PD-1 Blockade in Tumors with Mismatch-Repair Deficiency (original) (raw)
. Author manuscript; available in PMC: 2016 Jun 25.
Published in final edited form as: N Engl J Med. 2015 May 30;372(26):2509–2520. doi: 10.1056/NEJMoa1500596
Abstract
BACKGROUND
Somatic mutations have the potential to encode “non-self” immunogenic antigens. We hypothesized that tumors with a large number of somatic mutations due to mismatch-repair defects may be susceptible to immune checkpoint blockade.
METHODS
We conducted a phase 2 study to evaluate the clinical activity of pembrolizumab, an anti–programmed death 1 immune checkpoint inhibitor, in 41 patients with progressive metastatic carcinoma with or without mismatch-repair deficiency. Pembrolizumab was administered intravenously at a dose of 10 mg per kilogram of body weight every 14 days in patients with mismatch repair–deficient colorectal cancers, patients with mismatch repair–proficient colorectal cancers, and patients with mismatch repair–deficient cancers that were not colorectal. The coprimary end points were the immune-related objective response rate and the 20-week immune-related progression-free survival rate.
RESULTS
The immune-related objective response rate and immune-related progression-free survival rate were 40% (4 of 10 patients) and 78% (7 of 9 patients), respectively, for mismatch repair–deficient colorectal cancers and 0% (0 of 18 patients) and 11% (2 of 18 patients) for mismatch repair–proficient colorectal cancers. The median progression-free survival and overall survival were not reached in the cohort with mismatch repair–deficient colorectal cancer but were 2.2 and 5.0 months, respectively, in the cohort with mismatch repair–proficient colorectal cancer (hazard ratio for disease progression or death, 0.10 [P<0.001], and hazard ratio for death, 0.22 [P = 0.05]). Patients with mismatch repair–deficient noncolorectal cancer had responses similar to those of patients with mismatch repair–deficient colorectal cancer (immune-related objective response rate, 71% [5 of 7 patients]; immune-related progression-free survival rate, 67% [4 of 6 patients]). Whole-exome sequencing revealed a mean of 1782 somatic mutations per tumor in mismatch repair–deficient tumors, as compared with 73 in mismatch repair–proficient tumors (P = 0.007), and high somatic mutation loads were associated with prolonged progression-free survival (P = 0.02).
CONCLUSIONS
This study showed that mismatch-repair status predicted clinical benefit of immune checkpoint blockade with pembrolizumab. (Funded by Johns Hopkins University and others; ClinicalTrials.gov number, NCT01876511.)
The Programmed Death 1 (PD-1) PATHway is a negative feedback system that represses Th1 cytotoxic immune responses and that, if unregulated, can damage the host.1–3 It is up-regulated in many tumors and in their surrounding microenvironment. Blockade of this pathway with antibodies to PD-1 or its ligands has led to remarkable clinical responses in patients with many different types of cancer, including melanomas, non–small-cell lung cancer, renal-cell carcinoma, bladder cancer, and Hodgkin’s lymphoma.4–10 The expression of PD-1 ligands (PD-L1 or PD-L2) on the surface of tumor cells or immune cells is an important — but not a definitive — predictive biomarker of response to PD-1 blockade.4,6–8,11
In reports of the effects of PD-1 blockade in human tumors, only 1 of 33 patients with colorectal cancer had a response to this treatment, in contrast to substantial fractions of patients with melanomas, renal-cell cancers, and lung tumors who have a response.10,12 What was different about this single patient? We hypothesized that this patient had mismatch-repair deficiency, because mismatch-repair deficiency occurs in a small fraction of advanced colorectal cancers,13,14 somatic mutations found in tumors can be recognized by the patient’s own immune system,15 and mismatch repair–deficient colorectal cancers have 10 to 100 times as many somatic mutations as mismatch repair–proficient colorectal cancers.16–18 Moreover, mismatch repair–deficient cancers contain prominent lymphocyte infiltrates, a finding consistent with an immune response.19–22 In addition, two of the tumor types that were most responsive to PD-1 blockade in a study by Topalian et al.10 had high numbers of somatic mutations as a result of exposure to cigarette smoke (lung cancers) or ultraviolet radiation (melanomas).23,24 Our hypothesis was correct: the tumor of the single patient with colorectal cancer who had a response to PD-1 blockade was mismatch repair–deficient.25 Therefore, we hypothesized that mismatch repair–deficient tumors are more responsive to PD-1 blockade than are mismatch repair–proficient tumors.
To test this hypothesis, we initiated a phase 2 clinical trial to evaluate immune checkpoint blockade in patients whose tumors had or did not have mismatch-repair deficiency. Because mismatch-repair deficiency in tumors arises through two routes,26–28 we recruited patients with hereditary nonpolyposis colorectal cancer (also known as the Lynch syndrome), which results from an inherited germline defect in one of four mismatch-repair genes followed by a second inactivating somatic change in the remaining wild-type allele. We also recruited patients with sporadic mismatch repair–deficient tumors, in which both alleles of a mismatch-repair gene are inactivated by somatic mutations or by epigenetic silencing.29 In either case, the neoplasms that arise harbor hundreds or thousands of mutations.16,18
METHODS
PATIENTS
Patients with treatment-refractory progressive metastatic cancer were recruited from three centers for this phase 2 study (Table 1). Three cohorts were evaluated: cohort A included patients with mismatch repair–deficient colorectal adenocarcinomas, cohort B included patients with mismatch repair–proficient colorectal adenocarcinomas, and cohort C included patients with mismatch repair–deficient cancers of types other than colorectal.
Table 1.
Demographic and Baseline Characteristics of the Patients.*
| Characteristic | Mismatch Repair–Deficient Colorectal Cancer(N = 11) | Mismatch Repair–Proficient Colorectal Cancer(N = 21) | Mismatch Repair–Deficient Noncolorectal Cancer(N = 9) | P Value† |
|---|---|---|---|---|
| Median age (range) — yr | 46 (24–65) | 61 (32–79) | 57 (34–92) | 0.02 |
| Sex — no. (%) | 0.72 | |||
| Female | 5 (45) | 8 (38) | 4 (44) | |
| Male | 6 (55) | 13 (62) | 5 (56) | |
| Race — no. (%)‡ | 0.66 | |||
| White | 8 (73) | 17 (81) | 8 (89) | |
| Black | 1 (9) | 3 (14) | 0 | |
| Other | 2 (18) | 1 (5) | 1 (11) | |
| ECOG performance status — no. (%)§ | 0.07 | |||
| 0 | 0 | 6 (29) | 2 (22) | |
| 1 | 11 (100) | 15 (71) | 7 (78) | |
| Cancer type — no. (%) | >0.99 | |||
| Colon | 9 (82) | 18 (86) | 0 | |
| Rectal | 2 (18) | 3 (14) | 0 | |
| Ampullary or cholangiocarcinoma | 0 | NA | 4 (44) | |
| Endometrial | 0 | NA | 2 (22) | |
| Small bowel | 0 | NA | 2 (22) | |
| Gastric | 0 | NA | 1 (11) | |
| Histologic grade — no. (%) | 0.20 | |||
| Well or moderately differentiated | 7 (64) | 18 (86) | 4 (44) | |
| Poorly differentiated | 4 (36) | 3 (14) | 3 (33) | |
| Other | 0 | 0 | 2 (22) | |
| Stage IV cancer — no. (%) | 11(100) | 21 (100) | 9 (100) | >0.99 |
| Liver metastases — no. (%) | 6 (55) | 11 (52) | 6 (67) | >0.99 |
| Median time since initial diagnosis (range) — mo | 31 (6–95) | 58 (27–192) | 23 (2–105) | 0.07 |
| Previous therapies — no. (%) | 0.89 | |||
| 1 | 0 | 0 | 1 (11) | |
| 2 | 3 (27) | 4 (19) | 5 (56) | |
| 3 | 3 (27) | 5 (24) | 1 (11) | |
| >4 | 5 (45) | 12 (57) | 2 (22) | |
| Detected germline mutation or known Lynch syndrome — no. (%) | <0.001 | |||
| Yes | 9 (82) | 0 | 4 (44) | |
| No | 2 (18) | 21 (100) | 4 (44) | |
| Unknown | 0 | 0 | 1 (11) | |
| BRAF wild type — no. (%) | 0.64 | |||
| Yes | 8 (73) | 11 (52) | 4 (44) | |
| No | 0 | 1 (5) | 0 | |
| Unknown | 3 (27) | 9 (43) | 5 (56) | |
| KRAS wild type — no. (%) | 0.72 | |||
| Yes | 6 (55) | 13 (62) | 4 (44) | |
| No | 5 (45) | 8 (38) | 1 (11) | |
| Unknown | 0 | 0 | 4 (44) |
STUDY OVERSIGHT
The protocol, available with the full text of this article at NEJM.org, was approved by the institutional review board at each site, and the study was conducted in accordance with the provisions of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines. All the patients provided written informed consent before study entry. The first author (the principal investigator) and the last author (the Investigational New Drug sponsor) were responsible for oversight of the study. Merck donated the study drug and reviewed the final drafts of the protocol and of this manuscript before submission; they did not participate in the analysis of the data.
STUDY DESIGN
This phase 2 trial was conducted with the use of a Green–Dahlberg two-stage design and included the three parallel cohorts described above. The study agent, pembrolizumab, was administered intravenously at a dose of 10 mg per kilogram of body weight every 14 days (Fig. S1 in Supplementary Appendix 1, available at NEJM.org). Pembrolizumab is a humanized monoclonal anti–PD-1 antibody of the IgG4 kappa isotype that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2 (Fig. S1 in Supplementary Appendix 1).
Safety assessments were performed before each treatment. At the start of each treatment cycle, the total tumor burden was assessed by means of measurement of serum biomarkers. Radiographic assessments were performed at 12 weeks and every 8 weeks thereafter. Further details concerning the clinical protocol are available at NEJM.org.
ANALYSIS OF MISMATCH-REPAIR STATUS
Tumors with genetic defects in mismatch-repair pathways are known to harbor hundreds to thousands of somatic mutations, especially in regions of repetitive DNA known as microsatellites. The accumulation of mutations in these regions of the genome is termed microsatellite instability.26–28 Mismatch-repair status was assessed in tumors with the use of the MSI Analysis System (Promega), through the evaluation of selected microsatellite sequences that are particularly prone to copying errors when mismatch repair is compromised.26–28 Additional details are provided in Supplementary Appendix 1.
GENOMIC AND BIOINFORMATIC ANALYSES
Primary tumor samples and matched normal peripheral-blood specimens were obtained from a subgroup of patients with mismatch repair–deficient carcinomas and a subgroup with mismatch repair–proficient carcinomas, for whom sufficient tumor tissue was available for exome sequencing30 and HLA haplotyping. To assess the potential for mutant peptide binding, somatic exome data combined with each individual patient’s major histocompatibility complex (MHC) class I HLA haplotype were applied to an epitope prediction algorithm.31,32 This algorithm provided an estimate of the total number of mutation-associated neoantigens in each tumor. Additional details are provided in Supplementary Appendix 1.
STATISTICAL ANALYSIS
The primary end points for cohorts A and B were the immune-related objective response rate and the 20-week immune-related progression-free survival rate, assessed with the use of immune-related response criteria.33 The primary end point for cohort C was the immune-related progression-free survival rate at 20 weeks (Fig. S1 in Supplementary Appendix 1). Immune-related criteria (i.e., one of the types of criteria used to evaluate immune-based therapies) are based on radiographic responses, and unlike Response Evaluation Criteria in Solid Tumors (RECIST), they capture newly developed lesions detected on radiography in the measurement of tumor burden; these criteria are defined and compared with RECIST, version 1.1, in Table S1 in Supplementary Appendix 1. The response rate and 20-week progression-free survival rate were evaluated and reported in this study with the use of both RECIST, version 1.1, and immune-related response criteria. Progression-free survival and overall survival were summarized by means of the Kaplan–Meier method. Details of the hypothesis, the decision rules for the rejection of the null hypotheses, decision rules for early discontinuation of the study in a cohort because of efficacy or futility, and statistical methods are provided in Supplementary Appendix 1.
RESULTS
PATIENTS
A total of 41 consecutive patients were enrolled in the study and treated during the period from September 2013 through January 2015 (Table 1). Recruitment included patients in pursuit of a clinical trial option who were known to have tumors with mismatch-repair defects or who had tumors of unknown status who were then tested. One patient in the cohort with mismatch repair–deficient colorectal cancer was enrolled under an institutional review board eligibility waiver allowing a grade 3 bilirubin level (i.e., higher than the cutoff specified in the inclusion criteria). A total of 32 patients with colorectal cancer were enrolled in cohorts A and B. All patients with colorectal cancer had received two or more previous chemotherapy regimens (a median of four regimens), except for 1 patient with mismatch repair–proficient cancer who had received one chemotherapeutic and one (non–PD-1–based) immunotherapeutic regimen.
Nine patients with mismatch repair–deficient solid tumors other than colorectal cancer were enrolled in cohort C. All patients in cohort C had received one or more previous therapeutic regimens (a median of two regimens).
PRIMARY END POINT
The immune-related objective response rate in cohort A was 40% (4 of 10 patients; 95% confidence interval [CI], 12 to 74), and the immune-related progression-free survival rate at 20 weeks was 78% (7 of 9 patients; 95% CI, 40 to 97) (Table S2 in Supplementary Appendix 1); the corresponding rates in cohort C were 71% (5 of 7 patients; 95% CI, 29 to 96) and 67% (4 of 6 patients; 95% CI, 22 to 96). In cohort B, which included patients with mismatch repair–proficient colorectal cancers, the immune-related objective response rate was 0% (95% CI, 0 to 20), and the immune-related progression-free survival rate at 20 weeks was 11% (2 of 18 patients; 95% CI, 1 to 35). Both cohorts with mismatch repair–deficient cancers (cohorts A and C) reached the prespecified point at which the protocol indicated that the study reached its primary efficacy end point when 4 patients were free from disease progression at 20 weeks and objective responses on the basis of immune-related response criteria were observed in 4 patients (Table S2 and the Methods section in Supplementary Appendix 1).
The median follow-up was 36 weeks (range, 5 to 55) for patients with mismatch repair–deficient colorectal cancer (cohort A), 20 weeks (range, 4 to 52) for patients with mismatch repair–proficient colorectal cancer (cohort B), and 21 weeks (range, 0.1 to 49) for patients with mismatch repair–deficient noncolorectal cancer (cohort C). All patients for whom the 20-week immune-related progression-free survival rate could be evaluated were followed for at least 20 weeks.
RADIOGRAPHIC EVALUATION
Of the 10 patients with mismatch repair–deficient colorectal cancer (cohort A) who could be evaluated for RECIST, 4 (40%; 95% CI, 12 to 74) had objective responses according to these criteria (Table 2 and Fig. 1, and Fig. S2 in Supplementary Appendix 1). Patients were considered not to have been evaluated unless they underwent a radiographic scan at 12 weeks. The rate of disease control, which was defined as the percentage of patients who had an objective response or whose disease was stable, was 90% in cohort A (9 of 10 patients; 95% CI, 55 to 100). Of the 7 patients in cohort C who could be evaluated, 5 (71%; 95% CI, 29 to 96) had objective responses as defined by RECIST (Table 2 and Fig. 1, and Fig. S2 in Supplementary Appendix 1), and the rate of disease control was 71% (5 of 7 patients; 95% CI, 29 to 96).
Table 2.
Objective Responses According to RECIST Criteria.
| Type of Response | Mismatch Repair–Deficient Colorectal Cancer(N = 10) | Mismatch Repair–Proficient Colorectal Cancer(N = 18) | Mismatch Repair–Deficient Noncolorectal Cancer(N = 7) |
|---|---|---|---|
| Complete response — no. (%) | 0 | 0 | 1 (14)* |
| Partial response — no. (%) | 4 (40) | 0 | 4 (57)† |
| Stable disease at week 12 — no. (%) | 5 (50) | 2 (11) | 0 |
| Progressive disease — no. (%) | 1 (10) | 11 (61) | 2 (29) |
| Could not be evaluated — no. (%)‡ | 0 | 5 (28) | 0 |
| Objective response rate (95% CI) — % | 40 (12–74) | 0 (0–19) | 71 (29–96) |
| Disease control rate (95% CI) — %§ | 90 (55–100) | 11 (1–35) | 71 (29–96) |
| Median duration of response — wk | Not reached | NA¶ | Not reached |
| Median time to response (range) — wk | 28 (13–35) | NA¶ | 12 (10–13) |
Figure 1. Clinical Responses to Pembrolizumab Treatment.
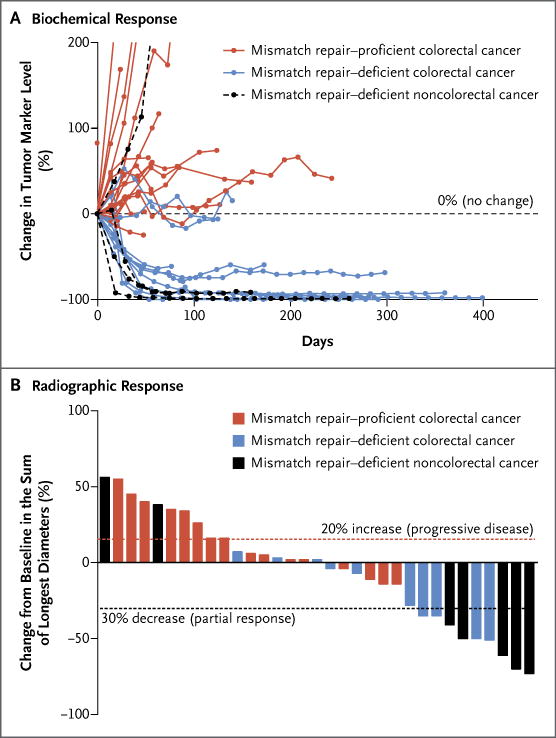
The biochemical responses to pembrolizumab treatment are shown in Panel A. Serum levels of protein biomarkers were measured at the start of each treatment cycle, and the values represent percentage changes from baseline. Each line represents one patient; patients were included if their baseline tumor marker values were higher than the upper limit of normal. CA-125 was used as the biomarker for one patient with endometrial cancer, CA19-9 was used for one patient with cholangiocarcinoma and one patient with ampullary cancer, and carcinoembryonic antigen (CEA) was used for all other patients. Radiographic responses to treatment with pembrolizumab, evaluated on the basis of Response Evaluation Criteria in Solid Tumors (RECIST), are shown in Panel B. Tumor responses were measured at regular intervals, and the values shown are the largest percentage change in the sum of longest diameters from the baseline measurements of each measurable tumor. Each bar represents one patient.
Patients in cohort C had faster responses than did patients in cohort A (median time to response according to RECIST, 12 weeks vs. 28 weeks; P = 0.03). Furthermore, all 6 patients (100%) with mismatch repair–deficient tumors that were not associated with the Lynch syndrome had an objective response, whereas only 3 of 11 patients (27%) with tumors associated with the Lynch syndrome had a response (Table S3 in Supplementary Appendix 2) (P = 0.009). No other baseline characteristics had a significant association with objective responses.
Among the 18 patients with mismatch repair–proficient colorectal cancers in cohort B, no objective responses as defined by RECIST were observed (Table 2 and Fig. 1, and Fig. S2 in Supplementary Appendix 1). In this group, the rate of disease control was 11% (2 of 18 patients; 95% CI, 1 to 35).
All the patients who had a response as defined by RECIST (Table 2) also had a response according to immune-related response criteria (Table S2 in Supplementary Appendix 1).
SURVIVAL
In the cohort of patients with mismatch repair–deficient colorectal cancer (cohort A), the median progression-free survival and median overall survival were not reached (Fig. 2). In contrast, among the patients with mismatch repair–proficient cancers (cohort B), the median progression-free survival was only 2.2 months (95% CI, 1.4 to 2.8), and the median overall survival was 5.0 months (95% CI, 3.0 to not estimable). In cohort C (patients with mismatch repair–deficient noncolorectal cancer), the median progression-free survival was 5.4 months (95% CI, 3 to not estimable), and the median overall survival was not reached. A post hoc comparison of the cohorts with mismatch repair–deficient and mismatch repair–proficient colorectal cancers showed hazard ratios for disease progression or death (0.10; 95% CI, 0.03 to 0.37; P<0.001) and for death (0.22; 95% CI, 0.05 to 1.00; P = 0.05) that favored patients with mismatch repair–deficient colorectal cancer (Fig. 2).
Figure 2. Clinical Benefit of Pembrolizumab Treatment According to Mismatch-Repair Status.
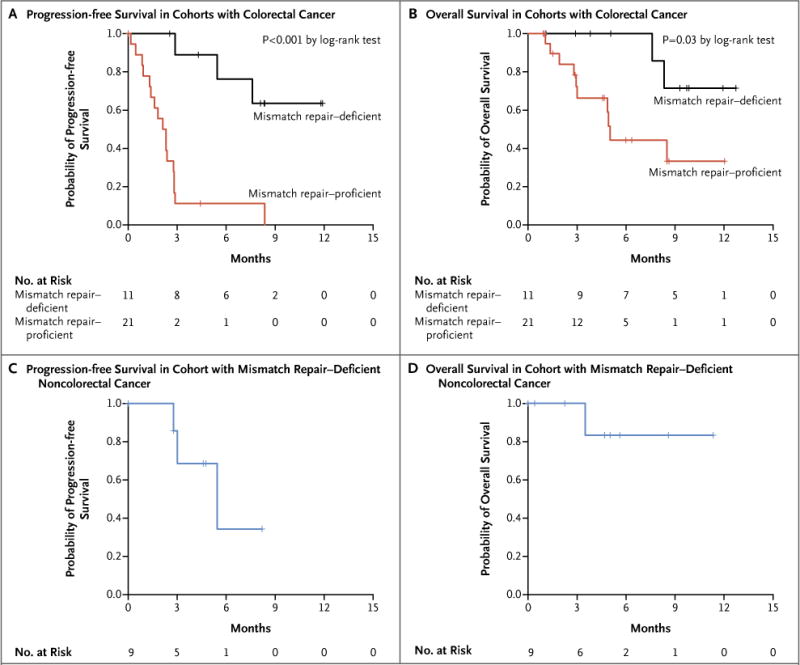
Kaplan–Meier curves are shown for progression-free survival in the cohorts with colorectal cancer (Panel A), overall survival in the cohorts with colorectal cancer (Panel B), progression-free survival among patients with mismatch repair–deficient noncolorectal cancers (Panel C), and overall survival among patients with mismatch repair–deficient noncolorectal cancers (Panel D). In both cohorts with mismatch repair–deficient tumors, median overall survival was not reached. Patients in the cohort with mismatch repair–proficient cancers had a median progression-free survival of 2.2 months (95% CI, 1.4 to 2.8) and a median overall survival of 5.0 months (95% CI, 3.0 to not estimable). Patients with mismatch repair–deficient noncolorectal cancers had a median progression-free survival of 5.4 months (95% CI, 3 to not estimable).
To evaluate whether the difference in survival might be due to prognostic differences, we measured the time since the diagnosis of metastatic disease and the clinical performance of the regimen that patients had received before enrollment. We found that there was no significant difference between patients with mismatch repair–deficient colorectal cancer and patients with mismatch repair–proficient colorectal cancer with respect to the duration of metastatic disease (P = 0.77 by the log-rank test) or the median progression-free survival while receiving their previous regimens (P = 0.60 by the log-rank test) (Fig. S3 in Supplementary Appendix 1). We also performed an additional multivariate analysis of progression-free and overall survival to examine the difference in outcomes between mismatch repair–deficient colorectal cancer and mismatch repair–proficient colorectal cancer, adjusting for elapsed time since the initial diagnosis. The magnitude of the hazard ratios for disease progression or death (hazard ratio, 0.04; 95% CI 0.01 to 0.21; P<0.001) and for death (hazard ratio, 0.18; 95% CI, 0.03 to 1.01; P = 0.05), representing the differing effects of pembrolizumab between mismatch repair–deficient tumors and mismatch repair–proficient tumors, was maintained after adjustment for this potential difference.
SAFETY ASSESSMENT
Adverse events occurring in more than 5% of patients are listed in Table 3. Events of clinical interest included rash or pruritus (24%); thyroiditis, hypothyroidism, or hypophysitis (10%); and asymptomatic pancreatitis (15%). Although the numbers were small, thyroid-function abnormalities were limited to the cohorts with mismatch repair–deficient cancer (Table 3).
Table 3.
Adverse Events.*
| Event | All Grades | Grade 3 or 4 |
|---|---|---|
| no. of patients (%) | ||
| Any | 40 (98) | 17 (41) |
| Blood or lymphatic | ||
| Anemia | 8 (20) | 7 (17) |
| Lymphopenia | 8 (20) | 8 (20) |
| Sinus tachycardia | 4 (10) | 0 |
| Dermatologic | ||
| Dry skin | 5 (12) | 0 |
| Rash or pruritus | 10 (24) | 0 |
| Thyroiditis, hypothyroidism, or hypophysitis | 4 (10) | 0 |
| Gastrointestinal | ||
| Abdominal pain | 10 (24) | 0 |
| Anorexia | 4 (10) | 0 |
| Constipation | 8 (20) | 0 |
| Diarrhea | 10 (24) | 2 (5) |
| Dry mouth | 5 (12) | 0 |
| Nausea | 5 (12) | 0 |
| Bowel obstruction | 3 (7) | 3 (7) |
| Hepatobiliary | ||
| Elevated alanine aminotransferase | 3 (7) | 2 (5) |
| Pancreatitis† | 6 (15) | 0 |
| Metabolism and nutrition | ||
| Hypoalbuminemia | 4 (10) | 4 (10) |
| Hyponatremia | 3 (7) | 3 (7) |
| Musculoskeletal | ||
| Arthralgia | 7 (17) | 0 |
| Myalgia | 6 (15) | 0 |
| Nervous system | ||
| Dizziness | 4 (10) | 0 |
| Headache | 7 (17) | 0 |
| Insomnia | 3 (7) | 0 |
| Respiratory‡ | ||
| Allergic rhinitis | 12 (29) | 0 |
| Cough | 4 (10) | 0 |
| Dyspnea | 6 (15) | 0 |
| Upper respiratory infection | 3 (7) | 0 |
| Cold intolerance | 6 (15) | 0 |
| Edema | 4 (10) | 0 |
| Fatigue | 13 (32) | 0 |
| Fever | 5 (12) | 0 |
| Pain | 14 (34) | 0 |
TUMOR MARKERS
In the two cohorts with colorectal cancer, it was possible to evaluate levels of carcinoembryonic antigen (CEA) before enrollment; in 29 of 32 patients, these levels were above the upper limit of normal (3 mg per deciliter). Substantial decreases in CEA level occurred in 7 of the 10 patients with mismatch repair–deficient colorectal cancer and in none of the 19 patients with mismatch repair–proficient colorectal cancer in whom CEA could be evaluated (Fig. 1, and Fig. S4 in Supplementary Appendix 1). Among patients with mismatch repair–deficient noncolorectal cancer, levels of tumor markers (CEA, CA19-9, or CA-125) were elevated above the upper limit of normal in 4 patients. Declines in CA19-9 or CA-125 of more than 70% occurred in 3 of these 4 patients. Tumor marker kinetics in all three cohorts are shown in Figure 1. The degree of CEA decline after one dose (between day 14 and day 28) of pembrolizumab was predictive of both progression-free survival (P = 0.01) and overall survival (P = 0.02). The CEA response occurred well in advance of radiographic confirmation of disease control (range, 10 to 35 weeks). In contrast, patients who had disease progression had rapid biomarker elevation within 30 days after the initiation of therapy. Thus, changes in CEA levels significantly preceded and correlated with ultimate radiographic changes.
GENOMIC ANALYSIS
The analysis of whole-exome sequences showed a mean of 1782 somatic mutations per tumor in patients with mismatch repair–deficient cancer (nine patients), as compared with 73 mutations per tumor in patients with mismatch repair–proficient cancer (six patients) (P = 0.007 by nonparametric Wilcoxon test) (Fig. S5 in Supplementary Appendix 1 and Table S3 in Supplementary Appendix 2). Most of these mutations (63%) are predicted to alter amino acids.
These mutations were then assessed for their immunogenic potential in the context of each patient’s MHC haplotype. We identified a mean of 578 potential mutation-associated neoantigens from the tumors of patients with mismatch repair–deficient cancers; 21 such neoantigens were identified in tumors from patients with mismatch repair–proficient cancers (Table S3 in Supplementary Appendix 2). The percentage of potential mutation-associated neoantigens among all somatic mutations was similar in the two cohorts (a mean of 32% in patients with mismatch repair–deficient cancer and 29% in patients with mismatch repair–proficient cancer). High numbers of somatic mutations and potential mutation-associated neoantigens were associated with longer progression-free survival and with a trend toward objective response (Fig. S5 and Table S4 in Supplementary Appendix 1).
IMMUNOHISTOCHEMICAL ANALYSIS
The expression of CD8 and PD-L1 was evaluated within the tumor and at the invasive fronts of the tumor in an immunohistochemical analysis in the 30 cases in which tumor tissue was available (Fig. S6 in Supplementary Appendix 1). Tumors from patients in cohorts A and C contained a greater density of CD8-positive lymphoid cells than did tumors from patients in cohort B (P = 0.10) (Fig. S7 in Supplementary Appendix 1), and CD8 labeling was associated with a trend toward objective response and stable disease (Fig. S8 and Table S5 in Supplementary Appendix 1). This CD8-positive lymphoid infiltrate was especially prominent at the invasive fronts of the tumors (P = 0.04) (Fig. S7 in Supplementary Appendix 1). Membranous PD-L1 expression occurred only in patients with mismatch repair–deficient cancer and was prominent on tumor-infiltrating lymphocytes and tumor-associated macrophages located at the invasive fronts of the tumor (P = 0.04) (Fig. S7 in Supplementary Appendix 1). The expression of CD8 and PD-L1 was not significantly associated with progression-free survival or overall survival (Table S5 in Supplementary Appendix 1).
DISCUSSION
The data from this small phase 2 trial of pembrolizumab for the treatment of tumors with and tumors without mismatch-repair deficiency support the hypothesis that mismatch repair–deficient tumors are more responsive to PD-1 blockade than are mismatch repair–proficient tumors. Mismatch-repair deficiency occurs in many cancers, including those of the colorectum, uterus, stomach, biliary tract, pancreas, ovary, prostate, and small intestine.18,34–42 It is possible that patients with mismatch repair–deficient tumors of these types may also benefit from anti–PD-1 therapy, as may patients whose tumors contain other DNA repair deficiencies, such as those with mutations in POLD, POLE, or MYH.18,43,44
The hypothesis that mismatch repair–deficient tumors stimulate the immune system is not a new idea45; it has been supported by observations of the dense immune infiltration and Th1-associated cytokine-rich environment in mismatch repair–deficient tumors.19–22,46 A recent study refined these classic observations by showing that the mismatch repair–deficient tumor microenvironment strongly expressed several immune checkpoint ligands, including PD-1, PD-L1, CTLA-4, LAG-3, and IDO, which indicates that their active immune microenvironment is counterbalanced by immune inhibitory signals that resist tumor elimination.47 The most likely explanation for both the old and new findings was that the immune infiltrate associated with mismatch repair–deficient carcinomas was directed at neoantigens. The correlation of a higher mutational load and a higher rate of response to anti–CTLA-4 in melanoma41 and anti–PD-1 in lung cancer48 provides further support for the idea that mutation-associated neoantigen recognition is an important component of the endogenous antitumor immune response.
On the basis of the results of the current and previous studies, we suggest that the greatly increased number of mutation-associated neoantigens resulting from mismatch-repair deficiency (more than 20 times higher than in tumors without this deficiency) (Table S4 in Supplementary Appendix 1) is the basis for the enhanced anti–PD-1 responsiveness of this genetically defined subset of cancers. Although our estimates of the number of mutation-associated neoantigens in tumors are based only on in silico predictions of binding affinity, this suggestion is consistent with the observation that mismatch repair–proficient tumors have far less infiltration of lymphocytes than do mismatch repair–deficient tumors (Fig. S6 and S7 and Table S5 in Supplementary Appendix 1). Recent studies49,50 have shown that only a tiny proportion of predicted neo-epitopes are actually presented on the cell surface with MHC and are targets of endogenous T cell responses. It seems likely, however, that the number of predicted mutation-associated neoantigens is proportionate to the number of actual mutation-associated neoantigens, and tumors with a high number of actual mutation-associated neoantigens are more likely to stimulate the immune system to react against the tumor.
Alternative mechanisms underlying the difference in anti–PD-1 responsiveness between mismatch repair–deficient tumors and mismatch repair–proficient tumors should also be considered. For example, different signaling pathways activated in the two types of tumors may result in differences in the secretion of soluble factors that could result in differential activation of the PD-1 pathway within the tumor microenvironment.26–28 Genetic differences could affect epigenetic differences that alter the expression of tumor-associated self-antigens; in turn, these could alter the antigenicity of the tumor. Experimental analyses of antigen-specific immune responses and of changes in immune microenvironments should help to define the relative contribution of these factors to the striking responsiveness of mismatch repair–deficient tumors to PD-1 antibodies.
Several other notable observations were made during the course of this study. First, changes in serum levels of protein biomarkers, such as CEA, corresponded with clinical benefit after a single dose of therapy. Declines in CEA levels preceded objective radiographic evidence of treatment benefit by several months; perhaps other biomarkers, such as circulating tumor DNA, would also be beneficial as surrogate markers of early response.51–53 Second, our results suggest that the evaluation of tumor genomes can help guide immunotherapy. They support the view that the number and type of alterations may prove to be valuable for judging the potential usefulness of immune checkpoint inhibitors, even in mismatch repair–proficient cancers.41,48,54 Most importantly, our results show an approach for the treatment of a specific class of tumors that is based solely on genetic status — that is, without regard to the underlying tumor type.
Supplementary Material
Supplemental Information
Acknowledgments
Supported by Swim Across America Laboratory at Johns Hopkins University, the Banyan Gate Foundation, the Lustgarten Foundation for Pancreatic Cancer Research, the Commonwealth Fund, the Virginia and D.K. Ludwig Fund for Cancer Research, the Sol Goldman Pancreatic Cancer Research Center, a Gastrointestinal Specialized Programs of Research Excellence (SPORE) grant (P50CA062924), and grants (P30CA006973, CA163672, CA43460, CA67941, CA16058, and CA57345) from the National Institutes of Health.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
We thank Kathy Helwig and Mary Savage for administrative support; Cheryl Blair, Katherine Judge, Tierra Brown, and Suping Chen for technical assistance; and Eric H. Rubin and Alise S. Reicin for review of an earlier version of the manuscript.
APPENDIX
The authors’ full names and academic degrees are as follows: Dung T. Le, M.D., Jennifer N. Uram, Ph.D., Hao Wang, Ph.D., Bjarne R. Bartlett, B.S., Holly Kemberling, R.N., Aleksandra D. Eyring, M.Pharm., Andrew D. Skora, Ph.D., Brandon S. Luber, Sc.M., Nilofer S. Azad, M.D., Dan Laheru, M.D., Barbara Biedrzycki, Ph.D., C.N.R.P., Ross C. Donehower, M.D., Atif Zaheer, M.D., George A. Fisher, M.D., Ph.D., Todd S. Crocenzi, M.D., James J. Lee, M.D., Ph.D., Steven M. Duffy, M.D., Richard M. Goldberg, M.D., Albert de la Chapelle, M.D., Ph.D., Minori Koshiji, M.D., Ph.D., Feriyl Bhaijee, M.D., Thomas Huebner, M.D., Ralph H. Hruban, M.D., Laura D. Wood, M.D., Ph.D., Nathan Cuka, M.D., Drew M. Pardoll, M.D., Ph.D., Nickolas Papadopoulos, Ph.D., Kenneth W. Kinzler, Ph.D., Shibin Zhou, M.D., Ph.D., Toby C. Cornish, M.D., Ph.D., Janis M. Taube, M.D., Robert A. Anders, M.D., Ph.D., James R. Eshleman, M.D., Ph.D., Bert Vogelstein, M.D., and Luis A. Diaz, Jr., M.D.
The authors’ affiliations are as follows: the Swim Across America Laboratory (D.T.L., J.N.U., B.R.B., L.A.D.), Sidney Kimmel Comprehensive Cancer Center (D.T.L., J.N.U., H.W., H.K., A.D.E., A.D.S., B.S.L., N.S.A., D.L., B.B., R.C.D., D.M.P., N.P., K.W.K., S.Z., B.V., L.A.D.), Ludwig Center and Howard Hughes Medical Institute (B.R.B., A.D.S., N.P., K.W.K., S.Z., B.V., L.A.D.), and the Departments of Radiology (A.Z.) and Pathology (F.B., T.H., R.H.H., L.D.W., N.C., T.C.C., J.M.T., R.A.A., J.R.E.), Johns Hopkins University School of Medicine, Baltimore; Department of Medicine, Stanford University School of Medicine, Stanford, CA (G.A.F.); Providence Cancer Center at Providence Health and Services, Portland, OR (T.S.C.); Department of Medicine, University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, Pittsburgh (J.J.L.); Bon Secours Cancer Institute, Richmond, VA (S.M.D.); Division of Medical Oncology, Ohio State University Comprehensive Cancer Center–James Cancer Center and Solove Research Institute, and Human Cancer Genetics Program, Ohio State University Comprehensive Cancer Center, Columbus (R.M.G., A.C.); and Merck, Kenilworth, NJ, and North Wales, PA (M.K.).
References
- 1.Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–22. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 2.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–47. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 3.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 4.Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti–PD-1) in melanoma. N Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powles T, Eder JP, Fine GD, et al. MP-DL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–62. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 8.Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–30. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20:5064–74. doi: 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–75. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koopman M, Kortman GA, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100:266–73. doi: 10.1038/sj.bjc.6604867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldstein J, Tran B, Ensor J, et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H) Ann Oncol. 2014;25:1032–8. doi: 10.1093/annonc/mdu100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segal NH, Parsons DW, Peggs KS, et al. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68:889–92. doi: 10.1158/0008-5472.CAN-07-3095. [DOI] [PubMed] [Google Scholar]
- 16.Timmermann B, Kerick M, Roehr C, et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One. 2010;5(12):e15661. doi: 10.1371/journal.pone.0015661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eshleman JR, Lang EZ, Bowerfind GK, et al. Increased mutation rate at the hprt locus accompanies microsatellite instability in colon cancer. Oncogene. 1995;10:33–7. [PubMed] [Google Scholar]
- 18.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolcetti R, Viel A, Doglioni C, et al. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am J Pathol. 1999;154:1805–13. doi: 10.1016/S0002-9440(10)65436-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexander J, Watanabe T, Wu TT, Rashid A, Li S, Hamilton SR. Histopathological identification of colon cancer with microsatellite instability. Am J Pathol. 2001;158:527–35. doi: 10.1016/S0002-9440(10)63994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001;91:2417–22. [PubMed] [Google Scholar]
- 22.Young J, Simms LA, Biden KG, et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. Am J Pathol. 2001;159:2107–16. doi: 10.1016/S0002-9440(10)63062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger MF, Hodis E, Heffernan TP, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–6. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee W, Jiang Z, Liu J, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–7. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 25.Lipson EJ, Sharfman WH, Drake CG, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19:462–8. doi: 10.1158/1078-0432.CCR-12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–87. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–32. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto H, Imai K, Perucho M. Gastrointestinal cancer of the microsatellite mutator phenotype pathway. J Gastroenterol. 2002;37:153–63. doi: 10.1007/s005350200015. [DOI] [PubMed] [Google Scholar]
- 29.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95:6870–5. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7:283ra53. doi: 10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate Web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008;36:W509–W512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008;24:1397–8. doi: 10.1093/bioinformatics/btn128. [DOI] [PubMed] [Google Scholar]
- 33.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 34.Maple JT, Smyrk TC, Boardman LA, Johnson RA, Thibodeau SN, Chari ST. Defective DNA mismatch repair in long-term (> or =3 years) survivors with pancreatic cancer. Pancreatology. 2005;5:220–7. doi: 10.1159/000085275. [DOI] [PubMed] [Google Scholar]
- 35.Meltzer SJ, Yin J, Manin B, et al. Microsatellite instability occurs frequently and in both diploid and aneuploid cell populations of Barrett’s-associated esophageal adenocarcinomas. Cancer Res. 1994;54:3379–82. [PubMed] [Google Scholar]
- 36.Nakata B, Wang YQ, Yashiro M, et al. Prognostic value of microsatellite instability in resectable pancreatic cancer. Clin Cancer Res. 2002;8:2536–40. [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agaram NP, Shia J, Tang LH, Klimstra DS. DNA mismatch repair deficiency in ampullary carcinoma: a morphologic and immunohistochemical study of 54 cases. Am J Clin Pathol. 2010;133:772–80. doi: 10.1309/AJCPGDDE8PLLDRCC. [DOI] [PubMed] [Google Scholar]
- 39.Cancer Genome Atlas Research Network. Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garg K, Leitao MM, Jr, Kauff ND, et al. Selection of endometrial carcinomas for DNA mismatch repair protein immunohistochemistry using patient age and tumor morphology enhances detection of mismatch repair abnormalities. Am J Surg Pathol. 2009;33:925–33. doi: 10.1097/PAS.0b013e318197a046. [DOI] [PubMed] [Google Scholar]
- 41.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams AS, Huang WY. The analysis of microsatellite instability in extracolonic gastrointestinal malignancy. Pathology. 2013;45:540–52. doi: 10.1097/PAT.0b013e3283653307. [DOI] [PubMed] [Google Scholar]
- 43.Jones S, Emmerson P, Maynard J, et al. Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C→T:A mutations. Hum Mol Genet. 2002;11:2961–7. doi: 10.1093/hmg/11.23.2961. [DOI] [PubMed] [Google Scholar]
- 44.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–44. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bodmer W, Bishop T, Karran P. Genetic steps in colorectal cancer. Nat Genet. 1994;6:217–9. doi: 10.1038/ng0394-217. [DOI] [PubMed] [Google Scholar]
- 46.Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994;145:148–56. [PMC free article] [PubMed] [Google Scholar]
- 47.Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology: mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–81. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Linnemann C, van Buuren MM, Bies L, et al. High-throughput epitope discovery reveals frequent recognition of neoantigens by CD4+ T cells in human melanoma. Nat Med. 2015;21:81–5. doi: 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- 51.Diehl F, Schmidt K, Choti MA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–86. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yadav M, Jhunjhunwala S, Phung QT, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572–6. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information