Biosynthetic Versatility and Coordinated Action of 5′-Deoxyadenosyl Radicals in Deazaflavin Biosynthesis (original) (raw)
Abstract

Coenzyme F420 is a redox cofactor found in methanogens and in various actinobacteria. Despite the major biological importance of this cofactor, the biosynthesis of its deazaflavin core (8-hydroxy-5-deazaflavin, Fo) is still poorly understood. Fo synthase, the enzyme involved, is an unusual multidomain radical SAM enzyme that uses two separate 5′-deoxyadenosyl radicals to catalyze Fo formation. In this paper, we report a detailed mechanistic study on this complex enzyme that led us to identify (1) the hydrogen atoms abstracted from the substrate by the two radical SAM domains, (2) the second tyrosine-derived product, (3) the reaction product of the CofH-catalyzed reaction, (4) the demonstration that this product is a substrate for CofG, and (5) a stereochemical study that is consistent with the formation of a _p_-hydroxybenzyl radical at the CofH active site. These results enable us to propose a mechanism for Fo synthase and uncover a new catalytic motif in radical SAM enzymology involving the use of two 5′-deoxyadenosyl radicals to mediate the formation of a complex heterocycle.
Introduction
Coenzyme F420 (4) is a redox cofactor found in methanogens and in various actinobacteria, while its biosynthetic precursor Fo (8-hydroxy-5-deazaflavin, 3) can also be found in certain cyanobacteria and eukaryotes.1−4 F420 was first isolated from Methanobacterium strain M.o.H. as a fluorescent cofactor involved in hydrogen metabolism and has subsequently been shown to be a key cofactor in methanogenesis.5−7 F420 is required for the breakdown of aflatoxin in Mycobacterium smegmatis.8 In addition, M. tuberculosis, the etiologic agent of tuberculosis, is predicted to contain a large, yet unexplored, number of F420-dependent enzymes, some implicated in nitrosative stress resistance.9,10 F420 is biosynthesized in Methanocaldococcus jannaschii by the action of eight enzymes with the formation of the deazaflavin chromophore (Fo) as the remaining unsolved step (Figure 1).11−17
Figure 1.
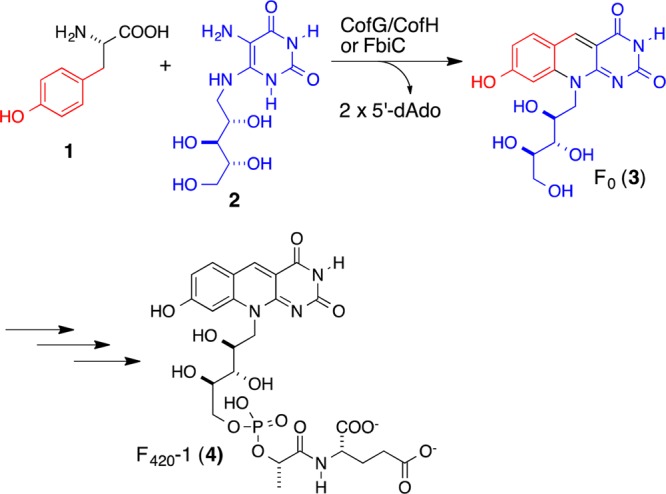
Biosynthesis of the deazaflavin chromophore of F420 (Fo, 3). The structure of F420 shown contains a single glutamic acid, which is designated as F420-1. The number of glutamic acid residues varies.
Despite the major biological importance of this cofactor, the biosynthesis of its deazaflavin core (Fo) remains only partially understood. The formation of Fo is mediated by two separate radical SAM active sites, one each in the CofG and CofH enzymes or both in the FbiC enzyme. These two radical SAM domains constitute the functional domains of Fo synthase as we recently demonstrated.18 While two [4Fe-4S] clusters have been found in other systems (MoaA, AlbA, HydG),19−24 Fo synthase is an unusual multidomain radical SAM enzyme in that it uses two separate 5′-deoxyadenosyl radicals to catalyze Fo formation.18
We recently reconstituted the Fo synthase and identified diaminouracil (5-amino-6-ribitylamino-2,4(1_H_,3_H_)-pyrimidine-dione) 2 and tyrosine 1 as the enzyme substrates.18 The detection of diaminouracil 2 bound to freshly purified CofH suggested that CofH catalyzes the early steps in deazaflavin formation.
The tyrosine lyase activity of Fo synthase is likely to be mechanistically analogous to the tyrosine lyase activity of HydG (involved in [FeFe]-hydrogenase maturation)22 and ThiH (involved in anaerobic thiamin thiazole biosynthesis).25 A recent structure of tryptophan lyase (NosL) demonstrated that the initial hydrogen atom abstraction was occurring from the amine NH,26 suggesting that the corresponding tyrosine lyases are also likely to occur by hydrogen atom abstraction from the amino group rather than the previously assumed phenolic hydroxyl. Based on these data, we elaborated the mechanism shown in Figure 2 as a starting point for our mechanistic investigations on Fo synthase.
Figure 2.

Mechanistic proposal for the Fo-synthase-catalyzed reaction. AdG and AdH correspond to the 5′-deoxyadenosyl radicals generated at the CofG and the CofH active sites by reduction of SAM using the active site [4Fe-4S]+1 cluster.23,24 Abstraction of the tyrosine amine hydrogen atom by the CofH 5′-deoxyadenosyl radical gives 5, which then undergoes fragmentation leading to the formation of the _p_-hydroxybenzyl radical 7. This then undergoes addition to the double bond of diaminouracil 2 followed by oxidation to give 9. Transfer of 9 from the CofH active site to the CofG active site gives 10. Hydrogen atom abstraction, by the CofG 5′-deoxyadenosyl radical, gives 11. Cyclization to 13, followed by oxidation by the [4Fe-4S]+2 cluster and elimination of ammonia completes the formation of the deazaflavin 3.
In this paper, we report a detailed mechanistic study on this complex enzyme that led us to identify (1) the hydrogen atoms abstracted from the substrate by the two radical SAM domains, (2) the second tyrosine-derived product, (3) the reaction product of the CofH-catalyzed reaction, (4) the demonstration that this product is a substrate for CofG, and (5) a stereochemical study that is consistent with the formation of a _p_-hydroxybenzyl radical at the CofH active site. Our study elucidates a new catalytic motif in radical SAM enzymology involving the use of two 5′-deoxyadenosyl radicals to mediate the formation of a complex heterocycle.
Results
Identification of the Two Sites of Hydrogen Atom Abstraction
Reconstitution of the CofG/CofH-catalyzed reaction in the presence of D7-tyrosine resulted in the incorporation of a single deuterium atom into 5′-deoxyadenosine as judged by mass spectrometry (MS) analysis, which showed an increase in the intensity of the 253.1 peak relative to the 252.1 peak (48.6% vs 13.4% in the reaction performed with D7-tyrosine compared with unlabeled substrate, respectively, Figure 3a,b, left panel). MS analysis of the Fo formed from D7-Tyr showed that it contained four deuteriums (Figure 3b, right panel). Reconstitution of the CofG/CofH reaction with other isotopologues of tyrosine ([2-D1]-, [3,3-D2]-, [2′,6′-D2]-, and [3′,5′-D2]-Tyr) localized the site of deuterium abstraction to the β-carbon of Tyr since deuterium incorporation in 5′-deoxyadenosine was detected only when using D7- or 3,3-D2-Tyr as substrate (Figure 3c).
Figure 3.

Deuterium incorporation into 5′-deoxyadenosine (left spectrum, [M + H]+ = 252.1 Da) and Fo (right spectrum, [M + H]+ = 364.1 Da, [M + K]+ = 402.1 Da) from various isotopologues of tyrosine: (a) tyrosine, (b) D7-tyrosine, or (c) 3,3-D2-tyrosine. The percentages written above the 253.1 Da peak indicate the relative height compared to the 252.1 peak. Each reaction mixture contained methyl viologen, SAM, diaminouracil, D_n_-tyrosine, CofH, and CofG.
MS analysis of the Fo formed in the reaction with [3,3-D2]-Tyr demonstrated that the second β-deuterium was retained in Fo. Using [3′,5′-D2]-Tyr led to the incorporation of both deuterium atoms into Fo while only one was incorporated when using [2′,6′-D2]_-_Tyr as substrate (Supplementary Figure 1). Conversely, no deuterium incorporation into the products was observed with [2-D1]-Tyr (Supplementary Figure 1). This confirmed that, in addition to one deuterium on the 3-position, a deuterium from a second nonexchangeable site (i.e., 2′/6′ position) is removed. Similar results were obtained using the full length FbiC enzyme demonstrating that both types of Fo synthase use an identical mechanism.
The observation that deuterium transfer from a second site on stably labeled tyrosine to the 5′-deoxyadenosyl radical was not occurring suggested that the second hydrogen atom transfer was from an exchangeable site. This was confirmed by demonstrating the incorporation of deuterium from solvent into 5′-deoxyadenosine when the CofH reaction was run in a buffer containing 80% D2O. In addition, noticeable peaks corresponding to [5′,5′-D2]-5′-deoxyadenosine and [5′,5′,5′-D3]-5′-deoxyadenosine were observed in the mass spectrum (Figure 4). The residual 252.1 peak in spectrum a is most likely due to the uncoupled formation of 5′-deoxyadenosine and to the presence of H2O in the reaction mixture.
Figure 4.

CofH-catalyzed deuterium incorporation into 5′-deoxyadenosine in reactions run in 80% D2O containing SAM, diaminouracil and methyl viologen. (a) Reaction run in the presence of tyrosine showing an enhanced 253.1 peak (calculated to be 69.4, 21.5 and 16.7% for the [M + D + H]+, [M + D2 + H]+, [M + D3 + H]+, respectively, in panel a and 30.8% for the [M + D + H]+ in panel b). (b) Reaction run in the absence of tyrosine showing the uncoupled production of 5′-deoxyadenosine. 5′-deoxyadenosine has an expected [M + H]+ of 252.1 m/z with the 253.1 peak calculated to be 12.9 ± 3%.
Characterization of the Second Tyrosine-Derived Product
Glyoxylate, derived from the hydrolysis of the glycine imine 6, was the most likely second product resulting from the fragmentation of the tyrosyl radical 5. Our initial attempts to trap glyoxylate as the 2-quinoxalinol 17, by treating the reaction mixture with _o_-phenylenediamine,27,28 failed because the glycerol used to stabilize the enzyme contained relatively large quantities of glyoxylate as an impurity. We therefore performed the reaction using [15N,13C9]-Tyr, which allowed for the selective LC-MS detection and quantitation of enzymatically produced glyoxylate (Figure 5). Comparison to standard curves showed that the glyoxylate:Fo ratio was 1.1:1. We were able to detect glyoxylate only in the presence of CofH (Figure 5), further supporting the proposal that CofH is the enzyme responsible for the tyrosine cleavage reaction.
Figure 5.
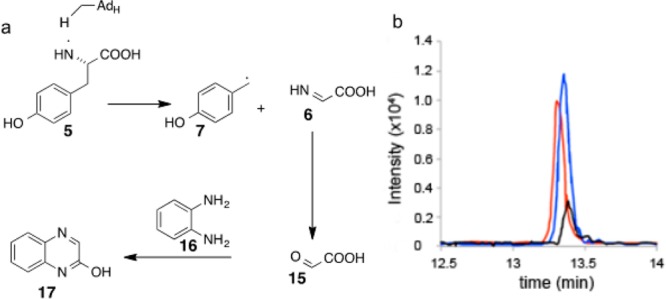
Trapping of the tyrosine-derived glyoxylate 15. (a) Proposed scheme for the generation of glyoxylate 15 by hydrolysis of the glycine imine 6 and its trapping using _o_-phenylenediamine 16. (b) Extracted ion chromatograms at m/z 148.05 ± 0.1 of labeled 2-quinoxalinol 17 generated from a reaction mixture containing CofG + CofH + SAM + sodium dithionite + [15N,13C9]-tyrosine + 2 (red trace); CofH + SAM + sodium dithionite + [15N,13C9]-tyrosine + 2 (blue trace). Control reactions lacking SAM, sodium dithionite, tyrosine, or CofH did not produce labeled glyoxylate.
Identification of the CofH Reaction Product
HPLC analysis of the CofH reaction mixture revealed the formation of a peak eluting at 11.2 min. This peak appeared only in reaction mixtures containing CofH, 1, 2, SAM and reduced methyl viologen (or dithionite) (Figure 6). LC-MS analysis yielded a protonated molecular ion at 383.1573 m/z when the reaction was reconstituted in the presence of tyrosine and 390.1775 m/z when the reaction was reconstituted in the presence of [15N,13C9]-Tyr (Figure 7). This demonstrated that CofH catalyzed the formation of a product containing seven tyrosine-derived carbons and suggested a molecular formula of C16H22N4O7 ([M + H]+ calcd 383.1561, 3.3 ppm error). CID fragmentation of the unlabeled product resulted in the formation of product ions at m/z 107.1 and 277.1, while the labeled product showed product ions at m/z 114.1 and 277.1 (Figure 7). This suggested that the smaller fragment contained all the tyrosine-derived carbon atoms (i.e., 7 carbon atoms) while the 277.1 fragment originated from diaminouracil.
Figure 6.
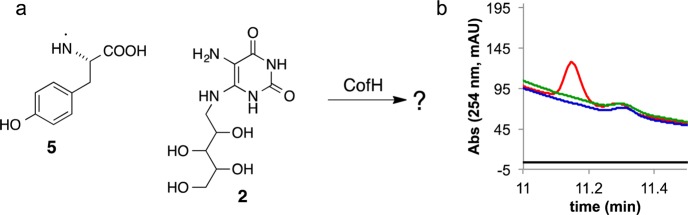
Detection of the CofH reaction product. (a) Reaction catalyzed by CofH. (b) HPLC chromatogram of the reaction mixture containing CofH + SAM + methyl viologen (reduced) + 1 + 2 (red) showing a new product eluting after 11.15 min. Reaction mixtures lacking reduced methyl viologen (green), SAM (black), or tyrosine (blue) did not show this product.
Figure 7.
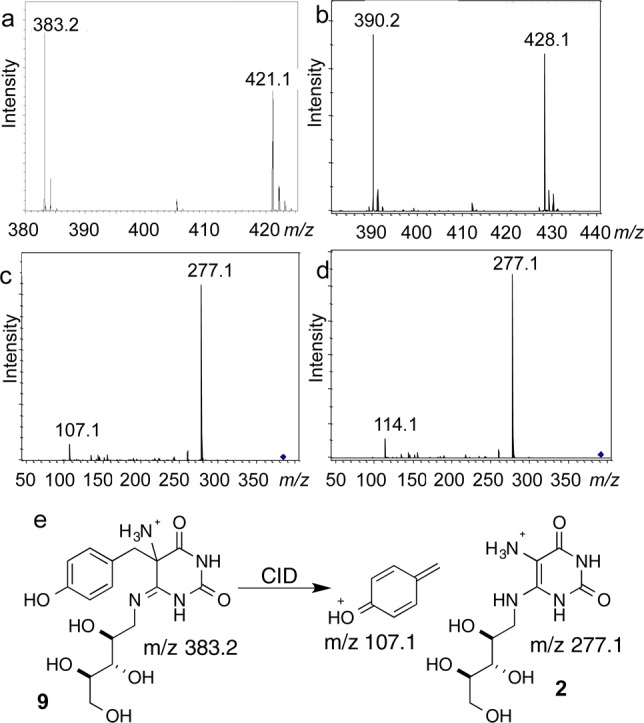
MS analysis of the CofH reaction product. (a) Mass spectrum of the product generated from tyrosine ([M + H]+ obs. 383.1573, calcd for 9, 383.1561, 3.3 ppm error, [M + K]+ obs. 421.1132, calcd for 9, 421.1120, 2.8 ppm error; (b) mass spectrum of the product generated from [15N,13C9]-Tyr ([M + H]+ obs. 390.1775, calcd for [13C7]-9, 390.1796, 5.4 ppm error, [M + K]+ obs. 428.1322, calcd for 9, 428.1355, 7.7 ppm error); (c) MS2 of the product from tyrosine (m/z 383.1); (d) MS2 of the product from [15N,13C9]-tyrosine (m/z 390.1). The isolation width was 10 m/z and the collision energy was 30 V. (e) Proposed CID fragmentation pattern of the CofH product 9.
The CofH reaction product was then produced on a larger scale, purified by HPLC and analyzed by nuclear magnetic resonance (NMR) spectroscopy. The 1H NMR spectrum had doublets at 6.77 and 6.57 ppm, consistent with a para-substituted benzene ring with electron-donating substituents, multiple signals in the 3–3.8 ppm region consistent with protons attached to oxygen-bound carbons, and two doublets at 2.87 ppm suggestive of a benzylic methylene group. Using 2D NMR techniques (1H–1H COSY, 1H–13C HSQC, and 1H–13C HMBC), the CofH reaction product was unambiguously identified as compound 9 (Supplementary Figures 2–10).
To confirm that 9 was an intermediate and not a shunt or a decomposition product, we produced D2-9 on a large scale using CofH and [3,3-D2]-Tyr. Treatment of the HPLC purified compound with reduced CofG resulted in the formation of Fo. MS analysis of the reaction products demonstrated deuterium incorporation into 5′-deoxyadenosine and Fo (Figure 8). The mixture of m/z 364/365 in the mass spectrum of Fo is due to the nonenzymatic reduction of [5-D1]-Fo by dithionite followed by nonenzymatic aerobic oxidation after removal of the reaction mixture from the anaerobic chamber as was previously observed during the initial characterization of F420/F0.5
Figure 8.

Incorporation of deuterium into 5′-deoxyadenosine in reactions consisting of SAM, [5,5-D2]-9 and dithionite reduced CofG. (a) Mass spectrum of 5′-deoxyadenosine (calcd 252.1 with a 253.1 peak calculated to be 12.9 ± 3% relative intensity; (b) mass spectrum of Fo (calcd 364.1). The presence of the 252.1 peak in the 5′-deoxyadenosine MS is most likely due to the uncoupled production of 5′-deoxyadenosine.
Identification of the Reaction Product of a FbiC Variant
Disruption of the conserved CXXXCXXC motifs of FbiC individually resulted in the variants FbiC-C1 (cluster is disrupted in the CofG homologous domain) and FbiC-C2 (cluster is disrupted in the CofH homologous domain). Both protein variants had all three cysteine residues substituted with alanine residues, thereby abolishing the [4Fe-4S] cluster. Incubation of each variant with tyrosine, diaminouracil, SAM, and flavodoxin/flavodoxin reductase led to the identification of a new peak only in the FbiC-C1 reaction (Figure 9). The intermediate was subjected to LC-MS/MS analysis and had an identical retention time, protonated ion and MS2 fragmentation pattern to intermediate 9 generated by CofH.
Figure 9.
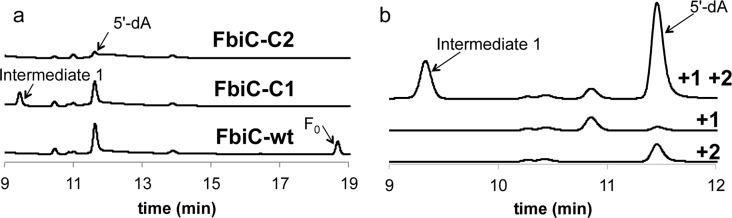
Detection of an intermediate produced by FbiC-C1. (a) HPLC analysis (Abs 257 nm) of the reaction mixtures containing SAM + tyrosine (1) + diaminouracil (2) + flavodoxin/flavodoxin reductase in the presence of FbiC (wild-type) (25 μM) or variants (FbiC-C1 or FbiC-C2, each at 50 μM). (b) HPLC analysis (Abs 257 nm) of the reaction of FbiC-C1 demonstrating that the production of the intermediate is dependent on both tyrosine (1) and diaminouracil (2).
Mixing of FbiC-C1 and FbiC-C2 resulted in the formation of Fo (Figure 10) demonstrating that both variants contain a successfully reconstituted [4Fe-4S] cluster and are fully active. Interestingly, this confirmed that a stable diffusible reaction intermediate is released from one radical SAM domain (CofH or the C-terminal part of FbiC) to the other one (CofG or the N-terminal part of FbiC). Surprisingly, the rate of product formation for the wild-type enzyme (25 μM enzyme = 50 μM [4Fe-4S]) was similar to the rate of product formation for the mixture of the two mutated enzymes (50 μM each = 50 μM [4Fe-4S]). This suggests that FbiC catalyzes two independent reactions where intermediate 9 is not transferred directly from the C-terminal domain to the N-terminal domain but most likely diffuses from one active site to the other.
Figure 10.
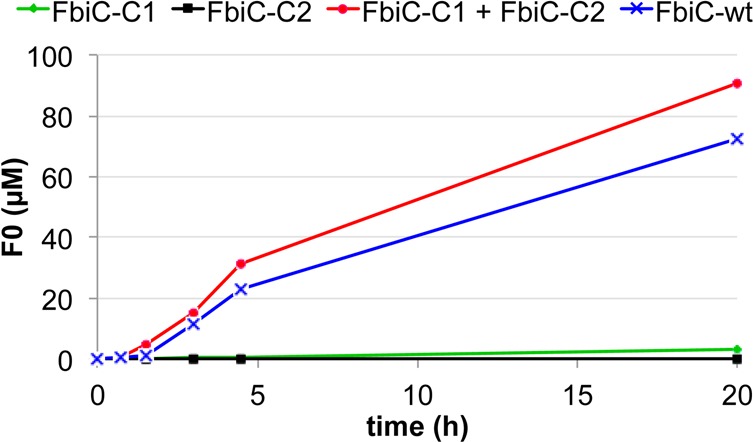
Fo production by FbiC-C1 and FbiC-C2 variants compared to FbiC-wt. The FbiC proteins were incubated with SAM, tyrosine (1) and diaminouracil (2), and the reaction was initiated with the flavodoxin/flavodoxin reductase system. The estimated rate of FbiC-catalyzed Fo formation is 2.4 × 10–5 s–1.
Stereochemistry of the C3 Hydrogen Atom Abstraction from Tyrosine
MS analysis of the product generated by treating [2,3-D2, 3_S_]-Tyr or [3-D, 3_R_]-Tyr with reduced CofG/CofH demonstrated the formation of deazaflavin as a 1:1 mixture of Fo and D1-Fo. This product ratio was independent of the stereochemistry at C3 of the starting tyrosine (Figure 11b,c) demonstrating that the stereochemical information at this carbon is lost during the formation of Fo. A control experiment using [3,3-D2]-Tyr demonstrated that Fo reduction/oxidation was not occurring under the reaction conditions (Figure 11a).
Figure 11.

Stereochemistry of deuterium transfer from C3 of tyrosine during CofG/CofH-catalyzed Fo formation. (a) Mass spectrum of Fo derived from [3,3-D2]-Tyr; (b) mass spectrum of Fo derived from [2,3-D2, 3_S_]-Tyr (1c); (c) mass spectrum of Fo derived from [3-D, 3_R_]-Tyr (1d). (Fo calcd [MH + H]+ 364.1, calcd [MD + H]+ 365.1, calcd [MH + K]+ 402.1, calcd [MD + K]+ 403.1.
Discussion
Fo synthase catalyzes the reductive condensation of tyrosine 1 and ribityl-diaminouracil 2 in a reaction catalyzed by two radical SAM enzymes (CofG and CofH) or the two-domain enzyme FibC.18 The central mechanistic question for this reaction is how two highly reactive 5′-deoxyadenosyl radicals cooperate to form the deazaflavin chromophore. A mechanistic proposal is outlined in Figure 2. In this paper, we report the isolation and characterization of the CofH reaction product and describe a series of experiments that enable us to test this mechanistic hypothesis.
Most radical SAM enzymes use the 5′-deoxyadenosyl radical to abstract a hydrogen atom from the substrate (Dph229 and MqnE30 are exceptions). Since Fo synthase uses two 5′-deoxyadenosyl radicals, two hydrogen atom abstractions are likely to occur during Fo formation. We determined that one of these takes place at the C3 position of tyrosine by characterizing the 5′-deoxyadenosine produced in the CofG/CofH reaction using various deuterated tyrosine isotopologues (Figure 3). This experiment also demonstrated the loss of a single deuterium in the Fo produced from [2′,6′-D2]-Tyr (Supplementary Figure 1d) as expected due to C–N bond formation at C2′/6′ of tyrosine.
Failure to observe a second hydrogen atom transfer from the stable deuterated tyrosine isotopologues suggested that the second hydrogen atom abstraction was occurring from an exchangeable position on tyrosine 1 or diaminouracil 2. Consistent with this prediction, treatment of unlabeled substrates 1 and 2 with CofH in D2O buffer resulted in deuterium transfer to 5′-deoxyadenosine (Figure 4). Phylogenetic analysis of HydG, ThiH, CofH, and CofG with Clustal Omega31 shows that CofH clusters with ThiH and HydG with CofG present as an out-group (Supplementary Figure 11). This sequence similarity between CofH, HydG, and ThiH suggested that these enzymes might catalyze a similar tyrosine fragmentation; hence we initially proposed that the second hydrogen atom abstraction is occurring from either the phenolic hydroxyl or the amino group of tyrosine (Figure 12). Since CofH catalyzes the formation of intermediate 9, generation of the radical at the exchangeable site must precede generation of the radical at the nonexchangeable site. This is consistent with the proposed order of the hydrogen atom abstraction events shown in Figure 2. We also observed significant [M + 2] and [M + 3] peaks for the 5′-deoxyadenosine produced in the CofH reaction (Figure 4). This observation suggests that the hydrogen atom abstraction from the exchangeable site is reversible (1 to 5 in Figure 2) and establishes that the rate of the back hydrogen atom transfer is competitive with the rate of the β-scission reaction. The relevant bond dissociation energies of phenol, methylamine and 5′-deoxyadenosine are 362, 425, and 433 kJ mol–1, respectively.33 These thermochemical data support hydrogen atom abstraction from the amine NH rather than from the phenolic OH because the phenoxy radical is not sufficiently reactive to abstract a hydrogen atom from 5′-deoxyadenosine. The reversibility also eliminates the possibility of a protein glycyl radical at the CofH active site because the bond dissociation energy of glycine is 358 kJ mol–1 demonstrating that the glycyl radical is also insufficiently reactive to abstract a hydrogen atom from 5′-deoxyadenosine.33
Figure 12.

Comparison of the reaction catalyzed by ThiH (thiamin biosynthesis) HydG (FeFe hydrogenase biosynthesis), NosL (Nosiheptide biosynthesis), and the proposed reaction catalyzed by CofH (F420 biosynthesis). Reversible abstraction of the exchangeable hydrogen atom has been observed for ThiH, (Begley,T. P. and Mehta A, unpublished) HydG,32 and CofH and structural studies on NosL26 clearly demonstrates abstraction of the amino hydrogen.
Our mechanism predicts that the tyrosine radical 5 will fragment to form glycine imine 6 which should then undergo hydrolysis to glyoxylate 15 (Figure 5a). Our search strategy for this putative product involved its conversion to a stable chromophoric 2-quinoxalinol 17 by derivatization with _o-_phenylenediamine 16 followed by detection and quantitation using LC-MS. The enzymatic reaction was run using [15N,13C9]-Tyr, and the resulting [2,3-13C]-2-quinoxalinol could be unambiguously differentiated from the unlabeled contaminant. In this way, we demonstrated that the amount of glyoxylate formed averaged 1.1 times the amount of Fo formed in two separate experiments. A similar amount of glyoxylate was present in the CofG/CofH coupled reaction and the CofH only reaction (Figure 5), demonstrating that glycine imine formation is catalyzed by CofH.
We previously demonstrated that CofH generated a product that served as a substrate for CofG.18 Here we characterized this product (Figure 6) and determined its structure as compound 9. To demonstrate that this compound was an intermediate, it was enzymatically synthesized on a large scale using [3,3-D2]-Tyr, purified by HPLC and then treated with dithionite reduced CofG and SAM. Analysis of the reaction by LC-MS showed CofG-dependent production of Fo and also showed the incorporation of deuterium into enzymatically generated 5′-deoxyadenosine and Fo (Figure 8). In this experiment we also observed washout of deuterium from [D1]-Fo, presumably due to its reduction by dithionite followed by aerobic oxidation with loss of deuterium.
In a complementary experiment, we created two variants of FbiC in which either the first or second CXXXCXXC motif was disrupted by alanine substitution (named FbiC-C1 or FbiC-C2, respectively). When the two variants were incubated with substrates, we observed a new peak only in the reaction mixture containing FbiC-C1 (Figure 9). This was identified as compound 9 by co-elution with an authentic standard, molecular mass determination and MS2 fragmentation. We then demonstrated that FbiC-C2 could also convert 9 to Fo and that the rate of Fo production by wild-type FbiC was approximately equal to the rate of Fo production by a FbiC-C1/FbiC-C2 mixture (Figure 10). This suggests that FbiC catalyzes two independent reactions where intermediate 9 is not transferred directly from the C-terminal domain to the N-terminal domain but instead follows diffusion from one active site to the other.
Our mechanism is in line with recent EPR results obtained with HydG, which show evidence for the formation of a dehydroglycine and _p_-hydroxylbenzyl radical during tyrosine Cα-Cβ bond cleavage.34 Our initial attempts to trap the proposed _p_-hydroxybenzyl radical 7 were unsuccessful because CofH did not catalyze the tyrosine lyase reaction in the absence of diaminouracil 2. Analysis of the stereochemistry of the C–C bond formation leading to 9, using tyrosine made chiral at C3 by deuterium substitution, suggested an alternative approach for the detection of 7 (Figure 13).
Figure 13.

Strategy to detect β bond cleavage involving loss of stereochemical information at C3 of tyrosine.
In this approach, 3-deuterio tyrosine, chiral at C2 and C3, would generate the protein-bound deuterated _p_-hydroxybenzyl radical 7a. We propose that this radical intermediate could scramble the stereochemical information originally present at C3 of tyrosine by C–C bond rotation or by flipping of the entire radical. It is well established that the ring of tyrosine and phenylalanine can undergo rapid flipping in the interior of a protein.35,36 Radical addition to 2 followed by oxidation would then give a mixture of 9a and 9b, the deuterated epimers of 9. Stereospecific hydrogen atom abstraction by the 5′-deoxyadenosyl radical at the active site of CofG would give 11 and 11a which would be converted to a mixture of 3 and 3a as shown in Figure 2. If 7 is not an intermediate, this mechanism of stereochemical scrambling could not operate, and the deazaflavin formed would be exclusively protonated or deuterated at C5 depending on the stereochemistry of the starting tyrosine. To test this, [2,3-D2, 3(S)]- and [3(R)-D1]-Tyr were synthesized37 and subjected to the enzymatic reaction. LC-MS analysis demonstrated that the Fo produced was a 1:1 mixture of 3 and 3a consistent with the intermediacy of the _p_-hydroxybenzyl radical 7.
In this paper we describe a set of experiments that explain how two 5′-deoxyadenosyl radicals cooperate to assemble the deazaflavin chromophore of the F420 cofactor. These experiments support the mechanistic proposal outlined in Figure 2. In this mechanism, abstraction of the tyrosine amine hydrogen atom by the CofH 5′-deoxyadenosyl radical gives 5, which then undergoes fragmentation leading to the formation of the _p_-hydroxybenzyl radical 7. Addition of this radical to diaminouracil 2 followed by oxidation gives intermediate 9. This intermediate diffuses to the CofG active site where a second hydrogen atom abstraction generates 11. Cyclization to 13 followed by oxidation and elimination of ammonia completes the formation of the deazaflavin 3.
This mechanism is consistent with the copurification of 2 with CofH, with the reversibility of the first hydrogen atom transfer (1 to 5), with the second hydrogen atom abstraction occurring from C3 of tyrosine (10 to 11) and with the formation of the glycine imine 6 as a byproduct. The scrambling of stereochemistry at the C3 of tyrosine during Fo formation also supports the formation of the _p_-hydroxybenzyl radical 7. Finally, it was possible to trap 9, the product of the CofH-catalyzed reaction, and to demonstrate that it was a substrate for CofG.
Finally, this paper reveals how a major cofactor of the prokaryotic world is assembled by an unprecedented mechanism and provides yet another example of the remarkably complex chemistry that nature uses in the assembly of the heterocyclic intermediates used in cofactor biosynthesis.38,39
Acknowledgments
We thank Dr. Howard Williams for help collecting the NMR spectra of the CofH product and for helpful discussions and Robert White (Virginia Polytechnic Institute and State University) for providing a sample of 5-amino-6-ribitylamino-2,4(1_H_,3_H_)-pyrimidinedione. Research was supported by the Robert A. Welch Foundation (A-0034 to T.P.B.), the National Institutes of Health (DK44083 to T.P.B.), Agence National de la Recherche (ANR-12-BSV8-0013-02 to O.B.) and the European Research Council (ERC) (Consolidator Grant 617053 to O.B.). The NMR console and cryoprobe were purchased with funds from NSF award number 0840464.
Supporting Information Available
Methods and materials, MS and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org
Author Present Address
∥ Oregon State University, College of Pharmacy, Corvallis, OR 97331
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kiener A.; Husain I.; Sancar A.; Walsh C. J. Biol. Chem. 1989, 264, 13880. [PubMed] [Google Scholar]
- Mayerl F.; Piret J.; Kiener A.; Walsh C. T.; Yasui A. J. Bacteriol. 1990, 172, 6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra K.; Kim S. T.; Walsh C.; Sancar A. J. Biol. Chem. 1992, 267, 15406. [PubMed] [Google Scholar]
- Glas A. F.; Maul M. J.; Cryle M.; Barends T. R.; Schneider S.; Kaya E.; Schlichting I.; Carell T. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman P.; Toms-Wood A.; Wolfe R. S. J. Bacteriol. 1972, 112, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eirich L. D.; Vogels G. D.; Wolfe R. S. Biochemistry 1978, 17, 4583. [DOI] [PubMed] [Google Scholar]
- Thauer R. K.; Kaster A.-K.; Goenrich M.; Schick M.; Hiromoto T.; Shima S. Annu. Rev. Biochem. 2010, 79, 507. [DOI] [PubMed] [Google Scholar]
- Taylor M. C.; Jackson C. J.; Tattersall D. B.; French N.; Peat T. S.; Newman J.; Briggs L. J.; Lapalikar G. V.; Campbell P. M.; Scott C.; Russell R. J.; Oakeshott J. G. Mol. Microbiol. 2010, 78, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selengut J. D.; Haft D. H. J. Bacteriol. 2010, 192, 5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwantini E.; Mukhopadhyay B. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupner M.; White R. H. Biochemistry 2001, 40, 10859. [DOI] [PubMed] [Google Scholar]
- Graupner M.; Xu H.; White R. H. Biochemistry 2002, 41, 3754. [DOI] [PubMed] [Google Scholar]
- Graham D. E.; Xu H.; White R. H. Arch. Microbiol. 2003, 180, 455. [DOI] [PubMed] [Google Scholar]
- Li H.; Graupner M.; Xu H.; White R. H. Biochemistry 2003, 42, 9771. [DOI] [PubMed] [Google Scholar]
- Li H.; Xu H.; Graham D. E.; White R. H. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 9785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochowski L. L.; Xu H.; White R. H. J. Bacteriol. 2006, 188, 2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochowski L. L.; Xu H.; White R. H. Biochemistry 2008, 47, 3033. [DOI] [PubMed] [Google Scholar]
- Decamps L.; Philmus B.; Benjdia A.; White R.; Begley T. P.; Berteau O. J. Am. Chem. Soc. 2012, 134, 18173. [DOI] [PubMed] [Google Scholar]
- Hanzelmann P.; Schindelin H. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann P.; Schindelin H. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluhe L.; Knappe T. A.; Gattner M. J.; Schafer A.; Burghaus O.; Linne U.; Marahiel M. A. Nat. Chem. Biol. 2012, 8, 350. [DOI] [PubMed] [Google Scholar]
- Driesener R. C.; Duffus B. R.; Shepard E. M.; Bruzas I. R.; Duschene K. S.; Coleman N. J. R.; Marrison A. P. G.; Salvadori E.; Kay C. W. M.; Peters J. W.; Broderick J. B.; Roach P. L. Biochemistry 2013, 52, 8696. [DOI] [PubMed] [Google Scholar]
- Vey J. L.; Drennan C. L. Chem. Rev. 2011, 111, 2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker S. J.; Grove T. L. F1000 biology reports 2010, 2, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriek M.; Martins F.; Challand M. R.; Croft A.; Roach P. L. Angew. Chem., Int. Ed. 2007, 46, 9223. [DOI] [PubMed] [Google Scholar]
- Nicolet Y.; Zeppieri L.; Amara P.; Fontecilla-Camps J. C. Angew. Chem., Int. Ed. Engl. 2014, 53, 11840. [DOI] [PubMed] [Google Scholar]
- Kriek M.; Martins F.; Challand M. R.; Croft A.; Roach P. L. Angew. Chem., Int. Ed. Engl. 2007, 46, 9223. [DOI] [PubMed] [Google Scholar]
- McNeill L. A.; Bethge L.; Hewitson K. S.; Schofield C. J. Anal. Biochem. 2005, 336, 125. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhu X.; Torelli A. T.; Lee M.; Dzikovski B.; Koralewski R. M.; Wang E.; Freed J.; Krebs C.; Ealick S. E.; Lin H. Nature 2010, 465, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanta N.; Fedoseyenko D.; Dairi T.; Begley T. P. J. Am. Chem. Soc. 2013, 135, 15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Soding J.; Thompson J. D.; Higgins D. G. Mol. Syst. Biol. 2011, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffus B. R.; Ghose S.; Peters J. W.; Broderick J. B. J. Am. Chem. Soc. 2014, 136, 13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hioe J.; Zipse H. Encycl. Radicals Chem., Biol. Mater. 2012, 1, 449. [Google Scholar]
- Kuchenreuther J. M.; Myers W. K.; Stich T. A.; George S. J.; NejatyJahromy Y.; Swartz J. R.; Britt R. D. Science 2013, 342, 472. [DOI] [PubMed] [Google Scholar]
- Weininger U.; Respondek M.; Loew C.; Akke M. J. Phys. Chem. B 2013, 117, 9241. [DOI] [PubMed] [Google Scholar]
- Wüthrich K.; Wagner G. FEBS Lett. 1975, 50, 265. [DOI] [PubMed] [Google Scholar]
- Kirby G. W.; Michael J. Chem. Commun. 1971, 187. [Google Scholar]
- Begley T. P.; Chatterjee A.; Hanes J. W.; Hazra A.; Ealick S. E. Curr. Opin. Chem. Biol. 2008, 12, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley T. P.; Ealick S. E.; McLafferty F. W. Biochem. Soc. Symp. 2012, 79, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.