An allosteric model for control of pore opening by substrate binding in the EutL microcompartment shell protein (original) (raw)
Abstract
The ethanolamine utilization (Eut) microcompartment is a protein-based metabolic organelle that is strongly associated with pathogenesis in bacteria that inhabit the human gut. The exterior shell of this elaborate protein complex is composed from a few thousand copies of BMC-domain shell proteins, which form a semi-permeable diffusion barrier that provides the interior enzymes with substrates and cofactors while simultaneously retaining metabolic intermediates. The ability of this protein shell to regulate passage of substrate and cofactor molecules is critical for microcompartment function, but the details of how this diffusion barrier can allow the passage of large cofactors while still retaining small intermediates remain unclear. Previous work has revealed two conformations of the EutL shell protein, providing substantial evidence for a gated pore that might allow the passage of large cofactors. Here we report structural and biophysical evidence to show that ethanolamine, the substrate of the Eut microcompartment, acts as a negative allosteric regulator of EutL pore opening. Specifically, a series of X-ray crystal structures of EutL from Clostridium perfringens, along with equilibrium binding studies, reveal that ethanolamine binds to EutL at a site that exists in the closed-pore conformation and which is incompatible with opening of the large pore for cofactor transport. The allosteric mechanism we propose is consistent with the cofactor requirements of the Eut microcompartment, leading to a new model for EutL function. Furthermore, our results suggest the possibility of redox modulation of the allosteric mechanism, opening potentially new lines of investigation.
Keywords: bacterial microcompartment, allostery, conformational change, ethanolamine metabolism, X-ray crystallography, ligand-binding
Introduction
The intestinal microbiota constitutes an intricate ecological community. Microorganisms, primarily bacteria, that colonize the human digestive tract are roughly an order of magnitude more numerous than human cells within a single human body,1–3 and they perform a number of functions that benefit their host. Various species of bacteria aid in carbohydrate digestion and produce vitamins that can be absorbed by the host.2–4 In addition, the ecology of the intestinal microbiota has been directly linked to human health, affecting many important aspects of physiology including immunological activity,5,6 tumor growth,2 obesity,7,8 and even cognitive function.9 Consequently, imbalance within this ecological community can lead to a variety of human diseases. Typically, the intestinal microbiome maintains a healthy distribution of species as a result of competitive exclusion; no single species can become too prolific due to constant competition for resources within the environment.2,5
To gain a competitive advantage over other microbes in the gut, certain bacterial species have evolved the ability to utilize ethanolamine as a metabolic resource.10–14 Genomic analysis revealed that over 100 sequenced bacterial genomes contain the “eut operon,” which contains genes required for utilization of this small molecule.11 Ethanolamine is produced mainly through degradation of phosphatidylethanolamine, a phospholipid that is abundant in cell membranes. Due to constant turnover of the intestinal epithelium, as well as dietary intake, ethanolamine is plentiful in the human intestine.10,15,16 The ethanolamine molecule enters bacterial cells through a transmembrane ethanolamine transporter,10 and is degraded by a well-characterized catabolic pathway.12,13,17 Initially, ethanolamine is deaminated by a vitamin-B12-dependent ethanolamine-ammonia lyase (EutBC), providing ammonia as a nitrogen source and acetylaldehyde as a carbon source. Subsequently, acetaldehyde is ligated to coenzymeA, forming acetyl-CoA, which can enter a variety of other metabolic pathways. For example, acetyl-CoA can be oxidized in the TCA cycle to produce NADH, or it can be used as a substrate for anabolic processes such as fatty-acid biosynthesis.
The presence of the eut operon in the genome and metabolic utilization of ethanolamine have both been linked to bacterial enteropathogenesis.10,11,18,19 Ethanolamine utilization allows pathogenic species to proliferate in the host digestive tract by giving them a nutritional advantage over other species, and also through potential modulation of the host innate immune response.10 A probabilistic analysis of phylogenetic patterns by Li et al. identified a strong correlation between the occurrence of eut genes and the “food poisoning” phenotype.18 Experimentally, it has been demonstrated that global virulence regulators induce expression of eut genes in several species,20,21 and it has also been shown that eut genes are down-regulated during commensal colonization of the host by Enterococcus faecalis.22 Furthermore, Winter and Thiennimitr et al. concluded that inflammation of the host intestine due to infection is critical for anaerobic ethanolamine catabolism, because it provides the necessary respiratory electron acceptor, tetrathionate.23–25
In most enteropathogenic facultative anaerobes that metabolize ethanolamine, the eut operon contains genes encoding BMC-domain proteins10,11,17,18 (We use the abbreviation BMC, for bacterial microcompartment, to refer to the small, roughly 100-amino acid protein domain that is present in most microcompartment shell proteins). The presence of these genes is diagnostic of encapsulation of the Eut metabolic enzymes within a bacterial microcompartment or MCP. Many organisms are known to make MCPs of the Eut type, which function to retain acetaldehyde, a volatile and toxic metabolic intermediate26,27 (Fig. 1). Organisms known to form Eut MCPs include Clostridium, Salmonella, E. coli, and Klebsiella, which can have a variety of negative effects on human health,2 ranging from food poisoning to increased risk of colon cancer, and it has been hypothesized that encapsulation of ethanolamine metabolism within a MCP is critical for pathogenesis in some species.10,11,18 Encapsulation of ethanolamine metabolism within an MCP allows the bacteria to turn over large amounts of this metabolic resource without elevating the intracellular acetaldehyde concentration, giving them a nutritional advantage over other microbes.10
Figure 1.
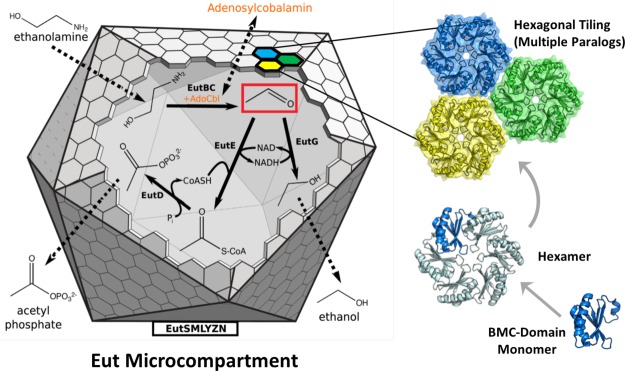
The Eut MCP is a bacterial organelle that functions to retain acetaldehyde (red box), a volatile and toxic metabolic intermediate. It consists of BMC-domain proteins, hexagonally tiled to form a polyhedral shell around a series of sequentially-acting enzymes that convert ethanolamine to ethanol and acetyl phosphate. Primarily small molecules, substrates and products, must traverse the shell, while the NAD(H) and acetyl CoA cofactors are recycled within the MCP. In contrast, the cobalamin cofactor (orange) required by the EutBC enzyme must be regenerated under certain conditions, requiring exchange of a large molecule between the MCP lumen and the cytosol.
The barrier surrounding the Eut MCP consists of a few thousand BMC-domain proteins. These BMC-domain proteins form two-dimensional, hexagonal layers, which fold and close to form a faceted, polyhedral shell (Fig. 1). From a functional perspective, this unique protein assembly is faced with a challenging problem. The reason for encapsulating ethanolamine catabolism is to retain a very small, volatile intermediate, acetaldehyde.26,27 At the same time, the biochemical system within the Eut MCP is believed to exchange at least one large cofactor with the cytoplasm. The nicotinamide cofactors, as well as coenzymeA, can be recycled within the MCP,28,29 but the ethanolamine-ammonia lyase enzyme requires an adenosylcobalamin cofactor for catalysis12,13 (Fig. 1). In the enzyme active site, the adenosylcobalamin coenzyme (vitamin B12) is subject to mechanism-based inactivation in the absence of substrate,30 and therefore needs to be replaced or regenerated. The Eut operon contains ATP-dependent enzymatic machinery for regenerating adenosylcobalamin,31,32 but it is unclear whether this set of enzymes is located inside or outside of the MCP structure. If the cofactor regeneration system is outside the MCP shell, the shell must support the transport of cobalamin compounds to replenish the enzyme with fresh cofactor. If the cofactor regeneration system is inside the MCP, then ATP must be able to enter. Cobalamin and ATP are both much bigger than acetaldehyde, so we expect the MCP shell to have a mechanism for promoting the passage of larger molecules without allowing the leakage of the small metabolic intermediate.
Previous X-ray crystallographic studies of the EutL shell protein from Escherichia coli have suggested an important transport role for this tandem BMC-domain pseudohexamer.33–35 A pair of X-ray crystal structures of E. coli EutL determined by Tanaka, et al. revealed two conformations of the protein35 (Fig. 2). In the “closed” conformation, the central region of the trimer is occluded by ordered loop segments. In the “open” conformation a wide, triangular-shaped pore is present at the center of the trimer. This pore—approximately 10Å across—is much wider than the pores in typical BMC homohexamers, and appears large enough for cofactor transport.35 The idea that EutL might provide a gated pore for transport of large molecules explains, in part, how the MCP shell might be able to provide interior enzymes with cofactors, while still retaining small intermediates like acetaldehyde. While previous crystallographic work has established that EutL can occupy two conformations, the regulation of the transition between the two states remains unclear.
Figure 2.
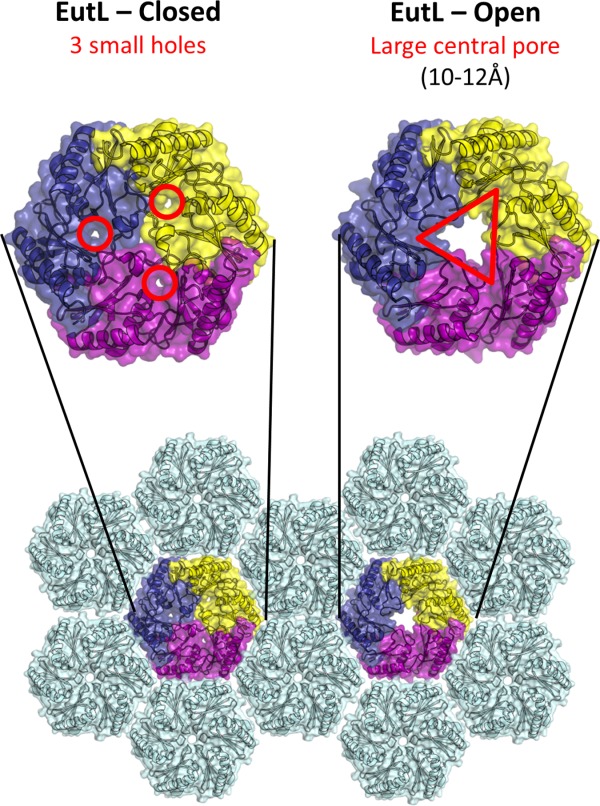
The closed and open structures of EutL, as established from previous studies.34,35 The comparison shown is of EutL from E. coli, which is the only homolog whose structure has been determined in both open and closed forms. EutL is shown in a hexagonal tiling with EutM hexamers (light blue). The expanded views show key features of the two conformations, including three narrow holes (channels) in the closed conformation, and a large central pore in the open conformation. The individual chains of the EutL pseudohexameric trimer are each colored differently (purple, yellow, dark blue).
Unraveling the mechanisms by which conformational changes are regulated in gated shell protein pores will be a key contribution to our understanding of how bacterial organelles maintain selective permeability. The present study is directed toward elucidating this regulation in the EutL shell protein, which serves as a model system for understanding these gated pores. Toward this goal, we present our study of an uncharacterized EutL homolog from Clostridium perfringens. Our results highlight the atomic interactions that govern the transition between the open and closed states. Based on our structural and biophysical data, we propose that ethanolamine, the substrate of the Eut MCP (which must transit the shell), acts as an allosteric regulator to promote EutL pore closing.
Results
Structure determination of untreated C. perfringens EutL
We determined the X-ray crystal structures of EutL from Clostridium perfringens (referred to hereafter as CpEutL) to elucidate structural features of the protein that contribute to its function. Using the hanging drop vapor-diffusion method, we grew crystals belonging to tetragonal space group _P_43212. These crystals diffracted to a resolution of 2.0 Å. Further details of the data collection and atomic refinement for this structure are provided in the Methods section and in Table1. A single trimer is present in the asymmetric unit of this crystal form. Continuous electron density could be traced for residues 1–216 out of the 217 native amino acids from each of the three polypeptide chains within the asymmetric unit, and additional residues, including portions of the C-terminal His6 affinity tag, could be modeled in two of the chains. The coordinates and diffraction data for this structure have been deposited in the protein data bank (PDB) under PDB ID 4EDI.
Table 1.
Data collection and Refinement Statistics
| Parameter | Tetragonal | Reduced | Ethanolamine | Room temperature |
|---|---|---|---|---|
| Wavelength | 0.9795Å | 0.9792Å | 0.9795Å | 0.9792Å |
| Resolution range | 61.34–2.00Å (2.07–2.00Å) | 19.8–1.8Å (1.85–1.8) | 83.04–1.07Å (1.74–1.70Å) | 72.87–1.90Å (1.95–1.90Å) |
| Unit cell | a = b = 86.7Å, c = 252Å | a = b = 88.0Å, c = 252Å | a = b = 87.96Å, c = 251.91Å | a = b = 88.89Å, c = 254.53Å |
| α = β = γ = 90° | α = β = γ = 90° | α = β = γ = 90° | α = β = γ = 90° | |
| Space group | _P_43212 | _P_43212 | _P_43212 | _P_43212 |
| Unique reflections | 65257 | 91132 | 109437 | 79392 |
| Multiplicity | 4.0 (4.0) | 4.7 (4.4) | 12.5 (8.4) | 4.5 (4.6) |
| Completeness | 98.6% (99.4%) | 98.7% (95.0%) | 100% (100%) | 97.6% (97.2%) |
| <_I_/_σI_> | 20.1 (6.4) | 18.4 (3.9) | 17.1 (1.8) | 10.10 (1.3) |
| Wilson B-factor | 25.7Å2 | 19.8Å2 | 25.3Å2 | 29.9Å2 |
| CC1/2a | 0.998 (0.951) | 0.999 (0.877) | 0.999 (0.693) | 0.998 (0.526) |
| CC*a | 0.999 (0.987) | 1.000 (0.967) | 1.000 (0.905) | 0.999 (0.830) |
| CCworka | 0.957 (0.939) | 0.969 (0.918) | 0.970 (0.863) | 0.981 (0.799) |
| CCfreea | 0.949 (0.914) | 0.960 (0.927) | 0.966 (0.833) | 0.956 (0.794) |
| _R_workb | 0.170 | 0.160 | 0.147 | 0.138 |
| _R_freeb | 0.193 | 0.177 | 0.162 | 0.159 |
| No. atoms | 5148 | 5638 | 5658 | 5401 |
| Protein residues | 659 | 663 | 663 | 661 |
| Solvent molecules | 245 | 619 | 524 | 221 |
| Average B-factor | 29.4Å2 | 24.3Å2 | 30.9Å2 | 35.8Å2 |
| RMSDbonds | 0.007Å | 0.006Å | 0.010Å | 0.009Å |
| RMSDangles | 1.1° | 1.0° | 1.246° | 1.137° |
| Rama. Plot: | ||||
| Favored | 98% | 98% | 98.8% | 99.6% |
| Allowed | 2% | 2% | 1.1% | 0.4% |
| Outliers | 0% | 0% | 0% | 0% |
| MolProbity Clashscorec | 3.6 | 3.8 | 0.49 | 0.39 |
| PDB ID | 4EDI | 4FDZ | 4TME | 4TM6 |
Structural overview of CpEutL
The structure of CpEutL recapitulates the typical topology and oligomerization reported previously for tandem BMC-domain proteins34–40 (Fig. 3). Within a single monomer, each of the two genetically fused BMC-domains adopts a permuted α/β BMC-domain fold. Domain 1 contains β-strands 1–4 (forming an antiparallel β-sheet) and α-helices A-C. Domain 2 contains β-strands 5–8 (also an antiparallel β-sheet) and α-helices D-F. The trimer observed in the asymmetric unit of the crystal corresponds to the expected functional assembly of EutL. Three polypeptide chains oligomerize in the C3-symmetric manner typical of the tandem BMC-domain proteins, giving the trimers their characteristic hexagonal shape required for incorporation into the two-dimensional lattice of the MCP shell. The structures of CpEutL superimpose upon the closed conformation of their E. coli homolog (PDB ID: 3I82, 57% sequence identity) with an RMSD of less than 1.0 Å on Cα atoms.
Figure 3.
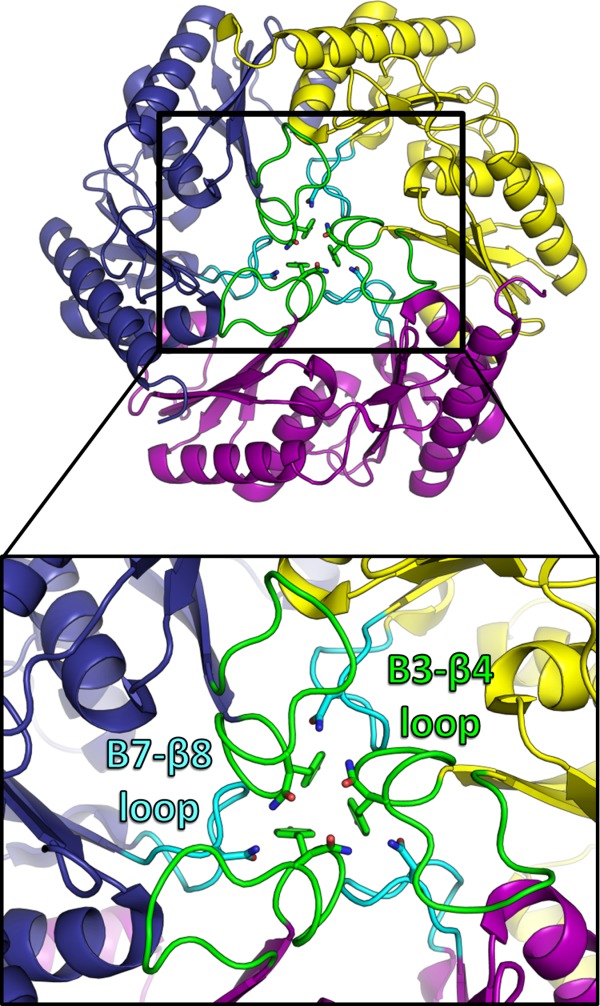
Crystal structure of EutL from Clostridium perfringens (CpEutL)(this work), colored as in Figure 2 but with the β3–β4 loops colored green, and the β7–β8 loops colored cyan. Three symmetry-related copies of each of these two loops pack tightly at the center of the trimer, resulting in a conformation that is essentially closed to transport. Three conserved amino acids that appear to be important for stabilizing this closed conformation (Y69, N74, N183) are shown as sticks.
Loop structures in the closed conformation
The crystal structures of CpEutL reveal the pseudohexameric trimer in a closed-pore conformation that is very similar to the closed conformation seen for E. coli EutL.33–35 The closed conformation of CpEutL, like the homologous E. coli protein, is characterized by a tight packing of three symmetry-related loops, one from each polypeptide chain in the trimer, about the three-fold molecular symmetry axis (Fig. 3). These loops consist of residues 68–82, which connect β-strands 3 and 4 in domain 1. In addition, the analogous loop region in domain 2, consisting of residues 176–185, which connects β-strands 7 and 8 in domain 2, also protrudes toward the center of the trimer. This β7–β8 loop makes contacts, both direct and water-mediated, with the β3–β4 loop within a single monomer, and between neighboring monomers in the trimer (Fig. 3). The packing of these loops at the center of the trimer is stabilized by the edge-to-face interaction of three aromatic rings, and by a hydrogen bonding network formed by polar residues. These interactions between the β7–β8 loops and the β3–β4 loops are likely important for stabilizing the closed conformation. Each monomer contributes several key residues to this symmetric interaction (Fig. 3), including Tyr69, Asn74, and Asn183, which are all well-conserved in EutL orthologs. While the conformations of both the β3–β4 and β7–β8 loops in CpEutL are consistent with the closed structure of EutL from E. coli (PDB IDs 3I82 and 3GFH), both of those loop regions are in different and/or disordered conformations in the open structures of the E. coli homolog (PDB IDs 3I87 and 3MPV).
Loose atomic packing in the monomers
In line with previous reports,33,35 we observe narrow holes or channels in areas of low packing density, one within each of the monomers, in the closed-pore conformation of the trimer (Fig. 4). These narrow holes (not to be confused with the large pore that is created in the center of the trimer in its open conformation), which are present also in previous structures of EutL homologs in the closed conformation (PDB IDs 3IO0, 3I82, 3GFH, 4FAY), are located at the N-terminal end of helix A between domains 1 and 2 within an individual monomer. The holes extend from one face of the trimer to the other, making three narrow, hourglass-shaped channels through the assembly [Fig. 4(a)]. The openings are lined with conserved acidic and polar residues on one side (D44, D45, T182), and hydrophobic residues on the other (F112, F184). The loose atomic packing in this region appears to provide space to accommodate conformational rearrangements to the open–pore conformation of the trimer [Fig. 4(b)].
Figure 4.
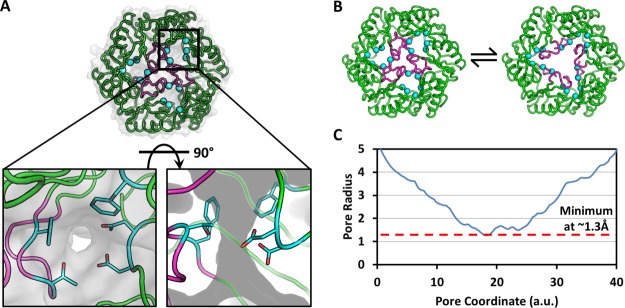
Visualization of narrow holes in the closed conformation of CpEutL. (A) Each CpEutL trimer presents three small holes, one in each monomer, which are also present in the closed structure of E. coli EutL. The gray surface serves to highlight these holes or channels, while the cyan spheres denote the Cα atoms of the residues that form the narrowest constriction points (D44, D45, V151, T182, and F184). (B) The open and closed structures of E. coli EutL demonstrate that opening of the central pore involves retraction of the central loops (magenta), into the small channels (cyan spheres). (C) Analysis of the narrow channels using the HOLE2 algorithm reveals that their narrowest constriction point has a radius of only 1.3Å.
We measured the minimum radius of the narrow channel using the HOLE2 algorithm,41 and found that the narrowest constriction along a traversal from one side of the trimer to the other has a radius of only 1.3 Å [Fig. 4(c)]. This measurement is in close agreement with the analysis of E. coli EutL performed by Sagermann, et al., who calculated a radius of 1.1 Å for the narrowest point.33 The narrow constriction points of the small channels consist of residues D44, D45, V151, T182, and F184 [Fig. 4(d)]. Whether these small holes could support molecular transport (perhaps upon further rearrangement) is an open question. In our view, the narrowness of the openings and the presence of larger and apparently constitutively open pores in other Eut shell proteins that are present in the MCP (e.g., the EutM shell protein), belie an alternate explanation.
Crystallographic evidence for disulfide bonding in CpEutL
Analysis of electron density maps calculated from CpEutL diffraction data revealed, unexpectedly, a disulfide bond in a tandem BMC-domain protein. The refined structure of CpEutL reveals an interesting triad of cysteine residues—Cys127, Cys196, and Cys200—poised within disulfide-bonding proximity. This triad, conserved among most EutL orthologs, consists of a helical CXXXC motif and a third Cys residue contributed by a neighboring β-strand, which creates a roughly equilateral triangle with the β-carbons of the three cysteine side chains at the vertices. In our structure of CpEutL, Cys127 of chain A appears in two alternate rotameric configurations, with one of those rotamers forming a disulfide bond with Cys200. Electron density corresponding to such a disulfide bond appears (at a contour level of 1.6_σ_) in a 2mFo − DFc map [Fig. 5(a)]. Initial refinement was performed using a model that did not contain a disulfide bond between those two residues. We modeled the disulfide-bonded rotamer of Cys127 only after positive density appeared in a difference (mFo − DFc) map. Subsequent refinement was carried out with Cys127 in two partially-occupied rotamers; the occupancies in the final refined model were 0.46 and 0.54 for the bonded and non-bonded conformations respectively. To minimize phase bias, we also calculated an omit map in which the two cysteine residues of the disulfide bond were excluded from the structure factor calculation and phasing model. This map also showed positive density (4.9_σ_) for both rotamers of Cys 127 as well as for the Cys 127–Cys200 disulfide bond [Fig. 5(b)].
Figure 5.
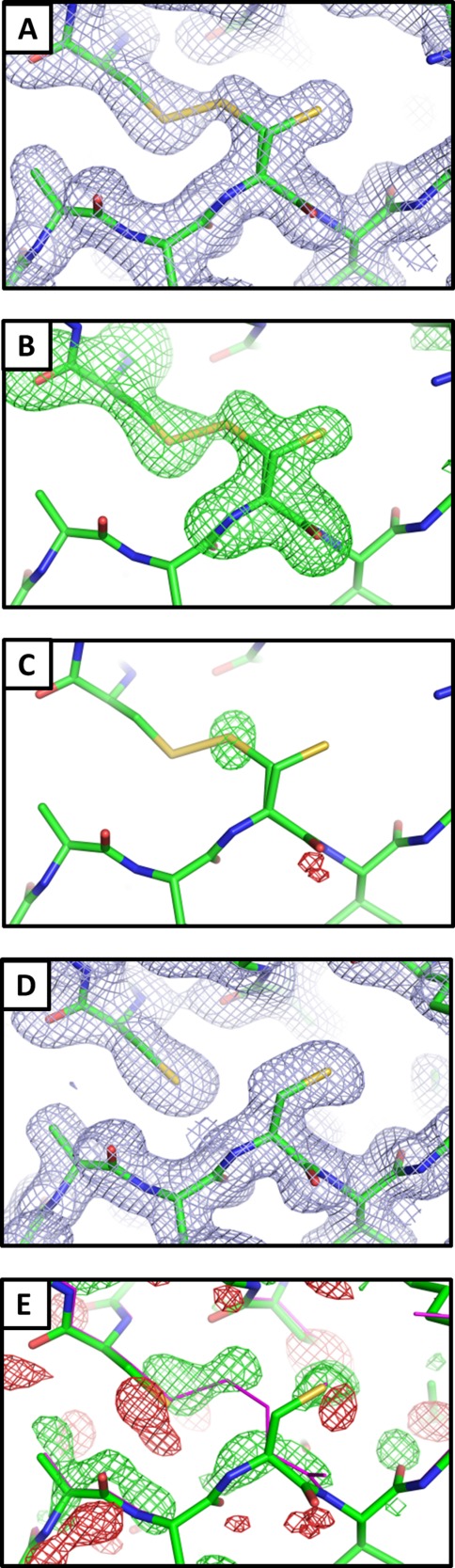
Visualization of a disulfide bond in CpEutL. (A) A 2mFo − DFc electron density map contoured at 1.0_σ_ shows contiguous electron density between the sulfur atoms of C127 and C200, indicating the presence of a partially occupied disulfide bond. (B) An omit (mFo − DFc, 3.0_σ_) electron density map, in which the atoms of Cys127 and Cys200 were excluded from the structure factor and phase calculations to eliminate model bias, shows strong positive density for both conformations of Cys127 and for the disulfide bond. (C) An _F_early − F_late (φ_model, 4.0_σ), electron density map, comparing data from the first 20° of Xray data collection with the last 20°, indicates that the Cys127–Cys200 disulfide bond is strongly populated prior to X-ray exposure, and is partially destroyed during the experiment. (D) Chemical reduction abolishes the disulfide bond in CpEutL, as evidenced by a lack of electron density between the cysteine side chains in the 2mFo − DFc (1.0_σ) electron density map derived from the reduced crystals. (E) An isomorphous difference map calculated using _F_oxidized − _F_reduced amplitudes (φ_model, 3.0_σ) show positive peaks between the sulfur atoms of C127 and C200, further verifying the existence of the disulfide bond. Experimental data (see text) shows that the disulfide bond is linked to substrate binding and allosteric modulation of the large central pore in EutL.
Because disulfide bonds are sometimes broken by intense synchrotron X-ray radiation,42–45 we also studied the possible effect of radiation damage on the electron density features corresponding to the disulfide bond. First, a 2mFo − DFc electron density map was recalculated from diffraction data collected during the first 20° of the X-ray experiment, which gave a data set that was approximately 82% complete. The density for the disulfide bond was stronger in this map than in the original map calculated from the full data set (2.2_σ_ versus 1.6_σ_), suggesting that the disulfide bond was partially broken during the course of data collection. To further demonstrate the radiation-sensitivity of the disulfide bond, we calculated a _F_o − _F_o isomorphous difference map comparing data from the first 20° of the X-ray experiment with data from the last 20° of the experiment (_F_early − F_late). This map revealed a strong peak of positive density (6.7_σ) surrounding the sulfur atom of the bonded rotamer of Cys127 [Fig. 5(c)]. These electron density calculations indicate that the disulfide bond was well populated in the crystallized protein, with partial loss occurring as a result of radiation damage.
X-ray crystal structure of the reduced state of EutL
The same crystallization conditions that yielded tetragonal crystals of the untreated protein also gave crystals of a protein sample that had been chemically reduced by treatment with 5 m_M_ tris-(2-carboxyethyl)phosphine (TCEP). Crystals of the TCEP-treated protein diffracted to a resolution of 1.8 Å. Further details of the data collection and atomic refinement for the reduced structure are also available in the Methods section and in Table1. Final atomic models derived from the untreated and reduced samples were strikingly similar to one another. These structures superimpose with an RMSD of only 0.2 Å on Cα atoms, and 2.6 Å on all non-hydrogen atoms. The disulfide bond observed in the untreated structure is completely absent in the electron density map derived from the reduced crystals [Fig. 5(d)]. The coordinates of the reduced structure were deposited in the PDB under PDB ID 4FDZ.
To further illustrate the potential for CpEutL to form disulfide bonds under oxidizing conditions, we calculated _F_untreated − F_reduced difference maps from the corresponding isomorphous datasets, using model phases. Those difference maps showed decisively strong positive density features (5.4_σ) situated between the sulfur atoms of Cys127 and Cys200, indicating that this disulfide bond is present in the untreated protein, but not in the reduced sample [Fig. 5(e)].
Calorimetric investigation of ligand binding to CpEutL
As a step toward defining the molecular interactions that govern the function of the EutL shell protein, we used isothermal titration calorimetry (ITC) to determine if CpEutL binds to any of the small molecules involved in the metabolism of ethanolamine. We tested the binding of four small molecules, including ethanolamine, ethanol, acetate, and acetyl-phosphate. (Acetaldehyde was not included in the analysis because it reacts with our buffered protein solution, producing a large amount of heat that obscures any potential ligand-binding signal present in the experiment.) The calorimetric behavior for the titration of ethanolamine was notably different from the behavior for the other titrations [Fig. 6(a)]. Specifically, plots of Δ_H_ vs. molar ratio of ligand to protein produce virtually flat lines for all of the ligands except ethanolamine, which produces a roughly hyperbolic curve indicative of a specific (i.e., saturatable) protein–ligand interaction.
Figure 6.
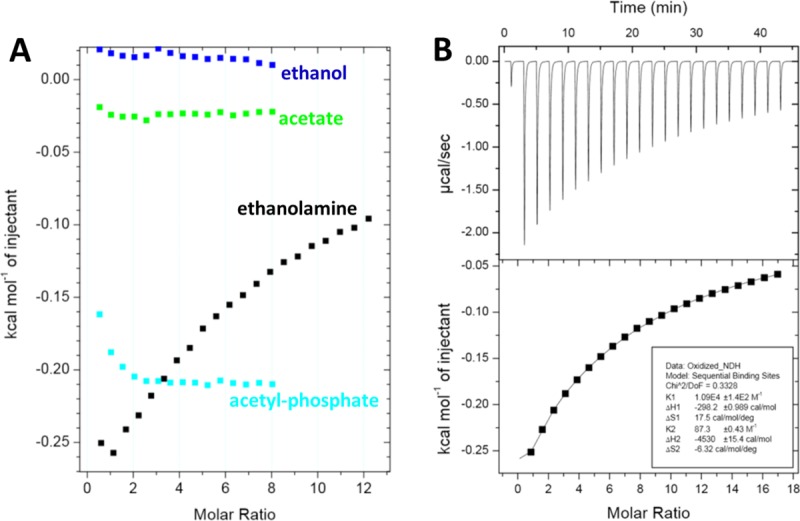
Thermodynamic characterization of ethanolamine binding to EutL. (A) A comparison of integrated ITC data for titrations of several small molecules into a CpEutL solution reveals that ethanolamine is the only titrant whose interaction with CpEutL releases heat in a manner consistent with a specific binding event, which appears to be highly selective for ethanolamine. (B) Plots of the raw (top panel) and integrated (bottom panel) ITC data measured for a titration of ethanolamine into CpEutL. The data shown were used to derive thermodynamic parameters for the ethanolamine-CpEutL interaction using a sequential, two-site binding model.
Our calorimetric data from the ethanolamine titration allowed us to generate a binding model that describes two ethanolamine binding sites per monomer with very different affinities for their ligand [Fig. 6(b)]. We fit the binding data to a sequential, two-site binding model to extract thermodynamic parameters for the interaction (Table2). The quality of the thermodynamic binding model was evaluated using a chi-square analysis, which revealed an excellent fit of the model to the data. The model describes a high-affinity site, where binding is only slightly favored by enthalpy but is accompanied by a significant increase in entropy, and a low-affinity site, where binding is strongly exothermic, but comes at an entropic cost.
Table 2.
Thermodynamic Parameters for CpEutL Binding to Ethanolamine
| Oxidized | Reduced | |||
|---|---|---|---|---|
| Site 1 | Site 2 | Site 1 | Site 2 | |
| K_d (µ_M) | 91.7 ± 1.1 | 11,500 ± 100 | 321 ± 3 | 97,000 ± 700 |
| Δ_H_ (cal mol−1) | −298.2 ± 0.99 | −4530 ± 15.4 | −247.9 ± 1.3 | −25,550 ± 174 |
| Δ_S_ (cal mol−1 K−1) | 17.5 | −6.3 | 15.2 | −81.1 |
| _χ_2/DoF | 0.33 | 0.27 |
We also performed ITC experiments using protein that had been chemically reduced with TCEP. We found that chemical reduction dramatically lowered the affinity of EutL for ethanolamine, nearly fourfold at the high-affinity binding site and nearly eightfold at the low-affinity binding site (Table2). In addition, the thermodynamic parameters (Δ_H_ and Δ_S_) extracted from the data differ significantly between the oxidized and reduced states.
X-ray crystal structure of ethanolamine-bound CpEutL
After confirming the interaction of ethanolamine with CpEutL, we determined the X-ray crystal structure of CpEutL bound to ethanolamine to elucidate the atomic details of the interaction. Using the hanging drop vapor-diffusion method, we grew CpEutL crystals belonging to space group _P_43212, as described above. To prepare crystals of the protein–ligand complex, we soaked our untreated crystals in mother liquor supplemented with ethanolamine. These crystals were subsequently cryoprotected, frozen at 100K, and used for the collection of X-ray diffraction data to a resolution of 1.7Å. Data collection and refinement statistics for the ethanolamine-bound structure are provided in Table1, and the final refined model was deposited in the PDB under PDB ID 4TME.
In the ethanolamine-bound CpEutL structure, two ligand molecules occupy each of the three narrow holes that perforate the trimeric assembly. The presence of two ethanolamine molecules per monomer is consistent with the binding model that was fit to the ITC data. One ethanolamine molecule is bound on the cytosolic side of the constriction point of the channel, while the second ethanolamine lies within the channel on the luminal side of the constriction [Fig. 7(a)]. The first ethanolamine molecule packs against Phe112 and Phe184, positioned so that its protonated amine group forms ionic interactions with acidic residues Asp44 and Glu82 and its hydroxyl group also makes a hydrogen bond to Glu82 [Fig. 7(b)]. The second ethanolamine molecule interacts less intimately with the protein, packing against Phe176, while forming an ion pair with Asp45, a hydrogen bond with Thr180, and several water-mediated contacts to other residues [Fig. 7(c)]. It is worth noting that both of these ethanolamine molecules lie at the interface between the two BMC-domains that make up a single CpEutL chain, and make contacts with both domains. The ethanolamine binding channel is highly conserved [Fig. 7(d)], and its acidic nature is illustrated by calculation of electrostatic surface potential [Fig. 7(e)]. The negative charge of the acidic residues within the channel complements the positive charge on the primary amine group of the protonated ethanolamine molecule.
Figure 7.
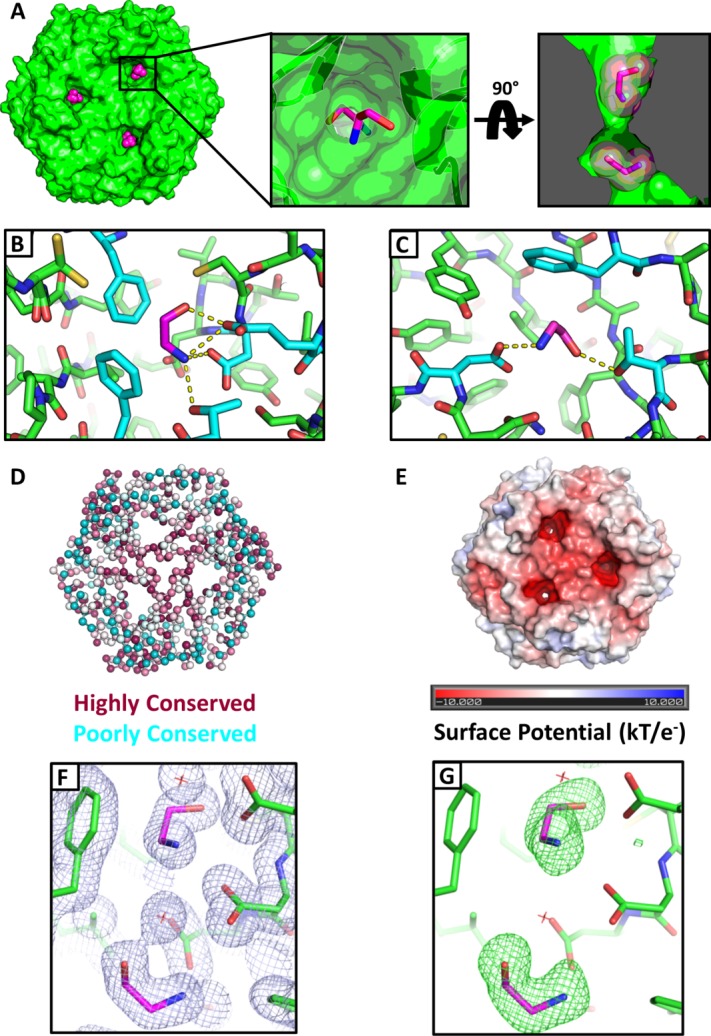
Crystallographic visualization of ethanolamine binding to EutL. (A) X-ray crystal structures reveal that ethanolamine molecules (magenta) bind to EutL in the narrow channels that perforate the trimers. Each monomer contains a single channel with two bound ethanolamine molecules. Within each channel, one ethanolamine molecule is bound on either side of the narrow, hourglass-shaped constriction point. Conserved amino acids (cyan) form hydrophobic and polar interactions with ethanolamine molecules (magenta) that occupy each of the two binding sites (panels B and C). (D) A cartoon diagram of CpEutL with Cα atoms depicted as spheres and colored according to sequence conservation scores, where dark magenta indicates highly conserved positions, and dark cyan indicates poorly conserved positions. Note the high degree of conservation among residues surrounding the ethanolamine binding sites. E) An electrostatic surface representation of CpEutL, calculated using the Poisson-Boltzmann equation, shows that the ethanolamine binding sites bear a strong negative charge, complementary to their ligand. (F) A 2mFo − DFc electron density map (φ_model, 1.0_σ) supports the placement of the ethanolamine ligands (magenta) in the crystallographic model. G) The presence of the ethanolamine ligands was confirmed by calculation of an omit (mFo − DFc, 3.0_σ_) electron density map, in which the ethanolamine molecules were excluded from the structure factor and phase calculations. This map shows exceptionally strong and well-resolved peaks corresponding to the ethanolamine molecules.
Placement of the bound ethanolamine molecules into the model was justified by strong features in both 2mFo − DFc and mFo − DFc (omit) electron density maps [Fig. 7(f,g)]. After modeling the bound ethanolamine and performing several cycles of atomic refinement, we generated an omit map using calculated structure factors and phases that did not include contributions from the ligand atoms. The resulting map showed very strong density corresponding to the omitted ligand molecules (average peak height of 13.7_σ_ in mFo − DFc map), validating their inclusion in the model [Fig. 7(f,g)].
Crystallographic examination of binding site dynamics
To explore the possibility that protein dynamics might allow for expansion of the ligand-binding cavities in EutL, enabling ethanolamine molecules to traverse the MCP shell, we performed several different crystallographic analyses which we thought might reveal evidence of such structural fluctuations.
Our initial attempt to detect molecular motions near the ethanolamine binding sites consisted of a straightforward analysis of individual atomic displacement parameters (B-factors), which provide an estimation of the harmonic displacement of each atom in the crystal structure due to vibrational motion, and/or static disorder. Our CpEutL structures were refined using a TLS model with isotropic B-factors, which were individually refined for all non-hydrogen atoms. We inspected the B-factors of atoms near the ethanolamine binding sites in both the unbound and ligand-bound structures, and found that in both structures these atoms tend to have lower than average total B-factors (Fig. 8). We took this as initial evidence that the ethanolamine binding sites do not undergo significant fluctuations that would be required for molecular transport of the substrate molecule.
Figure 8.
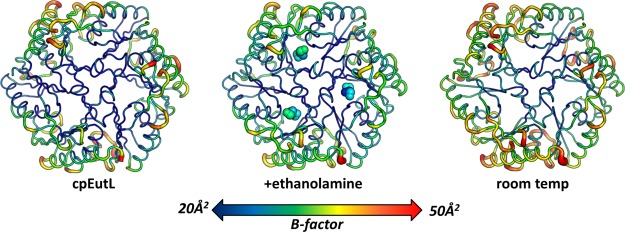
Evaluation of mobility in cryogenic and room temperature structures of EutL. Three structures of CpEutL, colored according to the average atomic B-factor for each residue, and also rendered such that the width of the cartoon diagram represents the average atomic B-factor of each residue, all show low B-factors for the ethanolamine binding channels, indicating these holes do not undergo fluctuations of sufficient amplitude to allow the passage of ethanolamine. The structure on the left is the untreated, cryogenic structure, the structure in the middle is the ethanolamine-bound, cryogenic structure, and the structure on the right is the untreated, room temperature structure.
Motivated by the idea that cryocooling can remodel loosely packed regions of protein structures46, we subsequently determined the structure of CpEutL using X-ray diffraction data collected under ambient temperature conditions (approximately 298K). The coordinates for the room temperature structure were deposited in the PDB under PDB ID 4TM6. Interestingly, by comparing the X-ray data from the room temperature and cryogenic studies, we found that cryocooling shrinks the tetragonal unit cell by 1.1% along the _c_-axis, and by 2.5% along the _a_- and _b_-axes. This unit cell shrinkage is in line with previous reports.46,47 Also, while the final structures are nearly identical, as were the refinement protocols used to obtain them, the room temperature structure gave considerably better refinement statistics, indicating the room temperature model fits the respective diffraction data better than the cryogenic model (Table1). The overall similarity of the room temperature and cryogenic structures, especially at the ethanolamine binding sites, proves that the geometry of these sites is not affected by cryocooling in a way that would complicate interpretation of ligand binding. Furthermore, we repeated our analysis of atomic B-factors, this time using the room temperature structure. Again, we found that the ethanolamine binding sites have low B-factors in general, arguing against the occurrence of large atomic fluctuations in this region of the protein (Fig. 8).
Next, as a complement to our B-factor analysis, we looked for evidence of binding site dynamics using time-averaged refinement of CpEutL structural ensembles. This method utilizes molecular dynamics simulation to create a time-averaged ensemble of structures that provides the best fit to the observed X-ray data,48 thereby capturing anisotropic and anharmonic structural fluctuations which cannot be described using traditional crystallographic B-factors. Starting from our cryogenic crystal structures, we created ensemble models for both the unbound and ethanolamine-bound states. The ensemble model of the unbound state consisted of 50 individual structures, and fit the diffraction data very well, with R/_R_free = 0.138/0.168. Similarly, the ensemble model of the ethanolamine-bound state consisted of 63 individual structures, with R/_R_free = 0.135/0.159. We also created an ensemble model using our room temperature X-ray data for CpEutL, which consisted of 50 individual structures, with R/_R_free = 0.129/0.157. Based on standard crystallographic _R_-factors, the ensemble models fit the diffraction data significantly better than the single-conformer models. In the ensemble models, the residues that form the narrow constriction points of the hourglass-shaped ligand binding channels show little structural fluctuation (Fig. 9). From the narrowness of these channels in which ethanolamine binds, and absent indications that dynamics would enable transit at these points (and given the existence of open routes for diffusion of small molecules through the pores of other shell proteins), the role of the observed ethanolamine binding in EutL is most likely to stabilize the protein in the bound conformation, which is essentially closed to transport.
Figure 9.

Examination of mobility based on ensemble refinement of EutL structures. The images show the ethanolamine binding channels in CpEutL as modeled using time-averaged ensemble refinement against cryogenic CpEutL diffraction data collected from both apo and ethanolaminebound (top and middle panels respectively) crystals, and also against CpEutL diffraction data collected from apo crystals at room temperature (bottom panel). The residues that form the constriction point of the hourglass-shaped channel are shown in magenta. The ensembles show that the channels do not undergo any significant dynamic expansion that might permit substrate transport.
Discussion
Studies of EutL have been critical to our understanding of molecular transport across the protein shells of MCP structures. Previous structures of E. coli EutL provided early evidence that the broken symmetry resulting from domain duplication allows for conformational change at the center of the trimer, which permits the opening of a large central pore.34,35 While the importance of this conformational change in facilitating molecular transport of large cofactor molecules seems clear, the details of how it takes place have yet to be fully elucidated. The crystallization conditions that initially yielded closed versus open structures left unanswered questions regarding the mechanistic details of the conformational transition. Takenoya, et al. showed that the open conformation of EutL can be induced by a high concentration (1_M_) of zinc ions, though they noted that such a high zinc concentration is unlikely to be the physiologically relevant signal for conformational change.34 The experiments presented here highlight features of EutL that appear to be important in allowing the structural rearrangement to take place, and suggest a biologically plausible allosteric mechanism for regulation of pore opening.
Key structural features of EutL facilitate conformational change
Our crystal structures of EutL form Clostridium perfringens (CpEutL) are all strikingly similar to the “closed” conformations of the E. coli homologs observed previously (Fig. 3). Critical interactions that stabilize the closed conformation are consistent between CpEutL and E. coli EutL. This includes important hydrogen-bond interactions within the β3–β4 and β7–β8 loop regions, and interactions between the aromatic rings at the center of the trimer (Fig. 3). Analysis of sequence conservation reveals that residues involved in these interactions (Y69, N74, N183) are strongly conserved among EutL orthologs, supporting their importance in stabilizing the closed conformation.
The closed CpEutL structure also reveals features that appear important for enabling an alternate open conformation, based on comparison with the E. coli structures. Each of the CpEutL monomers contains a narrow, elongated channel, which accommodates movement of the β3–β4 loop during opening of the central pore (Fig. 4a,b). This opening bears a strong negative charge, resulting from the presence of highly conserved acidic residues [Fig. 7(d,e)]. In the open conformation of E. coli EutL, these acidic residues make hydrogen bonds with the retracted β3–β4 loop. Finally, the presence of highly conserved glycine residues (G81, G186) at the ends of the β3–β4 and β7–β8 loops might be explained by a need for flexible movement of those segments.
EutL binds ethanolamine, the substrate of the Eut MCP
Recent work by Pang, et al. suggested that a EutL homolog, PduB, interacts with the substrate molecule of the Pdu MCP (namely 1,2-propanediol),40 prompting us to investigate the interaction of EutL with ethanolamine. Using isothermal titration calorimetry, we demonstrated that ethanolamine binds specifically to EutL, while other small molecules associated with the Eut MCP do not. Titrations of ethanol, acetate, and acetyl-phosphate into EutL release heat in a manner consistent with nonspecific interactions, whereas titration of ethanolamine into EutL produces a binding curve with hyperbolic character, indicative of a specific binding event [Fig. 6(a)]. The data for the titration of ethanolamine into EutL could be fit to a binding model that includes two binding sites per EutL monomer, with significantly different dissociation constants [Fig. 6(b) and Table2]. The dissociation constants calculated for the two binding sites are 92 µ_M_ and 11 m_M_. The concentration of ethanolamine in the human gut is in the range of 0.5–1 m_M_.24 This might indicate that only one of these two sites is physiologically relevant. However, the bacterial intracellular concentration of ethanolamine is unknown, and could fluctuate depending on metabolic conditions. Indeed, the intracellular concentration of ethanolamine could reach levels considerably higher than outside the cell given that bacteria that metabolize ethanolamine also express transmembrane ethanolamine transporters. With those considerations, both binding events could be relevant when bacteria inhabit the gut of their human host.
In addition to determining the thermodynamics of ethanolamine binding to CpEutL, we used X-ray crystallography to visualize, at relatively high resolution, the ligand molecules bound to the protein. The ethanolamine molecules bind in the narrow channels that perforate each monomer within the closed conformation of the CpEutL trimer [Fig. 7(a)]. Two ligand molecules bind in each channel, one on either side of the narrow constriction point. The ethanolamine molecule bound on the cytosolic side of the channel constriction point makes numerous direct interactions with amino acid side chains that line the channel, including ion pairing and hydrogen-bonding of the protonated amine with conserved acidic residues (D44 and E82), and van der Waals contacts with conserved aromatic residues (F112, F184) [Fig. 7(b)]. In contrast, the ethanolamine molecule bound on the luminal side of the channel appears to interact much less intimately with the protein molecule, forming only a few direct interactions (a hydrogen bond to T180, an ionic interaction with D45, and van der Waals interactions with Phe 176) and several indirect, water-mediated interactions [Fig. 7(c)].
The presence of two ethanolamine molecules bound to each monomer is consistent with the binding model derived from ITC experiments, but it is difficult to deduce from the crystal structure which binding site is the high-affinity site and which is the low-affinity site. Based on the observed binding interactions, it is tempting to speculate that the ethanolamine molecule bound to the cytosolic side of the channel represents the higher-affinity site, because it forms more direct interactions with the conserved residues of the binding site, and as a result, appears to be in a more stable orientation. Further experimentation will be required to confidently identify the high and low affinity binding sites from one another.
Ethanolamine binding channels do not conduct substrate transport
The closed conformation of EutL is a structural arrangement that leaves three small channels perforating the pseudohexameric homotrimer, one in each subunit [Fig. 4(a)]. While the possible movement of small molecules across these channels has been discussed,33,34,36 the opening and closing of the much larger central pore remains the most striking structural feature of EutL.34,35 We characterized the interaction of the substrate molecule, ethanolamine, with the EutL shell protein and evaluated the possibility of molecular transport through the narrow channels where it binds. Our results suggest that these holes in the effectively closed conformation of EutL are unlikely to transport ethanolamine, but that instead ethanolamine binding to EutL results in negative allosteric regulation of the conformational change that must occur to open the large pore in the center of the EutL trimer.
Based on analysis of our CpEutL crystal structures, we believe the ethanolamine-binding channels are too narrow to support the passage of substrates. Our geometric measurements reveal that the narrow constriction point of the hourglass-shaped channel has a radius of only 1.3Å [Fig. 4(c)], consistent with similar measurements performed by Sagermann et al. using the closed structure of E. coli EutL. For comparison, the van der Waals radius of a carbon atom is 1.7Å.49 Considering that ethanolamine has two _sp3_-hybridized carbon atoms, we think it is unlikely that this substrate molecule can diffuse through such a narrow pore. In addition, the shell of the Eut MCP also contains a homohexameric shell protein, EutM, whose central pore has a radius of roughly 2–3Å,35 making it a much more probable candidate for the transport of ethanolamine. The energetic barrier for ethanolamine crossing the EutM pore is likely to be so much smaller than for ethanolamine passing through the narrow EutL channel, that any potential flux of ethanolamine through EutL becomes irrelevant in the presence of EutM. Our conclusion that the small holes in the closed form of EutL are not the sites of ethanolamine transport is consistent with computational investigations of mobility based on static as well as time-averaged ensemble treatments of the X-ray data, which indicate these channels are rigid and do not expand to facilitate transport. We also examined the crystal structure at room temperature in addition to (typical) cryogenic temperature and again concluded that the small channels lack the kind of flexibility that would be required for transport of ethanolamine past the narrow constriction point.
Ethanolamine is a negative allosteric regulator of EutL pore opening
Rather than acting as channels for substrate transport, we propose instead that the ethanolamine binding sites within the individual EutL subunits are allosteric sites that negatively regulate opening of the large pore at the center of the trimer. Crystal structures of E. coli EutL determined by Tanaka, et al.35 demonstrated that the three channels through the EutL pseudohexamer are crucial for the conformational change of the pore, because the β3–β4 loops retract into this empty space as the pore at the center of the trimer opens [Fig. 4(b)]. By filling this empty space with atoms, the presence of ethanolamine molecules in these channels prevents the movement of the loop segments that must rearrange to open the large pore. The steric clash that prevents pore opening in the presence of bound ethanolamine is clearly illustrated by superimposing a model of the open conformation of EutL (based on the open structure of a homologous EutL protein reported earlier35) on the ethanolamine-bound structure (Fig. 10). The incompatibility of pore opening and ethanolamine binding strongly suggests that this small molecule serves as a negative allosteric regulator of the conformational rearrangement [Fig. 11(a)]. In addition, although there is some uncertainty about whether both ethanolamine binding sites are physiologically relevant, we note that both molecules bound in the crystal structure produce a steric clash with the open conformation, consistent with the allosteric model. Therefore, by mass action, the presence of ethanolamine favors the closed form of the shell protein.
Figure 10.
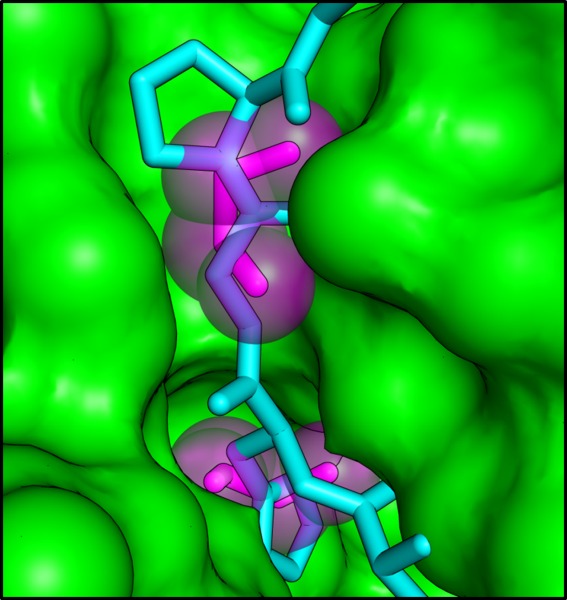
A steric clash forms the basis for allosteric regulation. The cutaway view depicts the ethanolamine binding channel as a molecular surface, with the ethanolamine molecules shown as magenta sticks surrounded by transparent spheres. The cyan polypeptide shows the open conformation of E. coli EutL superimposed on the CpEutL:ethanolamine complex. The overlay reveals that ethanolamine binding prevents a rearrangement from the closed conformation to the open conformation by blocking the space into which the β3–β4 loop moves during the transition.
Figure 11.
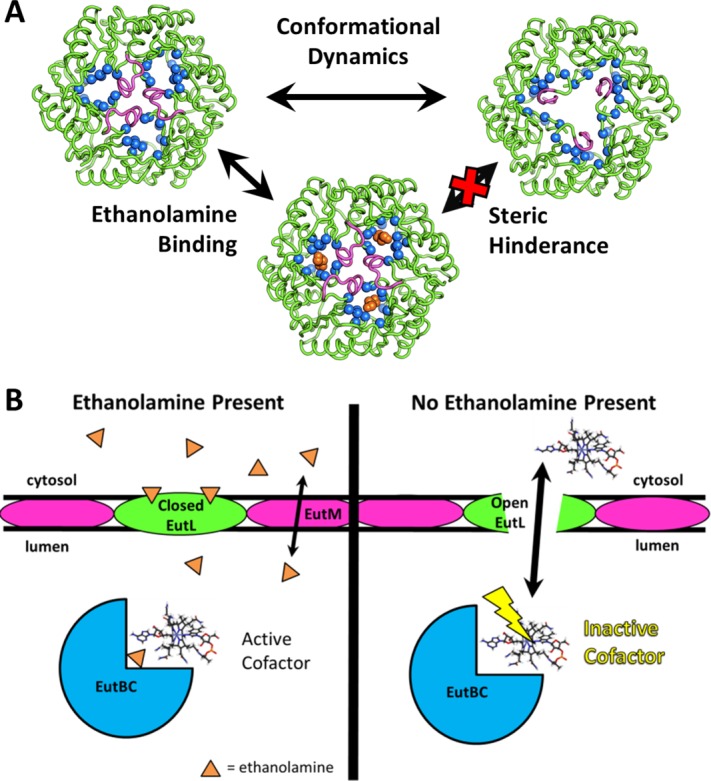
A model for allosteric regulation of cofactor transport in the Eut MCP. (A) Negative allosteric regulation of EutL pore opening is achieved by ethanolamine binding to the closed conformation of EutL, resulting in steric hindrance that prevents the transition to the open conformation. B) We propose a new model for EutL function in the context of the complex enzymology of the Eut MCP. The cobalamin cofactor of the encapsulated EutBC enzyme spontaneously inactivates over time. When the ethanolamine substrate is absent (or has been metabolized), the EutL shell protein remains in the open form so its large central pore can transport large cofactors for enzyme maintenance. Then, when ethanolamine is present, closure of the large EutL pore allows metabolism of ethanolamine to occur without release of the toxic acetaldehyde intermediate. Note that ethanolamine uptake is proposed to occur through a different shell protein paralog (EutM), whose central pore is narrow and selective, but constitutively open.
The binding of ethanolamine to EutL occurs at a position that is distinct from the open pore region, and the binding event stabilizes a particular protein conformation that has a defined effect on function. Therefore, we classify the observed interaction as an allosteric mechanism. This is notably different from some other classical allosteric mechanisms that have been described for oligomeric proteins, which often involve reorientation of subunits and cooperative transition between monomers.50,51 Although EutL is a trimer, the allosteric transition appears to occur within individual monomers, as cooperative binding was not detected by our ITC experiment. In addition, it is worth noting that the proposed allosteric mechanism relies on the existing equilibrium between open and closed states, with ethanolamine binding shifting this equilibrium by stabilizing the closed conformation. This assumption does not necessarily imply that the open conformation dominates the equilibrium in the absence of ethanolamine, only that the population of the open state is reduced by ligand binding, which would reduce the rate of diffusive transport of large molecules through the shell. The idea that the open state is prevalent even in the absence of ethanolamine might explain the difficulty in obtaining crystals of the open form of the protein, although this could also be explained by the crystal lattice being a poor model for the MCP shell, or by the need for an unknown positive allosteric factor for pore opening.
The idea that the EutL pore remains closed in the presence of physiological concentrations of ethanolamine is consistent with our current understanding of the cofactor requirements of the Eut MCP [Fig. 1(b)]. In the absence of substrate, the ethanolamine-ammonia lyase enzyme (EutBC) is susceptible to inactivation resulting from damage to the adenosylcobalamin cofactor.52 To reactivate the enzyme, the damaged cofactor needs to be replaced. While the details of this process are unclear, it appears that in order for it to take place either ATP or cobalamin compounds must pass through the MCP shell to become available to the interior enzymes of the MCP.31,32,52 Accordingly, recent findings suggest a direct interaction between EutL and cobalamin cofactors.53 Because of its capacity to open a large central pore, EutL appears to be the conduit for movement of these larger molecules across the shell. If the large cofactors are required to pass through the shell when EutBC is inactivated due to the absence of substrate, then EutL only needs to open its central pore at low ethanolamine concentrations. Furthermore, when the concentration of ethanolamine is low, then there is no threat of elevated acetaldehyde concentrations, and therefore the need for the MCP shell to maintain a low level of porosity is alleviated.
A potential role for disulfide bonding in EutL
Our crystallographic electron density for untreated CpEutL shows, for the first time, that EutL is able to form disulfide bonds upon air-oxidation under ambient atmospheric conditions. While the electron density features are not as clear as expected for fully formed disulfide bonds, we note that disulfide bonds are generally susceptible to radiation damage by X-rays used for structure determination.42–45 The Cys127–Cys200 disulfide might be especially prone to photochemical reduction because its dihedral angle, at approximately 124°, is outside the ideal range of 90°±12° (the full range of S—S dihedrals in the PDB is 27°–153°).54 Calculation of difference electron density maps comparing wedges of X-ray data from early and late in the course of data collection (_F_early − _F_late) show positive features that confirm the Cys127–Cys200 disulfide bond is more fully present in the freshly crystallized protein, and becomes partially cleaved by X-ray irradiation [Fig. 5(c)].
Motivated by the idea that disulfide bonds generally have a structural or functional significance,55 and by the generally high degree of conservation of the cysteine triad (Cys127, Cys196, and Cys200 in CpEutL) among EutL orthologs, we sought to determine if their presence in CpEutL might be coupled to the functionally relevant conformational change that opens the central pore. Interestingly, we observed that changing the electrochemical environment of the CpEutL protein affected its affinity for ethanolamine (Table2). Upon reduction, both binding sites decrease their affinity for the ethanolamine ligand. The finding that the disulfide-bonded form binds ethanolamine more tightly further argues against the possibility of the disulfide bond being an artifact. Because the ethanolamine binding channels are only present in the closed form of EutL, the observed change of the binding constant serves as the best evidence yet that there might be a coupling between electrochemistry and the conformational equilibrium of the EutL pore, possibly related to the observed disulfide bond. The high-affinity binding site, which is physiologically relevant, shows a nearly fourfold increase in the dissociation constant, from 92 µ_M_ to 321µ_M_, following chemical reduction. This decrease in binding could be explained by a shift in the conformational equilibrium toward the open conformation, which cannot bind the ligand.
Interestingly, a functional connection in enterobacteria has been noted before between ethanolamine metabolism and oxidative stress induced by the host immune response.23,24 This could provide a physiological basis for electrochemical regulation of MCP function. It could also help explain why this disulfide bond does not appear in structures of the E. coli EutL protein. The laboratory E. coli strain used for those structural studies is not pathogenic, nor does it utilize sulfur compounds as respiratory electron acceptors as do Clostridium and Salmonella,56 so ethanolamine metabolism may not be linked to intracellular redox state in certain bacteria. Additional experimentation will be necessary to describe the relationship between the electrochemical potential of the observed disulfide bond and that of the cellular environment, as well as to verify and characterize the interplay between allosteric and disulfide-based mechanisms of pore regulation in EutL.
A new model for EutL function
The work presented here provides a detailed analysis of the structural features within EutL that are critical for the conformational change that defines its function. Specifically, we have identified a conserved disulfide bond in EutL, and have provided initial evidence that this bond might be related to the function of EutL, possibly by shifting the conformational equilibrium in response to electrochemical environment. In addition, our analysis focused on three narrow (symmetry-related) channels that perforate the EutL trimer, which are essential for the conformational rearrangements that open the central pore. These holes are also the binding sites for ethanolamine molecules, but they are seemingly too narrow and rigid to support substrate transport. Instead, they appear to be allosteric sites, where bound ethanolamine molecules prevent transition to the open conformation of the oligomer by virtue of steric conflict, resulting in negative regulation of pore opening. Our new model for allosteric regulation of the EutL pore conformation by ethanolamine binding also provides new insight into how EutL functions within the context of the Eut MCP. The finding that ethanolamine serves as a negative allosteric regulator of EutL pore opening is consistent with the notion that at low ethanolamine concentrations the adenosylcobalamin cofactor required for EutBC activity is subject to inactivation, which would necessitate the opening of a large pore to facilitate the regeneration or exchange of cobalamin compounds across the MCP shell.
Materials and Methods
Cloning, expression, and protein purification
A detailed description of the cloning, expression, and purification protocols used to obtain CpEutL protein samples has been published recently.53 Briefly, we prepared two CpEutL constructs by amplifying the eutL gene from Clostridium perfringens chromosomal DNA and ligating it into the pET22b expression vector under control of the T7 promoter. One of these constructs contained a C-terminal His6 affinity tag, while the other contained an N-terminal, protease-cleavable (TEV) His6 affinity tag. We expressed recombinant protein using transformed Escherichia coli BL21 (DE3) Rosetta cells. The C-terminally tagged construct, which was used for crystallization, was purified by a single step of nickel affinity chromatography. The N-terminally tagged construct, which was used for ITC experiments, was purified by nickel affinity chromatography, followed by cleavage of the tag with TEV protease, and then by a second round of nickel affinity chromatography, anion-exchange chromatography, and finally by gel filtration.
Crystallization
A detailed description of the protocol used to obtain the tetragonal crystal form of CpEutL used for our studies was reported recently.53 In summary, protein samples were concentrated to approximately 20 mg/mL in buffer containing 20 m_M_ Tris pH 8.0 and 100 m_M_ sodium chloride. High quality crystals were grown by the vapor-diffusion method (hanging drop) from mother liquor containing 0.1_M_ HEPES Buffer pH 7.0, 5% PEG-8000, and 8% ethylene glycol.
Our modified structures (reduced and ethanolamine-bound) were obtained using the tetragonal crystal form of CpEutL. We grew the reduced crystals in the same fashion as the original tetragonal crystals, except we pre-treated the protein with 5 m_M_ TCEP. We obtained crystals of the ethanolamine-bound form of CpEutL by first growing the tetragonal crystals and subsequently soaking them overnight in mother liquor containing 20 m_M_ ethanolamine.
X-ray data collection and processing
Crystals used for collection of X-ray diffraction data under cryogenic conditions were harvested and cryoprotected using 50% mother liquor with 50% glycerol for the untreated specimens, or 50% mother liquor with 2_M_ trimethylamine-N-oxide57 for the reduced and ethanolamine-bound specimens. For room temperature data collection, crystals were not subjected to cryoprotectant, but were instead mounted inside of sealed polyester capillaries (MicroRT, Mitegen) along with a small plug of mother liquor to prevent crystal dehydration.
We collected single-crystal X-ray diffraction data at the Advanced Photon Source on beamline 24-ID-C. When we collected data from the untreated crystal, this beamline was equipped with an ADSC Quantum315 CCD detector. We note that with the large c-axis length for this particular crystal form, the resolution for the diffraction experiment was limited by the detector geometry when using this CCD detector. Following beamline upgrades, all other data sets were collected on a Pilatus 6M-F detector, whose larger size allowed the collection of higher resolution data. We maintained the crystals at cryogenic temperature (100K) throughout the course of the data collection, except during our room temperature studies which were carried out at approximately 298K. For the untreated crystal, we indexed and integrated the reflection data using DENZO, and performed scaling with SCALEPACK.58 Intensities were converted to structure factors using Ctruncate and the free set of reflections was assigned using Uniqueify, both within the CCP4 utility sca2mtz.59 For all other crystals, we performed indexing, integration, and scaling of the X-ray data using XDS and XSCALE, and then converted intensities to structure factors with XDSCONV.60 Diffraction data were routinely analyzed with Xtriage to check for crystal pathologies.61,62 Further information regarding data collection and processing is presented in Table1. It is important to note that the original selection of free reflections designated for the untreated data was maintained across all data sets used in our studies, to prevent cross-contamination of the free and working sets of reflections.
Structure determination
Using the program PHASER within the CCP4 suite,59,63 we implemented the method of molecular replacement to calculate phases for diffraction data from the untreated crystals. Initially, we prepared a molecular replacement search model based on the structures of E. coli EutL. This model consisted of the core regions of the protein, and excluded any parts of the polypeptide chain that occupy different positions in the open and closed structures of the E. coli protein. A solution was found in space group _P_43212. Following structure determination from the original CpEutL crystals, the tetragonal model (PDB ID 4EDI) was used for phase calculation by molecular replacement to solve subsequent crystal structures.
For all crystal structures described in this work, a similar model-building and refinement strategy was used. First, using electron density maps calculated with phases from molecular replacement, we rebuilt the missing or incorrect parts of the structures. In some cases, we performed initial atomic refinement with simulated annealing to remove residual model-bias. We then performed iterative steps of manual model rebuilding and atomic refinement of the model to convergence with TLS parameters, a riding hydrogen model, and automatic weight optimization. All model-building was performed using COOT,64 and refinement steps were performed with phenix.refine within the PHENIX suite.61,65 For refinement of the ethanolamine-bound structure, ligand restraints were prepared with phenix.elbow.66 Further information regarding model building and refinement is presented in Table1.
Calculation of difference electron density maps
Difference electron density maps of the _F_1 − _F_2 type were calculated by first scaling two sets of structure factors with Scaleit (contained within the CCP4 suite59(p4)). Those amplitudes were used, along with model phases, to calculate a fast Fourier transform67 with the PHENIX software.61
Measurement of channel dimensions
To analyze the dimensions of the molecular channels that traverse the CpEutL trimers, we used the HOLE2 software.41 We selected two points, one on either side of the channel present in chain A of our untreated structure, and used them to define a vector that passed through this channel. This vector was provided as input to HOLE2 to guide determination of the molecular channel.
Analysis of sequence conservation
We used the ConSurf algorithm68,69 to analyze the evolutionary conservation of critical amino acids in EutL orthologs.
Calculation of electrostatic surface potential
Electrostatic surface potentials were calculated using the Poisson-Boltzmann equation, as implemented in the APBS plugin for PyMOL.70,71 The calculations were performed using an ionic strength of 150 m_M_, a solvent dielectric of 78.54, and a protein dielectric of 2.0.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were performed using a protein sample from which the N-terminal affinity tag had been removed by protease cleavage. To very accurately measure the protein concentration of our ITC sample by UV absorption (λ = 280nm), we used a molar extinction coefficient derived from amino acid analysis. The protein sample was concentrated to 0.6 m_M_ in buffer containing 20 m_M_ Tris at pH 7.2, and 100 m_M_ NaCl. The titrant was prepared by dissolving ethanolamine hydrochloride in the identical buffer to a final concentration of 50 m_M_. We used the hydrochloride salt of ethanolamine to prevent pH differences between the protein sample and the ligand. Titrations were performed at 298K using an iTC-200 calorimeter (GE Healthcare/MicroCal). Similar titrations were performed using ethanol, acetate, and acetyl-phosphate as ligands.
We collected ITC data for the chemically reduced samples by adding 20 m_M_ TCEP to the protein and ligand solutions and repeating the titration described above.
Raw ITC data were processed using the Origin software. The raw data were integrated to give plots of Δ_H_ versus molar ratio of ligand to protein. For the ethanolamine titrations, we used Origin to fit the titration curves to a two-site, sequential binding model and extract thermodynamic parameters for the EutL-ethanolamine interaction.
Analysis of crystallographic B-factors
We used the PyMOL71 molecular graphics system to color the atoms in our CpEutL structures according to their individual atomic (isotropic) B-factors. Inferences about structural mobility were drawn from comparing atoms within the same crystal structure and not between structures, as overall B factors can reflect features of a crystal other than local mobility (such as the resolution limit).
Time-averaged X-ray refinement of structural ensembles
Time-averaged structural ensembles were refined against X-ray diffraction data using the method developed by Burnley et al. and implemented in phenix.ensemble_refinement.48 We used the original observed X-ray diffraction data and the final single-copy atomic models of specified CpEutL structures as inputs for the ensemble refinement procedure. We optimized the ensemble refinement protocol by first optimizing the ptls parameter, followed by the wxray_coupled_tbath_offset parameter, and then finally the tx parameter, choosing the models that gave the best _R_work and _R_free. For the final ensemble refinements, we used values of ptls = 0.90 for both the cryogenic and room temperature apo structures, and ptls = 1.0 for the cryogenic, ethanolamine-bound structure, and the default parameters for wxray_coupled_tbath_offset and tx were found to yield the best results. We note that this method is intended to capture anisotropic atomic motions that have relatively small amplitudes, and therefore is unable to describe large-scale conformational rearrangements such as loop motions that result in EutL pore opening.
Acknowledgments
The authors acknowledge Dr. Christopher Crowley for initiating work on CpEutL. They also thank Drs. Michael Sawaya, Julien Jorda, and Thomas Bobik for helpful suggestions regarding various aspects of this work, as well as Jason Navarro for his technical assistance with crystallization screening, and Margaret Condron for technical assistance with amino acid analysis. They thank the NE-CAT beamline staff for their technical assistance.
Glossary
BMC
bacterial microcompartment (in reference to the shell protein domain)
CpEutL
EutL protein from Clostridium perfringens
Eut
ethanolamine utilization
ITC
isothermal titration calorimetry
MCP
microcompartment
PDB
protein data bank
RMSD
root mean square deviation
TCEP
tris(2-carboxyethyl)phosphine
TLS
translation, libration, screw.
References
- Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–133. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- Guarner F, Malagelada J-R. Gut flora in health and disease. Lancet. 2003;361:512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- Sears CL. A dynamic partnership: celebrating our gut flora. Anaerobe. 2005;11:247–251. doi: 10.1016/j.anaerobe.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Cummings JH, Macfarlane GT. Role of intestinal bacteria in nutrient metabolism. Clin Nutr. 1997;16:3–11. doi: 10.1177/0148607197021006357. [DOI] [PubMed] [Google Scholar]
- Steinhoff U. Who controls the crowd? New findings and old questions about the intestinal microflora. Immunol Lett. 2005;99:12–16. doi: 10.1016/j.imlet.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Björkstén B, Sepp E, Julge K, Voor T, Mikelsaar M. Allergy development and the intestinal microflora during the first year of life. J Allergy Clin Immunol. 2001;108:516–520. doi: 10.1067/mai.2001.118130. [DOI] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Tillisch K, Labus J, Kilpatrick L, Jiang Z, Stains J, Ebrat B, Guyonnet D, Legrain–Raspaud S, Trotin B, Naliboff B, et al. Consumption of fermented milk product with probiotic modulates brain activity. Gastroenterology. 2013;144:1394–1401. doi: 10.1053/j.gastro.2013.02.043. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin DA. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat Rev Microbiol. 2010;8:290–295. doi: 10.1038/nrmicro2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoy O, Ravcheev D, Mushegian A. Comparative genomics of ethanolamine utilization. J Bacteriol. 2009;191:7157–7164. doi: 10.1128/JB.00838-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojiljkovic I, Bäumler AJ, Heffron F. Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J Bacteriol. 1995;177:1357–1366. doi: 10.1128/jb.177.5.1357-1366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof DM, Roth JR. Ethanolamine utilization in Salmonella typhimurium. J Bacteriol. 1988;170:3855–3863. doi: 10.1128/jb.170.9.3855-3863.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell CM, Scarlett FA, Turner JM. Ethanolamine catabolism by bacteria, including Escherichia coli. Biochem Soc Trans. 1976;4:495–497. doi: 10.1042/bst0040495. [DOI] [PubMed] [Google Scholar]
- Cotton PB. Non-dietary lipid in the intestinal lumen. Gut. 1972;13:675–681. doi: 10.1136/gut.13.9.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randle CL, Albro PW, Dittmer JC. The phosphoglyceride composition of Gram-negative bacteria and the changes in composition during growth. Biochim Biophys Acta. 1969;187:214–220. doi: 10.1016/0005-2760(69)90030-7. [DOI] [PubMed] [Google Scholar]
- Kofoid E, Rappleye C, Stojiljkovic I, Roth J. The 17-gene ethanolamine (eut) operon of Salmonella typhimurium encodes five homologues of carboxysome shell proteins. J Bacteriol. 1999;181:5317–5329. doi: 10.1128/jb.181.17.5317-5329.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Kristensen DM, Coleman MK, Mushegian A. Detection of biochemical pathways by probabilistic matching of phyletic vectors. PLoS ONE. 2009;4:e5326. doi: 10.1371/journal.pone.0005326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Doerks T, Jensen LJ, Perez-Iratxeta C, Kaczanowski S, Hooper SD, Andrade MA, Bork P. Systematic association of genes to phenotypes by genome and literature mining. PLoS Biol. 2005;3:e134. doi: 10.1371/journal.pbio.0030134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawhon SD, Frye JG, Suyemoto M, Porwollik S, McClelland M, Altier C. Global regulation by CsrA in Salmonella typhimurium. Mol Microbiol. 2003;48:1633–1645. doi: 10.1046/j.1365-2958.2003.03535.x. [DOI] [PubMed] [Google Scholar]
- Kelly A, Goldberg MD, Carroll RK, Danino V, Hinton JCD, Dorman CJ. A global role for Fis in the transcriptional control of metabolism and type III secretion in Salmonella enterica serovar Typhimurium. Microbiology. 2004;150:2037–2053. doi: 10.1099/mic.0.27209-0. [DOI] [PubMed] [Google Scholar]
- Bourgogne A, Hilsenbeck SG, Dunny GM, Murray BE. Comparison of OG1RF and an isogenic fsrB deletion mutant by transcriptional analysis: the Fsr System of Enterococcus faecalis is more than the activator of gelatinase and serine protease. J Bacteriol. 2006;188:2875–2884. doi: 10.1128/JB.188.8.2875-2884.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, JR Roth, AJ Bäumler. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010;467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, Bäumler AJ. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A. 2011;108:17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Bäumler AJ. A breathtaking feat: to compete with the gut microbiota, Salmonella drives its host to provide a respiratory electron acceptor. Gut Microbes. 2011;2:58–60. doi: 10.4161/gmic.2.1.14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon MR, Horswill AR, Escalante-Semerena JC. DNA polymerase I function is required for the utilization of ethanolamine, 1,2-propanediol, and propionate by Salmonella typhimurium LT2. J Bacteriol. 1995;177:7119–7124. doi: 10.1128/jb.177.24.7119-7124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penrod JT, Roth JR. Conserving a volatile metabolite: a role for carboxysome-like organelles in Salmonella enterica. J Bacteriol. 2006;188:2865–2874. doi: 10.1128/JB.188.8.2865-2874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huseby DL, Roth JR. Evidence that a metabolic microcompartment contains and recycles private cofactor pools. J Bacteriol. 2013;195:2864–2879. doi: 10.1128/JB.02179-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Fan C, Sinha S, Bobik TA. The PduQ enzyme Is an alcohol dehydrogenase used to recycle NAD+ internally within the Pdu microcompartment of Salmonella enterica. PLoS ONE. 2012;7:e47144. doi: 10.1371/journal.pone.0047144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan BH, Stadtman ER. Ethanolamine Deaminase, a Cobamide Coenzyme-dependent Enzyme II. Physical and chemical properties and interaction with cobamides and ethanolamine. J Biol Chem. 1968;243:1794–1803. [PubMed] [Google Scholar]
- Mori K, Bando R, Hieda N, Toraya T. Identification of a reactivating factor for adenosylcobalamin-dependent ethanolamine ammonia lyase. J Bacteriol. 2004;186:6845–6854. doi: 10.1128/JB.186.20.6845-6854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buan NR, Suh S-J, Escalante-Semerena JC. The eutT gene of Salmonella enterica encodes an oxygen-labile, metal-containing ATP:corrinoid adenosyltransferase enzyme. J Bacteriol. 2004;186:5708–5714. doi: 10.1128/JB.186.17.5708-5714.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagermann M, Ohtaki A, Nikolakakis K. Crystal structure of the EutL shell protein of the ethanolamine ammonia lyase microcompartment. Proc Natl Acad Sci U S A. 2009;106:8883–8887. doi: 10.1073/pnas.0902324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenoya M, Nikolakakis K, Sagermann M. Crystallographic insights into the pore structures and mechanisms of the EutL and EutM shell proteins of the ethanolamine-utilizing microcompartment of Escherichia coli. J Bacteriol. 2010;192:6056–6063. doi: 10.1128/JB.00652-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Sawaya MR, Yeates TO. Structure and mechanisms of a protein-based organelle in Escherichia coli. Science. 2010;327:81–84. doi: 10.1126/science.1179513. [DOI] [PubMed] [Google Scholar]
- Heldt D, Frank S, Seyedarabi A, Ladikis D, Parsons JB, Warren MJ, Pickersgill RW. Structure of a trimeric bacterial microcompartment shell protein, EtuB, associated with ethanol utilization in Clostridium kluyveri. Biochem J. 2009;423:199–207. doi: 10.1042/BJ20090780. [DOI] [PubMed] [Google Scholar]
- Pang A, Warren MJ, Pickersgill RW. Structure of PduT, a trimeric bacterial microcompartment protein with a 4Fe-4S cluster-binding site. Acta Crystallogr D Biol Crystallogr. 2011;67:91–96. doi: 10.1107/S0907444910050201. [DOI] [PubMed] [Google Scholar]
- Crowley CS, Cascio D, Sawaya MR, Kopstein JS, Bobik TA, Yeates TO. Structural insight into the mechanisms of transport across the Salmonella enterica Pdu microcompartment shell. J Biol Chem. 2010;285:37838–37846. doi: 10.1074/jbc.M110.160580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein MG, Zwart P, Bagby SC, Cai F, Chisholm SW, Heinhorst S, Cannon GC, Kerfeld CA. Identification and structural analysis of a novel carboxysome shell protein with implications for metabolite transport. J Mol Biol. 2009;392:319–333. doi: 10.1016/j.jmb.2009.03.056. [DOI] [PubMed] [Google Scholar]
- Pang A, Liang M, Prentice MB, Pickersgill RW. Substrate channels revealed in the trimeric Lactobacillus reuteri bacterial microcompartment shell protein PduB. Acta Crystallogr D Biol Crystallogr. 2012;68:1642–1652. doi: 10.1107/S0907444912039315. [DOI] [PubMed] [Google Scholar]
- Smart OS, Goodfellow JM, Wallace BA. The pore dimensions of gramicidin A. Biophys J. 1993;65:2455–2460. doi: 10.1016/S0006-3495(93)81293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister WP. Structural changes in a cryo-cooled protein crystal owing to radiation damage. Acta Crystallogr D Biol Crystallogr. 2000;56:328–341. doi: 10.1107/s0907444999016261. [DOI] [PubMed] [Google Scholar]
- Weik M, Ravelli RB, Kryger G, McSweeney S, Raves ML, Harel M, Gros P, Silman I, Kroon J, Sussman JL. Specific chemical and structural damage to proteins produced by synchrotron radiation. Proc Natl Acad Sci U S A. 2000;97:623–628. doi: 10.1073/pnas.97.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weik M, Bergès J, Raves ML, Gros P, McSweeney S, Silman I, Sussman JL, Houée-Levin C, Ravelli RBG. Evidence for the formation of disulfide radicals in protein crystals upon X-ray irradiation. J Synchrotron Radiat. 2002;9:342–346. doi: 10.1107/s0909049502014589. [DOI] [PubMed] [Google Scholar]
- Garman EF. Radiation damage in macromolecular crystallography: what is it and why should we care? Acta Crystallogr D Biol Crystallogr. 2010;66:339–351. doi: 10.1107/S0907444910008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proc Natl Acad Sci U S A. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juers DH, Matthews BW. Reversible lattice repacking illustrates the temperature dependence of macromolecular interactions. J Mol Biol. 2001;311:851–862. doi: 10.1006/jmbi.2001.4891. [DOI] [PubMed] [Google Scholar]
- Burnley BT, Afonine PV, Adams PD, Gros P. Modelling dynamics in protein crystal structures by ensemble refinement. eLife. 2012;1:e00311–e00311. doi: 10.7554/eLife.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- Blundell TL, Srinivasan N. Symmetry, stability, and dynamics of multidomain and multicomponent protein systems. Proc Natl Acad Sci. 1996;93:14243–14248. doi: 10.1073/pnas.93.25.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toraya T. Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem Rev. 2003;103:2095–2128. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- Thompson MC, Crowley CS, Kopstein J, Bobik TA, Yeates TO. Structure of a bacterial microcompartment shell protein bound to a cobalamin cofactor. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2014;70:1584–1590. doi: 10.1107/S2053230X1402158X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellequer J-L, Chen SW. Multi-template approach to modeling engineered disulfide bonds. Proteins. 2006;65:192–202. doi: 10.1002/prot.21059. [DOI] [PubMed] [Google Scholar]
- Marino SM, Gladyshev VN. Analysis and functional prediction of reactive cysteine residues. J Biol Chem. 2012;287:4419–4425. doi: 10.1074/jbc.R111.275578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EL, Clark MA. Tetrathionate reduction and production of hydrogen sulfide from thiosulfate. Microbiol Rev. 1987;51:192–205. doi: 10.1128/mr.51.2.192-205.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller-Dieckmann C, Kauffmann B, Weiss MS. Trimethylamine N-oxide as a versatile cryoprotective agent in macromolecular crystallography. J Appl Crystallogr. 2011;44:433–436. [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, SJ McNicholas, GN Murshudov, NS Pannu, EA Potterton, HR Powell, RJ Read, A Vagin, KS Wilson. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. 2010;66:125–132. doi: 10.1107/S0907444909047337. ) XDS. Acta Crystallogr D Biol Crystallogr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, AJ McCoy, NW Moriarty, R Oeffner, RJ Read, DC Richardson, JS Richardson, TC Terwilliger, PH Zwart. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart PH, Grosse-Kunstleve RW, Adams PD. 2005. ) Xtriage and Fest: automatic assessment of X-ray data and substructure structure factor estimation. CCP4 Newsl. [Internet] 43. Available at: https://publications.lbl.gov/islandora/object/ir%3A127478/
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty NW, Grosse-Kunstleve RW, Adams PD. Electronic ligand builder and optimization workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Eyck LF. Crystallographic fast Fourier transforms. Acta Crystallogr Sect A. 1973;29:183–191. [Google Scholar]
- Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anon. The PyMOL Molecular Graphics System. Schrödinger, LLC.
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2009;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]