The mechanism of mismatch repair and the functional analysis of mismatch repair defects in Lynch syndrome (original) (raw)
. Author manuscript; available in PMC: 2014 Nov 18.
Published in final edited form as: Fam Cancer. 2013 Jun;12(2):159–168. doi: 10.1007/s10689-013-9635-x
Abstract
The majority of Lynch syndrome (LS), also known as hereditary non-polyposis colorectal cancer (HNPCC), has been linked to heterozygous defects in DNA mismatch repair (MMR). MMR is a highly conserved pathway that recognizes and repairs polymerase misincorporation errors and nucleotide damage as well as functioning as a damage sensor that signals apoptosis. Loss-of-heterozygosity (LOH) that retains the mutant MMR allele and epigenetic silencing of MMR genes are associated with an increased mutation rate that drives carcinogenesis as well as microsatellite instability that is a hallmark of LS/HNPCC. Understanding the biophysical functions of the MMR components is crucial to elucidating the role of MMR in human tumorigenesis and determining the pathogenetic consequences of patients that present in the clinic with an uncharacterized variant of the MMR genes. We summarize the historical association between LS/HNPCC and MMR, discuss the mechanism of the MMR and finally examine the functional analysis of MMR defects found in LS/HNPCC patients and their relationship with the severity of the disease.
Keywords: DNA mismatch repair system, Lynch syndrome, Hereditary non polyposis colorectal cancer (HNPCC), Functional defects, hMSH2–hMSH6, hMLH1–hPMS2
Introduction
The history of the Lynch syndrome or hereditry nonpolyposis colorectal cancer (LS/HNPCC) dates back to 1895, when Dr. Aldred Warthin, intrigued by an increased onset of bowel and endometrial cancers in the family of his seamstress, performed a complete study of this family that was ultimately published in 1913 [1, 2]. The significance of early onset colon, gastric and endometrial carcinomas in the “Family G” of Warthin, could not be totally appreciated until two other families were described by Lynch [1, 3]. These families did not follow the diagnostic for Familial adenomatous polyposis (FAP) and presented with an absence of colon polyps in addition to other types of cancer, among them endometrial cancer.
Discovery of the genes involved in Lynch syndrome
In 1992, the APC gene was excluded as responsible for LS/HNPCC [4]. A year later, instability of simple repeated sequences (microsatellite instability or MSI) was detected in sporadic colorectal cancer (CRC) [5] and MSI was found to be associated with CRC tumors in LS/HNPCC patients [6, 7]. The observation of MSI implicated mismatch repair (MMR) processes that had been previously described in bacteria and yeast [8-10]. The MMR system is primarily responsible for the recognition and repair of nucleotide polymerase misincorporation errors introduced during replication [11-13]. Simple repeat sequences appear particularly prone to polymerase misincorporation errors and the resulting MSI appears to be a litmus for MMR defects [14]. The central players in Escherichia coli MMR are MutS, MutL and MutH.
In December of 1993, the human MutS homolog (MSH), hMSH2, was identified and associated with LS/HNPCC [15, 16]. In March of 1994, the human MutL homolog, hMLH1, was identified and associated with LS/HNPCC [17, 18]. Subsequently, the hPMS1 and hPMS2 (_p_ost-_meiotic s_egregation MutL homologs) genes were identified and suggested to be causative of LS/HNPCC [19]. Later genetic analysis of the hPMS1 gene excluded it as contributor to LS/HNPCC, while the hMSH6 and hMLH3 genes were ultimately included as causative genes in LS/HNPCC [20, 21].
Microsatellite instability
Vast majority of the cells deficient in MMR develop a “mutator phenotype” characterized by 102–103 fold increase in the spontaneous mutation rate [5, 22]. Elevated mutation rates affect the entire genome including DNA sequences that contain microsatellite repeats [23, 24]. A number of genes have been identified that include microsatellite sequences within their coding region [25, 26]. MSI in these genes results in altered signaling transduction, apoptosis, DNA repair, transcriptional regulation, protein translocation and modifications, and immune monitoring. For example, intragenic MSI results in inactivation of the TFGβ-RII tumor suppressor gene in ~80 % of MMR-defective tumors, while the remaining ~20 % appear to inactive the IGFRII tumor suppressor via intragenic MSI [27, 28]. Similarly, intragenic MSI also appears to inactivate the apoptosis promoter BAX [29].
The presence of high level of MSI (MSI-H) [14] is normally associated within a mutation of the hMLH1 and hMSH2 genes [30, 31]. A low level of MSI (MSI-L) [14] appears largely due to mutations in the hMSH6 gene (10 %), and the hPMS2 gene (5 %). The etiology of approximately 5 % of MSI tumors remains unknown [32]. More than 95 % of LS/HNPCC tumors show MSI, whereas only 10–15 % of the sporadic colorectal cancers display MSI [14, 30, 33]. Importantly, diagnostic MSI has become a dependable indicator of MMR defects in human tumors once reliable markers were established [14, 34].
Mismatch repair
The conversion of heteroduplex (mismatched) to homoduplex (nonmismatched) DNA following transformation into Pneumococcus began studies of MMR in the early 1970s [35, 36]. In 1975, Wildenberg and Meselson [37] demonstrated that E. coli differentially corrected λ DNA containing genetically defined mismatched nucleotides. Shortly thereafter, and based on observations of DNA adenine methylation biases in Okazaki fragments by Marinus [38], Radman and Meselson [39] suggested that E. coli MMR could correctly identify a polymerase nucleotide misincorporation error within double-stranded DNA (dsDNA) by uniquely excising a transiently unmethylated newly replicated strand. These seminal studies positioned E. coli as the paradigm for MMR where the previously identified mutator genes MutS [40], MutL [41], MutH [42], UvrD [42] and the DNA adenine methylase (Dam) [43] were determined to be required for the process. Interestingly, only MutS and MutL appear to be highly conserved throughout evolution, although there may be functional conservation of the other MMR activities (Table 1).
Table 1.
DNA mismatch repair protein functions
| E. coli | Yeast | Human | Overall function |
|---|---|---|---|
| MutS | Msh2–Msh3 | hMSH2–hMSH3 | Mismatch and small IDLs recognition. ATP-bound sliding clamp formation |
| Msh2–Msh6 | hMSH2–hMSH6 | ||
| MutL | Mlh1–Pms1 | hMLH1–hPMS2 | Coordinator of the downstream processes after mismatch recognition by MutS. GHRL ATPase. Cryptic endonuclease |
| Mlh1–Mlh2 | hMLH1–hPMS1 | ||
| Mlh1–Mlh3 | hMLH1–hMLH3 | ||
| MutH | – | – | Nick newly synthesized DNA strand in hemimethylated GATC sites |
| γ-δ complex | RFC complex | RFC complex | β-clamp loading |
| beta-clamp | PCNA | PCNA | Connects mismatch repair machinery to the replication fork |
| ExoI, ExoX | ExoI | hEXO1 | 5′–3′ DNA excision |
| RecJ ExoVII | ? | ? | 3′–5′ DNA excision |
The principal function of the MMR system is to correct DNA polymerase misincorporation errors that arising during DNA replication [44]. Overall the MMR system increases replication fidelity 100- to 1000-fold [45, 46]. MMR is also involved in ensuring the efficiency and fidelity of both mitotic and meiotic DNA recombination, class-switch recombination and somatic hypermutation of the variable regions of immunoglobulin genes, interstrand DNA cross-link repair, repair of aberrant triple-repeat expansions and in responses to DNA damage by participating in S and G2/M phase checkpoints as well as apoptosis [47].
The mechanism of mismatch repair
The DNA mismatch repair system is a bidirectional excision-resynthesis system that is initiated at a defined strand scission that is 3′- or 5′- of a mismatch and the excision tract extends to a nonspecific point just past the mismatch [12, 13]. The process can be divided into four main steps: (1) Recognition of a mismatch by the MSHs, (2) recruitment of the MLHs by ATP-bound MSHs that then connect the mismatch recognition signal to the distant DNA strand scission where excision begins, (3) excision of the DNA strand containing the wrong nucleotide and (4) resynthesis of the excision gap by the replicative DNA polymerase using the remaining DNA strand as a template. This latter step appears virtually identical to normal replicative DNA synthesis and will not be discussed in detail here. Clearly the unique aspect of MMR is the well-defined and targeted mismatch-dependent DNA strand excision that begins at a defined strand scission and extents to a non-specific point just beyond the mismatch.
The mechanism of MMR excision has been controversial and many of the detailed biophysical steps remain poorly understood. One of the most controversial issues was how the recognition of a mismatch is transmitted to a distant strand scission site along the DNA where the excision step begins. In bacteria, excision is initiated at a GATC site that has been incised (nicked) on the unmethylated strand by the MutH protein [48]. The location of this MutH incision can be several thousand nucleotides distant from the mismatch and on either the 5′- or 3′- side of the mismatch [49, 50]. While not absolutely demonstrated, it appears that the DNA strand scissions used to initiate excision in eukaryotes are likely to be the 3′-leading strand or the 5′-lagging strand ends. Thus the requirement of methylation and a MutH homolog appears to have been eliminated in eukaryotes as well as perhaps every organism except gram-negative enteric bacteria like E. coli. Regardless, it is clear that in all MMR systems, mismatch recognition by MSH proteins must be transmitted to a distant DNA strand scission.
Three conceptual mechanisms were developed to envision connecting mismatch binding to a distant strand incision [51]: (1) Static Transactivation [52], (2) Hydrolysis-Dependent Translocation [53, 54] and (3) Molecular Switch Sliding Clamp [55-58]. Static Transactivation suggests that a complex of the MMR proteins is assembled at the mismatch, which then awaits a random DNA looping collision event that brings the incision site in contact with this complex. This “trans-activation” model was largely eliminated by the observation that placing a block between the mismatch and the strand scission blocked MMR [59]. An intervening block should not affect a mechanism where DNA may loop to the distant strand scission site. Yet MMR excision was abolished when a variety or blocks were placed between the mismatch and the strand scission [59].
The remaining two mechanisms may be considered “cis-activation” models since they envision movement of a MMR complex along the DNA helix to the DNA strand scission; where an intervening block would stop MMR. These two models have been very difficult to distinguish since one requires ATP binding plus hydrolysis in order to “motor” down the DNA helix (Hydrolysis-Dependent Translocation), while the other only requires mismatch provoked ATP binding that results in the formation of an MSH hydrolysis-independent sliding clamp capable of 1-dimensional (1D) diffusion along the DNA helix (Molecular Switch Sliding Clamp). A growing body of evidence, including single molecule analysis, appears to largely underpin the Molecular Switch Sliding Clamp model [60, 61]. Our group developed a complete Molecular Switch Sliding Clamp model for the MMR excision reaction nearly a decade ago [55, 56, 62]. We will highlight several important features of MSH and MLH protein functions within the context of the Molecular Switch Sliding Clamp model where defects may be relevant to the pathobiology of LS/HNPCC.
MutS homologs
All MSH proteins appear to exist as an asymmetric homodimer or heterodimer where the dimeric/heterodimeric interaction domains are confined to the C-terminal regions (Fig. 1; [63]). In addition, MSH proteins are members of the ABC family of ATPases and contain a highly conserved Walker A/B nucleotide binding motif (Fig. 1; [52, 64]). The MSH ATPase domain is absolutely required for translocations from the mismatch to the distant strand scission. A fundamental feature of the Molecular Switch Sliding Clamp model envisions MSH proteins as a mismatch-provoked molecular switch that utilizes nucleotide binding and exchange mechanics similar to G proteins [56]. Most if not all MSH proteins display asymmetric ATP binding by the homodimeric subunits or the heterodimeric partners [65]. In eukaryotes the MSH6 subunit appears to bind and hydrolyze ATP very quickly [66]. The binding of ATP by MSH6 appears to enhance ATP binding by MSH2 [67]. Once the MSH6 subunit hydrolyzes ATP it appears to immediately release the ADP product [66-68]. In contrast, the MSH2 subunit retains the ADP product which appears to suppress further ATP binding by MSH6 [67, 68]. Interaction with a mismatch induces the release of the ADP bound by the MSH2 subunit allowing ATP binding by both MSH2 and MSH6, which results in the formation of a hydrolysis-independent sliding clamp capable of 1D diffusion along the DNA helix [57, 58, 60]. It is this mismatch-provoked nucleotide exchange that identified MSH proteins as a molecular switch [57, 69, 70]. The nature of bacterial MutS has precluded a similarly detailed subunit analysis of ATP bind-hydrolysis and nucleotide exchange. However, bacterial MutS clearly displays mismatch-induced ADP release that results in the formation of an ATP-bound hydrolysis-independent sliding clamp [55, 61]. These observations appear to underline a conserved biophysical mechanism for all MSH proteins [71].
Fig. 1.
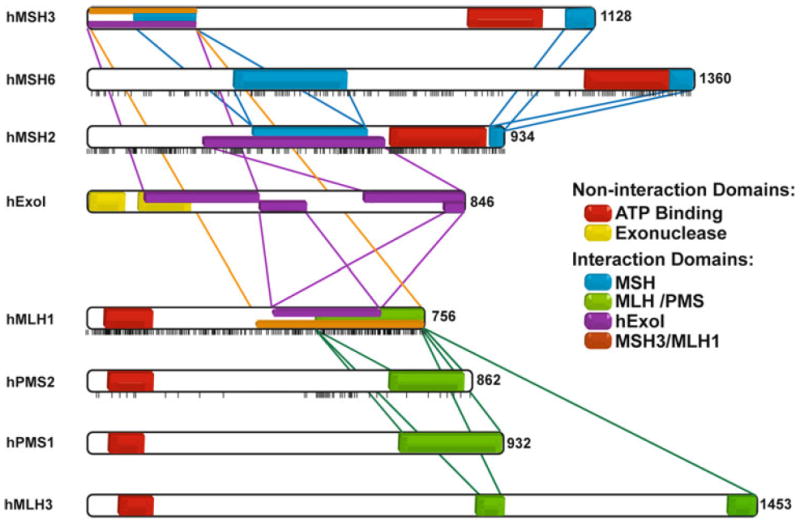
Interaction regions between MSH, MLH and EXO proteins and distribution of missense mutations in the main MMR proteins. Each vertical bar represents a missense mutation described in a patient and reported to the InSight database. Two important noninteracting regions of these proteins are also represented: the ATP binding domain and the exonuclease domain of hEXO1. Interaction regions among MSH proteins are represented in blue, among MLH/PMS proteins in green and between both groups of proteins in orange. Interactions with hExoI are represented in purple
While searching for a mismatch, MSH proteins appear to form an incipient clamp that tracks along the duplex DNA helix and undergoes rotational diffusion while in continuous contact with the backbone [60, 72]. At physiological salt the dwell time (lifetime) of MSH proteins on duplex DNA is approximately 1 s [61]. These same studies determined a diffusion coefficient that suggested a random-walk search distance capable of examining ~700 bp of naked DNA in that time. A diminished search area on chromatin is likely since nucleosomes will introduce a topological barrier to rotational diffusion [73]. In spite of these issues MSH proteins appear capable of recognizing a mismatch within a nucleosome [74].
Once a mismatch is encountered the MSH protein lingers for approximately 3 s [61]. While there have been a number of inferences regarding base-flipping and induced DNA bending [75, 76], it is more likely that MSH proteins linger at the mismatch because they encounter an altered intrinsic flexibility in the DNA surrounding a mismatch [77]. Such backbone flexibility would likely be distinct from the smooth helical backbone that would promote unhindered rotational diffusion during the mismatch search [77]. The fact that the MSH protein does not detect the mismatch pre se, but instead detects the intrinsic DNA flexibility of the mismatch region may explain the wide range of mismatches and lesions recognized by MSH proteins [77]. One imagines that the lingering process provides some time for the ordering of disordered or partially disordered peptides within the MSH dimer/heterodimer [78] that ultimately induces ADP release and ATP binding by both subunits [67, 68].
One of the dimeric bacterial MutS subunits, or in eukaryotes the MSH6 subunit, appears to directly interrogate the mismatched base through a conserved Phe-X-Glu motif in the N-terminal domain during this peptide ordering process [78-80]. The formation of an ATP-bound hydrolysis independent sliding clamp considerably increases the dwell time on the DNA to approximately 10 min and alters the diffusion mechanics such that the MSH freely rotates around the DNA backbone [60, 61]. The altered lifetime and diffusion mechanics suggests MSH proteins may diffuse away from the mismatch by a random-walk that could encompass hundreds of thousands of nucleotides along naked DNA. Moreover, multiple sliding clamps may be loaded once the MSH is released from the mismatch [58]. The long lifetime and multiple sliding clamps results in the ability to displace nucleosomes on model chromatin substrates by occupying transiently unwrapped DNA [81]. One might imagine a similar occupation/clearing mechanism for protein obstacles on DNA surrounding a mismatch that could be important for the MMR excision process.
MutL homologs
The detailed function(s) of the MLH proteins remains enigmatic. All MLH proteins studied to date exist as an asymmetric homodimer or heterodimer where the dimer/heterodimer interaction domains are located at the C-terminus of the respective proteins (Fig. 1; [82]). In addition, MLH proteins contain an N-terminal GHKL ATP binding cassette (Fig. 1; [83]). ATP binding by MLH appears to result in dimerization of the two ATPase-containing N-terminal domains [84, 85]. The role of this dimerization in MMR is unknown. It was reported that MLH proteins bind to single stranded DNA (ssDNA; [86, 87]. However, single molecule analysis has demonstrated that stable ssDNA binding does not occur in physiological salt [88]. These results suggest at best there may be a transient interaction with ssDNA which may have relevance for MMR. Interestingly, the human PMS2 and its yeast homolog Pms1 have been shown to contain an intrinsic endonuclease activity that is located in a largely disordered domain near the C-terminus [89-91]. It is interesting to note that E.coli MutL and all known gram negative enteric bacterial MLH’s do not appear to contain an intrinsic endonuclease, while most other bacteria including gram positiveB.subtilus MutL do contain an intrinsic endonuclease that is required for MMR [92, 93]. As outlined below, this cryptic endonuclease may account for an apparent lack of a 3′–5′ exonuclease in eukaryotes (and perhaps most other types of bacteria) that would be essential to facilitate bidirectional excision surrounding a mismatch.
A large body of evidence suggests that MLH proteins only interact stably with ATP-bound MSH sliding clamps [55, 94-96]. These results strongly suggest that MSH proteins may deliver MLH proteins to the site of excision and/or other functions. There is recent cellular evidence that suggests the MLH proteins may be left behind during or following repair [97]. A lack of clear evidence for MLH functions, once they have interacted with ATP-bound MSH proteins, leaves this part of the MMR mechanism substantially murky. It is likely that direct visualization on single molecules will help to clarify the role of MLH proteins in MMR.
Accessory MMR proteins
Major accessory proteins required for the bacterial MMR excision reaction include the single stranded DNA (ssDNA) binding protein SSB, the RecQ family DNA helicase UvrD, one of four exonucleases (the 5′ → 3′ exonucleases ExoI and ExoX, or the 3′ → 5′ exonucleases RecJ and Exo VII), and the replicative processivity factor β-clamp [98-101]. Similarly, the 3′ → 5′ exonuclease ExoI, the ssDNA binding heterotrimer RPA and PCNA are major accessory factors in eukaryotic MMR [49, 102, 103]. Interestingly, there have been no helicases shown to be essential for the eukaryotic MMR excision reaction, although there are many more apparently redundant RecQ family helicases in eukaryotes than prokaryotes.
The unique genetic requirement for a single 5′ → 3′ exonuclease (ExoI) in eukaryotes appeared inconsistent with bidirectional MMR excision until the intrinsic endonuclease in hPMS2 was identified and subsequently verified in Saccharomyces cerevisiae Pms1 as well as B.subtilus MutL [89, 90, 92, 93]. A simple mechanistic interpretation would suggest that MLH proteins containing the intrinsic endonuclease may occasionally nick the strand being excised in the 3′-direction, and its subsequent thermal or active displacement presents a 5′-end to ExoI for degradation. This result is consistent with biochemical studies that suggest hMLH1–hPMS2 is required for 3′-excision but not 5′-excision [49, 102, 103].
Assessing functional defects associated with MMR proteins
A major issue in the medical genetics of LS/HNPCC is assessing the functional implications of missense MMR gene alterations with unknown significance (Table 2). Such issues do not arise in large families where MMR alterations may be correlated with cancer predisposition. However, the advent of molecular diagnostic technologies where significant family history is either lacking or non-existent has presented genetic counselors and clinicians with diagnostic challenges that may only be resolved with some functional analysis. The problem becomes even more apparent when one localizes the missense variants along the length of the MMR genes (see black ticks below MMR genes in Fig. 1). In the case of both hMSH2 and hMLH1 the missense variants appear to be scattered over the entire length of the genes. This observation appears to preclude a simple bioinformatic analysis that might be based on the identification of important functional domains. Amusingly, for both hMSH6 and hPMS2 there appear to be clusters of mutations that may target important functional regions; although the statistical significance of such an interpretation appears limited by the reduced numbers of variants. Interestingly, an hPMS2 missense mutation cluster appears to be located near the N-terminal side of the hMLH1 interaction domain (Fig. 1). This is the general location of the metal binding domain that is associated with the cryptic endonuclease of hPMS2.
Table 2.
Mutations reported in the DNA MMR genes
| hMSH2 | hMSH6 | hMLH1 | hPMS2 | |
|---|---|---|---|---|
| Insertions | 28 (2) | 18 (6) | 34 (5) | 3 (2) |
| Deletions | 280 (37) | 118 (25) | 303 (42) | 34 (9) |
| Duplications | 74 (7) | 57 (18) | 778 (4) | 8 (1) |
| Missense | 221 | 161 | 289 | 29 |
| Nonsense | 107 | 55 | 92 | 18 |
| Splice variants | 16 | 8 | 51 | 9 |
Considering the protein mechanics outlined above one could envision a number of functional defects associated with missense mutation of the MMR genes. Both the MSH and MLH proteins contain heterodimeric interaction domains, essential ATP binding/hydrolysis domains and protein–protein interaction domains that might be susceptible to amino acid changes in the regions (Fig. 1). In addition, there are several important structural features such as the alpha helices in the MSH connector domains that link the mismatch binding domain with the ATP processing domain that might be susceptible to amino acid changes in these regions. It is not surprising that the first attempts at functional analysis focused on the MSH and MLH heterodimer interaction domains; a relatively easy test that used Glutathione-S-Transferase (GST) tagged MMR protein with in vitro transcribed-translated (IVTT) interacting partner [63, 82]. Precipitation of the GST-tagged MMR protein determined whether a missense mutation in the IVTT partner was capable of a stable interaction. Since these early studies functional analysis has fallen into two major catagories: (1) targeted functional studies (such as the GST-IVTT interaction analysis), and (2) analysis of the complete MMR reaction. All of the functional analysis assays have both strengths and weaknesses although none could be considered sufficient for a full-proof medical diagnostic.
Targeted functional analysis has identified missense variants that are defective in MMR heterodimer interactions as well as interactions between the MMR proteins [63, 82, 104-108]. These assays included both the GST-IVTT as well as yeast two-hybrid analysis. Immunohistochemical precipitation has provided a potential cellular variation of the interaction assay for hMLH1 that also included studies of protein stability and localization [109]. Combining yeast two hybrid with in vitro mismatch repair assays has similarly extended the interaction analysis [110].
The recently published structure of hMSH2–hMSH6 has allowed the localization of known missense variants of both these proteins (Fig. 2). As with missense mutation localization on the linear protein sequence (Fig. 1), the localization of missense mutations on the 3D structure does not appear to provide additional insights into a useful medical diagnostic (Fig. 2). The biochemical analysis of hMSH2 has examined heterodimer interactions as well as the ability of the protein to process ADP/ATP and form multiple sliding clamps on DNA [63, 111]. It is interesting that several of the ADP/ATP processing functions appear to be altered by missense variants that are distant from the Walker A/B nucleotide binding domain.
Fig. 2.
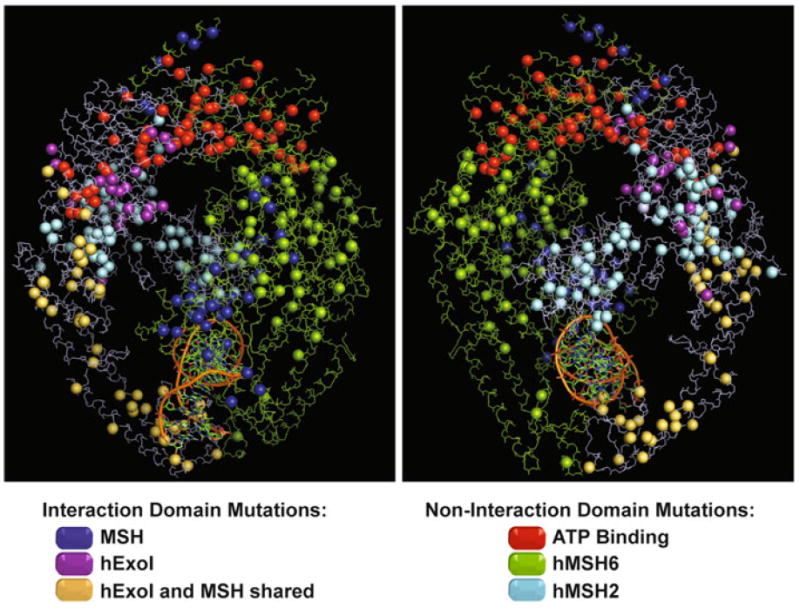
Spatial distribution of missense mutations described in the hMSH2-hMSH6 protein complex. The hMSH2 and hMSH6 proteins are delineated in blue and green respectively. Missense mutations are shown as spheres. Mutations located in the ATPase domain are represented in red. Mutations located in the interaction domain between both proteins are marked in dark blue. Mutations in hMSH2 protein located in the interaction region only with hEXO1 are purple and mutations located in the shared interaction region with hEXO1 and hMSH6 are represented in yellow (See Fig. 1). Other mutations are colored based on the protein color where it is found
Transient MMR protein expression has underpinned the analysis of the complete MMR reaction [112-115]. In these studies the MMR gene is transiently expressed in a complementary defective cell line and the resulting complete MMR activity is examined. A complete MMR reaction could be determined using one of several in vitro repair reactions. A similar cellular extract system may examine potential splice site variants that result in a defective MMR protein [116, 117]. The advantage to all of these assay systems is that if a functional defect is identified, it can often be interpreted as pathogenic. The disadvantage to of the assay systems is the scale of significance in any given defect. For example, if a protein interaction is reduced by 10 % is such a defect pathogenic? Moreover, the absence of a defect in any one of these assay systems does not necessarily eliminate a missense variant from being pathological.
Finally, the role of MMR genes in DNA damage sensing may require a completely different type in functional analysis [24, 118, 119]. Perhaps the DNA damage signaling functions of the MMR genes are the crux of tumorigenesis in LS/HNPCC and only a functional assay that examines such will be informative? If this is the case, then none of the functional assays developed to date is sufficient for a clinical diagnostic.
Prospects
Besides the disadvantages of the functional analysis outlined above, a major bottleneck in all of these assays is that each of the missense variants must be constructed de novo and then introduced onto the assay system for analysis. For several of the biochemical assays the proteins must be purified to near homogeneity to assure that the analysis is measuring the MMR protein function and not a sample contaminant. In fact, several of the published functional analysis for LS/HNPCC can be easily criticized as measuring indirect activities. Immunoprecipitation assay of MMR interactions would fall into this latter critique since non-specific protein interactions in these complex protein precipitates could affect the outcome. Ideally, one would like to examine a patient sample directly to determine MMR function. In essence this would require an assay that was extremely sensitive and could detect function from very small amounts of tissue samples.
Single molecule analysis may provide both the necessary sensitivity and ability to examine MMR functions with minute samples. As the name suggests, single molecule analysis examines single DNA molecules. Sample sizes are necessarily small and relevant target protein concentrations are usually in the low nanomolar. Moreover, recent studies have demonstrated that complex biological samples may be used in single molecule analysis as long as the read-out is specific [120]. Nearly all single-molecule systems utilize fluorescent probes that either tag the protein or the DNA [121, 122]. Combining two compatible fluorophores allows the detection of fluorescence resonance energy transfer (FRET), where the emission of one fluorophore may excite a second nearby fluorophore into an emission at a longer wavelength. FRET decays with the sixth power of the distance between fluorophors making it superbly sensitive to very small distance changes. A similar FRET system has been used by our group to examine the biophysical mechanism of MMR [60, 61, 88]. One could easily envision several combined FRET systems that examine mismatch recognition, protein interactions, and perhaps the entire MMR reaction with exceedingly small patient samples. Moreover, the system appears amenable to developing a DNA damage signaling assay that relies on MMR interactions [123].
Acknowledgments
The authors would like to thank Christopher Heinen and members of the Fishel Laboratory for helpful discussions. This work was supported by NIH Grant CA67007.
Footnotes
Conflict of interest The authors declare no conflicts of interest.
Contributor Information
Juana V. Martín-López, Department of Molecular Virology, Immunology and Medical Genetics, Human Cancer Genetics, The Ohio State University Wexner Medical Center, Columbus, OH 43210, USA
Richard Fishel, Email: rfishel@osu.edu, Department of Molecular Virology, Immunology and Medical Genetics, Human Cancer Genetics, The Ohio State University Wexner Medical Center, Columbus, OH 43210, USA; Physics Department, The Ohio State University, Columbus, OH 43210, USA.
References
- 1.Lynch HT. Classics in oncology. Aldred Scott Warthin, M.D., Ph.D. (1866–1931) CA Cancer J Clin. 1985;35(6):345–7. doi: 10.3322/canjclin.35.6.345. [DOI] [PubMed] [Google Scholar]
- 2.Classics in oncology. Heredity with reference to carcinoma as shown by the study of the cases examined in the pathological laboratory of the University of Michigan, 1895–1913. By Aldred Scott Warthin. 1913. CA Cancer J Clin. 1985 Nov-Dec;35(6):348–59. doi: 10.3322/canjclin.35.6.348. [DOI] [PubMed] [Google Scholar]
- 3.Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer. 1996;78(6):1149–1167. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1149::AID-CNCR1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 4.Peltomaki P, Sistonen P, Mecklin JP, et al. Evidence that the MCC-APC gene region in 5q21 is not the site for susceptibility to hereditary nonpolyposis colorectal carcinoma. Cancer Res. 1992;52(16):4530–4533. [PubMed] [Google Scholar]
- 5.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363(6429):558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 6.Peltomaki P, Lothe RA, Aaltonen LA, et al. Microsatellite instability is associated with tumors that characterize the hereditary non-polyposis colorectal carcinoma syndrome. Cancer Res. 1993;53(24):5853–5855. [PubMed] [Google Scholar]
- 7.Aaltonen LA, Peltomaki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260(5109):812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 8.Levinson G, Gutman GA. High frequencies of short frameshifts in poly-CA/TG tandem repeats borne by bacteriophage M13 in Escherichia coli K-12. Nucleic Acids Res. 1987;15(13):5323–5338. doi: 10.1093/nar/15.13.5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reenan RA, Kolodner RD. Isolation and characterization of two Saccharomyces cerevisiae genes encoding homologs of the bacterial HexA and MutS mismatch repair proteins. Genetics. 1992;132(4):963–973. doi: 10.1093/genetics/132.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strand M, Prolla TA, Liskay RM, Petes TD. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature. 1993;365(6443):274–276. doi: 10.1038/365274a0. [DOI] [PubMed] [Google Scholar]
- 11.Fishel R, Kolodner RD. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev. 1995;5(3):382–395. doi: 10.1016/0959-437x(95)80055-7. [Review] [158 refs] [DOI] [PubMed] [Google Scholar]
- 12.Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev. 1996;10(12):1433–1442. doi: 10.1101/gad.10.12.1433. [Review] [85 refs] [DOI] [PubMed] [Google Scholar]
- 13.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [Review] [225 refs] [DOI] [PubMed] [Google Scholar]
- 14.Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J. Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res. 1997;57(21):4749–4756. [PubMed] [Google Scholar]
- 15.Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary non-polyposis colon cancer. Cell. 1993;75(5):1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 16.Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75(6):1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 17.Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368(6468):258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 18.Papadopoulos N, Nicolaides NC, Wei YF, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263(5153):1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 19.Nicolaides NC, Papadopoulos N, Liu B, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371(6492):75–80. doi: 10.1038/371075a0. [DOI] [PubMed] [Google Scholar]
- 20.Papadopoulos N, Nicolaides NC, Liu B, et al. Mutations of GTBP in genetically unstable cells. Science. 1995;268:1915–1917. doi: 10.1126/science.7604266. [DOI] [PubMed] [Google Scholar]
- 21.Lipkin SM, Wang V, Jacoby R, et al. MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet. 2000;24(1):27–35. doi: 10.1038/71643. [DOI] [PubMed] [Google Scholar]
- 22.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51(12):3075–3079. [Review] [63 refs] [PubMed] [Google Scholar]
- 23.Shibata D, Peinado MA, Ionov Y, Malkhosyan S, Perucho M. Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet. 1994;6(3):273–281. doi: 10.1038/ng0394-273. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Richards B, Wilson T, et al. Apoptosis induced by overexpression of hMSH2 or hMLH1. Cancer Res. 1999;59(13):3021–3027. [PubMed] [Google Scholar]
- 25.Duval A, Hamelin R. Genetic instability in human mismatch repair deficient cancers. Ann Genet. 2002;45(2):71–75. doi: 10.1016/s0003-3995(02)01115-2. [DOI] [PubMed] [Google Scholar]
- 26.Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002;62(9):2447–2454. [PubMed] [Google Scholar]
- 27.Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268(5215):1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 28.Souza RF, Appel R, Yin J, et al. Microsatellite instability in the insulin-like growth factor II receptor gene in gastrointestinal tumours. Nat Genet. 1996;14(3):255–257. doi: 10.1038/ng1196-255. [DOI] [PubMed] [Google Scholar]
- 29.Rampino N, Yamamoto H, Ionov Y, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275(5302):967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 30.Boland CR, Thibodeau SN, Hamilton SR, et al. A national cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–5257. [PubMed] [Google Scholar]
- 31.Frayling IM. Microsatellite instability. Gut. 1999;45(1):1–4. doi: 10.1136/gut.45.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baudhuin LM, Burgart LJ, Leontovich O, Thibodeau SN. Use of microsatellite instability and immunohistochemistry testing for the identification of individuals at risk for Lynch syndrome. Fam Cancer. 2005;4(3):255–265. doi: 10.1007/s10689-004-1447-6. [DOI] [PubMed] [Google Scholar]
- 33.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bocker T, Diermann J, Friedl W, et al. Microsatellite instability analysis: a multicenter study for reliability and quality control. Cancer Res. 1997;57(21):4739–4743. [PubMed] [Google Scholar]
- 35.Roger M. Evidence for conversion of heteroduplex transforming DNAs to homoduplex by recipient pneumococcal cells. Proc Nat Acad Sci USA. 1972;69:466–470. doi: 10.1073/pnas.69.2.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tiraby J-G, Fox MS. Marker discrimination in transformation and mutation of pneumococcus. Proc Natl Acad Sci USA. 1973;70:3541–3545. doi: 10.1073/pnas.70.12.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wildenberg J, Meselson M. Mismatch repair in heteroduplex DNA. Proc Natl Acad Sci USA. 1975;72(6):2202–2206. doi: 10.1073/pnas.72.6.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marinus MG. Adenine methylation of Okazaki fragments in Escherichia coli. J Bacteriol. 1976;128(3):853–854. doi: 10.1128/jb.128.3.853-854.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radman M, Wagner RE, Glickman BW, Meselson M. DNA methylation, mismatch correction and genetic stability. In: Alacevic M, editor. Progress in environmental mutagenesis. Elsevier/North Holland Biomedical Press; Amsterdam: 1980. pp. 121–130. [Google Scholar]
- 40.Siegel EC, Bryson V. Mutator gene of Escherichia coli B. J Bacteriol. 1967;94:38–47. doi: 10.1128/jb.94.1.38-47.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldstein A, Smoot JS. A strain of Escherichia coli with an unusually high rate of auxotrophic mutation. J Bacteriol. 1955;70:588–595. doi: 10.1128/jb.70.5.588-595.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill RF. Location of genes controlling excision repair of UV damage and mutator activity in Escherichia coli WP2. Mutat Res. 1970;9(3):341–344. doi: 10.1016/0027-5107(70)90135-1. [DOI] [PubMed] [Google Scholar]
- 43.Marinus MG. Location of DNA methylation genes on the Escherichia coli K-12 genetic map. Mol Gen Genet. 1973;127(1):47–55. doi: 10.1007/BF00267782. [DOI] [PubMed] [Google Scholar]
- 44.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA repair and mutagenesis. 2. American Society of Microbiology; Washington: 2006. [Google Scholar]
- 45.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions, mechanisms. Chem Rev. 2006;106(2):302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 46.Jascur T, Boland CR. Structure and function of the components of the human DNA mismatch repair system. Int J Cancer. 2006;119(9):2030–2035. doi: 10.1002/ijc.22023. [DOI] [PubMed] [Google Scholar]
- 47.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7(5):335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 48.Welsh KM, Lu AL, Clark S, Modrich P. Isolation and characterization of the Escherichia coli mutH gene product. J Biol Chem. 1987;262(32):15624–15629. [PubMed] [Google Scholar]
- 49.Constantin N, Dzantiev L, Kadyrov FA, Modrich P. Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. J Biol Chem. 2005;280(48):39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grilley M, Griffith J, Modrich P. Bidirectional excision in methyl-directed mismatch repair. J Biol Chem. 1993;268(16):11830–11837. [PubMed] [Google Scholar]
- 51.Kolodner RD, Mendillo ML, Putnam CD. Coupling distant sites in DNA during DNA mismatch repair. Proc Natl Acad Sci USA. 2007;104(32):12953–12954. doi: 10.1073/pnas.0705698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Junop MS, Obmolova G, Rausch K, Hsieh P, Yang W. Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol Cell. 2001;7(1):1–12. doi: 10.1016/s1097-2765(01)00149-6. [DOI] [PubMed] [Google Scholar]
- 53.Allen DJ, Makhov A, Grilley M, et al. MutS mediates heteroduplex loop formation by a translocation mechanism. EMBO J. 1997;16(14):4467–4476. doi: 10.1093/emboj/16.14.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blackwell LJ, Bjornson KP, Modrich P. DNA-dependent activation of the hMutS alpha ATPase. J Biol Chem. 1998;273(48):32049–32054. doi: 10.1074/jbc.273.48.32049. [DOI] [PubMed] [Google Scholar]
- 55.Acharya S, Foster PL, Brooks P, Fishel R. The coordinated functions of the E coli MutS and MutL proteins in mismatch repair. Mol Cell. 2003;12(1):233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 56.Fishel R. Signaling mismatch repair in cancer. Nat Med. 1999;5(11):1239–1241. doi: 10.1038/15191. [DOI] [PubMed] [Google Scholar]
- 57.Gradia S, Acharya S, Fishel R. The human mismatch recognition complex hMSH2–hMSH6 functions as a novel molecular switch. Cell. 1997;91(7):995–1005. doi: 10.1016/s0092-8674(00)80490-0. [DOI] [PubMed] [Google Scholar]
- 58.Gradia S, Subramanian D, Wilson T, et al. hMSH2–hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell. 1999;3(2):255–261. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 59.Pluciennik A, Modrich P. Protein roadblocks and helix discontinuities are barriers to the initiation of mismatch repair. Proc Natl Acad Sci USA. 2007;104(31):12709–12713. doi: 10.1073/pnas.0705129104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cho WK, Jeong C, Kim D, et al. ATP alters the diffusion mechanics of MutS on mismatched DNA. Structure. 2012;20(7):1264–1274. doi: 10.1016/j.str.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeong C, Cho WK, Song KM, et al. MutS switches between two fundamentally distinct clamps during mismatch repair. Nat Struct Mol Biol. 2011;18(3):379–385. doi: 10.1038/nsmb.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fishel R, Acharya S, Berardini M, et al. Signaling mismatch repair: the mechanics of an adenosine-nucleotide molecular switch. Cold Spring Harb Symp Quant Biol. 2000;65:217–224. doi: 10.1101/sqb.2000.65.217. [DOI] [PubMed] [Google Scholar]
- 63.Guerrette S, Wilson T, Gradia S, Fishel R. Interactions of human hMSH2 with hMSH3 and hMSH2 with hMSH6: examination of mutations found in hereditary nonpolyposis colorectal cancer. Mol Cell Biol. 1998;18(11):6616–6623. doi: 10.1128/mcb.18.11.6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fishel R, Wilson T. MutS homologs in mammalian cells. Curr Opin Genet Dev. 1997;7(1):105–113. doi: 10.1016/s0959-437x(97)80117-7. [Review] [84 refs] [DOI] [PubMed] [Google Scholar]
- 65.Antony E, Hingorani MM. Asymmetric ATP binding and hydrolysis activity of the Thermus aquaticus MutS dimer is key to modulation of its interactions with mismatched DNA. Biochemistry. 2004;43:13115–13128. doi: 10.1021/bi049010t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Antony E, Khubchandani S, Chen S, Hingorani MM. Contribution of Msh2 and Msh6 subunits to the asymmetric ATPase and DNA mismatch binding activities of Saccharomyces cerevisiae Msh2–Msh6 mismatch repair protein. DNA Repair (Amst) 2006;5(2):153–162. doi: 10.1016/j.dnarep.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heinen CD, Cyr JL, Cook C, et al. Human MSH2 (hMSH2) protein controls ATP processing by hMSH2–hMSH6. J Biol Chem. 2011;286(46):40287–40295. doi: 10.1074/jbc.M111.297523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mazur DJ, Mendillo ML, Kolodner RD. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Mol Cell. 2006;22(1):39–49. doi: 10.1016/j.molcel.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 69.Fishel R. Mismatch repair, molecular switches, and signal transduction. Genes Dev. 1998;12(14):2096–2101. doi: 10.1101/gad.12.14.2096. [Review] [56 refs] [DOI] [PubMed] [Google Scholar]
- 70.Gradia S, Acharya S, Fishel R. The role of mismatched nucleotides in activating the hMSH2–hMSH6 molecular switch. J Biol Chem. 2000;275:3922–3930. doi: 10.1074/jbc.275.6.3922. [DOI] [PubMed] [Google Scholar]
- 71.Snowden T, Acharya S, Butz C, Berardini M, Fishel R. hMSH4–hMSH5 recognizes holliday junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol Cell. 2004;15(3):437–451. doi: 10.1016/j.molcel.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 72.Gorman J, Chowdhury A, Surtees JA, et al. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2–Msh6. Mol Cell. 2007;28(3):359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gorman J, Plys AJ, Visnapuu ML, Alani E, Greene EC. Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat Struct Mol Biol. 2010;17(8):932–938. doi: 10.1038/nsmb.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li F, Tian L, Gu L, Li GM. Evidence that nucleosomes inhibit mismatch repair in eukaryotic cells. J Biol Chem. 2009;284(48):33056–33061. doi: 10.1074/jbc.M109.049874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 76.Sass LE, Lanyi C, Weninger K, Erie DA. Single-molecule FRET TACKLE reveals highly dynamic mismatched DNA-MutS complexes. Biochemistry. 2011;49(14):3174–3190. doi: 10.1021/bi901871u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mazurek A, Johnson CN, Germann MW, Fishel R. Sequence context effect for hMSH2–hMSH6 mismatch-dependent activation. Proc Natl Acad Sci USA. 2009;106(11):4177–4182. doi: 10.1073/pnas.0808572106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS, its complex with a substrate DNA. Nature. 2000;407(6805):703–710. doi: 10.1038/35037509. see comments. [DOI] [PubMed] [Google Scholar]
- 79.Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, de Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G × T mismatch. Nature. 2000;407(6805):711–717. doi: 10.1038/35037523. see comments. [DOI] [PubMed] [Google Scholar]
- 80.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. 2007;26(4):579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 81.Javaid S, Manohar M, Punja N, et al. Nucleosome remodeling by hMSH2–hMSH6. Mol Cell. 2009;36(6):1086–1094. doi: 10.1016/j.molcel.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guerrette S, Acharya S, Fishel R. The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer. J Biol Chem. 1999;274(10):6336–6341. doi: 10.1074/jbc.274.10.6336. [DOI] [PubMed] [Google Scholar]
- 83.Dutta R, Inouye M. GHKL, An emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25(1):24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 84.Ban C, Yang W. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell. 1998;95(4):541–552. doi: 10.1016/s0092-8674(00)81621-9. [DOI] [PubMed] [Google Scholar]
- 85.Sacho EJ, Kadyrov FA, Modrich P, Kunkel TA, Erie DA. Direct visualization of asymmetric adenine nucleotide-induced conformational changes in Mutlalpha. Mol Cell. 2008;29(1):112–121. doi: 10.1016/j.molcel.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bende SM, Grafstrom RH. The DNA binding properties of the MutL protein isolated from Escherichia coli. Nucleic Acids Res. 1991;19:1549–1555. doi: 10.1093/nar/19.7.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Drotschmann K, Hall MC, Shcherbakova PV, et al. DNA binding properties of the yeast Msh2–Msh6, Mlh1-Pms1 heterodimers. Biol Chem. 2002;383(6):969–975. doi: 10.1515/BC.2002.103. [DOI] [PubMed] [Google Scholar]
- 88.Park J, Jeon Y, In D, Fishel R, Ban C, Lee JB. Single-molecule analysis reveals the kinetics and physiological relevance of MutL-ssDNA binding. PLoS ONE. 2010;5(11):e15496. doi: 10.1371/journal.pone.0015496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126(2):297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 90.Kadyrov FA, Holmes SF, Arana ME, et al. Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J Biol Chem. 2007;282(51):37181–37190. doi: 10.1074/jbc.M707617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kosinski J, Plotz G, Guarne A, Bujnicki JM, Friedhoff P. The PMS2 subunit of human MutLalpha contains a metal ion binding domain of the iron-dependent repressor protein family. J Mol Biol. 2008;382(3):610–627. doi: 10.1016/j.jmb.2008.06.056. [DOI] [PubMed] [Google Scholar]
- 92.Pillon MC, Lorenowicz JJ, Uckelmann M, et al. Structure of the endonuclease domain of MutL: unlicensed to cut. Mol Cell. 2010;39(1):145–151. doi: 10.1016/j.molcel.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pillon MC, Miller JH, Guarne A. The endonuclease domain of MutL interacts with the beta sliding clamp. DNA Repair (Amst) 2010;10(1):87–93. doi: 10.1016/j.dnarep.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grilley M, Welsh KM, Su SS, Modrich P. Isolation and characterization of the Escherichia coli mutL gene product. J Biol Chem. 1989;264(2):1000–1004. [PubMed] [Google Scholar]
- 95.Mendillo ML, Mazur DJ, Kolodner RD. Analysis of the interaction between the Saccharomyces cerevisiae MSH2–MSH6 and MLH1–PMS1 complexes with DNA using a reversible DNA end-blocking system. J Biol Chem. 2005;280(23):22245–22257. doi: 10.1074/jbc.M407545200. [DOI] [PubMed] [Google Scholar]
- 96.Schofield MJ, Nayak S, Scott TH, Du C, Hsieh P. Interaction of Escherichia coli MutS and MutL at a DNA mismatch. J Biol Chem. 2001;276(30):28291–28299. doi: 10.1074/jbc.M103148200. [DOI] [PubMed] [Google Scholar]
- 97.Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD. Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell. 2011;147(5):1040–1053. doi: 10.1016/j.cell.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lopez de Saro FJ, Marinus MG, Modrich P, O’Donnell M. The beta sliding clamp binds to multiple sites within MutL and MutS. J Biol Chem. 2006;281(20):14340–14349. doi: 10.1074/jbc.M601264200. [DOI] [PubMed] [Google Scholar]
- 99.Viswanathan M, Lovett ST. Single-strand DNA-specific exonucleases in Escherichia coli—roles in repair and mutation avoidance. Genetics. 1998;149(1):7–16. doi: 10.1093/genetics/149.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pluciennik A, Burdett V, Lukianova O, O’Donnell M, Modrich P. Involvement of the beta clamp in methyl-directed mismatch repair in vitro. J Biol Chem. 2009;284(47):32782–32791. doi: 10.1074/jbc.M109.054528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ramilo C, Gu L, Guo S, et al. Partial reconstitution of human DNA mismatch repair in vitro: characterization of the role of human replication protein A. Mol Cell Biol. 2002;22(7):2037–2046. doi: 10.1128/MCB.22.7.2037-2046.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pluciennik A, Dzantiev L, Iyer RR, Constantin N, Kadyrov FA, Modrich P. PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc Natl Acad Sci USA. 2010;107(37):16066–16071. doi: 10.1073/pnas.1010662107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang Y, Yuan F, Presnell SR, et al. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122(5):693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 104.Belvederesi L, Bianchi F, Galizia E, et al. MSH2 missense mutations, HNPCC syndrome: pathogenicity assessment in a human expression system. Hum Mutat. 2008;29(11):E296–E309. doi: 10.1002/humu.20875. [DOI] [PubMed] [Google Scholar]
- 105.Hardt K, Heick SB, Betz B, et al. Missense variants in hMLH1 identified in patients from the German HNPCC consortium, functional studies. Fam Cancer. 2011;10(2):273–284. doi: 10.1007/s10689-011-9431-4. [DOI] [PubMed] [Google Scholar]
- 106.Kondo E, Suzuki H, Horii A, Fukushige S. A yeast two-hybrid assay provides a simple way to evaluate the vast majority of hMLH1 germ-line mutations. Cancer Res. 2003;63(12):3302–3308. [PubMed] [Google Scholar]
- 107.Schmutte C, Marinescu RC, Sadoff MM, Guerrette S, Overhauser J, Fishel R. Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res. 1998;58(20):4537–4542. [PubMed] [Google Scholar]
- 108.Schmutte C, Sadoff MM, Shim KS, Acharya S, Fishel R. The interaction of DNA mismatch repair proteins with human exonuclease I. J Biol Chem. 2001;276(35):33011–33018. doi: 10.1074/jbc.M102670200. [DOI] [PubMed] [Google Scholar]
- 109.Raevaara TE, Korhonen MK, Lohi H, et al. Functional significance, clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129(2):537–549. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 110.Takahashi M, Shimodaira H, Andreutti-Zaugg C, Iggo R, Kolodner RD, Ishioka C. Functional analysis of human MLH1 variants using yeast and in vitro mismatch repair assays. Cancer Res. 2007;67(10):4595–4604. doi: 10.1158/0008-5472.CAN-06-3509. [DOI] [PubMed] [Google Scholar]
- 111.Heinen CD, Wilson T, Mazurek A, Berardini M, Butz C, Fishel R. HNPCC mutations in hMSH2 result in reduced hMSH2–hMSH6 molecular switch functions. Cancer Cell. 2002;1:469–478. doi: 10.1016/s1535-6108(02)00073-9. [DOI] [PubMed] [Google Scholar]
- 112.Brieger A, Plotz G, Raedle J, et al. Characterization of the nuclear import of human MutLalpha. Mol Carcinog. 2005;43(1):51–58. doi: 10.1002/mc.20081. [DOI] [PubMed] [Google Scholar]
- 113.Lei X, Zhu Y, Tomkinson A, Sun L. Measurement of DNA mismatch repair activity in live cells. Nucleic Acids Res. 2004;32(12):e100. doi: 10.1093/nar/gnh098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ollila S, Dermadi Bebek D, Jiricny J, Nystrom M. Mechanisms of pathogenicity in human MSH2 missense mutants. Hum Mutat. 2008;29(11):1355–1363. doi: 10.1002/humu.20893. [DOI] [PubMed] [Google Scholar]
- 115.Trojan J, Zeuzem S, Randolph A, et al. Functional analysis of hMLH1 variants, HNPCC-related mutations using a human expression system. Gastroenterology. 2002;122(1):211–219. doi: 10.1053/gast.2002.30296. [DOI] [PubMed] [Google Scholar]
- 116.Naruse H, Ikawa N, Yamaguchi K, et al. Determination of splice-site mutations in Lynch syndrome (hereditary non-polyposis colorectal cancer) patients using functional splicing assay. Fam Cancer. 2009;8(4):509–517. doi: 10.1007/s10689-009-9280-6. [DOI] [PubMed] [Google Scholar]
- 117.Tournier I, Vezain M, Martins A, et al. A large fraction of unclassified variants of the mismatch repair genes MLH1, MSH2 is associated with splicing defects. Hum Mutat. 2008;29(12):1412–1424. doi: 10.1002/humu.20796. [DOI] [PubMed] [Google Scholar]
- 118.Fishel R. The selection for mismatch repair defects in hereditary nonpolyposis colorectal cancer: revising the mutator hypothesis. Cancer Res. 2001;61(20):7369–7374. [PubMed] [Google Scholar]
- 119.Gong JG, Costanzo A, Yang HQ, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399(6738):806–809. doi: 10.1038/21690. see comments. [DOI] [PubMed] [Google Scholar]
- 120.Jain A, Liu R, Ramani B, et al. Probing cellular protein complexes using single-molecule pull-down. Nature. 2011;473(7348):484–488. doi: 10.1038/nature10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Joo C, Balci H, Ishitsuka Y, Buranachai C, Ha T. Advances in single-molecule fluorescence methods for molecular biology. Annu Rev Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- 122.Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5(6):507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22(4):501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]