 10_B_). The maximum fluorescence induced by U46619 in IP/TPα-HEK cells was significantly higher than that in TPα-HEK cells. U46619 did not increase fluorescence in nontransfected HEK293 cells (data not shown). In TPα-HEK cells, U46619 (10 nM)-induced fluorescence increases were inhibited by SQ29548 (0.01–1 μM) (Fig. 9_C_). The increase in fluorescence by U46619 (10 and 100 nM) in IP/TPα-HEK cells was inhibited by the higher concentration of SQ29548 (1 μM and 0.1–1 μM, respectively) (Fig. 10_C_), indicating that this event was TP receptor dependent. In both cell types, 12(S)-HETE and 12(R)-HETE inhibited the increase in fluorescence induced by U46619 (10 nM) in a concentration-related manner (Figs. 9_D_ and 10_D_). The inhibition caused by 12(R)-HETE was greater than the inhibition caused by 12(S)-HETE, which were consistent with the vascular studies. 12(S)-HETE or 12(R)-HETE alone (10 nM to 10 μM) did not stimulate fluorescence (data not shown). These data indicate that 12(S)-HETE and 12(R)-HETE antagonize U46619-induced increases in intracellular calcium.
Fig. 9.
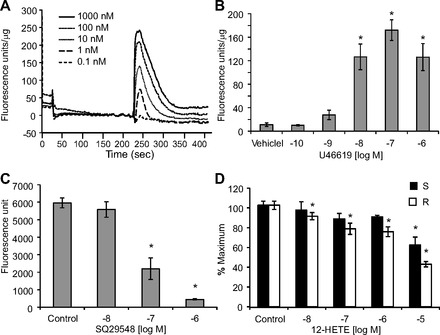
Fluorometric measurement of intracellular calcium in HEK293 cells overexpressing TPα receptor (TPα-HEK). A: representative fluorescence signals induced by increasing concentrations of U46619. B: average fluorescence increases induced by U46619. U46619 did not increase fluorescence in HEK293 cells not overexpressing TP receptors. C: effect of U46619 (10 nM) on fluorescence in the presence of SQ29548 (10 nM to 1 μM). D: U46619 (10 nM)-induced fluorescence in the presence of either 12(S)-HETE (black bar) or 12(R)-HETE (white bar). Percent maximum is the fluorescence signal stimulated by test compound compared with vehicle control as 100%. In A, B, and D, fluorescence was normalized to total protein (fluorescence units/μg). Each value represents the mean ± SE; n = 4–12. *P < 0.05 compared with vehicle control.
Fig. 10.
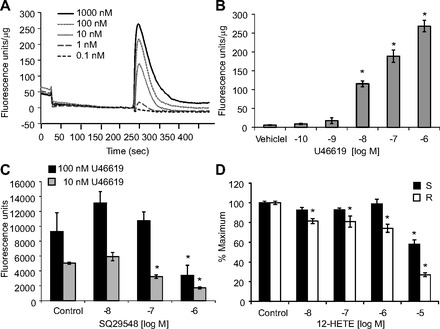
Fluorometric measurement of intracellular calcium in HEK293 coexpressing IP/TPα receptors (IP/TPα-HEK). A: representative fluorescence signals induced by increasing concentrations of U46619. B: average fluorescence increases induced by U46619. U46619 did not increase fluorescence in HEK293 cells not overexpressing IP/TP receptors. C: effect of U46619 (100 nM, black bar; 10 nM, gray bar) on calcium associated fluorescence in the presence of SQ29548 (10 nM to 1 μM) (D), U46619 (10 nM)-induced fluorescence in the presence of 12(S)-HETE (black bar) or 12(R)-HETE (white bar). Percent maximum is the fluorescence signal stimulated by test compound compared with vehicle control as 100%. In A, B, and D, fluorescence was normalized to total protein (fluorescence units/μg). Each value represents the mean ± SE; n = 4–8. *P < 0.05 compared with vehicle control.
Having identified the mechanism of exogenous 12(S)-HETE relaxation, we next investigated relaxation to endogenous 12(S)-HETE (Fig. 11). In U46619-preconstricted, indomethacin-treated mouse mesenteric arteries, AA caused PG-independent relaxations with maximum relaxations of 85.9 ± 3.3%. These relaxations to AA were significantly reduced by the LO inhibitor, NDGA (maximum relaxation of 47.6 ± 6.9%). Alternatively, in phenylephrine-preconstricted, indomethacin-treated arteries, AA relaxations were significantly reduced compared with the U46619-preconstricted arteries, and the relaxation response was not altered by NDGA (maximal relaxations = 40.7 ± 7.3% and 34.1 ± 3.5% for vehicle control and NDGA, respectively). Overall, our data indicate that both endogenous and exogenous 12(S)-HETE causes TP-receptor mediated relaxation of mouse mesenteric arteries.
Fig. 11.
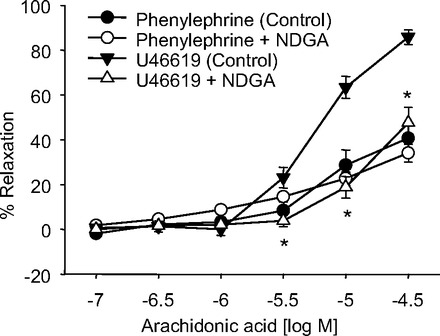
Effect of the LO inhibitor NDGA on arachidonic acid relaxations of mouse mesenteric arteries. Relaxation responses of arteries preconstricted with phenylephrine [vehicle control (●) or 10 μM NDGA (○)] or U46619 [vehicle control (▼) or 10 μM NDGA (△)]. Each value represents the mean ± SE; n = 8–10. *P < 0.05 compared with vehicle control (U46619-preconstricted arteries).
DISCUSSION
Endothelium-derived metabolites of AA modulate vascular tone and mediate the vascular effects of shear stress and hormones such as bradykinin, acetylcholine, substance P and others (2, 4, 19). CYP450 epoxygenase and lipoxygenase metabolites of AA relax arteries. While COX metabolites such as PGI2 relax arteries, other COX metabolites act as endothelium-derived constrictor factors. The effects of these AA metabolites and their mechanisms of action vary with artery and species.
12-HETE is the primary LO-derived AA metabolite produced by mouse aorta, carotid, femoral, mesenteric arteries (10), and cerebral microvessels (20, 21). Thus we continued to investigate the function and chiral properties of 12-HETE in these arteries. To establish the enzymatic source of 12-HETE, we determined its stereoisomer configuration. While 12(S)-HETE is a product of the 12(S)-lipoxygenase pathway, 12(R)-HETE is produced by either 12(R)-lipoxygenase or CYP450 (5, 17). HPLC chiral analysis of the mouse vascular 12-HETE demonstrated the presence of only the S isomer. This suggests that 12(S)- or 12/15-LO is responsible for the 12(S)-HETE production in mouse arteries. Consistent with our results, Moore et al. identified 12(S)-HETE as a product of mouse cerebral microvessels (20, 21).
12-HETE vascular activity varies with the specific stereoisomer, species and vascular bed. 12(R)-HETE, but not 12(S)-HETE, enhanced phenylephrine-induced contractions of rabbit thoracic aortic rings (16). Pretreatment of isolated hamster aorta with 12-HETE potentiated angiotensin II-induced contractions in a concentration-related manner (29). In contrast, 12(S)-HETE relaxed porcine coronary microvessels, but not conduit coronary arteries, by activating smooth muscle large-conductance Ca2+-activated K+ channels (35). Similar activity was described for 12(S)-HETE in rat small mesenteric arteries and rat basilar arteries (6, 18). Thus, in these arteries, 12(S)-HETE acted like an EDHF opening K+ channels and hyperpolarizing smooth muscle cells. Our findings that 12(S)- and 12(R)-HETE relax mouse mesenteric arteries supports a role of 12-HETE stereoisomers as a relaxing factors in smaller resistance size arteries. However, our data showed that 12(S)-HETE relaxations in the mouse arteries were not inhibited by K+ channel inhibitors and are therefore not acting as EDHFs. These findings suggested that 12-HETE was acting by a different mechanism to cause relaxation of mouse arteries.
The receptor(s) mediating 12-HETE-induced relaxation of mouse mesenteric arteries was further investigated. Guo et al. (11) demonstrated that the GPR31 receptor had high-affinity, stereospecific binding for [3H]-12(S)-HETE in cells overexpressing GPR31. Feinmark et al. (7) reported GPR31 mRNA in platelets and human umbilical vein endothelial cells. Our Western immunoblot confirmed GRP31 expression in mouse platelets. However, we did not observe GPR31 expression in mouse aorta and mesenteric arteries. Thus we eliminated GPR31 as the receptor mediating 12-HETE relaxations.
The BLT2 receptor is expressed in many tissues including human spleen, liver, intestine, leukocyte and kidney (30, 33). We demonstrated the BLT2 receptor was expressed in mouse aorta and mesenteric arteries. However, the BLT2 receptor agonists, CAY10583 or LTB4 did not relax either U46619- or phenylephrine-preconstricted arteries. Thus, although the BLT2 receptor is expressed in vascular tissue, it is not involved in relaxations to 12-HETE in mouse arteries. However, the BLT2 receptor has been implicated in VEGF-induced angiogenesis (14).
Our findings that 12(S)-HETE and 12(R)-HETE competed for binding of [3H]SQ29548 to TP receptors and 12(R)-HETE was more potent than 12(S)-HETE are consistent with a role of the TP receptor in 12-HETE relaxations. 12-HETE competed with TP receptor agonist radioligands ([125I]-BOP, [125I]-PTA-OH or [3H]-U44069) for binding to TP receptor in human platelets (8, 15). The IC50 value for 12(R)-HETE binding was ∼3 times greater than 12(S)-HETE for [125I]-BOP binding in human platelets (15). In the same study, 12(S)-HETE inhibited I-BOP-induced platelet aggregation with similar stereospecific potency. Interestingly, radioligand binding studies in cells overexpressing BLT2 or GPR31 receptors demonstrated that 12(R)-HETE was less potent than 12(S)-HETE (11, 32). Thus the relative potency of the 12(S)- and 12(R)-HETE isomers can be used as a tool to identify TP receptor-dependent activity and in our study, to link 12-HETE relaxation in mouse mesenteric arteries to the TP receptor.
The observation that 12(S)-HETE did not relax arteries preconstricted with I-BOP can be explained by the fact that I-BOP is a high-affinity TP receptor ligand (Kd = 2 nM) (22, 24). The high-affinity binding of I-BOP was so great that 12(S)-HETE was unable to compete for binding to the TP receptor. In contrast, U46619 has a lower affinity (_K_d = 44 nM) (23). Thus 12(S)-HETE was able to displace U46619, resulting in relaxation of U46619 preconstricted arteries. When the arteries were exposed to 12(S)-HETE (1 μM) prior to I-BOP, the contractions to I-BOP were reduced indicating that 12(S)-HETE acts as an inhibitor at the TP receptor.
The TP receptor exists in two isoforms, TPα and TPβ, which vary in their cytoplasmic COOH-terminal regions. The TPα isoform contains 343 amino acids and was first isolated from placenta (12). TPβ, with an extension of 64 amino acids at the COOH terminus, was cloned from endothelial cells.(27) Although TPα and TPβ mRNA can be found in many tissues, TPα is more commonly expressed. However, mice express only TPα, which is 76% identical to human TPα (25). Thus, for the FLIPR intracellular calcium assay, we used a stable cell line overexpressing the human TPα receptor. We demonstrated that 12(S)-HETE and 12(R)-HETE diminished U46619-induced calcium increases. 12(S)-HETE and 12(R)-HETE alone did not cause vasoconstriction of the mesenteric arteries or increase intracellular calcium in the TP receptor expressing HEK cells. Activity of the 12-HETE stereoisomers was evident only when the TP receptor agonist was present. Because calcium is crucial for vascular contraction, the calcium assay data support the role of 12-HETE in mediating vascular relaxation by inhibiting U46619-induced calcium increases and the subsequent vasoconstriction. Taken together, our results support a role of 12(S)-HETE and 12(R)-HETE as TP receptor antagonists in mouse arteries.
The prostacyclin receptor (IP receptor) mediates the action of prostacyclin (PGI2), a potent vasodilator and inhibitor of platelet aggregation whereas TxA2, acting through the TP receptor, is a vasoconstrictor and stimulates platelet aggregation. Smooth muscle cells and endothelial cells express both the TP and IP receptors. Interestingly, the IP receptor can interact with TPα receptors. As a result, we repeated studies on U46619-induced calcium release on cells overexpressing both the TP and IP receptors. We found that U46619-induced calcium increases in IP/TPα-HEK cells were significantly higher than TPα-HEK cells. This suggests a possible interaction of IP and TPα receptors in enhancing U46619-induced calcium responses. The physiological impact of the interaction of these two receptors requires further investigation. As with TP overexpressing cells, 12(S)- and 12(R)-HETE inhibited U46619-induced calcium release and did not affect calcium release in the absence of U46619. Furthermore, U46619 failed to stimulate calcium release in HEK293 cells not transfected and not expressing TP receptors. This indicates that the TP receptor mediates the calcium increase to U46619.
Epoxyeicosatrienoic acids (EETs), a CYP450 AA metabolite, relaxes coronary and renal arteries by K+ channel activation (2); however, in rodent arteries EETs cause vascular relaxation by interacting with the TP receptor. Behm et al. (1) reported that (EETs) relaxed rat and mouse arteries by reversing U46619 constriction. Therefore, 12-HETE is not unique in its ability to cause relaxation by inhibiting TP agonist constriction in mouse arteries.
We examined the vasoactive role of endogenous 12(S)-HETE by performing COX-independent AA relaxations with and without LO inhibition. Our findings of 12(S)-HETE as the major AA LO metabolite suggest a role for endogenous 12(S)-HETE in mediating AA relaxation. Previously, we demonstrated that NDGA nearly eliminated 12-HETE and other LO metabolite production in mouse arteries (10). Therefore, we used NDGA as a tool to evaluate the role of LO metabolites in AA relaxations. AA relaxations were greater in arteries preconstricted with the TP-receptor agonist, U46619 than in phenylephrine-constricted arteries. NDGA only inhibited AA relaxations in U46619-preconstricted arteries. This confirms the role of AA LO metabolites in mediating relaxation through TP-receptor inhibition. The mechanisms mediating AA relaxations of the phenylephrine-preconstricted arteries were not further investigated and must occur through LO-independent pathways.
The stereochemical composition of all 12(S)- and 12(R)-HETE standards used in this study were verified by chiral HPLC prior to use. This precaution was essential because two products that were purchased from Cayman Chemical were not the stated stereoisomer based on both biological activity and chiral chromatographic analysis.
In summary, we identified 12(S)-HETE as the major LO metabolite of AA in mouse arteries. This finding supports the role of 12(S)- or 12/15-LO as the predominant metabolic pathway for AA metabolism in mouse arteries. We further show that 12(S)-HETE causes relaxation of mouse arteries by acting as a TP receptor antagonist. This competitive binding decreases TP agonist-induced increases in intracellular calcium to cause vascular relaxation.
GRANTS
These studies were supported by grants from the National Heart, Lung, and Blood Institute (HL-37981, HL-103673, and HL-066233).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.S., K.M.G., S.L.P., E.M.S., and W.B.C. conception and design of research; L.S. performed experiments; L.S. analyzed data; L.S., K.M.G., and W.B.C. interpreted results of experiments; L.S. prepared figures; L.S. and K.M.G. drafted manuscript; L.S., K.M.G., S.L.P., E.M.S., and W.B.C. edited and revised manuscript; L.S., K.M.G., S.L.P., E.M.S., and W.B.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Gretchen Barg for secretarial assistance and Chuely X. Vang and Cody J. Cepura for technical assistance. We also thank Dr. Perry V. Halushka for helpful discussions.
REFERENCES
- 1.Behm DJ, Ogbonna A, Wu C, Burns-kurtis CL, Douglas SA. Epoxyeicosatrienoic acids function as selective, endogenous antagonists of native thromboxane receptors: identification of a novel mechanism of vasodilation. J Pharmacol Exp Ther 328: 231–239, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Campbell WB, Falck JR. Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension 49: 590–596, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Chawengsub Y, Aggarwal NT, Nithipatikom K, Gauthier KM, Anjaiah S, Hammock BD, Falck JR, Campbell WB. Identification of 15-hydroxy-11,12-epoxyeicosatrienoic acid as a vasoactive 15-lipoxygenase metabolite in rabbit aorta. Am J Physiol Heart Circ Physiol 294: H1348–H1356, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Chawengsub Y, Gauthier KM, Campbell WB. Role of arachidonic acid lipoxygenase metabolites in the regulation of vascular tone. Am J Physiol Heart Circ Physiol 297: H495–H507, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conners MS, Urbano F, Vafeas C, Stoltz RA, Dunn MW, Schwartzman ML. Alkali burn-induced synthesis of inflammatory eicosanoids in rabbit corneal epithelium. Invest Ophthalmol Vis Sci 38: 1963–1971, 1997 [PubMed] [Google Scholar]
- 6.Faraci FM, Sobey CG, Chrissobolis S, Lund DD, Heistad DD, Weintraub NL. Arachidonate dilates basilar artery by lipoxygenase-dependent mechanism and activation of K+ channels. Am J Physiol Regul Integr Comp Physiol 281: R246–R253, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Feinmark SJ, Nardi MA, Harleton E, Hu L, Pan R, Karpatkin S. The orphan receptor GPR31 is the platelet and HUVEC receptor for 12(S)-HETE. Proceedings of the 50th American Society of Hematology Annual Meeting and Exposition, San Francisco, CA, 2008 [Google Scholar]
- 8.Fonlupt P, Croset M, Lagarde M. 12-HETE inhibits the binding of PGH2/TXA2 receptor ligands in human platelets. Thromb Res 63: 239–248, 1991 [DOI] [PubMed] [Google Scholar]
- 9.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376, 1980 [DOI] [PubMed] [Google Scholar]
- 10.Gauthier KM, Goldman DH, Aggarwal NT, Chawengsub Y, Falck JR, Campbell WB. Role of arachidonic acid lipoxygenase metabolites in acetylcholine-induced relaxations of mouse arteries. Am J Physiol Heart Circ Physiol 300: H725–H735, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Y, Zhang W, Giroux C, Cai Y, Ekambaram P, Dilly AK, Hsu A, Zhou S, Maddipati KR, Liu J, Joshi S, Tucker SC, Lee MJ, Honn KV. Identification of the orphan G protein coupled receptor GPR31 as a receptor for 12(S)-hydroxyeicosatetraenoic acid. J Biol Chem 286: 33832–33840, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirata M, Hayashi Y, Ushikubi F, Yokota Y, Kageyama R, Nakanishi S, Narumiya S. Cloning and expression of cDNA for a human thromboxane A2 receptor. Nature 349: 617–620, 1991 [DOI] [PubMed] [Google Scholar]
- 13.Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res 61: 866–879, 1987 [DOI] [PubMed] [Google Scholar]
- 14.Kim GY, Lee JW, Cho SH, Seo JM, Kim JH. Role of the low-affinity leukotriene B4 receptor BLT2 in VEGF-induced angiogenesis. Artertio Thromb Vasc Biol 29: 915–920, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Mais DE, Saussy DL, Magee DE, Bowling NL. Interaction of 5-HETE, 12-HETE, 15-HETE and 5,12-diHETE at the human platelet thromboxane A2/prostaglandin H2 receptor. Eicosanoids 3: 121–124, 1990 [PubMed] [Google Scholar]
- 16.Masferrer J, Mullane KM. Modulation of vascular tone by 12(R)-, but not 12(S)-, hydroxyeicosatetraenoic acid. Eur J Pharmacol 151: 487–490, 1988 [DOI] [PubMed] [Google Scholar]
- 17.Mastyugin V, Aversa E, Bonazzi A, Vafaes C, Mieyal P, Schwartzman ML. Hypoxia-induced production of 12-hydroxyeicosanoids in the corneal epithelium: involvement of a cytochrome P-4504B1 isoform. J Pharmacol Exp Ther 289: 1611–1619, 1999 [PubMed] [Google Scholar]
- 18.Miller AW, Lee HC, Weintraub NL. Arachidonic acid-induced vasodilation of rat small mesenteric arteries is lipoxygenase-dependent. J Pharmacol Exp Ther 304: 139–144, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Moncada S, Vane JR. Pharmacology and endogenous roles of prostaglandin endoperoxides, thromboxane A2, and prostacyclin. Pharmacol Rev 30: 293–221, 1979 [PubMed] [Google Scholar]
- 20.Moore SA, Figard PH, Spector AA, Hart MN. Brain microvessels produce 12-hydroxyeicosatetraenoic acid. J Neurochem 53: 376–382, 1989 [DOI] [PubMed] [Google Scholar]
- 21.Moore SA, Giordano MJ, Kim HY, Salem N, Spector AA. Brain microvessel 12-hydroxyeicosatetraenoic acid is the (S) enantiomer and is lipoxygenase derived. J Neurochem 57: 922–929, 1991 [DOI] [PubMed] [Google Scholar]
- 22.Morinelli TA, Mais DE, Oatis JE, Jr, Crumbley AJ, 3rd, Halushka PV. Characterization of thromboxane A2/prostaglandin H2 receptors in human vascular smooth muscle cells. Life Sci 46: 1765–1772, 1990 [DOI] [PubMed] [Google Scholar]
- 23.Morinelli TA, Niewiarowski S, Daniel JL, Smith JB. Receptor-mediated effects of a PGH2 analogue (U46619) on human platelets. Am J Physiol Heart Circ Physiol 253: H1035–H1043, 1987 [DOI] [PubMed] [Google Scholar]
- 24.Morinelli TA, Oatis JE, Okwu AK, Mais DE, Mayeux PR, Masuda A, Knapp DR, Halushka PV. Characterization of an 125I-labeled thromboxane A2/prostaglandin H2 receptor agonist. J Pharmacol Exp Ther 251: 557–562, 1989 [PubMed] [Google Scholar]
- 25.Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther 118: 18–35, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Pfister SL, Falck JR, Campbell WB. Enhanced synthesis of epoxyeicosatrienoic acids by cholesterol-fed rabbit aorta. Am J Physiol Heart Circ Physiol 261: H843–H852, 1991 [DOI] [PubMed] [Google Scholar]
- 27.Raychowdhury MK, Yukawa M, Collins LJ, McGrail SH, Kent KC, Ware JA. Alternative splicing produces a divergent cytoplasmic tail in the human endothelial thromboxane A2 receptor. J Biol Chem 269: 19256–19261, 1994 [PubMed] [Google Scholar]
- 28.Rosolowsky M, Campbell WB. Role of PGI2 and epoxyeicosatrienoic acids in relaxation of bovine coronary arteries to arachidonic acid. Am J Physiol Heart Circ Physiol 264: H327–H335, 1993 [DOI] [PubMed] [Google Scholar]
- 29.Takai S, Jin D, Hara K, Takami H, Miyazaki M. 12-Hydroxyeicosatetraenoic acid directly potentiates angiotensin II-induced vascular contraction. Eur J Pharmacol 358: 161–164, 1998 [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Gustafson E, Pang L, Qiao X, Behan J, Maguire M, Bayne M, Laz T. A novel hepatointestinal leukotriene B4 receptor. Cloning and functional characterization. J Biol Chem 275: 40686–40694, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Wilson SJ, Roche AM, Kostetskaia E, Smyth EM. Dimerization of the human receptors for prostacyclin and thromboxane facilitates thromboxane receptor-mediated cAMP generation. J Biol Chem 279: 53036–53047, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Yokomizo T, Kato K, Hagiya H, Izumi T, Shimizu T. Hydroxyeicosanoids bind to and activate the low affinity leukotriene B4 receptor, BLT2. J Biol Chem 276: 12454–12459, 2001 [DOI] [PubMed] [Google Scholar]
- 33.Yokomizo T, Kato K, Terawaki K, Izumi T, Shimizu T. A second leukotriene B(4) receptor, BLT2. A new therapeutic target in inflammation and immunological disorders. J Exp Med 192: 421–432, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang DX, Gauthier KM, Chawengsub Y, Campbell WB. ACh-induced relaxations of rabbit small mesenteric arteries: role of arachidonic acid metabolites and K+. Am J Physiol Heart Circ Physiol 293: H152–H159, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Zink MH, Oltman CL, Lu T, Katakam PVG, Terry L, Lee Hc Dellsperger KC, Spector AA, Myers PR, Weintraub NL. 12-Lipoxygenase in porcine coronary microcirculation: implications for coronary vasoregulation. Am J Physiol Heart Circ Physiol 280: H693–H704, 2001 [DOI] [PubMed] [Google Scholar]