The Role of Inflammation in the Pathology of Acne (original) (raw)
Abstract
The conventional perspective of acne pathogenesis holds that Propionibacterium acnes colonizes the duct of the sebaceous follicle, causing an innate immune response and the progression from a so-called noninflammatory comedo to an inflammatory papule, pustule, or nodule. However, this viewpoint has come under increasing scrutiny over the last decade, as evidence has emerged supporting a role for inflammation at all stages of acne lesion development, perhaps subclinically even before comedo formation. The immunochemical pathways underlying the initiation and propagation of the inflammation in acne are complex and still being elucidated, but may involve Propionibacterium acnes as well as several inflammatory mediators and their target receptors, including cytokines, defensins, peptidases, sebum lipids, and neuropeptides. This review presents evidence to support the notion that acne is primarily an inflammatory disease, challenging the current nomenclature of noninflammatory versus inflammatory acne lesions and suggesting that the nomenclature is outdated and incorrect. The evidence in support of acne as an inflammatory disease also has clinical implications, in that anti-inflammatory drugs used to treat the disease can be expected to exert effects against all lesion stages, albeit via distinct mechanisms of anti-inflammation.
Acne vulgaris affects almost 80 percent of adolescents and young adults, often persists well into adulthood, and can result in scarring and hyperpigmentation.1,2 Acne lesions develop in the sebaceous follicles (the pilosebaceous unit), which are found on the cheek, forehead, chin, and back of both affected and unaffected individuals. A combination of increased sebum production and abnormal hyperproliferation of keratinocytes results in the formation of a small microscopic lesion called a microcomedo. Subsequent accumulation of sebum, enlargement of the follicle, and build-up of keratinous material within the microcomedo cause the formation of comedones.1 The conventional perspective of acne pathogenesis holds that Propionibacterium acnes, which is present on normal skin, colonizes the duct of the sebaceous follicle, causing an innate immune response and the progression from a so-called noninflammatory comedo to an inflammatory papule, pustule, or nodule.1 However, this viewpoint has come under increasing scrutiny over the last decade, as several lines of evidence have emerged suggesting that inflammation may exist throughout the lifecycle of the acne lesion, perhaps subclinically even before comedo formation. This evidence challenges the current nomenclature of noninflammatory versus inflammatory acne lesions and suggests that the nomenclature is both outdated and incorrect.
The purpose of this article is to review the current understanding of acne pathology, with a focus on the role of inflammation. Histological, immunological, and clinical evidence of the existence of subclinical inflammation during or even before comedogenesis is reviewed and applied to a reconsideration of acne lesion classification.
INFLAMMATION OCCURS IN EARLY AND LATE-STAGE ACNE LESIONS
There has been little debate about the involvement of inflammation in the later stages of acne, manifested as clinically inflamed papules and pustules. Accumulating histological and immunological evidence, but increasingly also evidence derived from a clinical platform, supports the notion of inflammation as a fundamental process in the early development of acne lesions (Table 1).3–28
TABLE 1.
Summary of evidence for early inflammatory events during acne lesion development
| INCREASED EXPRESSION AND BIOACTIVITY OF PROINFLAMMATORY MEDIATORS |
|---|
| Upregulation of inflammatory mediators in uninvolved skin and early lesions3,4 - E-selectin - Vascular adhesion molecule-1 - Interleukin-1 - Integrin Interleukin-1α bioactivity in open comedones5 Upregulation of defensin-2 immunoreactivity6,7 Elevation of CD3+ and CD4+ T cells and macrophages in uninvolved skin3 |
| PEPTIDASES |
| Expression of proinflammatory peptidases on sebocytes and keratinocytes8 Inhibition of peptidase activity leads to an anti-inflammatory profile8,9 |
| NEUROPEPTIDES |
| Upregulation of neuropeptides in uninvolved skin10,11 - Corticotropin-releasing hormone - Melanocortin-1 receptor - Substance P |
| TOLL-LIKE RECEPTORS |
| Activation by Propionibacterium acnes triggers inflammatory cytokine responses12,13- P. acnes is present in sebaceous follicles undergoing comedogenesis and may play a role in comedo formation14,15 Activation by endogenous ligands?16 |
| CHANGES IN LIPID BIOSYNTHESIS |
| Confirmed association between sebaceous lipid synthesis and inflammation17,18 Peroxidated lipids trigger inflammatory responses19 |
| CLINICAL TRIALS |
| The anti-inflammatory dapsone is effective in treatment of “noninflammatory” lesions20,21- Dapsone has multiple anti-inflammatory properties22,25 Retinoids are effective in treating “noninflammatory” lesions26- Retinoids downregulate toll-like receptor-2 and interleukin-10 expression27,28 |
Indeed, the concept that inflammation plays a role only in the late stages of acne vulgaris has been challenged repeatedly by emerging data, which are reshaping our understanding of acne pathology. Evidence for an early inflammatory response in the formation of acne lesions was provided by Norris and Cunliffe,29 who conducted a histological examination of early acne lesions, including lesions 6 to 72 hours old. The biopsied lesions had a morphology characteristic of small papules with minimal erythematous flare, and 52 percent were classified as microcomedones. One of the earliest histological changes identified was the appearance of a lymphoid perivascular infiltrate. Accumulation of polymorphonuclear leukocytes occurred in later stages, causing distension and pustule formation. Ultimately, this distension, accompanied by ongoing spongiosis, led to rupture of the lesion. This demonstration that inflammatory foci develop in early, apparently noninflamed, microcomedonal lesions before overt spongiosis or rupture of the pilosebaceous follicle wall, provides compelling evidence for the involvement of early inflammatory events in the development of acne lesions.29
Consistent with these findings, relatively recent studies have suggested that inflammatory events may also occur very early in the development of acne lesions (microcomedones), even before the initial hyperproliferative changes.3,4 In these studies, the uninvolved skin from acne patients was found to contain elevated levels of CD3+ and CD4+ T cells in the perifollicular and papillary dermis and increased macrophage numbers similar to those seen in papules.3,30 An example of the histological evidence for the presence of inflammatory cells in early acne is shown in Figure 1.31
Figure 1.

Inflammation in normal facial skin from a patient with acne (left); open comedo (right). Reprinted with permission from:
Plewig G, Kligman AM. Acne and Rosacea, 3rd ed. New York: Springer-Verlag; 2000.
Clinical evidence for inflammation during early acne development is derived from an investigation by Do et al,32 who conducted a serial photographic study of 25 acne patients. Patients were photographed every two weeks, and the digital photographs were spatially aligned and analyzed for acne lesions. A total of 3,099 lesions were counted, including closed comedones (37%), open comedones (12%), erythematous papules (26%), inflammatory papules (15%), pustules (2%), and nodules (1%). Of these 3,099 lesions, 219 (176 inflammatory papules, 35 pustules, 8 nodules) were not present at baseline and their origin was tracked. Notably, although 72 percent of these lesions were preceded by another acne lesion type or scar, 28 percent were preceded by normal-appearing skin. Thus, comedones were not detected at 28 percent of lesion sites before the appearance of the inflammatory lesions. These data support a model of acne pathogenesis in which the initiation of inflammatory events occurs early in the pathogenic process and before clinical detection of the acne lesion. However, although these studies have documented the occurrence of inflammation at the microcomedo stage and before any hyperproliferative changes, a definitive causal relationship between inflammation and the development of microcomedones from apparently normal skin still remains to be established.
Several comparative clinical trials also support the involvement of inflammation in early acne lesions. These studies investigated the effectiveness of agents with direct or indirect anti-inflammatory properties, including dapsone, retinoids, antibiotics, and benzoyl peroxide, to reduce comedonal lesions.
Dapsone is an anti-inflammatory agent with several potential mechanisms contributing to its clinical efficacy.33 It inhibits the release of interleukin (IL)–8 from human cultured keratinocytes25,34 and has been shown to prevent lipid peroxidation24 and neutrophil recruitment.22,23 In two vehicle-controlled, 12-week, double-blind, phase 3 acne clinical trials, dapsone significantly reduced both comedonal and clinically inflammatory lesions in patients with facial acne vulgaris.20
Topical retinoids, such as adapalene, are also known to have anti-inflammatory properties and are effective in treating patients with early-stage acne lesions.27,28 Adapalene causes reduced expression of toll-like receptor 2 (TLR2) and IL-10 both in normal skin and in acne explants, while all-trans retinoic acid has been shown to reduce macrophage TLR2 expression.27,28 In two large placebo-controlled, randomized, 12-week studies, significant reductions in both comedonal and clinically inflammatory lesions were experienced by acne patients treated with adapalene 0.1 percent lotion.26
Several studies have evaluated the response of comedonal acne to combination treatment with topical retinoids plus other topical agents having direct or indirect anti-inflammatory properties. In these studies, combination of a retinoid with either dapsone or clindamycin/benzoyl peroxide consistently elicited significantly greater reductions in comedonal lesion counts than retinoid monotherapy.21,35,36 Thus, combining a retinoid with a directly anti-inflammatory (dapsone) or indirectly anti-inflammatory (antibacterial) agent can effectively treat comedonal acne. Indeed, concurrent use of an anti-inflammatory or antibacterial agent with a retinoid appears to enhance the efficacy against comedonal acne relative to the use of the retinoid alone.
That inflammation can occur in all stages of acne is now increasingly clear. What has been unclear, however, is the mediators and events that are involved and their causal and temporal relationships. The sections that follow discuss this in more detail.
P. ACNES CAN CONTRIBUTE TO ACNE INFLAMMATION
The role of P. acnes in the etiology of inflammatory acne has been recognized for more than a century. Biopsy studies in patients with clinically inflamed acne vulgaris confirmed the association between P. acnes and clinically inflammatory acne by demonstrating that Propionibacteria are identifiable in 68 percent of one-day-old acne lesions and 79 percent of three-day-old lesions.37 Subsequent studies demonstrated that application of P. acnes to unaffected areas of skin in patients with acne results in clinical inflammation and pustule development and that injection of P. acnes into keratinous cysts causes rupture with accompanying profound inflammation, supporting the role of P. acnes as a causative agent of clinically inflammatory acne lesions.38,39
From a clinical perspective, the _P. acnes_–inflammation relationship was further supported by studies showing that antimicrobials, such as tetracycline and minocycline, suppress P. acnes in patients with moderately severe acne vulgaris, with an associated substantial reduction (up to 50% in one study40) in the number of clinically inflammatory lesions.40,41 Fulton et al42 also reported potent bacteriostatic effects of the antimicrobial agent benzoyl peroxide in patients with acne vulgaris. Thus, clinical investigations demonstrated the presence of P. acnes in late-stage acne lesions, the propensity for clinical inflammation and pustule development when P. acnes was applied to unaffected skin, and the alleviating properties of both topical and oral antimicrobials. Based on such evidence, P. acnes was identified as the causative agent of inflammation in acne, with inflammation accompanying P. acnes proliferation.
P. acnes and inflammatory mediators. P. acnes may trigger an innate immune reaction both in very early (microcomedogenic) and in late (inflammatory) acne lesions via the activation of TLR2. TLRs are a component of the innate immune system involved in host defense against invading micro-organisms,12,43 and their activation ultimately triggers the expression of immune response genes, including those coding for various cytokines and chemokines that stimulate recruitment of host immune cells.44
P. acnes has been shown to trigger proinflammatory cytokine release and expression of antimicrobial peptides. Colonization of P. acnes causes activation of monocyte TLR2, resulting in the production of IL-12 and IL-8.12 IL-12 is the major proinflammatory cytokine produced by monocytes in response to invading gram-positive organisms.12,43 Through its activation of TLRs, P. acnes can also induce human β-defensin-2 (and IL-8) expression in cultured human epidermal keratinocytes.13 The β-defensins (defensin-1, defensin-2, and defensin-3) are a family of antimicrobial peptides produced in the skin in response to microbial infection; they have been implicated in clinically inflammatory acne lesion formation. The β-defensins have a range of properties, including modification of cell migration and maturation, induction of cytokines, and chemoattraction of immunocompetent cells.45 In a gene array analysis conducted by Trivedi et al,46 β-defensin-2 gene expression was enhanced 33-fold in clinically inflammatory acne lesions relative to levels found in the unaffected skin of patients with acne or the skin of individuals without acne. β-defensin-2 protein levels were greater in the epidermis of inflammatory acne lesions than in the epidermis of uninvolved skin. Similarly, Philpott47 reported relatively low β-defensin-1 protein levels and intense upregulation of β-defensin-2 protein levels in pustules of patients with acne. Interestingly, fatty acids found within sebum may act as endogenous regulators of TLR signalling,16,45 and thus might influence the effects of P. acnes mediated by TLRs, but whether their effects would be proinflammatory or anti-inflammatory is not clear.
A recent study of biopsy samples from patients with acne vulgaris has suggested an alternative link between P. acnes and the release of inflammatory mediators.48 P. acnes is known to release various exogenous proteases, which, through the activation of the protease-activated receptor-2 (PAR-2) on keratinocytes, can enhance transcription of proinflammatory cytokines, including IL-1α, IL-8, and tumor necrosis factor-α, as well as various matrix metalloproteinases and LL-37, the only known human member of the cathelicidin group of antimicrobial peptides.48 Thus, in addition to stimulation of inflammatory processes through activation of TLRs, these data suggest another pathway, that involving PAR-2 activation, possibly linking P. acnes to proinflammatory events.12,48
Inflammation in the absence of P. acnes. Although several early reports implicated P. acnes as the central causative factor in the development of inflammation in acne vulgaris, numerous observations are inconsistent with this notion. Leyden et al30 found a much higher density of Propionibacteria among patients with acne vulgaris compared with persons without acne, but failed to show an association between P. acnes density and severity of inflammation. Indeed, in this study, there was a lower density of P. acnes on the foreheads of patients with numerous large papules or pustules with or without nodulocystic lesions, than in individuals with no inflammatory lesions.30 Similarly, Leeming et al37 found no significant difference in the proportion of inflamed pilosebaceous units containing P. acnes when biopsied after 1 or 3 days of development, again failing to establish an association between P. acnes density and the development or progression of inflammatory lesions. Additionally, several independent studies have also shown that comedones are not universally colonized by P. acnes, supporting the argument that the micro-organism is not required for comedogenesis.49-52 Lavker et al53 studied follicular casts (“incipient or potential comedones”) taken from prepuberal children and children aged 9 to 12 years with open and closed comedones. They found no evidence of P. acnes colonization, raising the possibility that early events in comedo formation may occur in the absence of P. acnes. Given that a substantial proportion of comedones have been found to be sterile,52 it has been suggested that the microorganisms found in inflammatory lesions are simply an extension of those already colonizing comedones and that their presence is not required for initiation of inflammation in acne.54
Considered collectively, these findings convincingly demonstrate that only a proportion of acne lesions, whether very early or clinically inflamed, contain micro-organisms. Thus, it appears that P. acnes is not required for the development of inflammation in acne lesions, regardless of the lesion type. That an inflammatory response can occur in the absence of P. acnes suggests that the inflammatory process is being driven via different immunochemical pathways independent of P. acnes. In this regard, accumulating evidence supports a significant role for the sebaceous gland in the development of inflammation in acne lesions.
SEBACEOUS GLAND ACTIVITY IS A DRIVER OF INFLAMMATION IN ACNE
Cytokines and inflammation in the pilosebaceous unit. IL-1 plays a central role in regulation of inflammatory and immune responses in general, and several studies have implicated a role for this cytokine in the pathogenesis of acne vulgaris. IL-1 is produced in and secreted by noninflamed skin, has been implicated in cutaneous homeostasis, and may serve as an initial reservoir for release under conditions of environmental challenge.55,56 There appears to be production of IL-1α and IL-1β in the sebaceous gland, reflected by the presence of both transcribed IL-1α and IL-1β mRNA and translated IL-1α and IL-1β immunoreactivity.55,56 Furthermore, cultured normal human keratinocytes maintain a constitutive release of IL-1α, suggesting an ongoing cycle of low-level expression and secretion.57 There is also evidence that IL-1 expression and secretion is dramatically increased during the early stages of acne lesion development. Bioactive IL-1α-like material has been detected and identified during comedogenesis in up to 76 percent of noninflamed open comedones.5 Furthermore, in 58 percent of these comedones, the levels of IL-1α-like material exceeded 100pg/mg, a level that surpasses that known to be capable of generating a visible proinflammatory response.5
These expression studies of IL-1 are supported by in vitro studies showing that proinflammatory cytokines are capable of triggering remodelling of the pilosebaceous unit and promotion of comedogenesis.58,59 Addition of IL-1α to pilosebaceous units maintained in vitro causes hypercornification similar to that seen in the development of comedones, and this effect is antagonized by IL-1 receptor blockade.58 Notably, addition of two other cytokines—epidermal growth factor or transforming growth factor-α—to these pilosebaceous units causes disorganization of the keratinocytes in the infundibulum, resulting in rupturing similar to that seen in more severe acne. Collectively, these data strongly suggest that inflammatory mediators are both present and capable of promoting comedogenesis within the pilosebaceous unit.58
In an effort to understand which inflammatory and matrix remodelling proteins may be involved in inflammatory acne, Trivedi et al46 biopsied inflammatory lesions from six acne patients and used gene array expression profiling to compare them to biopsies from uninvolved skin of the same patients, as well as with normal skin from six individuals not affected by acne. Within the inflammatory acne lesions, 211 genes were found to be upregulated relative to expression levels found in either uninvolved skin of acne patients or normal skin of individuals not affected by acne. Notably, the majority of these genes coded for proteins involved in matrix remodelling and inflammation. In regard to the latter, IL-8 expression was increased 52-fold. In an immuno-histochemical analysis of these same lesions, the investigators demonstrated the localization of IL-8 protein to the follicular and perifollicular sites of inflammation in the acne lesions, while IL-8 protein expression was “relatively absent” in normal skin. IL-8 is crucial in attracting neutrophils to the site of inflammation in acne vulgaris, the pilosebaceous unit.12,43 Release of lysosomal enzymes by these neutrophils leads to rupture of the follicular epithelium and further inflammation.60
Consistent with these findings, a separate cytokine mRNA analysis of biopsied inflammatory acne lesions revealed that facial acne lesions expressed significantly greater mRNA levels of tumor necrosis factor-α and the interleukins.61 In particular, levels of IL-8 were 3,000-fold higher in the acne lesions than in adjacent uninvolved skin from acne patients.59 Levels of IL-10 had a less dramatic 46-fold increase.61
Defensins and the sebaceous gland. The defensins have been implicated in early acne lesion formation. There is constitutive β-defensin mRNA and protein expression in the sebaceous glands of healthy skin, where the defensins may play a role in protecting the pilosebaceous unit from microbial invasion.6 Notably, there is a marked upregulation of β-defensin-1 and β-defensin-2 in most acne vulgaris lesions.6 β-defensin-1 protein expression, in particular, is markedly elevated in comedones, even more so than in pustules.6,47 In comedones, β-defensin-1 immunoreactivity was found in the suprabasal layers of lesional and perilesional epithelium, the pilosebaceous duct, sebaceous gland, and hair follicle inner root sheath.6,47 Given that proinflammatory cytokines can upregulate β-defensins, the observed upregulation of β-defensin-1 in comedones may be a secondary response to perilesional infiltrates.6,47
Changes in sebogenesis as a precursor to inflammation. Although seborrhea does not correlate directly with acne lesion development, it does affect lesion inflammatory changes. Synthesis of free fatty acids without bacterial involvement but accompanied by marked IL-1 expression has been observed in cultured sebocytes, leading investigators to suggest that sebocytes might initiate acne lesions by an intrinsic mechanism.62 Indeed, sebaceous gland lipids can have proinflammatory effects,63 and some lipids, such as oleic acid or palmitoleate, also have been shown to have antibacterial activity in vitro and in vivo.64 The mechanism by which lipids kill bacteria is not known, but as noted previously, may involve lipid-activated TLR-2. These data support the existence of an inducible lipid-based microbicidal effector pathway in the skin.64 Given the expression of TLRs in the sebaceous glands, and the antimicrobial activity of lipids, it has been postulated that the sebaceous gland plays a prominent role in the innate immune defense of the skin.64
Postsynthetic modifications in lipid composition also occur during acne lesion formation, and accumulation of lipid peroxides may be responsible for inflammatory changes in comedones.63 Lipid peroxides exist at significantly higher concentrations in comedones in patients with acne vulgaris than in healthy skin.18 Lipid peroxides stimulate the production of proinflammatory cytokines, and activate peroxisome proliferator activated receptors (PPARs).63 PPARs are nuclear transcription factors that regulate lipid metabolism and inflammation. Specifically, the peroxidation products of squalene, a characteristic human sebaceous lipid, are comedogenic and also possess proinflammatory activity: They can stimulate keratinocyte proliferation and lipo-oxygenase activity, induce PPAR expression, and increase expression and secretion of proinflammatory cytokines.19 This suggests the direct involvement of squalene peroxidation products in the onset of an inflammatory state in early acne lesions.19 Thus, although increased sebum per se has not been found to cause acne,65 it appears that changes in sebum lipid composition may initiate a previously unsuspected inflammatory cascade early in the development of acne lesions.
Proinflammatory peptidases in the sebaceous gland. Peptidases, such as dipeptidyl peptidase IV (DP-IV) and amino-peptidase N (APN), are ubiquitously expressed enzymes that affect many biological processes, including growth, differentiation, cell-cell interactions, and transformation.8 The pharmacological inhibition of these enzymes affects growth, cytokine activity, and T-cell function. Both DP-IV and APN are expressed on normal human keratinocytes and are upregulated in hyperproliferative skin disorders.8 Thielitz et al studied the expression and functional relevance of these enzymes in three cell types that exhibit an altered phenotype in early acne lesions. They found that these peptidases were expressed on human sebocytes and that their pharmacological inhibition suppressed proliferation, differentiation, and cytokine production in sebocytes and keratinocytes, which are involved in the initiation of acne.8,9 Furthermore, their inhibition also suppressed IL-3 production by _P. acnes_-stimulated T cells ex vivo and enhanced the expression of the immunosuppressive cytokine transforming growth factor-β18 These data suggest a functional role for DP-IV and APN in the sebaceous gland apparatus and a pro-inflammatory role in early acne pathogenesis.8,9
Proinflammatory neuropep-tides in the sebaceous gland.
Changes in the expression of neuropeptides have been identified during the early stages of acne, suggesting a potential neurogenic element to the early stages of lesion development. The expression of corticotropin-releasing hormone (CRH), a hormone involved in the stress response, is higher in the sebocytes of acne-involved skin (regardless of the stage of acne differentiation) than in those of uninvolved skin.10,11 Melanocortin-1 receptor (MC-1R) expression is also increased in the sebocytes and cells of the ductus seboglandularis, with MC-1R immunoreactivity being higher in skin with acne than in the skin of unaffected individuals.10,11 Both CRH and MC-1R possess proinflammatory properties.66,67 Substance P (SP) is also upregulated in the vicinity of sebaceous glands in patients with acne, but is rarely seen in skin samples from those without acne.68 SP is a potent initiator of neurogenic inflammation and it has been proposed that SP-induced neurogenic inflammation may represent a central process in stress-induced acne.11
CHRONIC INFLAMMATION IN ACNE: CLINICAL CONSEQUENCES
Several converging lines of evidence indicate that inflammation may be present throughout the development of acne lesions, both during the latter stages where inflammatory papules, pustules, and nodules are present, and also during the early stages of lesion development, in microcomedones and comedones. Indeed, many of the same classes of proinflammatory mediators have been implicated in the development of inflammation in both (Table 2, Figure 2). The evidence for inflammation during microcomedogenesis—a proinflammatory role for sebaceous lipids, the upregulation of proinflammatory mediators, early involvement of TLRs and PPARs, and a potential neurogenic component driven by neuropeptide upregulation in what have historically been regarded as noninflammatory lesions—is gaining credibility. Sufficient evidence for inflammation in early lesions now exists to reconsider acne lesion nomenclature. Based on the data presented within this review, it may be argued that classification of early stage lesions as noninflammatory is misleading and incorrect. Early stage lesions have inflammatory properties and are effectively treated with anti-inflammatory drugs. Thus, current lesion classification convention should be reconsidered in light of our expanding understanding of acne pathology.
TABLE 2.
Inflammatory mediators that have been associated with early- and late-stage acne lesions
| INFLAMMATORY MEDIATOR | FOUND IN ACNE LESIONS | |
|---|---|---|
| EARLY-STAGE ACNE LESIONS | LATE-STAGE ACNE LESIONS | |
| Propionibacterium acnes | ✓ | ✓ |
| Cytokines | ✓ | ✓ |
| Defensins | ✓ | ✓ |
| Peptidases | ✓ | |
| Neuropeptides | ✓ | |
| Immunocompetent cells | ✓ | ✓ |
Figure 2.
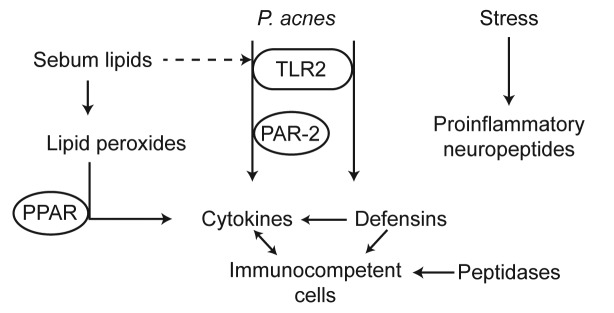
Elucidating the biochemistry of inflammation in early- and late-stage acne lesions.
Consideration of acne as a chronic inflammatory disease also raises important questions regarding acne treatment. Treatment guidelines have traditionally advocated the use of a topical retinoid for the treatment of comedonal acne.69 Given the findings regarding chronic inflammation in acne, combining a retinoid with an anti-inflammatory that has a nonoverlapping mechanism of action may provide an incremental gain in efficacy.21
DISCUSSION AND CONCLUSION
Emerging data indicate that acne vulgaris is a primary inflammatory disease, with histological, immunological, and clinical evidence suggesting that inflammation occurs at all stages of acne lesion development. The immunochemical pathways underlying the initiation and propagation of the inflammation in acne are complex and still being elucidated, but may involve P. acnes.19 However, given that an inflammatory response can also occur in the absence of P. acnes, in both early and clinically inflammatory lesions, other pathways of inflammation, in addition to those requiring P. acnes for activation or propagation, must exist. These include various inflammatory pathways activated within and around the sebaceous gland, and perhaps other as yet unidentified pathways. Thus, because inflammation is critical to all types of acne lesions and is multifactorial, anti-inflammatory drugs can be expected to exert effects against all lesion stages, albeit via distinct mechanisms of anti-inflammation.
ACKNOWLEDGMENT
ApotheCom provided suggestions for topic ideas and for authors of this manuscript to Allergan. Allergan was not involved in the development of the manuscript with the authors or the vendor. Allergan had the opportunity to review the final version of the manuscript and provide comments; however, the author maintained complete control over the content of the paper. The author determined final content and read and approved the final manuscript. No payments were made to the author for the writing of this manuscript. Third-party medical writing and editorial support for the development of this manuscript was provided by Bianca B. Ruzicka, PhD, of ApotheCom, San Francisco, California, and was funded by Allergan.
Footnotes
**DISCLOSURE:**Dr. Tanghetti is a consultant and speaker for Allergan, Galderma, Cynosure, and Palomar.
REFERENCES
- 1.Golnick H, Cunliffe W, Berson D, et al. Management of acne: a report from a Global Alliance to Improve Outcomes in Acne. J Am Acad Dermatol. 2003;49:S1–S38. doi: 10.1067/mjd.2003.618. [DOI] [PubMed] [Google Scholar]
- 2.Halder RM, Nootheti PK. Ethnic skin disorders overview. J Am Acad Dermatol. 2003;48:S143–S148. doi: 10.1067/mjd.2003.274. [DOI] [PubMed] [Google Scholar]
- 3.Jeremy AH, Holland DB, Roberts SG, et al. Inflammatory events are involved in acne lesion initiation. J Invest Dermatol. 2003;121:20–27. doi: 10.1046/j.1523-1747.2003.12321.x. [DOI] [PubMed] [Google Scholar]
- 4.Layton AM, Morris C, Cunliffe WJ, Ingham E. Immunohistochemical investigation of evolving inflammation in lesions of acne vulgaris. Exp Dermatol. 1998;7:191–197. doi: 10.1111/j.1600-0625.1998.tb00323.x. [DOI] [PubMed] [Google Scholar]
- 5.Ingham E, Eady EA, Goodwin CE, et al. Pro-inflammatory levels of interleukin-1 alpha-like bioactivity are present in the majority of open comedones in acne vulgaris. J Invest Dermatol. 1992;98:895–901. doi: 10.1111/1523-1747.ep12460324. [DOI] [PubMed] [Google Scholar]
- 6.Chronnell CM, Ghali LR, Ali RS, et al. Human beta defensin-1 and -2 expression in human pilosebaceous units: upregulation in acne vulgaris lesions. J Invest Dermatol. 2001;117:1120–1125. doi: 10.1046/j.0022-202x.2001.01569.x. [DOI] [PubMed] [Google Scholar]
- 7.Nakatsuji T, Kao MC, Zhang L, et al. Sebum free fatty acids enhance the innate immune defense of human sebocytes by upregulating beta-defensin-2 expression. J Invest Dermatol. 2010;130:985–994. doi: 10.1038/jid.2009.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thielitz A, Reinhold D, Vetter R, et al. Inhibitors of dipeptidyl peptidase IV and aminopeptidase N target major pathogenetic steps in acne initiation. J Invest Dermatol. 2007;127:1042–1051. doi: 10.1038/sj.jid.5700439. [DOI] [PubMed] [Google Scholar]
- 9.Thielitz A, Ansorge S, Bank U, et al. The ectopeptidases dipeptidyl peptidase IV (DP IV) and aminopeptidase N (APN) and their related enzymes as possible targets in the treatment of skin diseases. Front Biosci. 2008;13:2364–2375. doi: 10.2741/2850. [DOI] [PubMed] [Google Scholar]
- 10.Ganceviciene R, Bohm M, Fimmel S, Zouboulis CC. The role of neuropeptides in the multifactorial pathogenesis of acne vulgaris. Dermatoendocrinol. 2009;1:170–176. doi: 10.4161/derm.1.3.8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toyoda M, Nakamura M, Morohashi M. Neuropeptides and sebaceous glands. Eur J Dermatol. 2002;12:422–427. [PubMed] [Google Scholar]
- 12.Kim J, Ochoa MT, Krutzik SR, et al. Activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. J Immunol. 2002;169:1535–1541. doi: 10.4049/jimmunol.169.3.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagy I, Pivarcsi A, Koreck A, et al. Distinct strains of Propionibacterium acnes induce selective human beta-defensin-2 and interleukin-8 expression in human keratinocytes through toll-like receptors. J Invest Dermatol. 2005;124:931–938. doi: 10.1111/j.0022-202X.2005.23705.x. [DOI] [PubMed] [Google Scholar]
- 14.Leyden JJ, McGinley KJ, Vowels B. Propionibacterium acnes colonization in acne and nonacne. Dermatology. 1998;196:55–58. doi: 10.1159/000017868. [DOI] [PubMed] [Google Scholar]
- 15.Jarrousse V, Castex-Rizzi N, Khammari A, et al. Modulation of integrins and filaggrin expression by Propionibacterium acnes extracts on keratinocytes. Arch Dermatol Res. 2007;299:441–47. doi: 10.1007/s00403-007-0774-5. [DOI] [PubMed] [Google Scholar]
- 16.Lee JY, Plakidas A, Lee WH, et al. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res. 2003;44:479–486. doi: 10.1194/jlr.M200361-JLR200. [DOI] [PubMed] [Google Scholar]
- 17.Alestas T, Ganceviciene R, Fimmel S, et al. Enzymes involved in the biosynthesis of leukotriene B4 and prostaglandin E2 are active in sebaceous glands. J Mol Med. 2006;84:75–87. doi: 10.1007/s00109-005-0715-8. [DOI] [PubMed] [Google Scholar]
- 18.Tochio T, Tanaka H, Nakata S, Ikeno H. Accumulation of lipid peroxide in the content of comedones may be involved in the progression of comedogenesis and inflammatory changes in comedones. J Gosmet Dermatol. 2009;8:152–158. doi: 10.1111/j.1473-2165.2009.00437.x. [DOI] [PubMed] [Google Scholar]
- 19.Ottaviani M, Alestas T, Flori E, et al. Peroxidated squalene induces the production of inflammatory mediators in HaCaT keratinocytes: a possible role in acne vulgaris. J Invest Dermatol. 2006;126:2430–2437. doi: 10.1038/sj.jid.5700434. [DOI] [PubMed] [Google Scholar]
- 20.Draelos ZD, Carter E, Maloney JM, et al. Two randomized studies demonstrate the efficacy and safety of dapsone gel, 5% for the treatment of acne vulgaris. J Am Acad Dermatol. 2007;56:439–410. doi: 10.1016/j.jaad.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 21.Tanghetti E, Dhawan S, Green L, et al. Clinical evidence for the role of a topical anti-inflammatory agent in comedonal acne: findings from a study of dapsone 0.5% gel in combination with tazarotene 0.1% cream in patients with acne vulgaris. J Drugs Dermatol. 2011;10(7):783–792. [PubMed] [Google Scholar]
- 22.Booth SA, Moody CE, Dahl MV, et al. Dapsone suppresses integrin-mediated neutrophil adherence function. J Invest Dermatol. 1992;98:135–140. doi: 10.1111/1523-1747.ep12555654. [DOI] [PubMed] [Google Scholar]
- 23.Debol SM, Herron MJ, Nelson RD. Anti-inflammatory action of dapsone: inhibition of neutrophil adherence is associated with inhibition of chemoattractant-induced signal transduction. J Leukoc Biol. 1997;62:827–836. doi: 10.1002/jlb.62.6.827. [DOI] [PubMed] [Google Scholar]
- 24.Wozel G, Lehmann B. Dapsone inhibits the generation of 5-lipoxygenase products in human polymorphonuclear leukocytes. Skin Pharmacol. 1995;8:196–202. doi: 10.1159/000211346. [DOI] [PubMed] [Google Scholar]
- 25.Maloff BL, Fox D, Bruin E, Di Meo TM. Dapsone inhibits LTB4 binding and bioresponse at the cellular and physiologic levels. Eur J Pharmacol. 1988;158:85–89. doi: 10.1016/0014-2999(88)90256-7. [DOI] [PubMed] [Google Scholar]
- 26.Eichenfield LF, Jarratt M, Schlessinger J, et al. Adapalene 0.1% lotion in the treatment of acne vulgaris: results from two placebo-controlled, multicenter, randomized double-blind, clinical studies. J Drugs Dermatol. 2010;9:639–646. [PubMed] [Google Scholar]
- 27.Tenaud I, Khammari A, Dreno B. In vitro modulation of TLR-2, CDld and IL-10 by adapalene on normal human skin and acne inflammatory lesions. Exp Dermatol. 2007;16:500–506. doi: 10.1111/j.1600-0625.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 28.Liu PT, Krutzik SR, Kim J, Modlin RL. Cutting edge: all-trans retinoic acid down-regulates TLR2 expression and function. J Immunol. 2005;174:2467–2470. doi: 10.4049/jimmunol.174.5.2467. [DOI] [PubMed] [Google Scholar]
- 29.Norris JF, Cunliffe WJ. A histological and immuno-cytochemical study of early acne lesions. Br J Dermatol. 1988;118:651–659. doi: 10.1111/j.1365-2133.1988.tb02566.x. [DOI] [PubMed] [Google Scholar]
- 30.Leyden JJ, McGinley KJ, Mills OH, Kligman AM. Propionibacterium levels in patients with and without acne vulgaris. J Invest Dermatol. 1975;65:382–384. doi: 10.1111/1523-1747.ep12607634. [DOI] [PubMed] [Google Scholar]
- 31.Plewig G, Kligman AM. New York: Springer-Verlag; 2000. Acne and Rosacea, 3rd ed. [Google Scholar]
- 32.Do TT, Zarkhin S, Orringer JS, et al. Computer-assisted alignment and tracking of acne lesions indicate that most inflammatory lesions arise from comedones and. de novo. J Am Acad Dermatol. 2008;58:603–608. doi: 10.1016/j.jaad.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 33.Kircik LH. Harnessing the anti-inflammatory effects of topical dapsone for management of acne. J Drugs Dermatol. 2010;9:667–671. [PubMed] [Google Scholar]
- 34.Schmidt E, Reimer S, Kruse N, et al. The IL-8 release from cultured human keratinocytes, mediated by antibodies to bullous pemphigoid autoantigen 180, is inhibited by dapsone. Clin Exp Immunol. 2001;124:157–162. doi: 10.1046/j.1365-2249.2001.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanghetti E, Abramovits W, Solomon B, et al. Tazarotene versus tazarotene plus clindamycin/benzoyl peroxide in the treatment of acne vulgaris: a multicenter, double-blind, randomized parallel-group trial. J Drugs Dermatol. 2006;5:256–261. [PubMed] [Google Scholar]
- 36.Del Rosso JQ. Study results of benzoyl peroxide 5%/clindamycin 1% topical gel, adapalene 0.1% gel, and use in combination for acne vulgaris. J Drugs Dermatol. 2007;6:616–622. [PubMed] [Google Scholar]
- 37.Leeming JP, Holland KT, Cuncliffe WJ. The microbial colonization of inflamed acne vulgaris lesions. Br J Dermatol. 1988;118:203–208. doi: 10.1111/j.1365-2133.1988.tb01775.x. [DOI] [PubMed] [Google Scholar]
- 38.Heming A. On the etiology of acne vulgaris and its treatment by vaccines. Lancet. 1909;173:1035–1038. [Google Scholar]
- 39.Strauss JS, Kligman AM. The pathologic dynamics of acne vulgaris. Arch Dermatol. 1960;82:779–790. [Google Scholar]
- 40.Leyden JJ, McGinley KJ, Kligman AM. Tetracycline and minocycline treatment. Arch Dermatol. 1982;118:19–22. [PubMed] [Google Scholar]
- 41.Mills OH, Jr., Marples RR, Kligman AM. Acne vulgaris. Oral therapy with tetracycline and topical therapy with vitamin A. Arch Dermatol. 1972;106:200–203. doi: 10.1001/archderm.106.2.200. [DOI] [PubMed] [Google Scholar]
- 42.Fulton JE, Jr., Farzad-Bakshandeh A, Bradley S. Studies on the mechanism of action to topical benzoyl peroxide and vitamin A acid in acne vulgaris. J Gutan Pathol. 1974;1:191–200. doi: 10.1111/j.1600-0560.1974.tb00628.x. [DOI] [PubMed] [Google Scholar]
- 43.Kim J. Review of the innate immune response in acne vulgaris: activation of Toll-like receptor 2 in acne triggers inflammatory cytokine responses. Dermatology. 2005;211:193–198. doi: 10.1159/000087011. [DOI] [PubMed] [Google Scholar]
- 44.Mclnturff JE, Modlin RL, Kim J. The role of toll-like receptors in the pathogenesis and treatment of dermatological disease. J Invest Dermatol. 2005;125:1–8. doi: 10.1111/j.0022-202X.2004.23459.x. [DOI] [PubMed] [Google Scholar]
- 45.Wiesner J, Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence. 2010;1:440–464. doi: 10.4161/viru.1.5.12983. [DOI] [PubMed] [Google Scholar]
- 46.Trivedi NR, Gilliland KL, Zhao W, et al. Gene array expression profiling in acne lesions reveals marked upregulation of genes involved in inflammation and matrix remodeling. J Invest Dermatol. 2006;126:1071–1079. doi: 10.1038/sj.jid.5700213. [DOI] [PubMed] [Google Scholar]
- 47.Philpott MP. Defensins and acne. Mol Immunol. 2003;40:457–62. doi: 10.1016/s0161-5890(03)00154-8. [DOI] [PubMed] [Google Scholar]
- 48.Lee SE, Kim JM, Jeong SK, et al. Protease-activated receptor-2 mediates the expression of inflammatory cytokines, antimicrobial peptides, and matrix metalloproteinases in keratinocytes in response to. Propionibacterium acnes. Arch Dermatol Res. 2010;302:745–756. doi: 10.1007/s00403-010-1074-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shehadeh NH, Kligman AM. The bacteriology of acne. Arch Dermatol. 1963;88:829–831. doi: 10.1001/archderm.1963.01590240153025. [DOI] [PubMed] [Google Scholar]
- 50.Ganor S, Sacks TG. A comparison of the flora of the comedones of acne vulgaris and comedones in elderly people. Dermatologica. 1969;138:1–9. doi: 10.1159/000253959. [DOI] [PubMed] [Google Scholar]
- 51.Marples RR, McGinley KJ, Mills OH. Microbiology of comedones in acne vulgaris. J Invest Dermatol. 1973;60:80–83. doi: 10.1111/1523-1747.ep12724149. [DOI] [PubMed] [Google Scholar]
- 52.Puhvel SM, Amirian DA. Bacterial flora of comedones. Br J Dermatol. 1979;101:543–548. doi: 10.1111/j.1365-2133.1979.tb11883.x. [DOI] [PubMed] [Google Scholar]
- 53.Lavker RM, Leyden JJ, McGinley KJ. The relationship between bacteria and the abnormal follicular keratinization of acne. J Invest Dermatol. 1981;77:325–330. doi: 10.1111/1523-1747.ep12482524. [DOI] [PubMed] [Google Scholar]
- 54.Shaheen B, Gonzalez M. A microbial aetiology of acne: what is the evidence? Br J Dermatol. 2011;165:474–485. doi: 10.1111/j.1365-2133.2011.10375.x. [DOI] [PubMed] [Google Scholar]
- 55.Boehm KD, Yun JK, Strohl KP, Elmets CA. Messenger RNAs for the multifunctional cytokines interleukin-1 alpha, interleukin-1 beta and tumor necrosis factor-alpha are present in adnexal tissues and in dermis of normal human skin. Exp Dermatol. 1995;4:335–341. doi: 10.1111/j.1600-0625.1995.tb00057.x. [DOI] [PubMed] [Google Scholar]
- 56.Anttila HS, Reitamo S, Saurat JH. Interleukin 1 immunoreactivity in sebaceous glands. Br J Dermatol. 1992;127:585–588. doi: 10.1111/j.1365-2133.1992.tb14870.x. [DOI] [PubMed] [Google Scholar]
- 57.Ingham E, Walters CE, Eady EA, et al. Inflammation in acne vulgaris: failure of skin micro-organisms to modulate keratinocyte interleukin 1 alpha production. in vitro. Dermatology. 1998;196:86–88. doi: 10.1159/000017877. [DOI] [PubMed] [Google Scholar]
- 58.Guy R, Green MR, Kealey T. Modeling acne in vitro. J Invest Dermatol. 1996;106:176–182. doi: 10.1111/1523-1747.ep12329907. [DOI] [PubMed] [Google Scholar]
- 59.Guy R, Ridden C, Kealey T. The improved organ maintenance of the human sebaceous gland: modeling in vitro the effects of epidermal growth factor, androgens, estrogens, 13-cis retinoic acid, and phenol red. J Invest Dermatol. 1996;106:454–460. doi: 10.1111/1523-1747.ep12343608. [DOI] [PubMed] [Google Scholar]
- 60.Webster GF, Leyden JJ, Tsai CC, et al. Polymorphonuclear leukocyte lysosomal release in response to Propionibacterium acnes in vitro and its enhancement by sera from inflammatory acne patients. J Invest Dermatol. 1980;74:398–401. doi: 10.1111/1523-1747.ep12544494. [DOI] [PubMed] [Google Scholar]
- 61.Kang S, Cho S, Chung JH, et al. Inflammation and extracellular matrix degradation mediated by activated transcription factors nuclear factor-kappaB and activator protein-1 in inflammatory acne lesions in vivo. Am J Pathol. 2005;166:1691–1699. doi: 10.1016/s0002-9440(10)62479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zouboulis CC, Xia L, Akamatsu H, et al. The human sebocyte culture model provides new insights into development and management of seborrhoea and acne. Dermatology. 1998;196:21–31. doi: 10.1159/000017861. [DOI] [PubMed] [Google Scholar]
- 63.Ottaviani M, Camera E, Picardo M. Lipid mediators in acne. Mediators Inflamm. doi: 10.1155/2010/858176. 2010 August 25 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Georgel P, Crozat K, Lauth X, et al. A toll-like receptor 2-responsive lipid effector pathway protects mammals against skin infections with gram-positive bacteria. Infect Immun. 2005;73:4512–4521. doi: 10.1128/IAI.73.8.4512-4521.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Youn SW, Park ES, Lee DH, et al. Does facial sebum excretion really affect the development of acne? Br J Dermatol. 2005;153:919–924. doi: 10.1111/j.1365-2133.2005.06794.x. [DOI] [PubMed] [Google Scholar]
- 66.Webster EL, Torpy DJ, Elenkov IJ, Chrousos GP. Corticotropin-releasing hormone and inflammation. Ann N Y Acad Sci. 1998;840:21–32. doi: 10.1111/j.1749-6632.1998.tb09545.x. [DOI] [PubMed] [Google Scholar]
- 67.Hartmeyer M, Scholzen T, Becher E, et al. Human dermal microvascular endothelial cells express the melanocortin receptor type 1 and produce increased levels of IL-8 upon stimulation with alpha-melanocyte-stimulating hormone. J Immunol. 1997;159:1930–1937. [PubMed] [Google Scholar]
- 68.Toyoda M, Nakamura M, Makino T, et al. Sebaceous glands in acne patients express high levels of neutral endopeptidase. Exp Dermatol. 2002;11:241–247. doi: 10.1034/j.1600-0625.2002.110307.x. [DOI] [PubMed] [Google Scholar]
- 69.Thiboutot D, Gollnick H, Bettoli V, et al. New insights into the management of acne: an update from the Global Alliance to Improve Outcomes in Acne group. J Am Acad Dermatol. 2009;60:S1–S50. doi: 10.1016/j.jaad.2009.01.019. [DOI] [PubMed] [Google Scholar]