Repressing Notch Signaling and Expressing TNFα Are Sufficient to Mimic Retinal Regeneration by Inducing Müller Glial Proliferation to Generate Committed Progenitor Cells (original) (raw)
Abstract
Retinal damage in teleosts, unlike mammals, induces robust Müller glia-mediated regeneration of lost neurons. We examined whether Notch signaling regulates Müller glia proliferation in the adult zebrafish retina and demonstrated that Notch signaling maintains Müller glia in a quiescent state in the undamaged retina. Repressing Notch signaling, through injection of the γ-secretase inhibitor RO4929097, stimulates a subset of Müller glia to reenter the cell cycle without retinal damage. This RO4929097-induced Müller glia proliferation is mediated by repressing Notch signaling because inducible expression of the Notch Intracellular Domain (NICD) can reverse the effect. This RO4929097-induced proliferation requires Ascl1a expression and Jak1-mediated Stat3 phosphorylation/activation, analogous to the light-damaged retina. Moreover, coinjecting RO4929097 and TNFα, a previously identified damage signal, induced the majority of Müller glia to reenter the cell cycle and produced proliferating neuronal progenitor cells that committed to a neuronal lineage in the undamaged retina. This demonstrates that repressing Notch signaling and activating TNFα signaling are sufficient to induce Müller glia proliferation that generates neuronal progenitor cells that differentiate into retinal neurons, mimicking the responses observed in the regenerating retina.
Keywords: Ascl1, Müller glia, Notch signaling, quiescence, retinal regeneration, Stat3
Introduction
The damaged zebrafish retina undergoes a robust regeneration response that involves Müller glia dedifferentiation, cell cycle reentry, and production of neuronal progenitor cells that migrate to the correct retinal layer to regenerate lost neurons (Vihtelic and Hyde, 2000; Fimbel et al., 2007; Yurco and Cameron, 2007; Montgomery et al., 2010; Ramachandran et al., 2010, 2011; Nelson et al., 2012; Wan et al., 2012). In the damaged mammalian retina, however, Müller glia undergo reactive gliosis without regenerating retinal neurons (Ridet et al., 1997; Di Giovanni et al., 2005; Bringmann et al., 2006; Vázquez-Chona et al., 2011; de Melo et al., 2012; Liu et al., 2013). Elucidating the signaling pathways that induce zebrafish retinal regeneration may reveal why the damaged mammalian retina fails to regenerate and provide mechanisms that will induce a regenerative response in mammals to restore vision.
Notch signaling is an evolutionarily conserved pathway that regulates various steps during vertebrate development and is involved in regenerating the zebrafish nervous system (Bernardos et al., 2005; Raymond et al., 2006; Chapouton et al., 2010; Dias et al., 2012; Wan et al., 2012). Canonical Notch signaling occurs when a ligand binds a Notch receptor, leading to its cleavage to release the Notch intracellular domain (NICD), which relocates to the nucleus and activates transcription of downstream Notch target genes (Groot and Vooijs, 2012). The Notch receptor, its ligands, and downstream targets are differentially regulated in the damaged zebrafish retina (Raymond et al., 2006; Wan et al., 2012). Functionally, Notch signaling limits the number of proliferating Müller glia in the puncture-damaged zebrafish retina through a feedback loop that includes Ascl1a (Wan et al., 2012), an important regulator of Müller glia dedifferentiation and proliferation (Yurco and Cameron, 2007; Fausett et al., 2008; Ramachandran et al., 2010; Nelson et al., 2012). However, inhibiting Notch signaling by DAPT, a γ-secretase inhibitor, did not induce Müller glia proliferation in undamaged retinas (Wan et al., 2012). In contrast, two studies showed that high levels of Notch signaling maintain both endocrine progenitor cells and brain neural stem cells in a quiescent state, whereas reducing Notch signaling drives both cell populations to reenter the cell cycle (Chapouton et al., 2010; Ninov et al., 2012).
Here, we investigated the role of Notch signaling in maintaining Müller glia quiescence in the undamaged retina and its possible interaction with TNFα, which is produced by dying photoreceptors and is necessary to induce Müller glia proliferation (Nelson et al., 2013). Repressing Notch signaling in undamaged retinas stimulates a subset of Müller glia to express the dedifferentiation markers, Ascl1a and Stat3, reenter the cell cycle, and produce neuronal progenitor cells. Interestingly, in the undamaged Notch-repressed retina, applying TNFα induces the majority of Müller glia to reenter the cell cycle to produce progenitors that commit to the neuronal lineage. These data indicate that the combination of TNFα and repression of Notch signaling is sufficient to induce Müller glia and neuronal progenitor cell proliferation and subsequent differentiation of these progenitors into different neuronal subtypes in the undamaged zebrafish retina.
Materials and Methods
Fish lines and maintenance.
Male and female zebrafish (Danio rerio) used in this study were 6–12 months old and maintained at the Freimann Life Science Center at the University of Notre Dame as previously described (Vihtelic and Hyde, 2000). All animal care and protocols were approved by the University of Notre Dame Animal Care and Use Committee and are in compliance with the ARVO statement for the use of animals in vision research.
Light treatment protocol and γ-secretase inhibitor injections.
Adult albino Tg(gfap:EGFP)nt11 transgenic zebrafish were dark-adapted for 14 d and then immediately placed in constant light (18,000 lux) for 24 h, during which time maximal photoreceptor cell death occurs (Nelson et al., 2013). After light treatment, the fish were anesthetized in 1:1000 dilution of 2-phenoxyethanol (Sigma). A small incision was made in the cornea adjacent to the iris of each eye with a sapphire blade scalpel (World Precision Instruments) and ∼1 μl of 1% DMSO (as a vehicle control), 250 μm Compound E (Santa Cruz Biotechnology), or 100 μm RO4929097 (2,2-dimethyl_-N-_((S)-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-yl)-N′-(2,2,3,3,3-pentafluoro-propyl)-malonamide; SelleckBio) was intravitreally injected with a blunt-ended 30 gauge syringe (Hamilton Company) every 12 h (Nelson et al., 2012) until either 36 or 72 h after starting the light treatment. After each injection, the fish were kept in a dark incubator at ∼33°C, which corresponds to the temperature that the constant light-treated fish are maintained and allows for the initiation of Müller glia and neuronal progenitor cell proliferation in a relatively short time frame.
Control experiments were performed on either albino or albino Tg(atoh7:GFP) (formerly known as ath5) fish that were dark-adapted for 14 d and then placed in constant intense light for either 16 h (and labeled for TUNEL), 36 h (and labeled for TNFα expression), or 72 h (and immunolabeled for GFP expression).
Injections into undamaged fish.
Adult AB, Tg(gfap:EGFP)nt11, Tg(rho:Eco:NfsB-EGFP)nt19, _Tg(atoh7:GFP)_rw021, or Tg(nrd:egfp)nl1 fish that were not light treated were either injected intraperitoneally with 40 μl of 1 mm RO4929097 or 10% DMSO using a 30 gauge beveled needle or injected intravitreally with 1 μl of either 1% DMSO or 100 μm DAPT (Sigma) using a blunt-ended 30 gauge syringe every 12 h for up to 4 d. The intraperitoneal injections were performed to ensure that the injection needle did not induce any retinal damage, which would cause Müller glia proliferation. However, DAPT did not induce Müller glia proliferation when injected intraperitoneally, thus requiring it to be intravitreally injected. Fish that were treated with RO4929097 and either 1 mm Jak1/Jak2 inhibitor ruxolitnib (INCB018424, diluted in 10% DMSO/sterile water, SelleckBio) or 1 mm Stattic (diluted in 10% DMSO/sterile water, Tocris Bioscience) were coinjected intraperitoneally as a mixture every 12 h. In experiments that involved RO4929097 and TNFα coinjections, RO4929097 was injected intraperitoneally and TNFα was injected intravitreally because the TNFα failed to exhibit a proliferation effect when injected intraperitoneally. In these experiments, retinal sections were carefully analyzed for any potential retinal damage from the intravitreal injection, and those retinas that possessed potential signs of damage were excluded from the analysis. After each injection, the fish were kept in a dark incubator at ∼33°C, which corresponds to the temperature that the constant light-treated fish are maintained and allows for the initiation of Müller glia and neuronal progenitor cell proliferation in a relatively short time frame.
Heatshock-induced Notch activation.
Adult double-transgenic Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) fish (generous gift from R. Wingert, University of Notre Dame) were confirmed to have both transgenes by PCR amplification of transgene-specific fragments in genomic DNA. A _UAS:notch1a_-specific 450 bp fragment was amplified between the primers 5′-CATCGCGTCTCAGCCTCAC-3′ and 5′-CGGAATCGTTTATTGGTGTCG-3′, and a _hsp70l:Gal4_-specific 950 bp fragment was amplified with 5′-CGGGCATTTACTTTATGTTGC-3′ and 5′-CATCATTAGCGTCGGTGAG-3′ as published previously (Scheer and Campos-Ortega, 1999; Scheer et al., 2001, 2002). For heat shock-induced Notch activation, Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) fish and albino adult zebrafish were placed in a warm water bath at 28°C, the water temperature was elevated 1°C every 3–4 min up to 38.2°C, and maintained for 1 h. The fish were then slowly cooled and maintained at 31°C for 4 h before treatment with either RO4929097 or DMSO. Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) fish and albino fish were heat shocked three times (every 12 h) before the first injection and every 12 h during the previously described 3 d injection paradigm.
TNFα purification and injection.
The pQE30 plasmid containing recombinant zebrafish TNFα cDNA was a generous gift from the Drapeau lab (Knogler et al., 2010). The plasmid was transfected into M15 cells (QIAGEN), and recombinant TNFα protein was purified using the QIAExpressionist kit (QIAGEN). The purified TNFα was diluted to a working concentration of 0.5 mg/ml with sterile 1 × PBS and protease inhibitor mixture (tablets, Roche). The control lysate (CL) was an Escherichia coli protein lysate that was obtained from a bacterial culture that lacked the pQE30-_tnf_α recombinant plasmid but was induced and purified in the same manner as TNFα. We injected either 10% DMSO or 1 mm RO4929097 intraperitoneally and injected 0.5–1.0 μl of either the CL or the recombinant TNFα intravitreally as described above every 12 h for 4 d.
5-Ethynyl-2′-deoxyuridine (EdU) and BrdU labeling and detection.
We intraperitoneally injected 40 μl of either 1 mg/ml EdU (Invitrogen) or 2.5 mm BrdU (Sigma) (Vihtelic and Hyde, 2000; Bailey et al., 2010). EdU was detected with the Click-iT EdU AlexaFluor-647 Imaging Kit (Invitrogen) before immunohistochemistry. For BrdU detection, slides were washed in 0.05% PBS/Tween-20 (PBST) for 3 × 5 min, incubated in 2 m HCl for 30 min, and then rinsed 3 times in PBST before immunohistochemistry. For BrdU and EdU codetection, the BrdU protocol was performed first, followed by EdU detection and immunohistochemistry. The BrdU antibody (Invitrogen) used does not cross-react with EdU to ensure separate specific BrdU detection in EdU/BrdU-colabeled experiments.
Immunohistochemistry, antigen retrieval.
Enucleated eyes were either fixed in 9:1 ethanolic formaldehyde (100% ethanol/37% formaldehyde) or 4% PFA/5% sucrose/1 × PBS overnight at 4°C and immunolabeled as previously described (Vihtelic and Hyde, 2000; Kassen et al., 2007; Thummel et al., 2010; Nelson et al., 2012). Primary antibodies and dilutions used in this study were as follows: rabbit anti-GFP, 1:1000 (Abcam); mouse anti-GFP, 1:1000 (Abcam); chicken anti-GFP, 1:500 (Abcam); rabbit anti-Ascl1,1:25 (Sigma); rabbit anti-Stat3,1:50 (Kassen et al., 2007); mouse anti-proliferating cellular nuclear antigen (PCNA), 1:1000 (Sigma); mouse anti-BrdU, 1:1000 (Invitrogen); rabbit anti-TNFα, 1:25 (Anaspec); rabbit anti-Myc,1:1:00 (Millipore); mouse anti-zpr-1, 1:200 (Zebrafish International Resource Center); mouse anti-HuC/D, 1:300 (Millipore Bioscience Research Reagents); rabbit anti-PKCα, 1:300 (Santa Cruz Biotechnology); and rabbit anti-pERK, 1:200 (Cell Signaling Technology). For Ascl1a, Stat3, and TNFα, antigen retrieval was used before immunohistochemistry as previously described (Raymond et al., 2006; Nelson et al., 2012; Nelson et al., 2013).
Image acquisition and data analysis.
_Z_-stacked images of 5–10 μm were acquired on a Leica SP2 Confocal Microscope using a 40× oil-immersion lens. In light-damaged retinas, ∼350 μm of the central dorsal retina was quantified. In undamaged retinas, either an entire retinal section or the central dorsal and ventral regions (for the Müller glia activation rate counts in Fig. 6c) were quantified. To calculate the statistical difference between the control and one experimental value, we used the Student's t test. To determine the statistical difference between multiple samples at more than one time point, we used a two-way ANOVA followed by Tukey's post hoc test. The figure legends identify the statistical test used in each specific experiment, with error bars representing standard errors.
Figure 6.
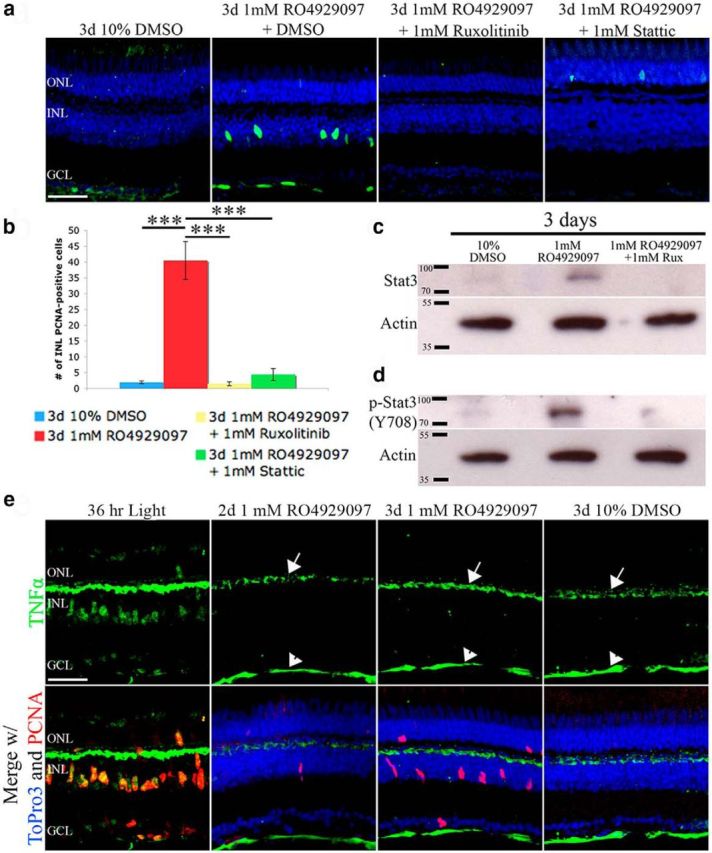
Jak-mediated Stat3 phosphorylation/activation is necessary for the Notch-inhibited undamaged retina to proliferate independent of TNFα. a, albino fish were intraperitoneally injected with DMSO, RO4929097 and DMSO, RO4929097 and ruxolitinib, or RO4929097 and Stattic for 3 d. Whereas RO4929097 and DMSO was sufficient to induce PCNA-positive INL cells, blocking the Stat3 signaling pathway with either ruxolitinib or Stattic inhibited the RO4929097-induced proliferation. b, Histogram depicting that the number of PCNA-positive INL cells in RO4929097-treated undamaged retinas was significantly reduced by cotreating with either ruxolitinib or Stattic (n = 8). c, d, Total protein lysates from DMSO, RO4929097 and DMSO, RO4929097 and ruxolitinib-treated retinas confirmed increased total Stat3 (c) and p-Stat3–708 (d) protein levels in the RO4929097-treated undamaged retinas and near control levels in the RO4929097 and ruxolitinib-cotreated retinas. β-Actin was used as a loading control. e, Confocal images of 36 h light-damaged retina (column 1) that was immunolabeled for TNFα (green) and PCNA (red) show robust TNFα expression in PCNA-positive INL cells. Undamaged albino zebrafish were intraperitoneally injected with either 1 mm RO4929097 or 10% DMSO vehicle control for either 2 or 3 d (columns 2, 3, and 4). The RO4929097 and DMSO control retinal sections were immunolabeled for TNFα (green, row 1) and merged with PCNA (red, row 2) and the nuclear marker ToPro3 (blue, row 2). Although TNFα expression was detected in both the outer plexiform layer and nerve fiber layer (arrow and arrowhead, respectively), there was no detectable TNFα expression in the INL or ONL of either the RO4929097 or DMSO-treated retinas. This demonstrates that RO4929097-induced PCNA expression in the INL is not the result of increased TNFα expression in the INL Müller glia. GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; PCNA, proliferating cellular nuclear antigen; Rux, ruxolitinib. Scale bars, 25 μm. ***p < 0.001 (one-way ANOVA and Tukey's post hoc test).
Western blotting.
Total protein collection and Western blotting techniques were used as previously described (Nelson et al., 2012, 2013), with minor modifications. Six retinas were pooled for each experimental replicate. Both rabbit anti-Stat3 (1:2000) (Kassen et al., 2007; Nelson et al., 2012) and rabbit anti-phospho-Stat3 (Tyr708) (1:1000; US Biological) polyclonal antisera were individually incubated on membranes that were blocked overnight at 4°C in 1× TBS/5% nonfat dry milk/0.1% Tween 20. Membranes were washed 4 × 20 min in 1× TBS/0.1% Tween 20. The membranes were incubated with the secondary antibody, washed, and detected as described previously (Nelson et al., 2012, 2013). However, the membranes were cut after the secondary antibody incubation and washed to separate either the Stat3 or phospho-Stat3 containing region from the actin region because of the very large difference in the levels of signal between the two detected proteins. The two portions of the same blot were then simultaneously incubated with the ECL-Prime detection system (GE Healthcare) and exposed separately to x-ray film to allow long exposures of either the Stat3 or phospho-Stat3 signals relative to the short exposures for the actin signal.
Cloning and in situ hybridization.
Zebrafish total RNA was isolated from embryos staged at 24 and 48 h after fertilization using TRIzol (Invitrogen) and reverse transcribed using random primers with the Superscript III Preamplification System (Invitrogen). Platinum TaqPCR (Invitrogen) using gene specific primer sets for notch1a (forward, TGGGAGTTTGTGTCAGGTGG; reverse, GCCGTTTTTACAGGGACGTG), notch1b (forward, CAACCCAGTCAACGGCAAAG; reverse, TTTGGGGCAGACACACAAGT), and notch3 (forward, ATTGGTTCGCTCTGTCAGCA; reverse, CAGGAGGGCACTGGCAATTA) with an annealing temperature of 55°C yielded expected size products at 577, 961, and 727 bp, respectively. The _her6_-containing plasmid was obtained from ZIRC EST (cb129) (Thisse et al., 2001). The notch1a, notch1b, and notch3 cDNAs were cloned into pCRII-TOPO (Invitrogen) vector following the manufacturer's suggested protocol and sequenced to confirm their identity. Antisense and sense digoxigenin (DIG)-labeled RNA probes were synthesized (Roche DIG RNA Labeling Kit SP6/T7) from the DNA-containing plasmids. notch1a, notch1b, and notch3 were linearized with SpeI, SpeI, and NotI and generated with T7, T7, and SP6 RNA polymerases, respectively. her6, in pBluescript, was linearized with XbaI and synthesized with SP6 RNA polymerase. In situ hybridization was performed on frozen adult retinal sections as previously described (Nelson et al., 2013) with minor modifications. The first ethanol wash series was replaced with a methanol series. The second ethanol series was replaced with a 10 min wash in 1 × PBS at room temperature, followed by incubation in prehybridization buffer (50% formamide, 10% 10× salt, and DEPC) at 55°C for 3–4 h.
qRT-PCR.
Total RNA was isolated from whole retinas using TRIzol reagent (Invitrogen) methods. cDNA was synthesized using SuperScript One-Step (Invitrogen) kit following the manufacturer's protocol and stored at −80°C. qRT-PCR was performed as previously described (Nelson et al., 2012). The ascl1a, stat3, and 18S primers were as previously described (Nelson et al., 2012). The following new primer sets were used: her4 (forward, 5′ CTTCTCTGAAGGTGTTTTCTGGTC; reverse, 5′ TATGGTGCAAAAACGCTCTGATT) and her6 (forward, 5′ AATGACCGCTGCCCTAAACA; reverse, 5′ GGCGTTGATCTGTGTCATGC).
Electroporation of morpholinos into zebrafish retina.
Lissamine-tagged morpholinos were reconstituted to a concentration of 3 mm using nuclease-free water and kept at 4°C. Morpholinos were electroporated as previously described (Thummel et al., 2011) into the dorsal portion of the retina, immediately following the initial RO4929097 intraperitoneal injection. The ascl1a morpholino (GeneTools) used to knock down Ascl1a was previously described (Fausett et al., 2008).
Results
Inhibition of Notch signaling induces increased numbers of Müller glia to reenter the cell cycle in light-damaged retinas
Notch signaling was previously shown to limit the number of proliferating Müller glia and resulting neuronal progenitor cells after retinal puncture injury (Wan et al., 2012). We were interested whether Notch signaling plays a similar role in the light-damaged retina, where damage is specific to photoreceptors, rather than all retinal cell types. To inhibit Notch signaling, we used the γ-secretase inhibitor RO4929097, which was previously shown to block Notch signaling in the regenerating zebrafish caudal fin (Münch et al., 2013). We intravitreally injected ∼1 μl of either RO4929097 (100 μm) or 1.0% DMSO after 24 h of constant light into Tg(gfap:EGFP)nt11 transgenic zebrafish, which express EGFP in Müller glia under the control of the gfap promoter (Kassen et al., 2007). Twelve hours after the injections (36 h after starting the constant light), there was a small, but significant, increase in the number of PCNA-positive Müller glia in the RO4929097-treated retinas relative to the DMSO-injected control retinas (Fig. 1a,b; 40.87 ± 2.05 [RO4929097] and 31.57 ± 2.22 [DMSO]; n = 8, p = 0.011), without a significant increase in the number of Müller glia present (Fig. 1a,b; 42.62 ± 1.82 [RO4929097] and 40.42 ± 1.42 [DMSO]; n = 8, p = 0.372). Furthermore, most of the PCNA-positive cells coexpressed Ascl1a (Fig. 1a), a marker for dedifferentiated Müller glia (Yurco and Cameron, 2007; Fausett et al., 2008; Ramachandran et al., 2010; Nelson et al., 2012). Also, the RO4929097-treated retinas contained significantly more Ascl1a-positive Müller glia than the control retinas (Fig. 1a,b; 38.0 ± 1.5 [RO4929097] and 30.4 ± 1.4 [DMSO]; n = 8, p = 0.003), which is consistent with a previous study (Wan et al., 2012).
Figure 1.
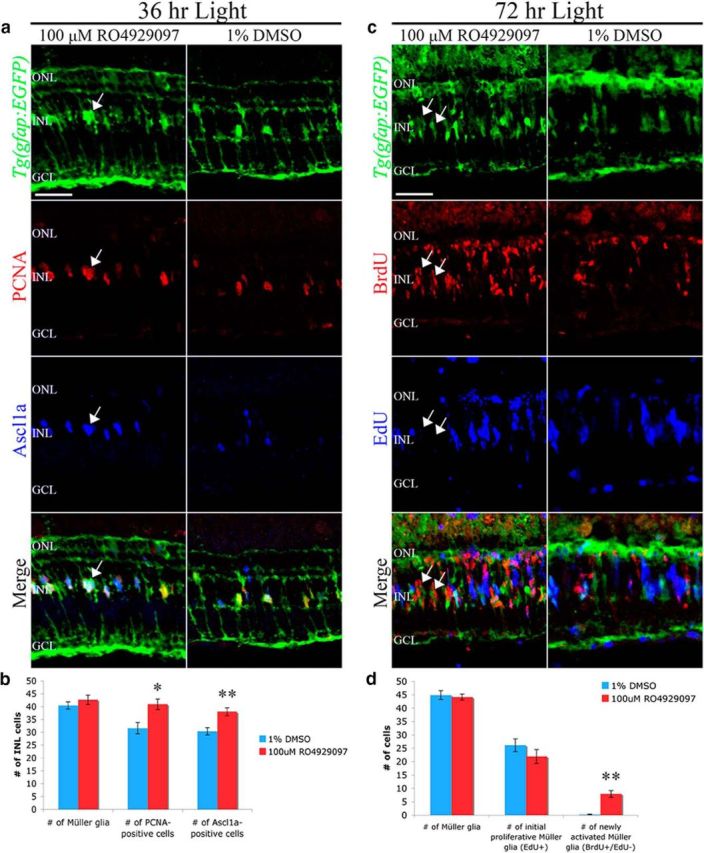
Notch-inhibited retinas exhibit increased Müller glia activation and proliferation after light damage. a, Adult Tg(gfap:EGFP)nt11 zebrafish (green, row 1) were light-damaged for 24 h and then intravitreally injected with either 100 μm RO4929097 or 1% DMSO. After 36 h of constant light, RO4929097-treated retinas (column 1) exhibited increased numbers of PCNA-positive (red, row 2) and Ascl1a-positive (blue, row 3) Müller glia relative to DMSO-treated retinas (column 2). Arrows (column 1) indicate Müller glia nuclei double-positive for Ascl1a and PCNA. b, Histogram depicting no change in the number of INL Müller glia but a significant increase in the number of PCNA-positive and Ascl1a-positive INL cells in the RO4929097-treated light-damaged Tg(gfap:EGFP)nt11 retinas (n = 8). c, Adult Tg(gfap:EGFP)nt11 zebrafish (green, row 1) were intraperitoneally injected with EdU at 36 h of constant light, intravitreally injected with either 100 μm RO4929097 (column 1) or 1% DMSO (column 2) at 48 and 60 h, pulsed intraperitoneally with BrdU at 63 and 66 h, and immunolabeled at 72 h of light for EGFP (green, row 1), BrdU (red, row 2), and EdU (blue, row 3). The RO4929097-treated retinas (column 1) contained increased numbers of BrdU-positive INL cell clusters that were EdU-negative (arrows) relative to the DMSO control (column 2). d, Histogram depicting no change in either the number of EGFP-positive or EdU-positive Müller glia at 72 h of light-damage in DMSO or RO492097-treated retinas. The number of newly activated BrdU-positive/EdU-negative cell clusters, however, were significantly increased by 72 h in RO492097-treated retinas relative to the DMSO control retinas (n = 8). GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; PCNA, proliferating cellular nuclear antigen. Scale bars, 25 μm. Student's t test, *p < 0.05. **p < 0.01.
To confirm these results were the result of inhibiting γ-secretase activity, we used a different γ-secretase inhibitor, Compound E. Following 24 h of constant light, retinas that were injected twice with Compound E yielded significantly greater numbers of PCNA-positive inner nuclear layer (INL) cells in the Compound E-injected eyes relative to the DMSO controls (64.5 ± 4.1 and 43.1 ± 5.6, respectively; n = 15, p = 0.005). Similarly, at 72 h of constant light, the Compound E-treated retinas contained significantly more PCNA-positive cells relative to the DMSO control (103 ± 9.1 and 74.4 ± 7.4, respectively; n = 15, p = 0.005). Thus, Compound E increased the numbers of PCNA-positive INL cells similar to RO4929097. Because RO4929097 exerted this increased proliferation effect when injected intraperitoneally and Compound E could not (data not shown), we used RO4929097 in all subsequent experiments in this manuscript.
The above data revealed that RO4929097-mediated inhibition of Notch signaling increased the percentage of proliferating Müller glia in the light-damaged retina relative to the DMSO control treatment (Fig. 1b; ∼95% [RO4929097] and ∼80% [DMSO]). To confirm that Notch signaling maintains some Müller glia in a quiescent state in the light-damaged retina, we EdU-labeled proliferating cells in the Tg(gfap:EGFP)nt11 transgenic line at 36 h of constant light, injected RO4929097 at 48 and 60 h, and followed with BrdU injections at 63 and 66 h (to label proliferating Müller glia induced by RO4929097 treatment). At 72 h, RO4929097- and DMSO-injected retinas were immunolabeled for EGFP, BrdU, and EdU (Fig. 1c). Thus, EdU-labeled cells were initially recruited into the cell cycle in response to the light damage, whereas BrdU-labeled cells were either continuing to proliferate in response to the light damage (EdU-positive and BrdU-positive) or were recruited into the cell cycle in response to the RO4929097 treatment (EdU-negative and BrdU-positive). Both the control and RO4929097-treated retinas contained equivalent numbers of EGFP-expressing Müller glia (Fig. 1c,d; 44.1 ± 1.1 [RO4929097] and 44.9 ± 1.7 [DMSO]) and clusters of EdU-labeled neuronal progenitor cells (Fig. 1d; 22.0 ± 2.6 [RO4929097] and 26.1 ± 2.4 [DMSO]). Each cluster represents a Müller glial cell that reentered the cell cycle to produce a neuronal progenitor cell that continued to proliferate. However, the RO4929097-treated retinas contained significantly greater numbers of INL cell clusters that contained BrdU-positive cells that lacked EdU-labeled cells relative to the control retinas (Fig. 1c,d; 7.9 ± 1.2 [RO4929097] and 0.3 ± 0.2 [DMSO]; n = 8, p = 0.008). These cell clusters represent Müller glia that remained quiescent in response to light-damage and only reentered the cell cycle after Notch signaling was blocked.
Loss of Notch signaling is sufficient to induce Müller glia reentry into the cell cycle in undamaged retinas
Because Notch signaling repressed some Müller glia from proliferating in the light-damaged retina, we hypothesized that it may maintain Müller glia quiescence in undamaged retinas. We intraperitoneally injected RO4929097 every 12 h (to prevent any potential retinal damage by the injection needle) and observed significantly greater numbers of PCNA-positive INL cells at 2, 3, and 4 d relative to the 10% DMSO controls (Fig. 2a,b; p < 0.001, n ≥ 7). Additionally, the increased number of PCNA-positive INL cells at 3 and 4 d of RO4929097 treatment relative to 2 d corresponded to small clusters of PCNA-positive cells (<3 cells; Fig. 2a, arrows), suggesting that this increase resulted from Müller glia undergoing cell divisions to generate proliferating neuronal progenitor cells. To verify that blocking Notch signaling in the undamaged retina induces proliferation, we tested the effects of another γ-secretase inhibitor, DAPT. Intraperitoneal injection of DAPT did not induce Müller glia proliferation in the undamaged retina. However, intravitreal injection of 100 μm DAPT resulted in a small, but significant, increase in the number of PCNA-positive Müller glia relative to the DMSO-injected control retinas after 4 d of injections twice daily (2.0 ± 1.4 and 0.6 ± 1.1, respectively, p = 0.012, n = 15). Although the two different γ-secretase inhibitors caused increased Müller glia proliferation in the undamaged retina, we continued to use RO4929097 because the intraperitoneal injections of RO4929097 ensures that the needle is not inducing any retinal damage and RO4929097 produced larger numbers of proliferating cells relative to the controls.
Figure 2.
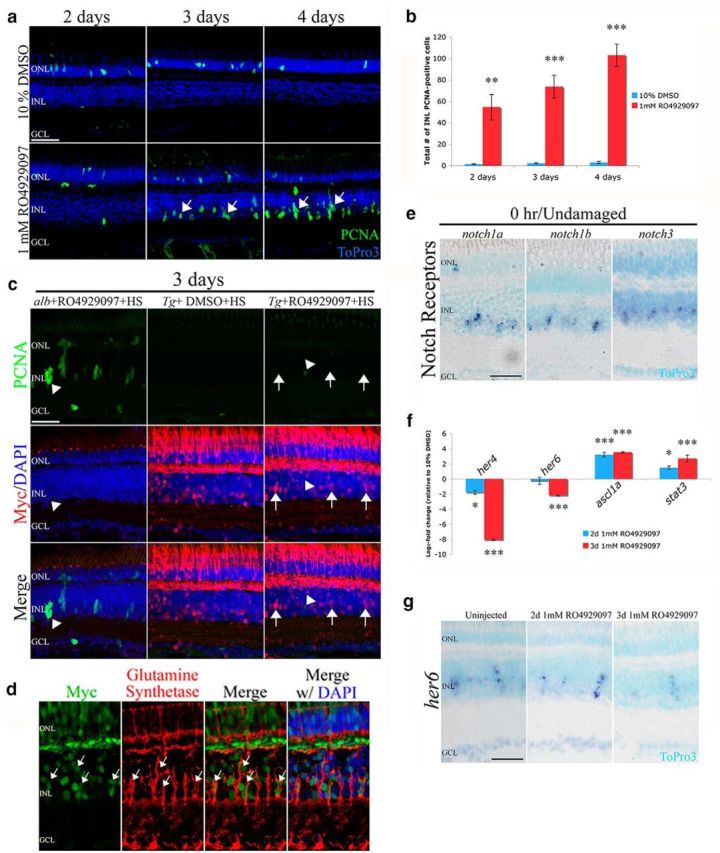
Inhibition of Notch signaling induces Müller glia proliferation in the absence of retinal damage. a, Intraperitoneal injection of 1 mm RO4929097 (bottom row) into AB zebrafish demonstrates an increased number of PCNA-positive INL cells (green, arrows) relative to the 1% DMSO-injected controls (top row). b, RO4929097 treatment results in a significant increase in the number of PCNA-positive INL cells relative to the DMSO control at 2, 3, and 4 d (n ≥ 7). c, Adult albino zebrafish were intraperitoneally injected with RO4929097 (alb+RO4929097+HS, column 1), and Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) transgenic zebrafish were intraperitoneally injected with either RO4929097 (Tg+RO4929097+HS, column 3) or DMSO (Tg+DMSO+HS, column 2) and heat-shocked for 3 d. NICD-Myc (red) was expressed in the heat-shocked Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) zebrafish and not in the heat-shocked albino fish. Expression of NICD-Myc reduced the number of PCNA-positive INL cells (green) that were induced by RO4929097. NICD-Myc-positive Müller glia (arrows) did not express PCNA. d, Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) zebrafish were intraperitoneally injected with RO4929097 and heat-shocked for 3 d. The NICD-Myc fusion protein was induced in the glutamine synthetase-positive Müller glia (arrows). e, notch1a, notch1b, and notch3 mRNA expression was localized to the basal INL of the undamaged retina by in situ hybridization. f, qRT-PCR revealed that RO4929097 inhibition of Notch signaling significantly reduced the transcript levels of the Notch-specific targets her4 and her6 at 2 and 3 d and increased expression of the activated Müller glia markers ascl1a and stat3 (n = 4). g, Tissue section in situ hybridization verified a downregulation of the Notch downstream target, her6, by 3 d of RO4929097 treatment. GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; PCNA, proliferating cellular nuclear antigen. Scale bars, 25 μm. ToPro3 (a, e, g) and DAPI staining (c, d) were used to distinguish the three nuclear layers. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
Next, we set out to verify that RO4929097 specifically acts via repression of Notch signaling rather than other γ-secretase-mediated targets by overexpressing Myc-tagged NICD in heat-shocked Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) fish (Scheer and Campos-Ortega, 1999; Scheer et al., 2001). RO4929097 was intraperitoneally injected for 3 d into Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) fish that had been heat-shocked before the start of the drug treatment. NICD overexpression was confirmed by immunocytochemistry showing broad upregulation of Myc expression throughout retinal sections from heat-shocked Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) (Fig. 2c). Labeling these heat-shocked sections with the Müller glial marker glutamine synthetase revealed that some Müller glia expressed the NICD-Myc fusion protein (Fig. 2d). Myc expression was not observed in heat-shocked albino control fish (Fig. 2c) or in non–heat-shocked Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) (data not shown). Heat-shocked RO4929097-injected Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) fish exhibited fewer PCNA-positive INL cells after 3 d relative to similarly treated albino fish (Fig. 2c; 1.28 ± 0.518 and 47.64 ± 10.85, respectively, p < 0.001, n = 14). Additionally, heat shock of DMSO-injected Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) fish yielded very few PCNA-positive INL cells (Fig. 2c). As NICD overexpression in the presence of RO4929097 suppressed the number of PCNA-positive INL cells, we concluded that the γ-secretase inhibitor RO4929097 exerts its effect on proliferation by repressing Notch signaling.
As RO4929097 inhibits the second cleavage event of the activated Notch receptor, we determined that three Notch receptors (notch1a, notch1b, and notch3) are expressed in the basal INL of the undamaged retina (Fig. 2e), consistent with the location of the Müller glia. Additionally, we determined the expression levels of two Notch target genes to further verify that RO4929097 treatment blocked Notch signaling. Transcript levels of her4 and her6 decreased significantly by 3 d of RO4929097 treatment relative to the DMSO control treatment (Fig. 2f), as assayed by qRT-PCR. Tissue in situ hybridization verified the reduction of her6 expression in INL cells between 2 and 3 d of RO4929097 treatment (Fig. 2g). Thus, these data revealed that RO4929097 repression of Notch signaling induces quiescent Müller glia to proliferate in undamaged retinas, suggesting that Notch signaling maintains Müller glia in a nonproliferating state.
Generally, Müller glia reenter the cell cycle in response to neuronal damage. RO4929097 could exert adverse effects on retinal cell survival that would initiate the observed proliferation response. Therefore, we used TUNEL to determine whether RO4929097 injections induced apoptosis. No TUNEL-positive cells were detected in any part of the retina at 2 and 3 d in the RO4929097-treated retina (Fig. 3b,c,e,f), suggesting that proliferation induced by RO4929097 does not occur in response to cell death.
Figure 3.

Cell death is not increased during RO4929097-induced inhibition of Notch signaling. a, d, Adult albino zebrafish that were treated with constant light for 16 h displayed TUNEL-positive cells (green, arrow) in the retina. Sections were counterstained with ToPro3 (blue) to label nuclei. b, c, e, f, Adult AB zebrafish that were intraperitoneally injected with 1 mm RO4929097 for either 2 d (b, e) or 3 d (c, f) exhibited no TUNEL-positive cells in the ONL, INL, or GCL. GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bars, 25 μm.
RO4929097-induced Müller glia proliferation requires Ascl1a and Stat3 expression in the undamaged retina
To further examine how Notch signaling affects Müller glia proliferation in the undamaged retina, we focused on two transcription factors (Ascl1a and Stat3) and the proinflammatory cytokine (TNFα), which were previously shown to be required for Müller glia reentry into the cell cycle after retinal damage (Kassen et al., 2007; Yurco and Cameron, 2007; Fausett et al., 2008; Ramachandran et al., 2010; Nelson et al., 2012, 2013; Wan et al., 2012). Tg(gfap:EGFP)nt11 transgenic retinas treated with RO4929097 for 3 and 4 d expressed Ascl1a in PCNA-positive Müller glia, but not in the 10% DMSO control retinas (Fig. 4a), verifying that RO4929097 induces Müller glia reentry into the cell cycle. We confirmed the increased ascl1a expression after 2 and 3 d of RO4929097 treatment using qRT-PCR (Fig. 2f), which is consistent with the increased Ascl1a expression observed in proliferating Müller glia in the damaged retina (Yurco and Cameron, 2007; Fausett et al., 2008; Ramachandran et al., 2010; Nelson et al., 2012; Wan et al., 2012).
Figure 4.

Inhibiting Notch signaling in the undamaged retina induces Ascl1a-dependent Müller glia proliferation. a, Undamaged adult Tg(gfap:EGFP)nt11 zebrafish (green, row 1) were intraperitoneally injected with either 1 mm RO4929097 or 10% DMSO. Confocal images of 1 mm RO4929097-treated retinas immunolabeled for PCNA (red, row 2) or Ascl1a (blue, row 3) at 3 and 4 d (columns 1 and 2, respectively) show increased numbers of both PCNA-positive and Ascl1a-positive Müller glia (merge) relative to the DMSO-injected controls (column 3). Additionally, the PCNA-positive cells colabeled (arrow) with the Ascl1a-positive activated Müller glia. b, Tg(gfap:EGFP)nt11 were either intraperitoneally injected with 1 mm RO4929097 (column 1) or intravitreally injected and electroporated with ascl1a MO (column 2) followed by intraperitoneal injection with 1 mm RO4929097 for 3 d. Knockdown of Ascl1a expression decreased the number of PCNA-positive (red, row 1) and Ascl1a-expressing (blue row 2) cells. Although Ascl1a levels were clearly decreased in the morphant retina, a very small number of Ascl1a-positive (single arrow) and Ascl1a-negative (double arrow) PCNA-expressing cells remained. c, Histogram depicting a significant decrease in the number of RO4929097-induced PCNA-positive cells in the INL following ascl1a morpholino knockdown (n = 8). GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; PCNA, proliferating cellular nuclear antigen; MO, morpholino. Scale bars, 25 μm. **p < 0.01 (Student's t test).
Next, we examined the requirement of Ascl1a for Müller glia proliferation in the RO4929097-treated undamaged retinas. To knockdown Ascl1a expression, we intravitreally injected and electroporated a lissamine-tagged ascl1a morpholino into the retina (Thummel et al., 2008, 2011; Fausett et al., 2008; Nelson et al., 2012). Knockdown of Ascl1a in the RO4929097-treated retina resulted in significantly fewer PCNA-positive Müller glia after 3 d relative to retinas treated with only RO4929097 (Fig. 4b,c; 13.1 ± 3.4 and 28.3 ± 3.9, respectively; p = 0.005, n ≥ 13), suggesting that Ascl1a is required for Müller glia proliferation in the RO4929097-treated retina similar to the damaged retina.
Stat3 is expressed in all Müller glia of the light-damaged zebrafish retina (Kassen et al., 2007; Nelson et al., 2012). When RO4929097-treated and DMSO-injected control Tg(gfap:EGFP)nt11 transgenic retinas were immunolabeled at 3 and 4 d, Stat3 was detected in both PCNA-positive and PCNA-negative Müller glia in the RO4929097-treated undamaged retinas, but not in the 10% DMSO controls (Fig. 5). We confirmed the increased stat3 expression after 2 and 3 d of RO4929097 treatment using qRT-PCR (Fig. 2f). Thus, RO4929097 induces increased Stat3 expression in nearly all Müller glia, but Ascl1a only in the proliferating Müller glia, a pattern that mimics the light-damaged retina (Nelson et al., 2012).
Figure 5.
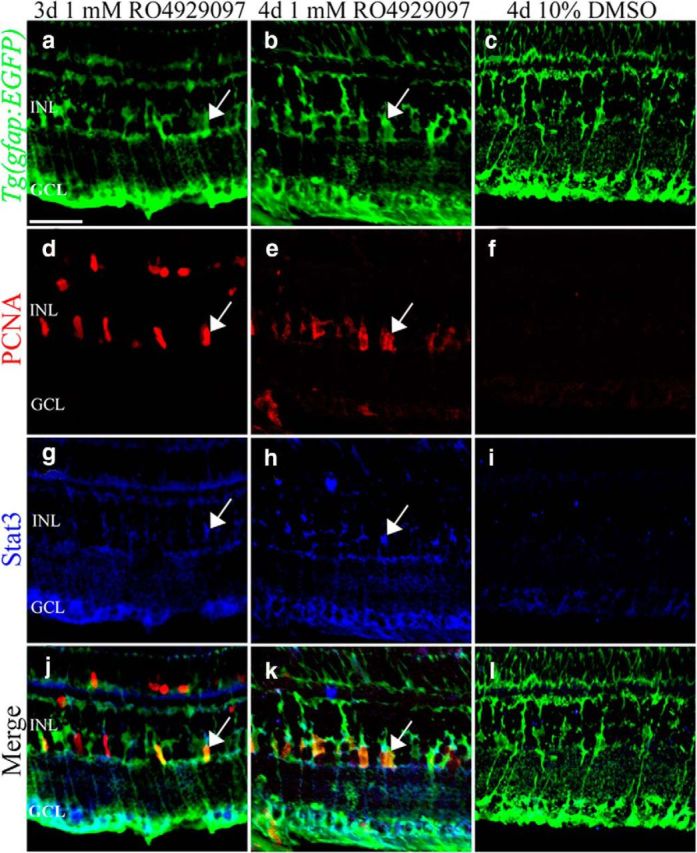
Inhibiting Notch signaling in the undamaged retina induces Stat3 expression in proliferating Müller glia. Undamaged Tg(gfap:EGFP)nt11 zebrafish were intraperitoneally injected with either 1 mm RO4929097 or 10% DMSO for 3 or 4 d and immunolabeled for EGFP (a–c, green), PCNA (d–f, red), and Stat3 (g–i, blue). 1 mm RO4929097-treated Tg(gfap:EGFP)nt11 retinas at 3 d (column 1) and 4 d (column 2) showed increased numbers of PCNA-positive (d, e, red) and Stat3-positive (g, h, blue) cells that colabeled EGFP-positive Müller glia (j, k, l, merge arrows) relative to the DMSO-injected controls (column 3). GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; PCNA, proliferating cellular nuclear antigen. Scale bars, 25 μm.
By conditional morpholino-mediated knockdown, Stat3 was previously shown to be necessary for Müller glia proliferation in the damaged retina (Nelson et al., 2012). To disrupt Stat3 activation, we used ruxolitinib (INCB018424), which preferentially inhibits Jak1/Jak2 activity. Jak1 is required for Stat3 activation/phosphorylation and Müller glia proliferation in the light-damaged retina (Raycroft and Hyde, unpublished data). Intraperitoneal injection of 1 mm ruxolitinib significantly reduced the number of PCNA-positive cells relative to the DMSO-injected control retina after 72 h of constant light (images not shown; 90 ± 19.96 [ruxolitinib] and 308 ± 21.5 [DMSO]; p < 0.001, n = 7).
To determine the requirement for Stat3 activation/phosphorylation in RO4929097-induced Müller glia proliferation, we intraperitoneally injected DMSO, RO4929097 with DMSO, or RO4929097 with 1 mm ruxolitinib into undamaged zebrafish. The RO4929097/ruxolitinib coinjected fish possessed significantly fewer PCNA-positive INL cells relative to the RO4929097/DMSO-coinjected fish (Fig. 6a,b; 1.5 ± 0.7 [ruxolitinib] and 40.4 ± 5.9 [DMSO]; p = 0.0002, n = 8). To verify that inhibiting Notch signaling stimulated Stat3 expression and phosphorylation in the undamaged retina, we analyzed the undamaged RO4929097-treated zebrafish using immunoblots. The immunoblots confirmed that RO4929097 treatment of undamaged retinas yielded increased amounts of both total and phosphorylated (p-Stat3-Y708) Stat3 relative to DMSO-treated retinas (Fig. 6c,d), whereas RO4929097/ruxolitinib coinjection reduced both total Stat3 and p-Stat3–708 to near control levels (Fig. 6c,d). Thus, RO4929097 treatment was sufficient to induce Stat3 phosphorylation and increased Stat3 expression.
To confirm that ruxolitinib inhibition of Jak1 acted specifically on Stat3 in the RO4929097-treated retinas, we intraperitoneally coinjected RO4929097 with Stattic, which inhibits the activation, dimerization, and nuclear translocation of Stat3 (Schust et al., 2006). The RO4929097/Stattic coinjected undamaged retinas possessed significantly fewer PCNA-positive Müller glia than the RO4929097-treated retinas (Fig. 6a,b; 4.4 ± 1.9 [RO4929097/Stattic] and 40.4 ± 5.9 [RO4929097]; p = 0.001, n = 8). Thus, Stat3 activation is required for Müller glia proliferation induced by repressing Notch signaling.
In the light-damaged retina, we showed that TNFα is upregulated in dying neurons (Nelson et al., 2013). Additionally, this increased TNFα expression is required for increased Ascl1a and Stat3 expression in the Müller glia and their induced proliferation (Nelson et al., 2013). We did not detect significant TNFα expression in the INL or outer nuclear layer (ONL) at 2, 3, or 4 d after RO4929097 treatment (Fig. 6e). The low level of TNFα expression in the outer plexiform and the nerve fiber layers of the RO4929097-treated retinas (Fig. 6e, arrows and arrowheads, respectively) are consistent with the previously described low levels of TNFα in these layers in the undamaged retina (Nelson et al., 2013). These data suggest that repression of Notch signaling induced Müller glia proliferation through increased Ascl1a expression and Stat3 phosphorylation/activation in a TNFα-independent manner.
EdU-positive cells produced by loss of Notch signaling fail to commit to an Atoh7-expressing neuronal progenitor fate and die
To investigate the fate of the RO4929097-induced proliferating cells in the undamaged retina, we EdU-labeled the proliferating cells in the RO4929097-treated transgenic Tg(atoh7:GFP) line (Poggi et al., 2005). Atoh7 is a bHLH transcription factor that restricts the progenitor cell fate to retinal ganglion cells, amacrine cells, horizontal cells, and photoreceptors in the retina (Vitorino et al., 2009). After 72 h of constant light, nearly all PCNA-positive cells expressed the atoh7:GFP transgene (Fig. 7a). In contrast, undamaged Tg(atoh7:GFP) retinas that were intraperitoneally injected with RO4929097 possessed less than five GFP-positive cells per retinal section between 5 and 9 d of treatment (Fig. 7a), suggesting that at least one additional signal must be necessary for these neuronal progenitor cells to commit to a neuronal lineage.
Figure 7.
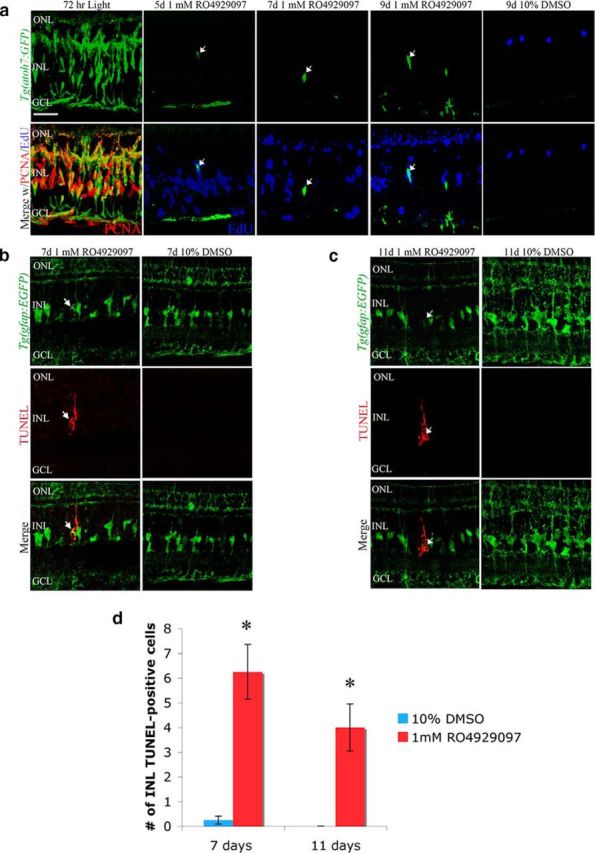
The proliferating neuronal progenitors produced by inhibiting Notch signaling in the undamaged retina do not differentiate and undergo cell death. Undamaged albino Tg(atoh7:GFP) zebrafish (a) and _albino Tg_[_gfap:EGFP_]nt11 transgenic zebrafish (b, c) were intraperitoneally injected with either 1 mm RO4929097 or 10% DMSO every 12 h for 4 d, EdU was coinjected at 1.5, 2.5, and 3.5 d, and retinas were collected at 5, 7, 9, or 11 d. a, Confocal image of Tg(atoh7:GFP) (green, column 1) retinas that were light-damaged for 72 h displayed robust PCNA expression (red, column 1) in the GFP-positive INL clusters. RO4929097-treated Tg(atoh7:GFP) zebrafish (columns 2–4) displayed large numbers of EdU-positive INL cells (blue, lower row) relative to the DMSO control (column 5). Although limited, there was also an increase in the number of GFP-positive INL cells that colabel with EdU-positive INL cells (arrow) in the RO4929097-treated retinas relative to the DMSO-treated retinas. b, c, Confocal images of _Tg_[_gfap:EGFP_]nt11 transgenic zebrafish (which express EGFP in the Müller glia from the gfap promoter) treated with either 1 mm RO4929097 (column 1) or 10% DMSO (column 2) for 7 d (b) and 11 d (c) and labeled with anti-GFP to identify the Müller glia (green, row 1) and TUNEL (red, row 2). Arrows indicate Müller glia that are TUNEL-positive. d, Histogram depicting an increased number of TUNEL-positive INL cells at both 7 and 11 d in RO4929097-injected retinas compared with 10% DMSO (n = 5). GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; PCNA, proliferating cellular nuclear antigen. Scale bars, 25 μm. Student's t test, *p < 0.05.
Large numbers of EdU-positive cells were observed in the retina between 5 and 9 d after starting the RO4929097 treatments relative to the control retina (Fig. 7a). Furthermore, the EdU-labeled cells appeared primarily in the INL at 5 d and in the ONL at 9 d (Fig. 7a), suggesting that the EdU-positive neuronal progenitors are either migrating to the ONL or are dying. As the number of EdU-positive cells decreased from 5 to 9 d after RO4929097 treatment, we examined whether the RO4929097-induced neuronal progenitors are lost shortly after being generated. RO4929097-treated retinas contained significantly more TUNEL-positive INL cells at 7 and 11 d (Fig. 7b–d; 6.25 ± 1.1 and 4.0 ± 0.9, respectively) relative to the DMSO-treated control retinas (Fig. 7b–d; 0.25 ± 0.16 and 0 ± 0; n = 5, p = 0.01). The TUNEL-positive INL cells often possessed a Müller glial-fusiform morphology, suggesting that either a subset of Müller glia or neuronal progenitor cells were dying. However, there did not appear to be fewer EGFP-positive Müller glia in the RO4929097-treated Tg(gfap:EGFP)nt11 transgenic retinas relative to the DMSO control retinas (Fig. 7b,c). Rather than dying, the TUNEL-positive Müller glia could be phagocytosing the remnants of dying cells, as described previously (Bailey et al., 2010).
Loss of Notch signaling and TNFα act synergistically to induce Müller glia and neuronal progenitor cell proliferation in undamaged retinas
Because TNFα is required for Müller glia proliferation in the light-damaged retina (Nelson et al., 2013), we examined whether application of recombinant soluble TNFα is sufficient to induce Müller glia to reenter the cell cycle in undamaged retinas. Intravitreal injection of immunopurified bacterial recombinant TNFα induced a small but statistically significant increase in the number of PCNA-positive Müller glia relative to the induced and purified CL-injected Tg(gfap:EGFP)nt11 transgenic retinas at 2, 3, and 4 d (Fig. 8a,b; 7.2 ± 1.7 [2 d], 15.7 ± 3.3 [3 d], and 24.1 ± 6.8 [4 d]; p < 0.01, n = 6). Intraperitoneal injection of RO4929097 induced a statistically significant larger increase in the number of PCNA-positive Müller glia relative to the DMSO-injected Tg(gfap:EGFP)nt11 transgenic retinas at 2, 3, and 4 d (Fig. 8a,b; 60.7 ± 9.0 [2 d], 111.6 ± 22.7 [3 d], and 112.6 ± 17.0 [4 d]; p < 0.01, n = 7). However, intraperitoneal injection of RO4929097 combined with intravitreal injection of recombinant TNFα yielded significantly greater numbers of PCNA-positive INL cells than either single treatment (Fig. 8a,b; 186.1 ± 28.3 [2 d], 290.9 ± 14.1 [3 d], and 386.2 ± 44.3 [4 d]; p < 0.005, n = 7). Not only did coinjection of TNFα and RO4929097 result in significantly more Müller glia reentering the cell cycle at 2–4 d relative to either the RO4929097-treated or the TNFα-injected retinas (Fig. 8c; 78.5% [TNFα and RO4929097], 31.5% [RO4929097], 7.3% [TNFα], and 2.7% [control] at 4 d), but the coinjection also induced proliferation of the neuronal progenitor cells to generate columns of PCNA-positive cells rather than the single or doublet cells observed in either single treatment (Fig. 8a). These data suggest that both repression of Notch signaling and exposure to TNFα are sufficient for Müller glia to reenter the cell cycle and to synergistically stimulate proliferation of neuronal progenitors to generate columns of PCNA-positive cells.
Figure 8.

Inhibition of Notch signaling in combination with exogenous TNFα act synergistically to induce robust Müller glia proliferation. a, Adult Tg(gfap:EGFP)nt11 transgenic zebrafish were intraperitoneally injected with either 1 mm RO4929097 or 10% DMSO in combination with an intravitreal injection of either purified recombinant TNFα or purified control bacterial lysate (CL) for 2–4 d. The TNFα/DMSO, RO4929097/CL, and TNFα/RO4929097 coinjected retinas possessed signifcantly more PCNA-positive cells (red) at 2, 3, and 4 d of injections relative to the CL/DMSO coinjected control. The fourth column (4 d PCNA) contains higher-magnification images of PCNA labeling to highlight the single INL cells in the TNFα/DMSO coinjected retina, the single and double cells in the RO4929097/CL coinjected retina, and the columns containing several PCNA-positive cells in the TNFα/RO4929097 coinjected retina. b, Histogram depicting a significant increase in the number of PCNA-positive INL cells in the TNFα/DMSO, RO4929097/CL, and RO4929097/TNFα-coinjected retinas compared with the CL/DMSO coinjected control (n ≥ 6). c, Histogram showing a significant increase in the number of dorsal and ventral PCNA-positive Müller glia in the TNFα/DMSO, RO4929097/CL, and RO4929097/TNFα-coinjected retinas compared with the CL/DMSO coinjected control (n ≥ 6), whereas the total number of Müller glia remained unchanged in all four treatment groups. GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; PCNA, proliferating cellular nuclear antigen. Scale bars, 25 μm. **p < 0.01 (two-way ANOVA and Tukey's post hoc test). ***p < 0.001 (two-way ANOVA and Tukey's post hoc test).
Loss of Notch signaling and TNFα expression act synergistically to induce neuronal progenitor cells to commit to a neuronal lineage
As described above, inhibition of Notch signaling alone was sufficient to initiate a proliferative response but was unable to direct those Müller glia-derived neuronal progenitors to commit into a neuronal fate. We used the Tg(atoh7:GFP) transgenic line to determine whether the neuronal progenitor cells generated by Müller glia in the RO4929097 and TNFα-cotreated retinas could commit to a neuronal lineage. Injection of either RO4929097 or TNFα alone into the undamaged Tg(atoh7:GFP) transgenic retina yielded less than five GFP-positive cells per retinal section after 4 and 5 d (Fig. 9a,b; 3.0 ± 1.57 [RO4929097 4 d], 1.44 ± 0.44 [TNFα 4 d], 3.43 ± 1.19 [RO4929097 5 d], 1.33 ± 0.41 [TNFα 5 d]). This suggests that the majority of proliferating Müller glia and neuronal progenitor cells are not committing to a neuronal lineage. In contrast, coinjection of both RO4929097 and TNFα resulted in significantly increased numbers of PCNA- and GFP-positive committed neuronal progenitor cell clusters in the Tg(atoh7:GFP) transgenic line relative to either of the individual treatments (Fig. 9a,b; 46.86 ± 9.21 [4 d] and 144.17 ± 7.09 [5 d]).
Figure 9.

Robust proliferation stimulated by coinjection of RO4929097 and TNFα stimulates neuronal progenitors to commit to a neuronal fate. Tg(atoh7:GFP) or Tg(nrd:GFP) zebrafish were coinjected with TNFα/DMSO, RO4929097/CL, and RO4929097/TNFα for 4 d. a, The Tg(atoh7:GFP) zebrafish were collected at 4 and 5 d and immunolabeled for PCNA (red). The RO4929097/TNFα coinjected retinas had significantly more GFP-positive cell clusters that robustly colabeled with PCNA than either of the other two treatments. b, Histogram depicting the increased number of GFP-positive INL clusters present in the RO4929097/TNFα cotreated retinas relative to the other two treatments (n = 7). c, The Tg(nrd:egfp) zebrafish were collected at 5 d and immunolabeled for PCNA (red). The RO4929097/TNFα coinjected retinas had more diffuse GFP-positive cells (representing committed neuronal progenitor cells) that robustly colabeled with PCNA (arrows) than either of the other two treatments. GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; PCNA, proliferating cellular nuclear antigen. Scale bars, 25 μm. **p < 0.01 (two-way ANOVA and Tukey's post hoc test). ***p < 0.001 (two-way ANOVA and Tukey's post hoc test).
To further support that the neuronal progenitor cells in the RO4929097 and TNFα-coinjected retinas were committing to a neuronal lineage, we repeated the experiment in the Tg(nrd:egfp) transgenic line that expresses EGFP in a subset of amacrine cells, bipolar cells, rod photoreceptors, and photoreceptor progenitor cells as they migrate to the ONL during retinal regeneration (Thomas et al., 2012). The photoreceptor progenitor cells in the regenerating retina exhibit a weak and diffuse EGFP expression pattern (Thomas et al., 2012). After 4 d of injections, we found no EGFP-positive progenitor cells in the RO4929097, TNFα, or the coinjected undamaged transgenic retinas (data not shown). After 5 d, a few progenitor cells (diffuse EGFP expression in the INL) coexpressed PCNA in either the TNFα- or RO4929097-injected undamaged retinas (Fig. 9c, arrows). In contrast, coinjection of both RO4929097 and TNFα resulted in an increased number of progenitor cells that coexpressed diffuse EGFP and PCNA (Fig. 9c, arrows). Thus, coinjection of RO4929097 and TNFα was sufficient to induce an increased number of neuronal progenitors that committed to a neuronal lineage in the undamaged retina.
Having observed that proliferating cells in the RO4929097-treated retinas underwent apoptosis at 7 and 11 d (Fig. 7b,c), we examined whether the committed neuronal progenitor cells induced by exposure to both RO4929097 and TNFα in the undamaged Tg(atoh7:GFP) transgenic line also underwent apoptosis. At 7 and 11 d, we did not observe any TUNEL-positive cells in the RO4929097 and TNFα-treated retinas (Fig. 10). Furthermore, at these time points, EdU-positive cells persisted in the RO4929097 and TNFα-exposed retina, and a few of these were GFP-positive. Together, our data suggest that both repression of Notch signaling and exposure to TNFα are sufficient to induce the Müller glia to reenter the cell cycle and to initiate the commitment of the resulting neuronal progenitor cells into a neuronal lineage.
Figure 10.
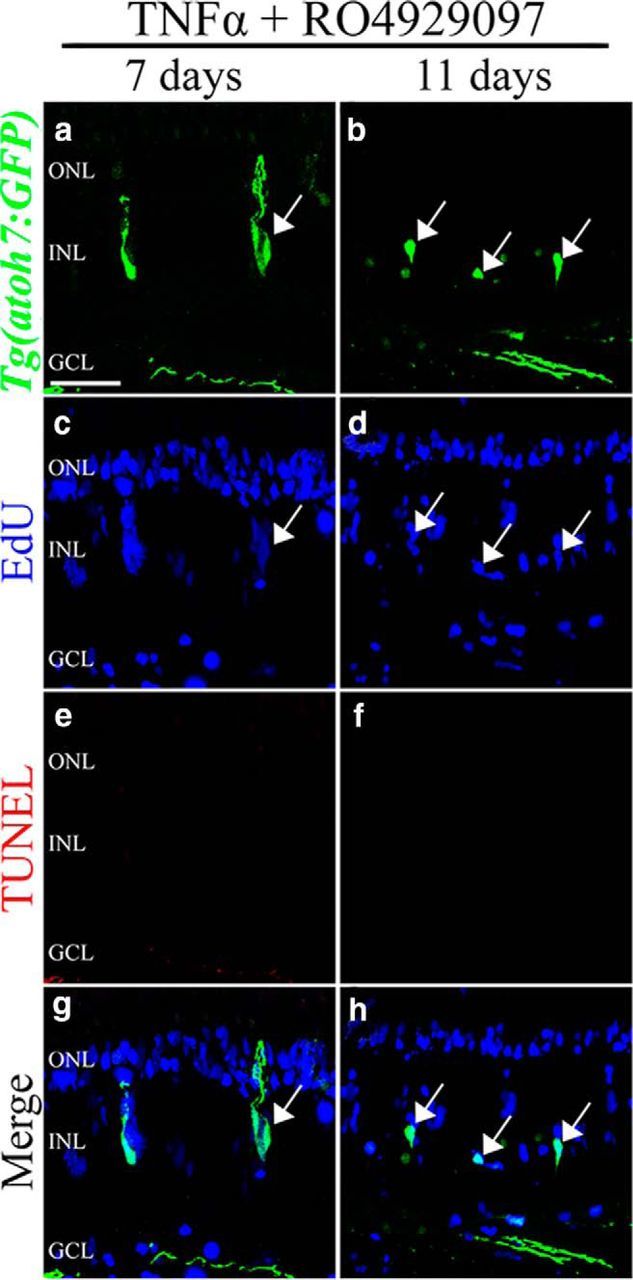
Coinjection of RO4929097 and TNFα reduces neuronal progenitor cell death. Tg(atoh7:GFP) zebrafish were coinjected with TNFα and RO4929097 and labeled with EdU. After 7 d (a, c, e, g) and 11 d (b, d, f, h), the retinas were immunolabeled for GFP (a, b, g, h), EdU (c, d, g, h), and TUNEL (e–h). The merged images of the three different labels are shown (g, h). After 7 d, the retina possessed EdU-labeled cells that were GFP-positive (arrows). No TUNEL-positive cells were detected in the retinas at either day. GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bars, 25 μm.
To determine whether these EdU-labeled neuronal progenitor cells ultimately differentiated into retinal neurons, we coinjected RO4929097 and TNFα into either wild-type or transgenic zebrafish lines [Tg(rho:Eco.NfsB-EGFP)nt19 or Tg(nrd:egfp)] and performed EdU labeling. After 11 d, we immunostained the retinas for Zpr-1, which labels double-cone cells (Zou et al., 2008), EGFP in the transgenic lines, HuC/D, to identify amacrine and ganglion cells (Wei et al., 2006), PKCα, a marker for on-bipolar cells (Haverkamp et al., 2003; Wei et al., 2006), and pERK, which labels Müller glia (Ghai et al., 2010). We found EdU-labeled double-cone cells (Fig. 11a–c, arrowheads), rod photoreceptors (Fig. 11d–i, arrowheads), bipolar cells (Fig. 11g–i, arrows), amacrine cells (Fig. 11j–l, arrows), and Müller glia (Fig. 11p–r, arrows). However, we did not detect EdU and HuC/D colabeled ganglion cells (Fig. 11j–l) or PKCα and EdU colabeled on-bipolar cells (Fig. 11m–o). This demonstrates that coinjection of RO4929097 and TNFα into an undamaged retina is not only sufficient to induce Müller glia to reenter the cell cycle and stimulate continued proliferation of neuronal progenitor cells, but also to induce differentiation of the neuronal progenitor cells into most of the retinal cell types.
Figure 11.
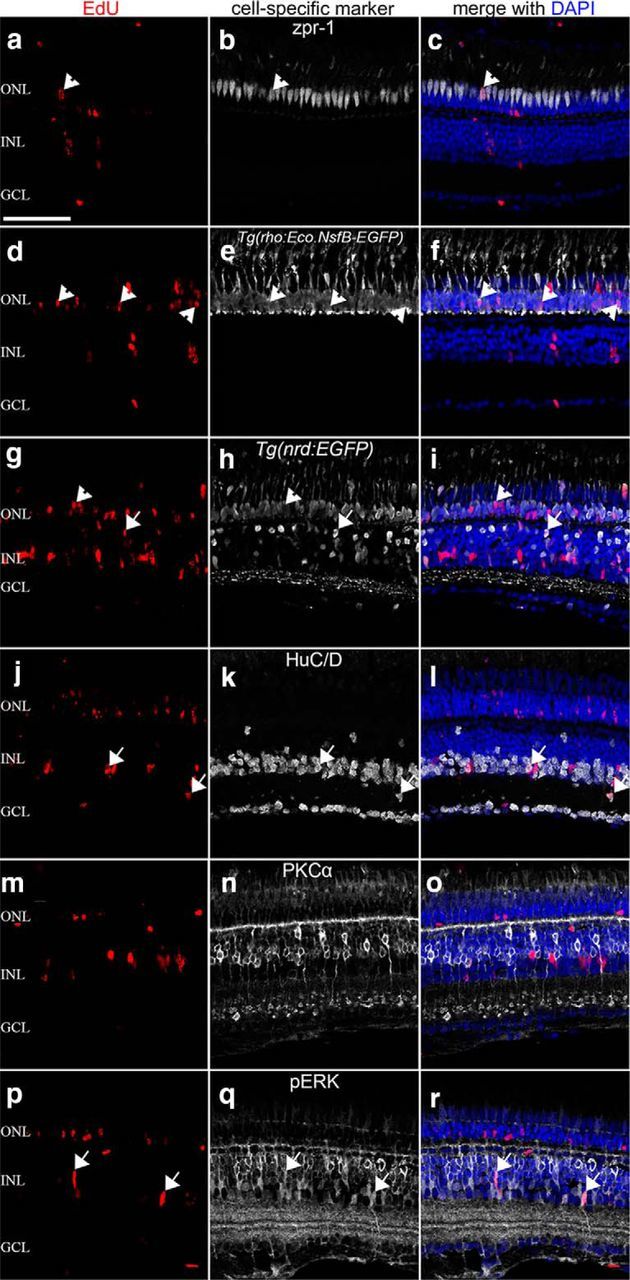
Coinjection of RO4929097 and TNFα is sufficient to induce neuronal progenitor cells to differentiate into most types of retinal neurons. Wild-type, Tg(nrd:egfp), or Tg(rho:Eco.NfsB-EGFP)nt19 zebrafish were coinjected with TNFα and RO4929097 and labeled with EdU. After 11 d, the retinas were immunolabeled for EdU (a, c, d, f, g, i, j, l, m, o, p, r), Zpr-1 (b, c), EGFP (e, f, h, i), HuC/D (k, l), PKCα (n, o), pERK (q, r), and DAPI (c, f, i, l, o, r). EdU-labeled cone (a–c) and rod (d–i) are marked with arrowheads. EdU-labeled bipolar cells (g–i), amacrine cells (j–l), and Müller glia (p–r) are indicated with arrows. There are no detectable EdU-labeled ganglion cells (j–l) or on-bipolar cells (m–o). GCL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bars, 25 μm.
Discussion
This study demonstrates, for the first time, that Notch signaling is required to repress Müller glia dedifferentiation and proliferation in the undamaged zebrafish retina. Furthermore, repressing Notch signaling in the undamaged retina is sufficient to upregulate both Ascl1a and Stat3 expression, which are both required for Müller glia proliferation, mimicking the activation mechanisms observed in the light-damaged retina (Ramachandran et al., 2010; Nelson et al., 2012; Wan et al., 2012). Surprisingly, in the absence of detectable retinal damage or TNFα upregulation, which is required for damage-induced Müller glial proliferation (Nelson et al., 2013), inhibition of Notch signaling through an unidentified mechanism stimulates Jak-mediated phosphorylation/activation of Stat3, which is necessary for Müller glial proliferation. Thus, blocking Notch signaling in the undamaged retina is sufficient to induce crucial signaling pathways that are similar to those required for Müller glia activation and proliferation in the light-damaged retina. Interestingly, exposing undamaged retinas to exogenous TNFα and repressing Notch signaling resulted in a synergistic increase in the numbers of Müller glia that reentered the cell cycle. Therefore, both an activating stimulus (TNFα) and elimination of an inhibitory signal (Notch) are necessary for maximal proliferation in the light-damaged retina. Moreover, neuronal lineage commitment and differentiation into retinal neurons were also dependent on both of these mechanisms, as loss of Notch signaling alone was insufficient to promote lineage specification of neuronal progenitor cells.
Previously, Notch signaling was shown to play a critical role in zebrafish retinal regeneration (Raymond et al., 2006; Yurco and Cameron, 2007; Wan et al., 2012). In puncture-damaged retinas, Notch signaling defines a zone of responding Müller glia (Wan et al., 2012), which proliferate in an Ascl1a-dependent manner and loss of Notch signaling expands this proliferative zone, similar to our observations in the RO4929097-treated light-damaged retina. This indicates that Notch signaling acts independent of the damage stimulus to regulate the number of Müller glia recruited into the cell cycle. Exposing the undamaged retina to RO4929097 induced Müller glia to dedifferentiate and reenter the cell cycle, which was inhibited by conditionally overexpressing NICD in the Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) zebrafish line. This demonstrated that Müller glia proliferation is specifically mediated by repressing Notch signaling rather than other potential γ-secretase targets. In contrast, inhibition of Notch signaling with the γ-secretase inhibitor DAPT was previously reported to be insufficient to induce Müller glia dedifferentiation and proliferation in the undamaged retina (Wan et al., 2012). Similarly, when we exposed undamaged retinas to either DAPT or Compound E, only a small number of Müller glia proliferated. Possible explanations for relatively low numbers of proliferating Müller glia following either DAPT or Compound E injections compared with the RO4929097-treated undamaged retinas may be the stability of each γ-secretase inhibitor, the relative ability of each inhibitor to diffuse into the retina, and the relative efficiency of each molecule to inhibit the zebrafish γ-secretase enzyme rather than the mammalian γ-secretase.
In the ventricular zone of the adult zebrafish telencephalon, Notch signaling maintains radial glia in a quiescent state (Chapouton et al., 2010). Loss of Notch signaling causes these radial glia to reenter the cell cycle and act as neural stem cells (Chapouton et al., 2010), which is analogous to our observations in the undamaged zebrafish retina. Similar mechanisms act in the injured adult zebrafish spinal cord where Notch signaling also serves as a negative regulator of neuronal progenitor cell proliferation during motor neuron regeneration (Dias et al., 2012). This suggests that Notch signaling maintains multipotent cells in a quiescent state throughout the zebrafish nervous system. It remains to be determined how Notch signaling is terminated upon damage and whether loss of Notch signaling induces Ascl1a expression and Stat3 phosphorylation/activation in these other regenerating tissues as in the retina.
Similar to light damage, repression of Notch signaling by RO4929097 in the undamaged retina resulted in increased expression of the dedifferentiation markers Ascl1a and Stat3, as well as Stat3 phosphorylation, which are required for RO4929097-induced proliferation. In the light-damage model, expression of these transcription factors is also induced by the damage signal TNFα, whereas in the RO4929097-treated undamaged retinas, TNFα is not expressed and is thus not upstream of ascl1a and stat3 signaling. Interactions between Notch signaling and both Ascl1a and Stat3 have been previously described (Sriuranpong et al., 2002; Kamakura et al., 2004; Somasundaram et al., 2005; Bhattacharya et al., 2008), but the exact mechanisms causing their upregulation upon Notch repression are currently unknown. It was previously shown that Notch targets, such as hes1, can suppress ascl1 expression in the spinal cord (Jacob et al., 2013). Thus, repression of Notch signaling could release the negative regulation of Ascl1a and result in increased Ascl1a expression in Müller glia. Surprisingly, in the undamaged retina, Notch repression results in Müller glia proliferation through Jak1-mediated phosphorylation of Stat3, but how these two signaling pathways are linked is currently unknown. Previously in Drosophila, it was demonstrated that Notch negatively regulates the expression of the Jak ligand, Unpaired, thereby suppressing Jak-Stat-mediated proliferation in intestinal stem cells (Liu et al., 2010). Although an Unpaired zebrafish homolog has not been identified, other Jak ligands may also be negatively regulated by Notch signaling.
Stat3 and Ascl1a form a central pathway leading to proliferation in both the light-damaged and RO4929097-induced undamaged retina. However, the activation mechanism is different in that TNFα, released from dying photoreceptors, is a critical signal in the light-damaged retina but is absent in the undamaged RO4929097-exposed retina. Furthermore, in the light-damaged retina, TNFα-mediated Stat3 signaling regulates TNFα expression in Müller glia that functions to recruit neighboring Müller glia into the cell cycle (Nelson et al., 2013). In contrast, Stat3 signaling in the RO4929097-induced undamaged retina is insufficient to upregulate TNFα in Müller glia, suggesting that the concentration, the subcellular localization, or temporal expression pattern of Stat3 is different. Interpreting these data obtained from the undamaged model in the context of light damage would indicate that two parallel pathways exist that result in Stat3-Ascl1a-induced proliferation. Moreover, repression of Notch signaling and exogenous application of TNFα in the undamaged retina induces Müller glia proliferation to a similar extent as in the damaged retina, likely through the same Stat3 and Ascl1a-dependent pathways (Fausett et al., 2008; Nelson et al., 2012; Nelson et al., 2013).
Attempts to induce Müller glia proliferation in the damaged mammalian retina produced limited numbers of neuronal progenitor cells that rarely differentiated (Ooto et al., 2004; Osakada et al., 2007; Karl et al., 2008). Similarly, either inhibiting Notch signaling or exogenous application of TNFα in the undamaged zebrafish retina resulted in Müller glia reentering the cell cycle without producing neuronal progenitor cells that would commit to a neuronal lineage. However, repressing Notch signaling combined with TNFα exposure caused a significant increase in the number of proliferating Müller glia and neuronal progenitors, similar to the light-damaged retina, and these neuronal progenitor cells committed to the neuronal lineage and differentiated into retinal neurons. These data suggest that proliferation and commitment/differentiation in the damaged zebrafish retina necessitate both an activating signal (TNFα) and removal of a repressing signal (Notch). However, in the context of both mouse and chick retinal regeneration research, Notch repression was shown to inhibit proliferation (Das et al., 2006; Hayes et al., 2007; Del Debbio et al., 2010; Ghai et al., 2010). Therefore, to attempt regeneration in higher vertebrates, induction of both TNFα and Notch-signaling may be required.
Recently, it was demonstrated in mouse retinal explants that viral-mediated ectopic expression of Ascl1 reprogrammed Müller glia to proliferate and generate new bipolar cells, but not mature photoreceptors (Pollak et al., 2013). Similar to the requirement of Ascl1a during zebrafish retinal regeneration (Fausett et al., 2008; Ramachandran et al., 2010), ectopic Ascl1 expression in Müller glia is sufficient to induce dedifferentiation but increased expression of only limited differentiation markers. Thus, our data showing that the combination of repression of Notch signaling and exposure to TNFα induces Ascl1a-dependent Müller glia proliferation and commitment of neuronal progenitors in the undamaged zebrafish retina is a crucial step in identifying upstream pathways that regulate Ascl1 expression and Stat3 activation. Controlling the temporal and quantitative amounts of Ascl1 through upstream signaling cascades (e.g., TNFα expression and Notch repression) might overcome limitations that ectopic Ascl1 overexpression might have on Müller glia-dependent regeneration in mammals. It was shown that Notch signaling represses Ascl1 expression in mouse neuronal progenitor cells (Jacob et al., 2013) and that the graded expression of Ascl1 is critical for specifying different neuronal fates from the same progenitor cell pool (Jacob et al., 2013). Thus, increased Ascl1 expression may be required for Müller glia and neuronal progenitor cell proliferation, but it may then need to be downregulated to render the progenitor cells amenable for commitment and differentiation into retinal neurons. It remains to be determined whether repressing or inducing Notch signaling, in combination with exposure to TNFα, is sufficient to induce a regeneration response in the damaged mammalian retina or if additional regulatory molecules are necessary.
Footnotes
This work was supported by National Eye Institute of National Institutes of Health Grant R01-EY018417 to D.R.H. and the Center for Zebrafish Research, University of Notre Dame. We thank Freimann Life Sciences technicians for their care and husbandry of the zebrafish; the Zebrafish International Resource Center (supported by National Institutes of Health Grant P40-RR12546) for providing the her6 cDNA clones; Rebecca Wingert (University of Notre Dame) for the gift of the Tg(hsp70l:Gal4);Tg(UAS:myc-notch1a-intra) transgenic line; and members of the D.R.H. laboratory for thoughtful discussions.
The authors declare no competing financial interests.
References
- Bailey TJ, Fossum SL, Fimbel SM, Montgomery JE, Hyde DR. The inhibitor of phagocytosis, O-phospho-L-serine, suppresses Müller glia proliferation and cone cell regeneration in the light-damaged zebrafish retina. Exp Eye Res. 2010;91:601–612. doi: 10.1016/j.exer.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardos RL, Lentz SI, Wolfe MS, Raymond PA. Notch-Delta signaling is required for spatial patterning and Müller glia differentiation in the zebrafish retina. Dev Biol. 2005;278:381–395. doi: 10.1016/j.ydbio.2004.11.018. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Das AV, Mallya KB, Ahmad I. Ciliary neurotrophic factor-mediated signaling regulates neuronal versus glial differentiation of retinal stem cells/progenitors by concentration-dependent recruitment of mitogen-activated protein kinase and Janus kinase-signal transducer and activator of transcription pathways in conjunction with Notch signaling. Stem Cells. 2008;26:2611–2624. doi: 10.1634/stemcells.2008-0222. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Müller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Chapouton P, Skupien P, Hesl B, Coolen M, Moore JC, Madelaine R, Kremmer E, Faus-Kessler T, Blader P, Lawson ND, Bally-Cuif L. Notch activity levels control the balance between quiescence and recruitment of adult neural stem cells. J Neurosci. 2010;30:7961–7974. doi: 10.1523/JNEUROSCI.6170-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AV, Mallya KB, Zhao X, Ahmad F, Bhattacharya S, Thoreson WB, Hegde GV, Ahmad I. Neural stem cell properties of Müller glia in the mammalian retina: regulation by Notch and Wnt signaling. Dev Biol. 2006;299:283–302. doi: 10.1016/j.ydbio.2006.07.029. [DOI] [PubMed] [Google Scholar]
- Del Debbio CB, Balasubramanian S, Parameswaran S, Chaudhuri A, Qiu F, Ahmad I. Notch and Wnt signaling mediated rod photoreceptor regeneration by Müller cells in adult mammalian retina. PLoS One. 2010;5:e12425. doi: 10.1371/journal.pone.0012425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo J, Miki K, Rattner A, Smallwood P, Zibetti C, Hirokawa K, Monuki ES, Campochiaro PA, Blackshaw S. Injury-independent induction of reactive gliosis in retina by loss of function of the LIM homeodomain transcription factor Lhx2. Proc Natl Acad Sci U S A. 2012;109:4657–4662. doi: 10.1073/pnas.1107488109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias TB, Yang YJ, Ogai K, Becker T, Becker CG. Notch signaling controls generation of motor neurons in the lesioned spinal cord of adult zebrafish. J Neurosci. 2012;32:3245–3452. doi: 10.1523/JNEUROSCI.6398-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A. 2005;102:8333–8338. doi: 10.1073/pnas.0500989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausett BV, Gumerson JD, Goldman D. The proneural bHLH gene ascl1a is required for retina regeneration. J Neurosci. 2008;28:1109–1117. doi: 10.1523/JNEUROSCI.4853-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimbel SM, Montgomery JE, Burket CT, Hyde DR. Regeneration of inner retinal neurons after intravitreal injection of ouabain in zebrafish. J Neurosci. 2007;27:1712–1724. doi: 10.1523/JNEUROSCI.5317-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghai K, Zelinka C, Fischer AJ. Notch signaling influences neuroprotective and proliferative properties of mature Müller glia. J Neurosci. 2010;30:3101–3112. doi: 10.1523/JNEUROSCI.4919-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot AJ, Vooijs MA. The role of Adams in Notch signaling. Adv Exp Med Biol. 2012;727:15–36. doi: 10.1007/978-1-4614-0899-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp S, Haeseleer F, Hendrickson A. A comparison of immunocytochemical markers to identify bipolar cell types in human and monkey retina. Vis Neurosci. 2003;20:589–600. doi: 10.1017/S0952523803206015. [DOI] [PubMed] [Google Scholar]
- Hayes S, Nelson BR, Buckingham B, Reh TA. Notch signaling regulates regeneration in the avian retina. Dev Biol. 2007;312:300–311. doi: 10.1016/j.ydbio.2007.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J, Kong J, Moore S, Milton C, Sasai N, Gonzalez-Quevedo R, Terriente J, Imayoshi I, Kageyama R, Wilkinson DG, Novitch BG, Briscoe J. Retinoid acid specifies neuronal identity through graded expression of Ascl1. Curr Biol. 2013;23:412–418. doi: 10.1016/j.cub.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamakura S, Oishi K, Yoshimatsu T, Nakafuku M, Masuyama N, Gotoh Y. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signaling. Nat Cell Biol. 2004;6:547–554. doi: 10.1038/ncb1138. [DOI] [PubMed] [Google Scholar]
- Karl MO, Hayes S, Nelson BR, Tan K, Buckingham B, Reh TA. Stimulation of neural regeneration in the mouse retina. Proc Natl Acad Sci U S A. 2008;105:19508–19513. doi: 10.1073/pnas.0807453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassen SC, Ramanan V, Montgomery JE, Burket CT, Liu CG, Vihtelic TS, Hyde DR. Time course analysis of gene expression during light-induced photoreceptor cell death and regeneration in albino zebrafish. Dev Neurobiol. 2007;67:1009–1031. doi: 10.1002/dneu.20362. [DOI] [PubMed] [Google Scholar]
- Knogler LD, Liao M, Drapeau P. Synaptic scaling and the development of a motor network. J Neurosci. 2010;30:8871–8881. doi: 10.1523/JNEUROSCI.0880-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Hunter DJ, Rooker S, Chan A, Paulus YM, Leucht P, Nusse Y, Nomoto H, Helms JA. Wnt signaling promotes Müller cell proliferation and survival after injury. Invest Ophthalmol Vis Sci. 2013;54:444–453. doi: 10.1167/iovs.12-10774. [DOI] [PubMed] [Google Scholar]
- Liu W, Singh SR, Hou SX. JAK-STAT is restrained by Notch to control cell proliferation of the Drosophila intestinal stem cells. J Chem Biochem. 2010;109:992–999. doi: 10.1002/jcb.22482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery JE, Parsons MJ, Hyde DR. A novel model of retinal ablation demonstrates that the extent of rod cell death regulates the origin of the regenerated zebrafish rod photoreceptors. J Comp Neurol. 2010;518:800–814. doi: 10.1002/cne.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch J, González-Rajal A, de la Pompa JL. Notch regulates blastema proliferation and prevents differentiation during adult zebrafish fin regeneration. Development. 2013;140:1402–1411. doi: 10.1242/dev.087346. [DOI] [PubMed] [Google Scholar]
- Nelson CM, Gorsuch RA, Bailey TJ, Ackerman KM, Kassen SC, Hyde DR. Stat3 defines three populations of Müller glia and is required for initiating maximal Müller glia proliferation in the regenerating zebrafish retina. J Comp Neurol. 2012;520:4294–4311. doi: 10.1002/cne.23213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Ackerman KM, O'Hayer P, Bailey TJ, Gorsuch RA, Hyde DR. Tumor necrosis factor-alpha is produced by dying retinal neurons and is required for Müller glia proliferation during zebrafish retina regeneration. J Neurosci. 2013;33:6524–6539. doi: 10.1523/JNEUROSCI.3838-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninov N, Borius M, Stainier DY. Different levels of Notch signaling regulate quiescence, renewal and differentiation in pancreatic endocrine progenitors. Development. 2012;139:1557–1567. doi: 10.1242/dev.076000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooto S, Akagi T, Kageyama R, Akita J, Mandai M, Honda Y, Takahashi M. Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. Proc Natl Acad Sci U S A. 2004;101:13654–13659. doi: 10.1073/pnas.0402129101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakada F, Ooto S, Akagi T, Mandai M, Akaike A, Takahashi M. Wnt signaling promotes regeneration in the retina of adult mammals. J Neurosci. 2007;27:4210–4219. doi: 10.1523/JNEUROSCI.4193-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggi L, Vitorino M, Masai I, Harris WA. Influences on neural lineage and mode of division in the zebrafish retina in vivo. J Cell Biol. 2005;19:991–999. doi: 10.1083/jcb.200509098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak J, Wilken MS, Ueki Y, Cox KE, Sullivan JM, Taylor RJ, Levine EM, Reh TA. ASCL1 reprograms mouse Muller glia into neurogenic retinal progenitors. Development. 2013;140:2619–2631. doi: 10.1242/dev.091355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Fausett BV, Goldman D. Ascl1a regulates Müller glia dedifferentiation and retinal regeneration through a Lin-28-dependent let-7 microRNA signaling pathway. Nat Cell Biol. 2010;12:1101–1117. doi: 10.1038/ncb2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Zhao XF, Goldman D. Ascl1a/Dkk/β-catenin signaling pathway is necessary and glycogen synthase kinase-3β inhibition is sufficient for zebrafish retina regeneration. Proc Natl Acad Sci U S A. 2011;108:15858–15863. doi: 10.1073/pnas.1107220108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond PA, Barthel LK, Bernardos RL, Perkowski JJ. Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Dev Biol. 2006;6:36. doi: 10.1186/1471-213X-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/S0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Scheer N, Campos-Ortega JA. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mech Dev. 1999;80:153–158. doi: 10.1016/S0925-4773(98)00209-3. [DOI] [PubMed] [Google Scholar]
- Scheer N, Groth A, Hans S, Campos-Ortega JA. An instructive function for Notch in promoting gliogenesis in the zebrafish retina. Development. 2001;128:1099–1107. doi: 10.1242/dev.128.7.1099. [DOI] [PubMed] [Google Scholar]
- Scheer N, Riedl I, Warren JT, Kuwada JY, Campos-Ortega JA. A quantitative analysis of the kinetics of Gal4 activator and effector gene expression in the zebrafish. Mech Dev. 2002;112:9–14. doi: 10.1016/S0925-4773(01)00621-9. [DOI] [PubMed] [Google Scholar]
- Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Somasundaram K, Reddy SP, Vinnakota K, Britto R, Subbarayan M, Nambiar S, Hebbar A, Samuel C, Shetty M, Sreepathi HK, Santosh V, Hegde AS, Hegde S, Kondaiah P, Rao MR. Upregulation of ASCL1 and inhibition of Notch signaling pathway characterize progressive astrocytoma. Oncogene. 2005;24:7073–7083. doi: 10.1038/sj.onc.1208865. [DOI] [PubMed] [Google Scholar]
- Sriuranpong V, Borges MW, Strock CL, Nakakura EK, Watkins DN, Blaumueller CM, Nelkin BD, Ball DW. Notch signaling induces rapid degradation of achaete-scute homolog 1. Mol Cell Biol. 2002;22:3129–3139. doi: 10.1128/MCB.22.9.3129-3139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse B, Pfumio S, Fürthauer M, Loppin B, Heyer V, Degrave A, Woehl R, Lux A, Steffan T, Charbonnier XQ, Thisse C. Expression of the zebrafish genome during embryogenesis (NIH R01 RR15402) ZFIN Direct Data Submission. 2001. http://zfin.org.
- Thomas JL, Ochocinska MJ, Hitchcock PF, Thummel R. Using the Tg(nrd:egfp)/albino zebrafish line to characterize in vivo expression of neurod. PLoS One. 2012;7:e29128. doi: 10.1371/journal.pone.0029128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Kassen SC, Montgomery JE, Enright JM, Hyde DR. Inhibition of Müller glial cell division blocks regeneration of the light-damaged zebrafish retina. Dev Neurobiol. 2008;68:392–408. doi: 10.1002/dneu.20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Enright JM, Kassen SC, Montgomery JE, Bailey TJ, Hyde DR. Pax6a and Pax6b are required at different points in neuronal progenitor cell proliferation during zebrafish photoreceptor regeneration. Exp Eye Res. 2010;90:572–582. doi: 10.1016/j.exer.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Bailey TJ, Hyde DR. In vivo electroporation of morpholinos into the adult zebrafish retina. J Vis Exp. 2011;58:e3608. doi: 10.3791/3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Chona FR, Swan A, Ferrell WD, Jiang L, Baehr W, Chien WM, Fero M, Marc RE, Levine EM. Proliferative reactive gliosis is compatible with glial metabolic support and neuronal function. BMC Neurosci. 2011;12:98. doi: 10.1186/1471-2202-12-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihtelic TS, Hyde DR. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J Neurobiol. 2000;44:289–307. doi: 10.1002/1097-4695(20000905)44:3<289::AID-NEU1>3.0.CO%3B2-H. [DOI] [PubMed] [Google Scholar]
- Vitorino M, Jusuf PR, Maurus D, Kimura Y, Higashijima S, Harris WA. Vsx2 in the zebrafish retina: restricted lineages through derepression. Neural Dev. 2009;4:14. doi: 10.1186/1749-8104-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Ramachandran R, Goldman D. HB-EGF is necessary and sufficient for Müller glia dedifferentiation and retina regeneration. Dev Cell. 2012;22:334–347. doi: 10.1016/j.devcel.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Luo Y, Hyde DR. Molecular cloning of three zebrafish lin7 genes and their expression patterns in the retina. Exp Eye Res. 2006;82:122–131. doi: 10.1016/j.exer.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Yurco P, Cameron DA. Cellular correlates of proneural and notch-δ gene expression in the regenerating zebrafish retina. Vis Neurosci. 2007;24:437–443. doi: 10.1017/S0952523807070496. [DOI] [PubMed] [Google Scholar]
- Zou J, Lathrop KL, Sun M, Wei X. Intact retinal epithelium maintained by Nok is essential for retinal epithelial polarity and cellular patterning in zebrafish. J Neurosci. 2008;28:13684–13695. doi: 10.1523/JNEUROSCI.4333-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]