Protein Quality Control and Metabolism: Bidirectional Control in the Heart (original) (raw)
. Author manuscript; available in PMC: 2016 Feb 3.
Abstract
The prevalence of heart disease, especially heart failure, continues to increase, and cardiovascular disease remains the leading cause of death worldwide. As cardiomyocytes are essentially irreplaceable, protein quality control is pivotal to cellular homeostasis and, ultimately, cardiac performance. Three evolutionarily conserved mechanisms – autophagy, the unfolded protein response, and the ubiquitin-proteasome system– act in concert to degrade misfolded proteins and eliminate defective organelles. Recent advances have revealed that these mechanisms are intimately associated with cellular metabolism. Going forward, comprehensive understanding of the role of protein quality control mechanisms in cardiac pathology will require integration of metabolic pathways and metabolic control.
Age-adjusted mortality from cardiovascular disease (CVD) has decreased an astonishing 75% over the past 40 years (Nabel and Braunwald, 2012). Many patients who would have died previously now survive with an injured heart, predisposing them to the clinical syndrome of heart failure. Life-saving successes such as these, coupled with deterioration in lifestyle-related issues and the epidemic of obesity, have culminated in dramatic increases in the prevalence of heart failure. CVD accounted for 30% of all deaths in 2008, and this is projected to rise to 35% in 2030 (Mathers and Loncar, 2006). By 2030, more than 40% of the US population is projected to suffer from some form of CVD, with estimated direct and indirect costs exceeding $1 trillion. Clearly, the spectrum of CVD continues to evolve, posing ever-new challenges to deciphering mechanisms of pathogenesis in hopes of discovering new life-preserving therapies.
Protein quality control in heart disease
Protein quality control is vital to cellular homeostasis and integrity. Loss of protein homeostasis leads to accumulation of misfolded proteins and protein aggregates with consequent proteotoxicity. Intricate adaptive responses have evolved to clear aberrantly folded proteins and defective organelles. Multiple, interdependent mechanisms are orchestrated to ensure protein homeostasis and to respond to stress.
Loss or imbalance of protein quality control has been implicated in numerous diseases, including neurodegenerative disease, cardiovascular disease, diabetes, and cancer. As a post-mitotic cell, cardiomyocytes possess little, if any, replicative capacity in adult life (Bergmann et al., 2009). Thus, maintaining balanced protein quality is fundamental to minimize cellular dysfunction and death (Willis and Patterson, 2013). At the same time, it has been firmly established that metabolic derangements contribute to the pathogenesis of heart disease (Lopaschuk et al., 2010). While protein quality control is essential to maintain protein homeostasis, recent studies have revealed that these mechanisms are also actively engaged in the regulation of cardiac metabolism (Figure 1). Here, we provide an overview of protein quality control in heart under various stresses, placing emphasis on the interplay with metabolism.
Figure 1. Protein quality control and cardiac metabolism in heart disease.
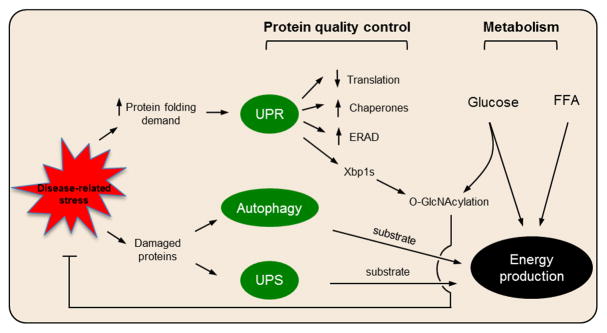
Disease-related stress on the myocardium triggers increased demand for protein folding and protein damage. These events, in turn, activate the UPR, autophagy, and the UPS. Glucose and free fatty acid (FFA) are the major nutrients supporting energy production in the myocardium. Recent insights have uncovered complex interplay between these two major cellular processing, including bidirectional signaling, transcriptional control, and substrate provision.
Cellular mechanisms of protein homeostasis
Protein homeostasis involves gene transcription, mRNA translation, protein post-translational modification, high-order complex assembly, and protein clearance, all of which participate in every aspect of cardiac physiology and pathology. While considerable progress has emerged in our understanding of the regulation of gene expression in the heart, advances in elucidating protein quality control are relatively limited.
Multiple cellular mechanisms serve as surveillance and monitoring systems to ensure protein homeostasis in the cardiomyocyte (Mearini et al., 2008; Wang and Robbins, 2014). Autophagy and the ubiquitin-proteasome system (UPS) are the most important proteolytic cascades to eliminate misfolded proteins and defective organelles. These mechanisms involve the lysosome and proteasome, respectively. The unfolded protein response (UPR) is an adaptive process to accommodate protein-folding stress in the endoplasmic reticulum (ER) (Hetz et al., 2013; Walter and Ron, 2011).
A characteristic feature of each of these 3 processes is inherent dichotomy: each is capable of beneficial changes, and each is capable of destructive, even lethal, harm. Whether the dichotomy intrinsic to those processes stems from quantitative differences (too much or too little), as opposed to qualitative distinctions, remains an ongoing puzzle. Either way, defects in each of these regulatory mechanisms have been reported in heart disease, and modulation of their activities holds promise in combating disease.
Autophagy, a self-eating process
Autophagy is an evolutionarily conserved process in which a cell cannibalizes small portions of its cytoplasm (Mizushima et al., 2008). At least three forms of autophagy have been described, all of which involve delivery of cargo to the lysosome for degradation. Among them, macroautophagy (herein referred to as autophagy) is the most highly regulated and best studied with functional implications in virtually every cell type.
Autophagy is a regulated, highly dynamic process, governed by more than 30 autophagy-related proteins (ATGs) (Figure 2) (Feng et al., 2014). Initiation of autophagy is triggered by nutrient deprivation or lack of growth factor support. mTOR (mammalian target of rapamycin), the master regulator of cell growth, is associated with the ULK1-ATG13-FIP200 protein complex, inhibiting it by phosphorylation. Starvation leads to inactivation and dissociation of mTOR from ULK1. Liberated ULK1, in turn, is activated to phosphorylate and recruit a class III PI3K complex to the site of formation of an isolation membrane, the nascent organelle for cargo engulfment. The class III PI3K complex consists of VPS34, Beclin 1, VPS15 and ATG14L, and its primary function is to generate PI3P and attract more ATGs to the nucleation site.
Figure 2. The autophagy pathway.

Autophagy is an evolutionarily conserved self-eating process. Autophagy is dynamic, involving multiple steps of initiation, vesicle nucleation, elongation/expansion and fusion. Engulfed cargo is digested by acid hydrolases from the lysosome, and degraded components are released into the cytosol to meet energetic demand. Several complexes participate in autophagosome formation at various steps, including an initiation complex, the class III PI3K complex, and two ubiquitin-like systems.
Two ubiquitin-like conjugation systems, working in parallel, then participate in vesicle expansion and elongation. In the first one, ATG12 is activated by an E1-like enzyme, ATG7, transferred to an E2-like enzyme, ATG10, and finally to ATG5. After interacting with ATG16L, the ATG12-ATG5-ATG16L complex functions similarly to an E3 ligase. In the second conjugation system, ATG8 is initially processed by ATG4, a cysteine protease, to expose a C-terminal glycine. ATG8 is then activated by ATG7, transferred to an E2-like molecule, ATG3, and finally conjugated by the ATG12-ATG5-ATG16L complex to phosphatidylethanolamine (PE) to become ATG8-PE. The double-membrane vesicle then engulfs portions of cytoplasm, generating a discrete structure termed an autophagosome. The autophagosome ultimately delivers its cytosolic cargo to the lysosome, forming an autolysosome. Acid hydrolases within the lysosome digest macromolecules into amino acids, lipids, nucleotides, and carbohydrates, along with the autolysosome inner membrane, to yield cellular building blocks to refuel the cell.
Autophagy in starvation
As a continuously working pump, the heart consumes robust quantities of ATP, more than any other organ in the body. That said, myocardial energy reserves in human myocardium are remarkably low, sufficient for fewer than 10 contractions (Stanley et al., 2005). As a result, cardiomyocytes must generate energy continuously, consuming nutrients constantly and without interruption (Lopaschuk et al., 2010).
During mammalian fetal development, nutrients are available continuously. At birth, however, nutrient supply is abruptly interrupted, and neonates experience an energy crisis before nutrients are restored in the form of mother’s milk. During this critical window, autophagy is activated in numerous cell types; in mouse heart, autophagy is activated within 30 minutes post-delivery and remains high for at least 24 hours (Kuma et al., 2004). Genetic suppression of this autophagic response dramatically reduces survival in nutrient-deprived neonates as compared to wild-type controls. Conversely, forced feeding of milk enhances survival in autophagy-deficient neonates, highlighting the role of autophagy in supplying energy at this stage. More recent studies demonstrate that restoration of nutrient supply rapidly suppresses autophagy through insulin signaling, thereby obviating excessive “self-eating” and promoting normal development (Riehle et al., 2013).
Likewise, in adult mice exposed to starvation conditions, cardiomyocyte autophagy plays a pivotal role to maintain cellular homeostasis and sustain pump function. Indeed, autophagy is significantly induced in heart by as little as 12 hours of nutrient deprivation (Kanamori et al., 2009). Normally, heart function is preserved over the course of 3 days of food deprivation. However, when autophagy is inhibited by administration of Bafilomycin A1, cardiac performance is impaired, coupled with significant decreases in myocardial concentration of amino acids and ATP. These in vivo findings are corroborated by in vitro observations. In cultured adult cardiomyocytes, nutrient deprivation triggers necrotic cell death by day 4 (Maruyama et al., 2008). Inhibition of autophagy pharmacologically or by means of RNAi knockdown of key ATGs accelerates cell death, while stimulation of autophagy decelerates death. Similar findings have been reported in neonatal rat ventricular myocytes (NRVM) and HL-1 cells, pointing to a common protective role for autophagy in the metabolic stress of starvation (Hariharan et al., 2010; Xu et al., 2013).
Adult heart derives 50–70% of its energy from oxidation of free fatty acids (FFA), while the rest comes from oxidation of glucose, lactate, and other nutrients. Given its incessant requirement for energy, the myocardium is an obligate omnivore, consuming any nutrient available to generate ATP. When nutrient supplies are limiting, autophagy fuels the heart through several routes (Rabinowitz and White, 2010). Nucleic acid degradation yields ribose, which can feed into glycolysis through the pentose phosphate pathway. Sugars and lipids liberated by lysosomal digestion can be channeled into glucose and FFA oxidation pathways, respectively. Proteins are initially broken down into peptides and ultimately, amino acids. Following deamination, degradation products derived from various amino acids serve as key intermediates supporting major metabolic pathways. For example, glutamate and alanine can be converted into alpha-ketoglutarate and pyruvate, respectively, both of which feed directly into the TCA cycle. Collectively, cardiac autophagy not only “takes out the trash” but it recycles that trash to support metabolism.
Autophagy as a housekeeping process
Autophagy is the primary means of degrading long-lived proteins and defective organelles. In post-mitotic cells, such as cardiac myocytes and neurons, which have very limited capacity for regeneration, mechanical and oxidative stress over the course of a lifetime must be repaired. Here, the housekeeping function of autophagy is critical to sustain cellular viability and performance.
Deficiency of autophagy by genetic silencing of ATG5 under basal conditions triggers cardiac hypertrophy and ventricular dilatation, possibly due to toxic accumulation of defective organelles (Nakai et al., 2007). This is a cell autonomous response, as knockdown of ATG7 in NRVM is sufficient to recapitulate these phenotypes. Danon disease is a lysosomal glycogen storage disease caused by mutation of LAMP2, a principal lysosome membrane protein (Nishino et al., 2000). Loss of LAMP2 by targeted silencing in mice leads to accumulation of autophagic vacuoles, extension of the half-life of long-lived proteins, and deterioration of cardiac contractility, again highlighting the critical role of autophagy in maintaining cardiomyocyte homeostasis (Tanaka et al., 2000).
The housekeeping role of autophagy is further exemplified in its actions to antagonize cardiac proteotoxicity. In cardiomyocytes, alpha-B crystallin (CryAB) is a molecular chaperone serving to prevent aggregation of highly abundant proteins, such as desmin. Expression of a dominant-negative mutant of CryAB (CryABR120G) elicits protein aggregates, myofibril disarray, and cardiomyopathy (Rajasekaran et al., 2007; Sanbe et al., 2004). Our lab has shown that over-expression of this mutant human CryAB in NRVM is sufficient to trigger autophagy (Tannous et al., 2008). Inhibition of autophagy by 3-MA leads to increases in aggresome size and insolubility. Moreover, blunting autophagy in vivo accelerates pathological remodeling in CryABR120G-expressing heart and leads to heart failure (Tannous et al., 2008). Consistently, boosting autophagy by over-expressing ATG7 reduces aggregate content and cytotoxicity both in vivo and in vitro (Bhuiyan et al., 2013; Pattison et al., 2011).
Autophagy in cardiac hypertrophy
Hypertension affects 1 in 3 Americans (Mozaffarian et al., 2014). In response to high blood pressure and consequent increases in ventricular wall stress, the heart undergoes a transformation that involves hypertrophic growth and incorporation of new sarcomeres in parallel. Held to be an acute, adaptive response initially, cardiac hypertrophy, when persistent, is maladaptive (Frey et al., 2004). In epidemiological studies, left ventricular hypertrophy is an independent risk factor for systolic dysfunction and heart failure, a clinical syndrome wherein the hearts in unable to maintain adequate perfusion of peripheral organs (Mozaffarian et al., 2014). Extensive efforts have been marshaled to decipher mechanisms governing the transition from hypertrophy to heart failure, which however remain elusive (Heineke and Molkentin, 2006) (Burchfield et al., 2013).
Hypertrophic growth of the heart is more than simple addition of sarcomeres within cardiomyocytes. It involves profound structural, electrophysiological, functional, and metabolic remodeling (Hill and Olson, 2008). Numerous reports demonstrate that autophagy participates in this process. In early phases of hypertrophic growth, autophagy may be acutely suppressed to favor anabolism and rapid cardiomyocyte growth. Indeed, early studies reported that short-term (10-minute) infusion of isoproterenol, a beta-adrenergic agonist which elicits cardiac hypertrophy, in rat heart leads to a decrease of autophagic vacuoles (Pfeifer et al., 1987). Thoracic aortic constriction (TAC) is a well-established procedure in rodent models wherein the aortic arch is constricted surgically to increase afterload. TAC in mice has been suggested to lead to suppression of autophagy 1 week after surgery (Nakai et al., 2007). However, steady-state levels of LC3-I and p62 were emphasized in this study, which may not provide a reliable evaluation of autophagic flux. Ultimately, anabolism and catabolism achieve equipoise at higher levels.
Using a mouse model of severe TAC (sTAC), we reported that autophagy is induced as early as 24 hours after surgery, along with augmentation of lysosomal markers (Zhu et al., 2007). Blunting autophagy in a model of Beclin 1 haploinsufficiency (Beclin 1+/− mice) significantly reduced hypertrophic growth and cardiac remodeling, whereas cardiomyocyte-restricted over-expression of Beclin 1 exacerbated hypertrophy and heart failure triggered by pressure overload. Further, angiotensin II infusion for 4 weeks in mice triggers significant hypertrophy and autophagy in heart (Dai et al., 2011). And these in vivo findings have been confirmed in vitro; angiotensin II treatment for 48 hours elicits increases in autophagy in NRVM (Porrello et al., 2009).
Intriguingly, pharmacological intervention to block autophagy is sufficient to blunt hypertrophic growth. Chronic inhibition of histone deacetylase (HDAC) activity by small molecules elicits a reduction in autophagic activity. HDAC inhibition in mice effectively suppresses TAC-induced hypertrophy (Antos et al., 2003; Kong et al., 2006). Importantly, when treatment is initiated after establishment of cardiac hypertrophy, suppression of autophagy can reverse hypertrophic growth and restore systolic performance (Cao et al., 2011). Together, these results highlight the importance of autophagy in afterload-induced cardiac hypertrophic growth and point to a novel therapeutic strategy to tackle maladaptive cardiac remodeling.
Some findings, however, are seemingly contradictory (Nakai et al., 2007). These investigators engineered mice harboring cardiomyocyte-specific deficiency of ATG5. When subjected to TAC, ATG5 knockout heart manifested more profound hypertrophy, exacerbated pathological remodeling, and systolic dysfunction. Whereas the reasons underlying these apparent discrepancies remain unclear, we suggest that differences in the models being employed contribute. Autophagy, like many other biological processes, exists as a continuum (Rothermel and Hill, 2008). Basal autophagy is essential to maintain cellular homeostasis. Complete abrogation of autophagy in ATG5 knockout hearts likely predisposes cardiomyocytes to load-induced stress. In that context, TAC triggers a severe maladaptive response. In Beclin 1+/− mice, however, where autophagic flux is essentially halved, pressure overload induces autophagy within an adaptive range, and cardiac performance is well preserved. We suggest that increased levels of autophagic flux in wild-type mice subjected to pressure stress are maladaptive. When autophagic flux is amplified by Beclin 1 over-expression, maladaptive remodeling is further increased. Thus, optimal levels of autophagic flux are critical to maintain cellular homeostasis and cardiac performance.
It is firmly established that cardiac hypertrophy is associated with profound metabolic remodeling. The main source of ATP production in healthy adult heart is FFA. During hypertrophic growth, utilization of glucose increases, ultimately becoming an important fuel source for ATP generation. Early studies showed that ATP derived from glycolysis increases from 7% in control to 19% in hypertrophied heart (Allard et al., 1994). Concomitantly, ATP production from FFA decreases from 70% to 55%. Increases in glycolysis are, however, uncoupled from further oxidative phosphorylation, raising the prospect of inefficient energetics.
It is also possible that the increase in glycolysis, uncoupled from oxidative phosphorylation, may provide critical metabolic intermediates for hypertrophic growth. To achieve significant enlargement of the cardiomyocyte, nutrients may not be completely oxidized to ATP, CO2 and H2O. Rather, metabolites from glycolysis may be channeled to biosynthesis of macromolecules (Locasale and Cantley, 2011). Consistent with this notion, intermediate metabolites, such as glucose-6-phosphate, fructose-6-phosphate, 3-phosphoglycerate, 2-phosphoglycerate and phosphoenoylpyruvate are all elevated in hypertrophied heart of Dahl salt-sensitive rats (Kato et al., 2010). Likewise, NADPH, a metabolite of the pentose phosphate pathway, is significantly increased as well. However, this hypothesis of selective diversion from downstream substrate oxidation to support growth remains to be conclusively verified.
Cardiac metabolism is further altered during the transition from hypertrophy to heart failure (Lopaschuk et al., 2010). Glucose uptake is enhanced yet more, while uncoupling from oxidative phosphorylation persists. At the same time, FFA oxidation is reduced. To maintain normal TCA flux, key intermediates are replenished via anaplerosis from glycolytic pyruvate and up-regulation of malic enzyme activity (Sorokina et al., 2007). These metabolic alterations, however, bypass energy-yielding reactions and contribute to reduced energetic efficiency in heart failure.
Studies from the cancer field may shed light on mechanisms whereby autophagy affects cardiomyocyte metabolism (Kenific and Debnath, 2014; Rabinowitz and White, 2010). In oncogenic H-Ras-transformed mouse embryonic fibroblasts, autophagy is induced following extracellular matrix detachment. Inhibition of autophagy by knockout of ATG5 leads to suppression of anchorage-independent transformation, attenuation of proliferation, and inhibition of glycolysis (Lock et al., 2011). Similar findings have been reported in mammary tumor cells silenced for FIP200 (Wei et al., 2011). Although the specific target(s) of autophagy within the glycolytic pathway remain to be uncovered, these results highlight the intrinsic connections between autophagy and glycolysis.
Excessive activation of autophagy accelerates the decompensation process and promotes heart failure. Over-activation of autophagy may non-selectively degrade essential metabolic enzymes and mitochondria, exacerbating metabolic derangements. We have reported that sTAC in mice stimulates robust increases in autophagy in heart, and attenuation of autophagy can decelerate cardiac remodeling (Zhu et al., 2007). Chaanine et al reported excessive autophagic elimination of mitochondria in heart failure triggered by pressure overload (Chaanine et al., 2012). Inhibition of autophagy by 3-MA led to improvements in contractility and blunting of pathological remodeling (Chaanine et al., 2012). Collectively, we suggest that autophagy in the initial phase of hypertrophic transformation is adaptive by modulating cardiac metabolism though induction of glycolysis. Excessive autophagy, however, promotes maladaptive remodeling and accelerates disease progression.
Autophagy in ischemic heart disease
Limitations in the delivery of oxygen and nutrients to the myocardium are common, typically resulting from atherosclerotic narrowing of coronary arteries. Complete occlusion of a coronary artery ignites a process culminating in irreversible cell death. In most instances of ischemic disease, blood supply is restored, spontaneously or therapeutically. Whereas cell death is rescued by the restoration of blood flow, a second wave of toxic events accompanies this response, termed reperfusion injury (Hausenloy and Yellon, 2013; Turer and Hill, 2010).
The hypoxic/anoxic environment of ischemia is akin to starvation and triggers potent induction of autophagy. Studies in the late 1970’s demonstrated that autophagic vacuoles are strongly induced in rabbit heart after only 40 minutes of hypoxia (Decker and Wildenthal, 1980). In a swine model of ischemic cardiomyopathy, widespread up-regulation of autophagic markers, including LC3-II (Atg8), Beclin 1, and cathepsins are seen (Yan et al., 2005). More recently, Sadoshima and colleagues reported significant autophagy induction in rodent heart from only 30 minutes of ischemia (Matsui et al., 2007).
It is widely agreed that autophagy elicited by myocardial infarction protects the heart from ischemic injury. Due to abrupt interruption of exogenous nutrient supply, metabolites from autophagic digestion become a major source for energy production. Moreover, autophagy-stimulated glycolysis may be an adaptive feature during hypoxia, as glycolysis can generate ATP in the absence of oxygen. Some evidence suggests that peak autophagy induction in ischemia is coincident with suppression of apoptosis, highlighting a cardioprotective role (Yan et al., 2005). Stimulation of autophagy in mice using TAT-p27 reduces infarct size and improves cardiac performance after myocardial infarction (Sun et al., 2014). Moreover, autophagy may be protective by eliminating defective mitochondria, thereby limiting accumulation of toxic reactive oxygen species (ROS) (Zhang et al., 2008).
Timely and effective reperfusion of ischemic myocardium is the most effective means of mitigating cardiac damage. Again, in the late 1970’s it was shown that reperfusion after hypoxia in rabbit heart elicits a second wave of autophagy (Decker and Wildenthal, 1980). Similar findings have been reported in mouse heart (Matsui et al., 2007), HL-1 cells (Hamacher-Brady et al., 2006) and NRVM (Sengupta et al., 2011). Although induction of autophagy by ischemia/reperfusion (I/R) is firmly established, its functional role is likely complex and time-dependent. In the setting of suppressed autophagic activation due to Beclin 1+/− haploinsufficiency, I/R injury is attenuated (Matsui et al., 2007). Likewise, suppression of autophagy in vitro using 3-MA increases cell viability post I/R (Valentim et al., 2006). Together, these results points to a detrimental role of autophagy in I/R.
In contrast, there is evidence that enhanced autophagy protects cardiomyocytes from I/R damage (Hamacher-Brady et al., 2006). Stimulation of autophagy by chloramphenicol succinate in pigs is cardioprotective against I/R (Sala-Mercado et al., 2010). Further, elevated basal levels of autophagy in high-altitude residents is associated with diminished I/R injury (Hu et al., 2014). In aggregate, these findings point to a beneficial role of autophagy in I/R.
This apparent discrepancy may be reconciled by understanding the dynamic nature of autophagy. A static snapshot of LC3-II levels does not provide an accurate picture of autophagic flux, because either an increase in autophagy initiation (increased autophagic flux) or blockage of downstream processing (decreased autophagic flux) generate the same result. Some evidence of impaired autophagic flux was reported in HL-1 cells exposed to I/R (Hamacher-Brady et al., 2006). Working with both murine and rabbit models of I/R injury, we have reported that autophagic flux is suppressed in heart, and stimulating flux is cardioprotective (Xie et al., 2014). The blockage of downstream processing is likely due to decreases in the key lysosomal protein LAMP-2 (Ma et al., 2012). Beclin 1+/− haploinsufficient mice may manifest less cardiac injury from I/R due to attenuated accumulation of unprocessed autophagosomes (Matsui et al., 2007). In aggregate, modulation of autophagic flux, with effects on both protein quality control and metabolism, represents a novel and attractive means to ameliorate I/R injury to the myocardium.
Unfolded protein response
Up to 35% proteins require concerted processing by translation, folding, assembly, and secretion in the endoplasmic reticulum (ER) (Blobel, 2000). When these secretory or transmembrane molecules are not processed properly, accumulating in the ER, an evolutionarily conserved process is activated, termed the unfolded protein response (UPR) (Walter and Ron, 2011). The UPR serves to relieve ER stress by diminishing the translation of new protein, promoting expression of protein chaperones, and degrading excess protein through ER-associated degradation (ERAD) and via autophagy. Further, cytosolic chaperones and mitochondrial chaperones play important roles in cardioprotection in the settings of several heart diseases (Tarone and Brancaccio, 2014; Willis and Patterson, 2010).
There are three arms of the UPR that transmit signals to the nucleus to elicit downstream events (Figure 3). PERK (PKR-like ER kinase) is a transmembrane protein kinase in the ER. In the basal state, PERK is sequestered in an inactive state by interacting with BiP, an ER-resident chaperone. In the setting of increased demand for protein folding, BiP preferentially binds hydrophobic domains of folding proteins, liberating PERK to oligomerize and autophosphorylate. Active PERK phosphorylates eIF2α, a translational initiation factor, suppressing its activity and effectively reducing global mRNA translation to create a “time window for repair” in the ER. Phosphorylated eIF2α, however, can selectively promote translation of certain mRNAs harboring a small open reading frame in the 5′-UTR, including ATF4. ATF4 mediates expression of ER chaperones to facilitate protein folding.
Figure 3. The unfolded protein response.

The unfolded protein response (UPR) is an adaptive mechanism to cope with protein folding stress in the endoplasmic reticulum (ER). Misfolded proteins in the ER occupy the ER-resident chaperone BiP. Three signal transducers of the UPR are induced by a variety of mechanisms to attenuate global protein translation, and stimulate expression of genes coding for proteins involved in protein chaperoning, autophagy, ER-associated protein degradation, and metabolic regulation. In the setting of unremitting stress, cell death ensues to eliminate terminally defective cells. nATF6, nuclear ATF6.
IRE1 (inositol-requiring 1) is the most highly conserved branch of the UPR, present from yeast to mammals. IRE1, upon dissociation from BiP, autophosphorylates and manifests endoribonuclease activity. One prominent target of IRE1 is X-box binding protein 1 (Xbp1). Cleavage of a cryptic 26bp intron creates a frame shift, and the resultant spliced Xbp1 (Xbp1s) is a potent transcriptional activator, driving expression of genes coding for multiple chaperones and elements of ERAD.
In contrast to PERK and IRE1, the third element of the UPR, ATF6 (activating transcription factor 6), is translocated from the ER to Golgi when its binding partner within the ER is occupied by protein clients. Then, ATF6 is cleaved by 2 proteases, and the cytoplasmic domain of ATF6 is released and transported to the nucleus where it stimulates transcription of genes coding for ER-resident chaperones (Ye et al., 2000).
As noted, activation of the UPR attenuates global protein translation, promotes gene expression of ER chaperones, and enhances ERAD to degrade terminally misfolded proteins. If all these adaptive attempts fail in the face of persistent stress, apoptosis ensues for the benefit of whole organism (Tabas and Ron, 2011). Recent studies indicate the three branches also exert partially overlapping but distinct functions in the determination of cell fate (Lin et al., 2007). Given the widespread and diverse events triggered by the UPR, it is not surprising that perturbations of the UPR have been implicated in the pathogenesis of multiple diseases, including cancer (Wang and Kaufman, 2014), cardiovascular disease (Doroudgar and Glembotski, 2013), neurodegenerative disease (Hetz and Mollereau, 2014), and diabetes (Back and Kaufman, 2012; Fu et al., 2012).
UPR in cardiac hypertrophy
Cardiac hypertrophic growth requires biosynthesis of numerous proteins and lipids. As a consequence, the protein folding load within the ER is high. Surprisingly, there are relatively few studies of UPR induction in load-induced heart growth. Early work reported that markers of the UPR are strongly induced at 1 and 4 weeks after TAC in mice, which is accompanied by increases in apoptosis and deterioration of contractility (Okada et al., 2004). This group went further to present evidence that CHOP, a downstream pro-apoptotic target of the UPR, is responsible for the detrimental effects of the UPR during pressure overload. Accordingly, deletion of CHOP is associated with blunting of cardiac hypertrophy and improvements in cardiac performance (Fu et al., 2010). Whereas the specific UPR branches responsible for these events were not elucidated, PERK is likely involved based on stimulation of eIF2α phosphorylation and CHOP induction. Other work, however, found that ATF6 activation suppresses hypertrophic growth in vitro by directly targeting RCAN1 (Belmont et al., 2008).
Cardiac hypertrophy also entails profound shifts in glucose utilization and FFA oxidation. Cardiac insulin signaling is crucial for load-induced hypertrophic growth, as reductions in plasma insulin suppress cardiac enlargement and systolic dysfunction (Shimizu et al., 2010). This finding is reminiscent of the action of Xbp1s to sensitize hepatocytes to the action of insulin (Deng et al., 2013). Further, Taegtmeyer and colleagues have reported that glucose flux through the hexosamine biosynthetic pathway is enhanced in cardiac hypertrophy (Young et al., 2007). We recently showed that Xbp1s is the upstream transcription factor that directly activates expression of multiple enzymes of hexosamine biosynthesis, and Xbp1s over-expression in heart leads to elevations of UDP-N-acetylglucosamine (Wang et al., 2014). However, the precise role of Xbp1s in cardiac hypertrophy remains elusive.
UPR in ischemia/reperfusion injury
In contrast to the dearth of studies of the UPR in cardiac hypertrophy, numerous reports have demonstrated strong induction of the UPR in ischemic heart disease. PDI, a marker of the UPR, is induced 3-fold in the viable peri-infarct region of human myocardium (Severino et al., 2007). Likewise, we and others have found that Xbp1s is up-regulated in human heart failure of ischemic etiology, suggesting that the UPR is an intrinsic component of the cardiac stress response (Sawada et al., 2010; Wang et al., 2014). Coronary occlusion in pigs induces all three branches of the UPR (Xin et al., 2011). Consistently, activation of ER stress has been observed in rodent models of myocardial infarction. Glembotski and colleagues showed that BiP levels are increased in the infarct zone after permanent coronary ligation in mice (Thuerauf et al., 2006). Using NRVM, the same group reported potent induction of BiP, Xbp1s, and CHOP by hypoxia. A dominant negative mutant of ATF6 inhibited BiP promoter activation by hypoxia (Doroudgar et al., 2009). Further, knockdown of ATF6 by siRNA leads to blunting of hypoxia-induced BiP induction, contributing to NRVM cell death. In contrast, activation of the PERK pathway by hypoxia has not been widely reported in NRVM. However, PERK activation and downstream eIF2α phosphorylation in hypoxia have been reported in human carcinoma cells and fibroblasts. The consequent inhibition of protein translation is required for cell survival (Koumenis et al., 2002). PERK deficiency or expression of a dominant-negative mutant of PERK leads to reduction of eIF2α phosphorylation and diminished cell viability, while forced activation of PERK promotes survival in hypoxia (Lu et al., 2004). Collectively, these results point to robust induction of the UPR by I/R in heart, a response which contributes to the maintenance of cellular homeostasis and cardiomyocyte survival.
Mechanistically, the cardioprotective effects of the UPR have been attributed largely to induction of ER chaperones and consequent enhanced protein folding. Indeed, over-expression of wild-type, but not mutant, PDI protects HL-1 cells from apoptosis under hypoxia conditions (Severino et al., 2007). Moreover, adenoviral-mediated expression of PDI in vivo is strongly cardioprotective in acute myocardial infarction, with improvements in contractility and reductions in infarct size. Hypoxia provokes elevations of ROS and deterioration of oxidative protein folding in the ER. Over-expression of chaperones enhances ER folding capacity, prevents accumulation of misfolded proteins, and improves survival.
Additionally, hypoxia is accompanied by impairment of oxidative phosphorylation, reduction in nutrient supply due to deficiency of growth factor signaling, and defects in energetics. In this state, enhancements in insulin signaling, glucose uptake, and glycolysis provide significant survival advantage. In fact, recent studies have delineated a significant metabolic role of the UPR, especially the Xbp1s pathway (Back and Kaufman, 2012). Thus, the relative contributions to UPR-mediated cardioprotection of promoting the ER folding environment by chaperone induction versus metabolic improvements by Xbp1s remain to be determined.
Ample evidence shows that reperfusion after ischemic insult (I/R) triggers strong UPR activation in heart. Extensive efforts have been directed at dissecting pathological mechanisms of I/R injury, with calcium mishandling, over-production of ROS, metabolic derangements and inflammation as widely cited culprits (Hausenloy and Yellon, 2013; Murphy and Steenbergen, 2008; Turer and Hill, 2010). Intriguingly, these alterations are well established triggers of the UPR (Schroder and Kaufman, 2005). Reperfusion of mouse heart after 50 minutes of ischemia leads to potent induction of Xbp1s, BiP and eIF2α phosphorylation in a time-dependent manner (Miyazaki et al., 2011). Similarly, elicitation of the UPR in heart in vivo occurs early (Wang et al., 2014). Splicing of Xbp1 occurs as early as 5 minutes into the reperfusion phase of I/R in mouse. These findings have been corroborated in vitro with NRVM, highlighting a cardiomyocyte-autonomous phenomenon (Thuerauf et al., 2006; Wang et al., 2014).
Functionally, up-regulation of the UPR confers strong cardioprotective effects. Das and colleagues reported a significant beneficial effect of brief exposure to tunicamycin or thapsigargin to elicit ER stress in rat (Petrovski et al., 2011). Likewise, multiple studies from the Glembotski group suggest that induction of ATF6 is cardioprotective. In a tamoxifen-inducible system, cardiomyocyte-specific over-expression of active ATF6 triggers significant protection against I/R damage ex vivo and in vivo (Martindale et al., 2006), a finding consistent with improvements in survival in vitro by simulated I/R (sI/R) (Doroudgar et al., 2009).
We have reported that over-expression of Xbp1s in cardiomyocytes blunts I/R injury and improves cardiac performance in mice (Wang et al., 2014). Conversely, cardiomyocyte-specific knockout of Xbp1 amplifies I/R damage and exacerbates deterioration in cardiac function. Importantly, further work using Xbp1s gain- and loss-of-function strategies in vitro yielded consistent results, suggesting that Xbp1s is necessary and sufficient to protect cardiomyocyte from I/R insult. In addition, activation of the pro-apoptotic branch of the UPR contributes to cardiac injury, as highlighted by improvements in cardiac function in CHOP-deficient animals (Miyazaki et al., 2011). In aggregate, the UPR is acutely induced by I/R in heart, which helps to restore cellular homeostasis and accelerate functional recovery from reperfusion.
UPR and the hexosamine biosynthetic pathway
The intimate association between the UPR and carbohydrate metabolism prompted us to investigate the potential role of the UPR in regulating glucose utilization. Up to 5% of glucose is metabolized via the hexosamine biosynthetic pathway (HBP). Under certain conditions, the contribution may be significantly greater (Zachara, 2012). Glucose is initially phosphorylated to glucose-6-phosphate and then converted to fructose-6-phosphate. Glutamine fructose-6-phosphate aminotransferase (GFAT), the rate-limiting enzyme of the HBP, converts fructose-6-phosphate to glucosamine-6-phosphate. After that, an acetyl group is conjugated by glucosamine-phosphate N-acetyltransferase (GNPNAT), and N-acetylglucosamine-6-phosphate is formed, which is then converted to N-acetylglucosamine-1-phosphate by phosphoglucomutase 3 (PGM3). After activation, UDP-GlcNAc is generated, which is an important donor for biosynthesis of glycans, proteoglycans, glycolipids, and O-GlcNAc modifications.
Protein O-GlcNAc modification (O-GlcNAcylation) is a prominent post-translational modification of thousands of cytosolic and nuclear proteins. This event governs protein subcellular localization, protein-protein interactions, enzymatic activity, and protein function (Bond and Hanover, 2013; Hart et al., 2011). The O-GlcNAcylation reaction is catalyzed by O-GlcNAc transferase (OGT) and antagonized by O-GlcNAcase (OGA). UDP-GlcNAc, a cellular sugar derivative, is the donor molecule conjugated to Ser/Thr residues on target proteins. Studies have shown that the enzymatic activity of OGT is largely driven by the concentration of the UDP-GlcNAc substrate, the end-product of a series of biochemical reactions collectively named the hexosamine biosynthetic pathway.
Although chronic activation of the HBP and protein O-GlcNAcylation may be pathological, early studies firmly established that acute stimulation of O-GlcNAcylation in the heart is strongly cardioprotective under various stress conditions including I/R (Bond and Hanover, 2013; Marsh et al., 2014). Cardiomyocyte-specific deletion of OGT, and consequent suppression of O-GlcNAcylation, leads to deteriorations in the response to myocardial infarction, with increased fibrosis, apoptosis and pathological remodeling (Watson et al., 2010). Importantly, elevations in O-GlcNAc in heart are associated with ischemic pre-conditioning, a classical cardioprotective maneuver (Jones et al., 2008). Pharmacological increases in O-GlcNAc in cardiac myocytes improve cell survival in hydrogen peroxide treatment, which is potentially linked to maintenance of mitochondrial integrity. Further, over-expression of OGT and consequent elevation of O-GlcNAcylation protects NRVM from sI/R damage (Champattanachai et al., 2008). Conversely, knockdown of OGT leads to decreased cell survival. Moreover, pharmacological treatment (PUGNAc) to inhibit OGA and thereby increase O-GlcNAcylation confers strong protection against I/R damage ex vivo (Liu et al., 2007). Collectively, these data suggest that acute increases in protein O-GlcNAc modification in cardiomyocytes are cardioprotective, possibly mediated by inhibition of apoptosis and enhanced mitochondrial function.
At least two questions remain regarding O-GlcNAcylation and cardioprotection. What stimulates the HBP and O-GlcNAc modification? How does O-GlcNAcylation lead to cardioprotection? We recently unveiled an intrinsic link between the UPR and HBP, which provides mechanistic insight into the first question (Wang et al., 2014). We first observed that Xbp1s is potently and acutely stimulated by I/R in heart as early as 5 minutes into the reperfusion stage. To investigate the relationship between the UPR and HBP, we focused on the rate-limiting enzyme, GFAT1. We found that Xbp1s is a direct activator of the gene coding for Xbp1s, based on the presence of an Xbp1s binding site in its promoter, coupled with luciferase reporter assays and chromatin immunoprecipitation analysis. Further, we found that two additional genes within the HBP pathway, as well as GalE, are also under the control of Xbp1s in vitro and in vivo. Using a doxycycline-inducible mouse model, we found that over-expression of Xbp1s in cardiomyocytes leads to robust activation of HBP activity, as documented by increases in UDP-GlcNAc levels and protein O-GlcNAcylation. Additionally, we found that the causal link between the UPR and HBP is a generalized phenomenon beyond myocardial I/R injury. Original work by Zachara et al revealed that various stress conditions in COS-7 cells elicit up-regulation of O-GlcNAcylation, which we found to be associated with induction of Xbp1s and the UPR (Zachara et al., 2004). We also reported correlation between Xbp1s and O-GlcNAcylation in cardiomyocytes exposed to starvation, consistent with prior work (Zou et al., 2012). Xbp1s over-expression protected heart from I/R damage in vivo (Wang et al., 2014). In contrast, cardiomyocyte-specific deletion of Xbp1 amplified I/R-induced pathological cardiac remodeling. GFAT1 and O-GlcNAcylation are involved, at least partially, in Xbp1s-mediated cardioprotection ex vivo and in vitro. Thus, our recent report brings together two large areas of biology and unveils a mechanistic link between the UPR and HBP, which may have broad implications in multiple fields (Glembotski, 2014; Vincenz and Hartl, 2014).
Insight into the mechanism of O-GlcNAcylation-mediated cardioprotection remains elusive, with several possible explanations suggested by prior studies. Elevations in O-GlcNAc protein modification protect mitochondria from loss of membrane potential elicited by exposure to hydrogen peroxide (Champattanachai et al., 2008; Jones et al., 2008). In addition, increases in O-GlcNAcylation may suppress expression of the pro-apoptotic branch of the UPR by suppressing CHOP protein levels (Ngoh et al., 2009). Recently, it was reported that feeding C. elegans O-GlcNAc significantly increased life span by inducing autophagy and improving protein folding (Denzel et al., 2014).
O-GlcNAc itself is a unique molecule with a sugar backbone, an amine group, and an acetyl group. In light of this, it is tempting to speculate that O-GlcNAc per se may be an ideal source of nutrients during energy crises or a storage vehicle when nutrients are plenty. This way, O-GlcNAc can serve as a metabolic buffer between feast and famine. Further work is warranted to test this hypothesis. Collectively, O-GlcNAcylation may protect the cell from stress-induced demise by modulating functions of essential proteins, maintaining integrity of mitochondria, or possibly regulating carbohydrate metabolism.
Ubiquitin-proteasome system and cardiac hypertrophy
The ubiquitin-proteasome system (UPS) is a principal mechanism to degrade misfolded proteins and facilitate cellular homeostasis (Schlossarek et al., 2014; Wang and Robbins, 2014). It is a highly regulated, multistep process, commencing with ATP-dependent activation of ubiquitin by activating enzymes, termed E1’s. Activated ubiquitin is then transferred to a conjugating enzyme, termed E2. Ubiquitin ligases (E3’s), in turn, bind both the E2 and target substrates and catalyze the ubiquitin modification on lysine(s) of target proteins as well as the subsequent elongation of the poly-ubiquitin chain. There are two E1’s and approximately 40 E2’s in the human genome, but more than 600 E3’s, conferring precise selectivity of protein recognition and degradation.
The presence of attached poly-ubiquitin chains targets a protein to the 26S proteasome, a protein complex comprising a 20S catalytic core and one or two 19S regulatory cap particles. Ubiquitinated proteins are recognized, deubiquitinated, unfolded by the 19S cap, and translocated into the interior of the 20S core for degradation by proteasomal peptidases. Finally, small peptides are released and quickly hydrolyzed to amino acids. In some specialized cases, such as processing of the precursor of the transcription factor NFκB, degradation can yield a biologically active protein fragment (Rape and Jentsch, 2002). Beyond degradation of misfolded proteins, the target turnover of key regulatory molecules by the UPS has been implicated in cell cycle control, inflammation, and cell death.
Cardiac hypertrophic growth is exemplified by increased stress on the myocardium, with consequent turnover and oxidative damage of cardiac proteins. Therefore, the UPS plays an important role in maintaining cellular function and viability, and dysfunction of the UPS may contribute to disease pathogenesis. Proteasome function, measured as both chymotrypsin-like and caspase-like activities, is significantly diminished in patients with hypertrophic cardiomyopathy (Predmore et al., 2010). Whereas alterations in UPS protein content have not been reported, post-translational modifications are significantly increased in hypertrophic human hearts. Failing human hearts manifest similar reductions in proteasome function. Subsequent studies showed that reductions in proteasome activity in failing hearts stem from decreases in ATPase activity and diminished docking of the 19S regulatory cap to the 20S proteolytic core (Day et al., 2013). Consistently, failing human hearts are marked by accumulation of ubiquitinated proteins, cardiac hypertrophy, and fibrosis (Tsukamoto et al., 2006). Moreover, malfunction of the UPS precedes cardiac dysfunction in a rodent model of cardiac hypertrophy (Tsukamoto et al., 2006), and chronic exposure to a proteasome inhibitor provokes deterioration in cardiac performance in pigs, accompanied by increases in apoptosis and fibrosis (Herrmann et al., 2013).
Mechanistically, insufficiency of proteasome function may lead directly to activation of NFAT and consequent hypertrophic growth (Tang et al., 2010). However, it has been reported that pressure overload provokes increases in proteasome activity in both chronic canine and acute rodent models (Depre et al., 2006). Consistently, inhibition of proteasome activity leads to suppression of cardiac hypertrophy when a proteasome inhibitor is administrated concomitantly with isoproterenol (Stansfield et al., 2008). Further, even established hypertrophy can be regressed by proteasome inhibition. An explanation for this apparent discrepancy has not emerged, although these studies involve distinct models, different stages of hypertrophy and heart failure, and different degrees of proteasome inhibition.
UPS and myocardial infarction
Reperfusion following myocardial infarction is associated with profound remodeling of cellular architecture and machinery. In this setting, the UPS is critically involved. Inflammation is one of the most important contributors to I/R injury. Sudden increases in ROS elicit protein misfolding and damage. Activation of NFκB, a master regulator of inflammatory responses, relies on proteolytic degradation of IkBα, the inhibitor of IKK and NFκB activity. Pharmacological inhibition of global UPS activity with a 20S proteasome inhibitor suppresses NFκB activation and leads to strong cardioprotective effects in pigs (Pye et al., 2003). Consistently, administration of the same inhibitor before myocardial infarction, or even during reperfusion, elicits significant protection against I/R damage in mice (Stansfield et al., 2007). In both cases, suppression of NFκB activity is involved.
Manipulation of UPS activity specifically in cardiomyocytes, however, provokes different results. Three enzymatic activities of the UPS are reportedly suppressed by I/R in mice (Tian et al., 2012). Working with a model of cardiomyocyte-restricted over-expression of a peptidase-disabled mutant of the 20S complex β5 subunit, thereby inhibiting endogenous chymotrypsin-like activity in 26S proteasome, Tian et al concluded that inhibition of the UPS in cardiomyocytes leads to deterioration of the cardiac response to I/R and increases in apoptosis. In contrast, over-expression of the PA28α (an alternative regulatory subunit independent of ubiquitin chain targeting) in cardiomyocytes to enhance proteasomal protease activity protects mouse hearts from I/R damage and prevents accumulation of misfolded proteins (Li et al., 2011). This finding may be elucidated by a recent report that PA28α can play a special role in the removal of oxidatively damaged proteins (Hernebring et al., 2013). Thus, modulation of the UPS within cardiomyocytes versus non-cardiomyocytes results in distinct phenotypes, calling for caution when addressing the function of UPS in cardiac I/R injury.
Conclusion and perspectives
Even as we have witnessed robust advances in our understanding and treatment of CVD, the disease continues to evolve and expand. As a consequence, effective treatments are still lacking, and CVD remains the leading cause of morbidity and mortality worldwide. Due to its minimal capacity for replication, the cardiomyocyte is uniquely reliant on mechanisms of protein quality control. Owing to its incessant requirement of high levels of energy, metabolic processes in the myocyte are similarly paramount. At some level, then, it is not surprising that these two processes – protein quality control and cellular metabolism – are intimately linked.
Autophagy, the UPR, and the UPS are the principal mechanisms to degrade misfolded proteins and defective organelles, serving to maintain a functional cellular environment. Malfunction and dysregulation of any of these processes culminates in disease, such as cardiac hypertrophy, myocardial infarction, and heart failure. Further, each of these mechanisms is intimately linked to the interlacing cascades of metabolism. Precise dissection of the complex interplay between protein quality control systems and their metabolic context and consequences is mandatory. It is our expectation that coordinated understanding of this fascinating biology will propel us toward novel therapeutic targets with clinical relevance.
Acknowledgments
We thank members of the Hill lab for constructive comments.
Sources of Funding
This work was supported by grants from the NIH (HL-120732; HL-100401), AHA (14SFRN20740000), CPRIT (RP110486P3), and the Leducq Foundation (11CVD04). Zhao V. Wang was supported by a Scientist Development Grant from the American Heart Association (14SDG18440002).
Footnotes
Conflicts of Interest
We declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267:H742–750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. The Journal of biological chemistry. 2003;278:28930–28937. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–793. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont PJ, Tadimalla A, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, Gude N, Sussman MA, Glembotski CC. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J Biol Chem. 2008;283:14012–14021. doi: 10.1074/jbc.M709776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel G. Protein targeting. Biosci Rep. 2000;20:303–344. doi: 10.1023/a:1010318832604. [DOI] [PubMed] [Google Scholar]
- Bond MR, Hanover JA. O-GlcNAc cycling: a link between metabolism and chronic disease. Annu Rev Nutr. 2013;33:205–229. doi: 10.1146/annurev-nutr-071812-161240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaanine AH, Jeong D, Liang L, Chemaly ER, Fish K, Gordon RE, Hajjar RJ. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012;3:265. doi: 10.1038/cddis.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am J Physiol Cell Physiol. 2008;294:C1509–1520. doi: 10.1152/ajpcell.00456.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW, 2nd, Kang YJ, Prolla TA, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res. 2011;108:837–846. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day SM, Divald A, Wang P, Davis F, Bartolone S, Jones R, Powell SR. Impaired assembly and post-translational regulation of 26S proteasome in human end-stage heart failure. Circ Heart Fail. 2013;6:544–549. doi: 10.1161/CIRCHEARTFAILURE.112.000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I Ultrastructural and cytochemical changes. The American journal of pathology. 1980;98:425–444. [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Wang ZV, Tao C, Gao N, Holland WL, Ferdous A, Repa JJ, Liang G, Ye J, Lehrman MA, et al. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. The Journal of clinical investigation. 2013;123:455–468. doi: 10.1172/JCI62819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzel MS, Storm NJ, Gutschmidt A, Baddi R, Hinze Y, Jarosch E, Sommer T, Hoppe T, Antebi A. Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell. 2014;156:1167–1178. doi: 10.1016/j.cell.2014.01.061. [DOI] [PubMed] [Google Scholar]
- Depre C, Wang Q, Yan L, Hedhli N, Peter P, Chen L, Hong C, Hittinger L, Ghaleh B, Sadoshima J, et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114:1821–1828. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: programmed to conserve. J Mol Cell Cardiol. 2013;55:85–91. doi: 10.1016/j.yjmcc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem. 2009;284:29735–29745. doi: 10.1074/jbc.M109.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- Fu HY, Okada K, Liao Y, Tsukamoto O, Isomura T, Asai M, Sawada T, Okuda K, Asano Y, Sanada S, et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122:361–369. doi: 10.1161/CIRCULATIONAHA.109.917914. [DOI] [PubMed] [Google Scholar]
- Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell metabolism. 2012;15:623–634. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Glembotski CC. Finding the missing link between the unfolded protein response and O-GlcNAcylation in the heart. Circ Res. 2014;115:546–548. doi: 10.1161/CIRCRESAHA.114.304855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nature reviews Molecular cell biology. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Hernebring M, Fredriksson A, Liljevald M, Cvijovic M, Norrman K, Wiseman J, Semb H, Nystrom T. Removal of damaged proteins during ES cell fate specification requires the proteasome activator PA28. Scientific reports. 2013;3:1381. doi: 10.1038/srep01381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann J, Wohlert C, Saguner AM, Flores A, Nesbitt LL, Chade A, Lerman LO, Lerman A. Primary proteasome inhibition results in cardiac dysfunction. Eur J Heart Fail. 2013;15:614–623. doi: 10.1093/eurjhf/hft034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- Hill JA, Olson EN. Cardiac plasticity. The New England journal of medicine. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- Hu Y, Sun Q, Li Z, Chen J, Shen C, Song Y, Zhong Q. High basal level of autophagy in high-altitude residents attenuates myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2014;148:1674–1680. doi: 10.1016/j.jtcvs.2014.03.038. [DOI] [PubMed] [Google Scholar]
- Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, Hart GW, Marban E. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation. 2008;117:1172–1182. doi: 10.1161/CIRCULATIONAHA.107.730515. [DOI] [PubMed] [Google Scholar]
- Kanamori H, Takemura G, Maruyama R, Goto K, Tsujimoto A, Ogino A, Li L, Kawamura I, Takeyama T, Kawaguchi T, et al. Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am J Pathol. 2009;174:1705–1714. doi: 10.2353/ajpath.2009.080875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. 2010;3:420–430. doi: 10.1161/CIRCHEARTFAILURE.109.888479. [DOI] [PubMed] [Google Scholar]
- Kenific CM, Debnath J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2014 doi: 10.1016/j.tcb.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, Hill JA. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation. 2006;113:2579–2588. doi: 10.1161/CIRCULATIONAHA.106.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest. 2011;121:3689–3700. doi: 10.1172/JCI45709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Marchase RB, Chatham JC. Increased O-GlcNAc levels during reperfusion lead to improved functional recovery and reduced calpain proteolysis. Am J Physiol Heart Circ Physiol. 2007;293:H1391–1399. doi: 10.1152/ajpheart.00285.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell metabolism. 2011;14:443–451. doi: 10.1016/j.cmet.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004;23:169–179. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SA, Collins HE, Chatham JC. Protein O-GlcNAcylation and Cardiovascular (Patho)physiology. J Biol Chem. 2014 doi: 10.1074/jbc.R114.585984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, Glembotski CC. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- Maruyama R, Goto K, Takemura G, Ono K, Nagao K, Horie T, Tsujimoto A, Kanamori H, Miyata S, Ushikoshi H, et al. Morphological and biochemical characterization of basal and starvation-induced autophagy in isolated adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008;295:H1599–1607. doi: 10.1152/ajpheart.91449.2007. [DOI] [PubMed] [Google Scholar]
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circulation research. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- Mearini G, Schlossarek S, Willis MS, Carrier L. The ubiquitin-proteasome system in cardiac dysfunction. Biochim Biophys Acta. 2008;1782:749–763. doi: 10.1016/j.bbadis.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K, Hokimoto S, Yamamuro M, Iwawaki T, Gotoh T, et al. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1124–1132. doi: 10.1161/ATVBAHA.111.224519. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres J, Fullerton HJ, Howard VJ, et al. Heart Disease and Stroke Statistics-2015 Update: A Report From the American Heart Association. Circulation. 2014 doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. The New England journal of medicine. 2012;366:54–63. doi: 10.1056/NEJMra1112570. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nature medicine. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Ngoh GA, Hamid T, Prabhu SD, Jones SP. O-GlcNAc signaling attenuates ER stress-induced cardiomyocyte death. Am J Physiol Heart Circ Physiol. 2009;297:H1711–1719. doi: 10.1152/ajpheart.00553.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109:151–160. doi: 10.1161/CIRCRESAHA.110.237339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski G, Das S, Juhasz B, Kertesz A, Tosaki A, Das DK. Cardioprotection by endoplasmic reticulum stress-induced autophagy. Antioxid Redox Signal. 2011;14:2191–2200. doi: 10.1089/ars.2010.3486. [DOI] [PubMed] [Google Scholar]
- Pfeifer U, Fohr J, Wilhelm W, Dammrich J. Short-term inhibition of cardiac cellular autophagy by isoproterenol. J Mol Cell Cardiol. 1987;19:1179–1184. doi: 10.1016/s0022-2828(87)80528-x. [DOI] [PubMed] [Google Scholar]
- Porrello ER, D’Amore A, Curl CL, Allen AM, Harrap SB, Thomas WG, Delbridge LM. Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy. Hypertension. 2009;53:1032–1040. doi: 10.1161/HYPERTENSIONAHA.108.128488. [DOI] [PubMed] [Google Scholar]
- Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;121:997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye J, Ardeshirpour F, McCain A, Bellinger DA, Merricks E, Adams J, Elliott PJ, Pien C, Fischer TH, Baldwin AS, Jr, et al. Proteasome inhibition ablates activation of NF-kappa B in myocardial reperfusion and reduces reperfusion injury. Am J Physiol Heart Circ Physiol. 2003;284:H919–926. doi: 10.1152/ajpheart.00851.2002. [DOI] [PubMed] [Google Scholar]
- Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rape M, Jentsch S. Taking a bite: proteasomal protein processing. Nature cell biology. 2002;4:E113–116. doi: 10.1038/ncb0502-e113. [DOI] [PubMed] [Google Scholar]
- Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, Bugger H, Frank D, Bevins J, Chen D, et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. The Journal of clinical investigation. 2013;123:5319–5333. doi: 10.1172/JCI71171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermel BA, Hill JA. Autophagy in load-induced heart disease. Circ Res. 2008;103:1363–1369. doi: 10.1161/CIRCRESAHA.108.186551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala-Mercado JA, Wider J, Undyala VV, Jahania S, Yoo W, Mentzer RM, Jr, Gottlieb RA, Przyklenk K. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation. 2010;122:S179–184. doi: 10.1161/CIRCULATIONAHA.109.928242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10132–10136. doi: 10.1073/pnas.0401900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada T, Minamino T, Fu HY, Asai M, Okuda K, Isomura T, Yamazaki S, Asano Y, Okada K, Tsukamoto O, et al. X-box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE-like element in cardiomyocytes. J Mol Cell Cardiol. 2010;48:1280–1289. doi: 10.1016/j.yjmcc.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Schlossarek S, Frey N, Carrier L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J Mol Cell Cardiol. 2014;71:25–31. doi: 10.1016/j.yjmcc.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Sengupta A, Molkentin JD, Paik JH, DePinho RA, Yutzey KE. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. The Journal of biological chemistry. 2011;286:7468–7478. doi: 10.1074/jbc.M110.179242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U, Frede S, Toietta G, Di Rocco G, Bussani R, et al. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol. 2007;50:1029–1037. doi: 10.1016/j.jacc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, Tateno K, Moriya J, Yokoyama M, Nojima A, et al. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. The Journal of clinical investigation. 2010;120:1506–1514. doi: 10.1172/JCI40096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokina N, O’Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF, Ballal K, Taegtmeyer H, Buttrick PM, Lewandowski ED. Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation. 2007;115:2033–2041. doi: 10.1161/CIRCULATIONAHA.106.668665. [DOI] [PubMed] [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- Stansfield WE, Moss NC, Willis MS, Tang R, Selzman CH. Proteasome inhibition attenuates infarct size and preserves cardiac function in a murine model of myocardial ischemia-reperfusion injury. Ann Thorac Surg. 2007;84:120–125. doi: 10.1016/j.athoracsur.2007.02.049. [DOI] [PubMed] [Google Scholar]
- Stansfield WE, Tang RH, Moss NC, Baldwin AS, Willis MS, Selzman CH. Proteasome inhibition promotes regression of left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2008;294:H645–650. doi: 10.1152/ajpheart.00196.2007. [DOI] [PubMed] [Google Scholar]
- Sun X, Momen A, Wu J, Noyan H, Li R, von Harsdorf R, Husain M. p27 protein protects metabolically stressed cardiomyocytes from apoptosis by promoting autophagy. J Biol Chem. 2014;289:16924–16935. doi: 10.1074/jbc.M113.542795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- Tang M, Li J, Huang W, Su H, Liang Q, Tian Z, Horak KM, Molkentin JD, Wang X. Proteasome functional insufficiency activates the calcineurin-NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc Res. 2010;88:424–433. doi: 10.1093/cvr/cvq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2008;105:9745–9750. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarone G, Brancaccio M. Keep your heart in shape: molecular chaperone networks for treating heart disease. Cardiovasc Res. 2014;102:346–361. doi: 10.1093/cvr/cvu049. [DOI] [PubMed] [Google Scholar]
- Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circulation research. 2006;99:275–282. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- Tian Z, Zheng H, Li J, Li Y, Su H, Wang X. Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia-reperfusion injury in mice. Circ Res. 2012;111:532–542. doi: 10.1161/CIRCRESAHA.112.270983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto O, Minamino T, Okada K, Shintani Y, Takashima S, Kato H, Liao Y, Okazaki H, Asai M, Hirata A, et al. Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem Biophys Res Commun. 2006;340:1125–1133. doi: 10.1016/j.bbrc.2005.12.120. [DOI] [PubMed] [Google Scholar]
- Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. The American journal of cardiology. 2010;106:360–368. doi: 10.1016/j.amjcard.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS, Stephanou A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. Journal of molecular and cellular cardiology. 2006;40:846–852. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]
- Vincenz L, Hartl FU. Sugarcoating ER Stress. Cell. 2014;156:1125–1127. doi: 10.1016/j.cell.2014.02.035. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- Wang X, Robbins J. Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol. 2014;71:16–24. doi: 10.1016/j.yjmcc.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell. 2014;156:1179–1192. doi: 10.1016/j.cell.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan YT, Jones SP. O-linked beta-N-acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci U S A. 2010;107:17797–17802. doi: 10.1073/pnas.1001907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510–1527. doi: 10.1101/gad.2051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis MS, Patterson C. Hold me tight: Role of the heat shock protein family of chaperones in cardiac disease. Circulation. 2010;122:1740–1751. doi: 10.1161/CIRCULATIONAHA.110.942250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis MS, Patterson C. Proteotoxicity and cardiac dysfunction. N Engl J Med. 2013;368:1755. doi: 10.1056/NEJMc1302511. [DOI] [PubMed] [Google Scholar]
- Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, Wang ZV, Morales C, Luo X, Cho G, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–1151. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin W, Lu X, Li X, Niu K, Cai J. Attenuation of endoplasmic reticulum stress-related myocardial apoptosis by SERCA2a gene delivery in ischemic heart disease. Mol Med. 2011;17:201–210. doi: 10.2119/molmed.2010.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Pacheco BD, Leng L, Bucala R, Ren J. Macrophage migration inhibitory factor plays a permissive role in the maintenance of cardiac contractile function under starvation through regulation of autophagy. Cardiovasc Res. 2013;99:412–421. doi: 10.1093/cvr/cvt116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Young ME, Yan J, Razeghi P, Cooksey RC, Guthrie PH, Stepkowski SM, McClain DA, Tian R, Taegtmeyer H. Proposed regulation of gene expression by glucose in rodent heart. Gene Regul Syst Bio. 2007;1:251–262. doi: 10.4137/grsb.s222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachara NE. The roles of O-linked beta-N-acetylglucosamine in cardiovascular physiology and disease. Am J Physiol Heart Circ Physiol. 2012;302:H1905–1918. doi: 10.1152/ajpheart.00445.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279:30133–30142. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Zhu-Mauldin X, Marchase RB, Paterson AJ, Liu J, Yang Q, Chatham JC. Glucose Deprivation-induced Increase in Protein O-GlcNAcylation in Cardiomyocytes Is Calcium-dependent. J Biol Chem. 2012;287:34419–34431. doi: 10.1074/jbc.M112.393207. [DOI] [PMC free article] [PubMed] [Google Scholar]