MECP2 Duplication Syndrome: Evidence of Enhanced Oxidative Stress. A Comparison with Rett Syndrome (original) (raw)
Abstract
Rett syndrome (RTT) and MECP2 duplication syndrome (MDS) are neurodevelopmental disorders caused by alterations in the methyl-CpG binding protein 2 (MECP2) gene expression. A relationship between MECP2 loss-of-function mutations and oxidative stress has been previously documented in RTT patients and murine models. To date, no data on oxidative stress have been reported for the MECP2 gain-of-function mutations in patients with MDS. In the present work, the pro-oxidant status and oxidative fatty acid damage in MDS was investigated (subjects n = 6) and compared to RTT (subjects n = 24) and healthy condition (subjects n = 12). Patients with MECP2 gain-of-function mutations showed increased oxidative stress marker levels (plasma non-protein bound iron, intraerythrocyte non-protein bound iron, F2-isoprostanes, and F4-neuroprostanes), as compared to healthy controls (P ≤ 0.05). Such increases were similar to those observed in RTT patients except for higher plasma F2-isoprostanes levels (P < 0.0196). Moreover, plasma levels of F2-isoprostanes were significantly correlated (P = 0.0098) with the size of the amplified region. The present work shows unique data in patients affected by MDS. For the first time MECP2 gain-of-function mutations are indicated to be linked to an oxidative damage and related clinical symptoms overlapping with those of MECP2 loss-of-function mutations. A finely tuned balance of MECP2 expression appears to be critical to oxidative stress homeostasis, thus shedding light on the relevance of the redox balance in the central nervous system integrity.
Introduction
Methylated-CpG binding protein 2 (MeCP2) is a nuclear protein encoded by the X-linked MECP2 gene (OMIM*300005). MeCP2 can be defined as a multifunctional protein, due to its involvement in chromatin architecture, regulation of RNA splicing, and its role both as transcriptional repressor or activator [1]. MECP2 appears to be universally expressed in all cell types with few exceptions, including microglia and rod photoreceptors [2]. Loss-of-function mutations in MECP2 is the main cause of Rett syndrome (RTT), which is a neurodevelopmental disease with severe cognitive impairment occurring at a ratio of approximately 1:10,000 girls [3,4].
However, RTT is not the only known pathological condition related to MECP2 mutations, as a wide series of conditions, collectively termed as _MECP2_-related disorders [5], has been reported. These disorders include asymptomatic female carriers, boys with MECP2 mutations typically causing a RTT phenotype in girls, and rare individuals with mutations in MECP2 showing other neurodevelopmental disorders [5]. Interestingly, intellectual disability (ID) is a common feature between RTT as well as _MECP2_-related disorders. Gain-of-function mutations in relation to MECP2 also lead to a severe neurodevelopmental disorder, named MECP2 duplication syndrome (MDS) or MECP2 triplication syndrome [6–8]. The phenotypes include major cognitive and motor deficits, stunted motor development, early onset hypotonia, epilepsy, and progressive spasticity, clinical features which are overlapping with some of those seen in RTT [6,7].
Although the prevalence of MDS is unknown, it has been estimated that MDS could be responsible for 1 to 2% of all X-linked ID cases [9]. MDS is well documented in males, with 150 affected individuals reported in the literature, while it rarely occurs in females [9], as female carriers of MECP2/Xq28 duplications are almost always asymptomatic due to extremely skewed X-inactivation [9]. This is in contrast to females with classical RTT harboring a loss-of-function mutations of the MECP2 [10,11].
Interestingly, a genotype-phenotype correlation in relation to the size of the Xq28 duplicated region has emerged. The minimal region of duplication that is sufficient to cause the core MDS phenotype involves the MECP2 and IRAK1 [12–14]. Evaluating previously data and data from mouse models, Ramocki and colleagues posit that MECP2 is the primary dosage-sensitive gene responsible for the neurological phenotypes in the Xq28 duplications [7].
Overall, MeCP2 appears to play a key role in the brain as a regulator of synaptic and neuronal plasticity as well as an etiological role in the development of RTT and MDS [15].
Oxidative stress (OS) is a nonspecific pathological condition that has frequently been associated with neurological disorders, including several diseases linked to cognitive impairment [16]. Compelling evidence between MECP2 loss-of-function and aberrant redox homeostasis has been shown by our and other research groups [17,18]. Specifically, a cause-effect relationship between oxidative brain damage and Mecp2 loss-of-function has been reported by our group in several murine models of the disease [19].
Since both a critical role for MECP2 in central nervous system integrity [20], and an involvement of OS in brain synaptic plasticity have been reported [21–23], MDS can represent a clinical model for testing the hypothesis that a finely tuned balance of MECP2 expression is critical to the control of redox homeostasis. We hypothesize that overexpression of MECP2 would lead to a status of enhanced OS.
Materials and Methods
Subjects
The study included a total of 42 subjects. Six subjects carrying an Xq28 duplication/triplication (ranging from 0.426 to 3.9 Mb) all-encompassing MECP2 (female n = 1, males n = 5; mean age 10.7 ± 4.3 years, range: 2.5–14) were enrolled in the study. One patient (case #6) was admitted at the Danish Epilepsy Centre, Dianalund, Denmark, while five patients (cases #1–5) were followed at the Child Neuropsychiatry Unit, Azienda Ospedaliera Universitaria Senese, Siena Italy. For comparative purpose, “positive” controls (RTT patients with proven MECP2 loss-of-function mutations; n = 24; all females; mean age 10.1 ± 3.3 years, range 3–14) were also analyzed, as a part of cohort of patients on regular follow-up at the Child Neuropsychiatry Unit, University Hospital, Siena Italy. All the RTT patients were identified with a MECP2 mutation and clinically evaluated according to the revised diagnostic criteria by Neul et al. [24]. The patients were all diagnosed with the classical form of RTT [4]. In addition, “negative” controls were newly recruited (healthy subjects n = 12; males 4, females 8; mean age 10.3 ± 3.7 years, range 3–14). Age differences were not statistically significant (P = 0.927).
The work was carried out in accordance to the rules expressed in the Declaration of Helsinki, and written informed consent was obtained by the parents of the enrolled subjects. This study was approved by the competent institutions: Ethics Committee of Western Zealand, Denmark (for the patient admitted at the Danish Epilepsy Centre), and Ethics Committee of the Tuscan Region, Azienda Ospedaliera Universitaria Senese, Siena, Italy (for the subjects recruited at the Child Neuropsychiatry Unit).
Microarray-comparative genomic hybridisation (array-CGH) analysis
The Xq28 duplications were identified by Array-CGH analysis using Agilent oligonucleotide array kit 44B (Human Genome CGH Microarray Kit 44B; Agilent Technologies, Santa Clara, CA), with an average resolution of about 75 kb or by a sub-megabase resolution whole genome tiling path BAC array (http://www.molgen.mpg.de/~abt_rop/molecular_cytogenetics/). The duplications were confirmed by real-time quantitative polymerase chain reaction (qPCR) [25].
Blood sampling
Blood sampling was carried out after the overnight fast. Blood samples were collected in heparinized tubes, and centrifuged at 2,400Xg for 15 min at room temperature. The platelet poor plasma was saved, and the buffy coat was removed by aspiration. Erythrocytes were washed twice with physiological solution, suspended in Ringer solution, pH 7.4 as a 50% (vol/vol) suspension, and then used for the determination of intraerythrocyte non-protein bound iron (IE-NPBI). Plasma was used for free F2-isoprostanes (F2-IsoPs), F4-neuroprostanes (F4-NeuroPs), F2-dihomo-isoprostanes (F2-dihomo-IsoPs), and plasma non-protein bound iron (p-NPBI). For all isoprostane determinations, butylated hydroxytoluene (BHT) (90 μM) was added to plasma as an antioxidant.
Oxidative stress marker evaluations
The examined markers included non-protein bound iron (NPBI) (pro-oxidant factor), F2-IsoPs (the oxidized products from arachidonic acid and markers of OS/systemic lipoperoxidation), F4-NeuroPs (the oxidized products from docosahexaenoic acid and markers of neuronal membrane damage/brain gray matter), and F2-dihomo-IsoPs (the oxidized products from adrenic acid and markers of glia membrane damage/brain white matter) [17]. OS markers were measured as previously reported. Briefly, IE-NPBI and p-NPBI were determined as a desferrioxamine-iron complex by high-performance liquid chromatography [26], whereas F2-IsoPs, F4-NeuroPs, and F2-dihomo-IsoPs were determined by a gas chromatography/negative ion chemical ionization tandem mass spectrometry (GC/NICI-MS/MS) analysis [26–28].
Routine blood tests
Serum concentrations of total cholesterol, high density lipoproteins (HDL)-cholesterol, triglycerides, immunoglobulin class G, class A and class M (IgG, IgA, IgM), were performed by a fully automatic analyzer (Cobas 6000 System; Roche Diagnostics). Total cholesterol, HDL-cholesterol, and triglycerides were assayed by using an enzymatic methods [29–31]. Serum fibrinogen concentration was determined by using Thromborel Reagent on BCS XP coagulation analyzer (Siemens Healthcare) [32]. Erythrocyte Sedimentation Rate (ESR) was assayed by using an automated system [33]. Blood cells counts were assayed on Sysmex XT-2100 system [34].
Data analysis
All variables were tested for normal distribution (D’Agostino-Pearson test). Differences between groups were evaluated by either one-way analysis of variance (ANOVA), or the Kruskal–Wallis test, as appropriate. Associations between variables were tested using linear regression analyses (for continuous normally distributed data) or the Spearman rank correlation (for continuous non normally distributed variables). A two-tailed P < 0.05 was considered to indicate statistical significance. The MedCalc ver. 12.0 statistical software package (MedCalc. Software, Mariakerke, Belgium) was used for data analysis.
Results
All the enrolled patients showed a MECP2 duplication/triplication whose position and size was defined according to the GRCh37/hg19 human genome annotation (Fig 1 and Table 1).
Fig 1. Graphical view of the MECP2 duplications/triplication.

Graphical view of the MECP2 duplications/triplication was created with custom tracks in the UCSC genome browser (GRCh37/hg19), (Patient 1 was identified with a triplication). The involved regions are shown in blue and MECP2 is marked by a red circle.
Table 1. MECP2 duplication syndrome: clinical features and genetic details.
| Clinical features | Patient #1 | Patient #2 | Patient #3 | Patient #4 | Patient #5 | Patient #6 |
|---|---|---|---|---|---|---|
| Age (years) | 14 | 8 | 2.5 | 9 | 13 | 12 |
| Gender | Male | Female | Male | Male | Male | Male |
| Microcephaly | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) | No |
| Hand stereotypies | Yes (++) | Yes (+) | Yes (+) | Yes (++) | Yes (+) | No |
| Abnormal breathing | Yes (+) | Yes (+) | No | Yes (+) | Yes (+) | No |
| Bruxism | Yes (++) | Yes (+) | Yes (+) | Yes (++) | Yes (+) | No |
| Laryngomalacia | Yes | No | Yes | No | No | No |
| Sleep disturbances | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+++) |
| GERD, drooling | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) |
| Constipation | Yes (++) | Yes (++) | Yes (++) | Yes (+++) | Yes (++) | Yes (++) |
| Genital abnormalities | No | No | No | Yes (hypospadias) | No | No |
| Facial dysmorphism | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) | No |
| Facial hypotonia | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) |
| Dysphagia | Yes (+++) | Yes (++) | No | Yes (+++) | Yes (++) | Yes (+) |
| Intellectual Disability | Yes (+++) | Yes (+++) | Yes (+++) | Yes (+++) | Yes (+++) | Yes (+++) |
| Developmental regression | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) |
| Epilepsy | Yes (+++) | Yes (+++) | No | Yes (+++) | Yes (+++) | Yes (+++) |
| MRI abnormalities | Yes* | Yes*†¶‡ | Yes*¶‡** | Yes** | Yes* | Yes* |
| Hypotonia/ spasticity | Yes (+++) | Yes (+) | Yes (+) | Yes (+) | Yes (+++) | Yes |
| Feeding difficulties | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes (+) | Yes |
| Recurrent Infections | Yes§ | No | Yes§ | Yes§ | Yes§ | Yes |
| Endocrine abnormalities | No | Yes (hypothyroidism) | No | No | No | No |
| MECP2 duplication/triplication• position and size (GRCh37/hg19) | chrX:151198447–155190224• (~4Mb) | chrX:152370000–153410000 (1Mb) | chrX:153049224–153877929 (~828kb) | chrX: 153101077–153713921 (~613kb) | chrX: 153043806–154294453 (~1,2Mb) | chrX: 152982000–153408000 (~426kb) |
The typical features of the MDS, including developmental regression, epilepsy, magnetic resonance imaging abnormalities, hypotonia/spasticity were present in all the examined patients with the single exception of epilepsy, reported in five out of six subjects (Table 1).
All the MDS patients showed relevant differences in four out of five of the investigated OS markers, as compared to the healthy controls (Fig 2). In particular, significantly increased plasma levels of p-NPBI and IE-NPBI, forms of redox active iron, and both F2-IsoPs and F4-NeuroPs, non-enzymatic oxidized products from polyunsaturated fatty acids (i.e., arachidonic and docosahexaenoic acid, respectively), were detected. No significant differences were evidenced for F2-dihomo-IsoPs, biomarkers of adrenic acid oxidation. Both examined forms of iron (i.e., p-NPBI and IE-NPBI) are considered pro-oxidant agents [26], whereas the investigated isoprostanoids are indexes of lipid peroxidation (i.e., F2-IsoPs) [26], and brain gray (i.e., F4-NeuroPs) or white (i.e., F2-dihomo-IsoPs) matter oxidative damage [27, 28].
Fig 2. Oxidative stress marker plasma levels in MDS and RTT.

Levels of NPBI, plasma free F2-IsoPs, F4-NeuroPs, and F2-dihomo-IsoPs in MDS are compared with those of RTT and healthy control subjects. All the statistical significant differences were reported. Legend: ANOVA, analysis of variance; F2-dihomo-isoPs, F2-dihomo-isoprostanes; F2-IsoPs, F2-isoprostanes; F4-NeuroPs, F4-neuroprostanes; IE-NPBI, intraerythrocyte non protein bound iron; MDS, MECP2 Duplication Syndrome; p-NPBI, plasma non protein bound; RTT, Rett syndrome.
A comparison of the OS status in MDS patients and RTT patients vs the healthy control population evidenced similarities, with the exceptions of significantly higher plasma levels of F2-IsoPs and lower plasma levels of F2-dihomo-IsoPs in MDS. Indeed, the levels of p-NPBI, IE-NPBI, and F4-NeuroPs in MDS were found to be comparable to those observed in RTT (Fig 2). Moreover, the levels of all the examined plasma and intraerythrocyte biomarkers were significantly higher in RTT as compared to the control group (Fig 2).
A significant positive correlation between the Xq28 region duplication/triplication size and plasma levels of F2-IsoPs (r = 0.9181; 95% C.I.: 0.4181 to 0.9912; P = 0.0098) was evidenced (Fig 3). Additionally, a correlation trend between the Xq28 region duplication size and the erythrocyte sedimentation rate (ESR) (r = 0.732; P = 0.1598) was observed. On the other hand, no significant correlations were detected between OS markers and age, thus reinforcing the hypothesis of a link between the MECP2 gain-of-function mutations and OS derangement.
Fig 3. Relationship between plasma F2-IsoPs and Xq28 size (univariate regression analysis).
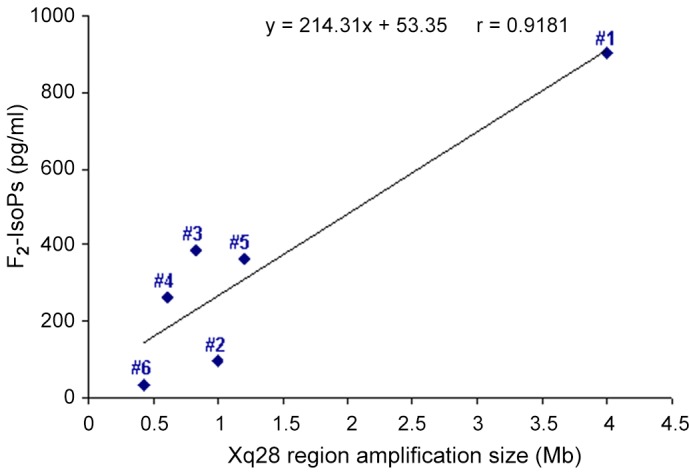
A positive linear relationship of plasma F2-IsoPs vs. Xq28 duplication/triplication size is showed. The strength of the relationship is indicated by the correlation coefficient (r = 0.9181, P = 0.0098). The linear regression equation was reported.
Significant differences were observed for total cholesterol, ESR, and ALC (post-hoc comparison: RTT > MDS ≈ Control Subjects; MDS ≈ RTT > Control Subjects; MDS ≈ RTT > Control Subjects, respectively) (Table 2). In contrast, no significant differences were reported for all the other parameters of the typical clinical-chemical pattern (Table 2). No significant differences were present for immunoglobulin class G and class M, high density lipoproteins-cholesterol, white blood cells, absolute counts for neutrophils, eosinophils, or basophils. (data not shown).
Table 2. Routine chemistry biomarkers in patients with MECP2 duplication syndrome, Rett syndrome, and control subjects.
| MECP2 duplication syndrome (n = 5)* | Rett syndrome (n = 24) | Control subjects (n = 12) | ANOVA P value | |
|---|---|---|---|---|
| Total cholesterol (mg/dl) | 141.8 ± 39.80 | 172.6 ± 27.3 | 144.6 ± 16.0 | 0.007 |
| ESR (mm/h) | 30.4 ± 21.43 | 32.68 ± 15.58 | 9.16 ± 5.85 | <0.001 |
| ALC (cells x103/mm3) | 3.32 ± 2.04 | 3.8 ± 0.85 | 2.68 ± 0.62 | 0.019 |
| AMC (cells x103/mm3) | 0.394 ± 0.21 | 0.737 ± 0.37 | 0.55 ± 0.15 | 0.052 |
| Fibrinogen (mg/dl) | 332.2 ± 90.8 | 404.2 ± 92.1 | 334.7 ± 60.78 | 0.055 |
| Triglycerides (mg/dl) | 89.2 ± 27.29 | 66.5 ± 15.26 | 67.8 ± 28.47 | 0.111 |
| IgA (mg/dl) | 201.0 ± 157 | 117.2 ± 33.4 | 140.9 ± 34.1 | 0.101 |
Discussion
Enhanced OS has been claimed to be involved in several pathological processes [35], including RTT, a rare genetic disease in which a deficiency of MeCP2 is demonstrated [36, 37]. In particular, we have previously shown that OS in brains of mice with _MECP_2 loss-of-function mutations takes place and can be rescued, along with neurological signs, by Mecp2 brain specific gene reactivation [19]. However, to date, no hints on the opposite being true exist, i.e., no evidence is available for supporting the paradoxical concept that “too much” MeCP2 could lead to similar biochemical events as observed in conditions of “too little” MeCP2.
In the present study, we demonstrate, for the first time, the occurrence of a redox imbalance in patients with MECP2 overexpression. Moreover, our data, while adding new evidence with regard to the _MECP2_-OS link, suggest that a fine tuning of the MECP2 dosage may play a key role in regulating redox homeostasis in humans.
Whether mitochondria would be the main, or the only major, intracellular sources for the abnormal redox status in MDS and RTT is still to be ascertained [19, 38–41]. To this regard, an ultrastructural analysis of primary cultures of skin fibroblasts from RTT patients has shown no major morphological changes in the mitochondria [42]. At the same time, to the best of our knowledge, no studies on mitochondrial function in MDS have been published to date.
Oxidized products from polyunsaturated fatty acids appear to be promising molecules to be investigated in MDS, as they could mirror an ongoing oxidative brain damage. To this regard, in RTT mouse models, the presence of isoprostanes was evidenced in brain concomitant to elevated isoprostane plasma levels [19]. The reason for the strongly increased F2-IsoPs plasma levels in patients presenting MECP2 gain-of-function mutations remains to be elucidated. Nevertheless, the evidenced dose-effect relationship between Xq28 duplication size and F2-IsoPs production further supports the existence of a close link between MECP2 and redox homeostasis control. A close link between MECP2 gene expression and F2-IsoPs formation has been reported in RTT where the MECP2 mutations associated to more severe phenotypes exhibited higher F2-IsoPs plasma levels [17].
Interestingly, increased F4-NeuroP plasma levels in both MDS and RTT (Fig 2), suggest the involvement of the gray matter oxidative damage in both conditions. Currently, F4-NeuroPs are investigated as potential biomarker for neurological disease. In human RTT, F4-NeuroPs has been related to neurological symptoms severity, mutation type and clinical presentation [27], whereas their levels were significantly elevated in the brain of RTT symptomatic null mice [19]. The relevance of F4-NeuroPs in the neurological disease is also reported for the pathogenesis of Alzheimer’s disease [43, 44].
Gain-of-function and loss-of-function MECP2 mutations were found to generate distinct F2-dihomo-IsoPs patterns, oxidized fatty acid products considered as biomarkers of free radical damage to myelin [28, 45]. Although a reduced volume of the brain white matter has been recently reported in patients affected by Xq28 duplication involving the MECP2 gene [46], our data might suggest a differential involvement of the brain white matter damage for RTT and MDS, on the base of the recovered F2-dihomo-IsoPs plasma levels.
This peculiar OS markers pattern further reinforces the specificity of the _MECP2_-OS link, which, far from being an epiphenomenon, appears to be intimately related to the specific _MECP2_-related disorders [17]. For some of those molecules known to be implicated in the general regulation of the redox homeostasis, an epigenetic modulation by MeCP2 has already been demonstrated [20]. To this regard, a possible role for contiguous genes to MECP2, i.e., IRAK1 (OMIM *300283), cannot be ruled out, as a compensatory upregulation of IRAK1 has been reported in Mecp2 loss-of-function mutations associated with experimental RTT murine models [47, 48].
Given that MDS and RTT both lead to severe neurodevelopmental disorders sharing several similar features (Table 3), our data, beyond stressing the critical role of the MECP2 function for the OS status, shed further light on the relevance of redox homeostasis in the central nervous system integrity.
Table 3. Commonalities and differences between MECP2 duplication syndrome and Rett syndrome.
| MECP2 duplication syndrome | Rett syndrome | |
|---|---|---|
| MeCP2 | ↑ | ↓ |
| Irak1 | ↑ | ↑/(↓ in deletions) |
| p-NPBI | ↑ ** | ↑ * |
| F2-IsoPs | ↑*** | ↑ ** |
| F2-dihomo-IsoPs (supposed white matter involvement) | ↔ / ↑* | ↑*** |
| F4-NeuroPs (supposed gray matter involvement) | ↑*** | ↑*** |
| IE-NPBI | ↑• | ↑• |
| Inflammation | Yes | Yes |
| Gender | Male/Female | Female |
| Microcephaly | Yes (83.3%) | Yes |
| Hand stereotypies | Yes (83.3%) | Yes (+++) |
| Abnormal breathing | Yes (66.6%) | Yes (+++) |
| Bruxism | Yes (83.3%) | Yes (+++) |
| Laryngomalacia | Yes (16.6%) | No |
| Sleep disturbances | Yes (100%) | Yes (+++) |
| Gastro esophageal reflux disease (GERD), drooling | Yes (100%) | Yes |
| Constipation | Yes (100%) | Yes |
| Genital abnormalities | Yes (16.6%) | No |
| Facial dysmorphism | Yes (83.3%) | No |
| Facial hypotonia | Yes (100%) | No |
| Dysphagia | Yes (83.3%) | Yes |
| Intellectual disability | Yes (100%) | Yes |
| Developmental regression | Yes (100%) | Yes |
| Epilepsy | Yes (83.3%) | Yes |
| Magnetic resonance imaging (MRI) abnormalities | Yes (100%) | Rare |
| Hypotonia/ spasticity | Yes (100%) | Yes |
| Feeding difficulties | Yes (100%) | Yes |
| Recurrent Infections | Yes (66.6%) | Yes/No |
| Endocrine abnormalities | Yes (16.6%) | Yes/No |
| Hypoxia | Not evaluated | Yes (mild chronic) |
Alike RTT, MDS seems to elicit an inflammatory response. This condition could be linked to either imbalanced gene control or abnormal redox status. Although recurrent infections are a quite common feature of MDS [49], present in the clinical history of five out of our six patients, no signs of ongoing infection were detectable at the time of the blood sampling. However, it is also possible that the elevated ESR values are not directly related to the underlying genetic abnormality. Nevertheless, the inflammatory similarities observed in MDS and RTT well comply with the potential role for MeCP2 in regulating cytokine-dependent T helper cell differentiation [20]. Thus, _MECP_2 seems to exert a finely tuned regulation of major biological processes.
Conclusion
Taken together, our data indicate that a MECP2 disequilibrium, either due to loss-of-function or gain-of-function mutation, leads to similar phenotypes in terms of OS status, thus adding new evidence on the relationship between OS and MECP2.
Acknowledgments
We thank the families for their collaboration. We acknowledge Medical Genetics-AOUS, Siena (Head: Prof. Alessandra Renieri) for characterization of the some of the Xq28 duplication. We thank Dr. Pierluigi Tosi, Dr. Silvia Briani and Dr. Roberta Croci from the Administrative Direction of the Azienda Ospedaliera Senese for continued support to our studies and for prior purchasing of the gas spectrometry instrumentation. We also thank professional singer Matteo Setti for his artistic contributions to the exploration of Rett syndrome and his many charity concerts dedicated to Rett syndrome.
Data Availability
All relevant data are within the paper.
Funding Statement
The present work was funded by the Tuscan Region (Bando Salute 2009; “Antioxidants—omega-3 polyunsaturated Fatty Acids, lipoic acid—supplementation in Rett syndrome: A novel approach to therapy,” RT no. 142). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hite KC, Adams VH, Hansen JC. Recent advances in MeCP2 structure and function. Biochem Cell Biol. 2009; 87: 219–227. 10.1139/O08-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song C, Feodorova Y, Guy J, Peichl L, Jost KL, Kimura H, et al. DNA methylation reader MECP2: cell type- and differentiation stage-specific protein distribution. Epigenetics Chromatin 2014; 7: 17 Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4148084/10.1186/1756-8935-7-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999; 23: 185–188. [DOI] [PubMed] [Google Scholar]
- 4.Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010; 68: 944–950. 10.1002/ana.22124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christodoulou J, Ho G. MECP2-Related Disorders In: Pagon R. A.; Adam M. P.; Ardinger H.H. et al. , editors. GeneReviews [Internet], 2012, Seattle (WA), University of Washington, Seattle; 1993–2014, 2001 Oct 03 [updated 2012 Jun 28]. [Google Scholar]
- 6.Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 2005; 77: 442–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A 2010; 152A: 1079–88. 10.1002/ajmg.a.33184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Esch H. MECP2 Duplication Syndrome. Mol Syndromol. 2012; 2: 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimada S, Okamoto N, Ito M, Arai Y, Momosaki K, Togawa M, et al. MECP2 duplication syndrome in both genders. Brain Dev 2013; 35: 411–9. 10.1016/j.braindev.2012.07.010 [DOI] [PubMed] [Google Scholar]
- 10.Bao X, Jiang S, Song F, Pan H, Li M, Wu XR. X chromosome inactivation in Rett Syndrome and its correlations with MECP2 mutations and phenotype. J Child Neurol. 2008; 23: 22–25. 10.1177/0883073807307077 [DOI] [PubMed] [Google Scholar]
- 11.Ravn K, Roende G, Duno M, Fuglsang K, Eiklid KL, Tümer Z, et al. Two new Rett syndrome families and review of the literature: expanding the knowledge of MECP2 frameshift mutations. Orphanet J Rare Dis. 2011; 6:58 Available: http://www.ojrd.com/content/6/1/5810.1186/1750-1172-6-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.del Gaudio D, Fang P, Scaglia F, Ward PA, Craigen WJ, Glaze DG, et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med 2006; 8: 784–792. [DOI] [PubMed] [Google Scholar]
- 13.Carvalho CM, Zhang F, Liu P, Patel A, Sahoo T, Bacino CA, et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet 2009; 18: 2188–2203. 10.1093/hmg/ddp151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, Yntema HG, Tzschach A, et al. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet 2009; 17: 444–453. 10.1038/ejhg.2008.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Na ES, Nelson ED, Kavalali ET, Monteggia LM. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology 2013; 38: 212–219. 10.1038/npp.2012.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shichiri M. The role of lipid peroxidation in neurological disorders. J Clin Biochem Nutr 2014; 54: 151–160. 10.3164/jcbn.14-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Felice C, Signorini C, Leoncini S, Pecorelli A, Durand T, Valacchi G, et al. The role of oxidative stress in Rett syndrome: an overview. Ann NY Acad Sci 2012; 1259: 121–135. 10.1111/j.1749-6632.2012.06611.x [DOI] [PubMed] [Google Scholar]
- 18.Müller M, Can K. Aberrant redox homoeostasis and mitochondrial dysfunction in Rett syndrome. Biochem Soc Trans 2014; 42: 959–964. 10.1042/BST20140071 [DOI] [PubMed] [Google Scholar]
- 19.De Felice C, Della Ragione F, Signorini C, Leoncini S, Pecorelli A, Ciccoli L, et al. Oxidative brain damage in Mecp2-mutant murine models of Rett syndrome. Neurobiol Dis 2014; 68: 66–77. 10.1016/j.nbd.2014.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang S, Li C, McRae G, Lykken E, Sevilla J, Liu SQ, et al. MeCP2 reinforces STAT3 signaling and the generation of effector CD4+ T cells by promoting miR-124-mediated suppression of SOCS5. Sci Signal 2014; 7(316):ra25 Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4053436/10.1126/scisignal.2004824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janc OA, Müller M. The free radical scavenger Trolox dampens neuronal hyperexcitability, reinstates synaptic plasticity, and improves hypoxia tolerance in a mouse model of Rett syndrome. Front Cell Neurosci 2014; 8:56 Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3932407/. 10.3389/fncel.2014.00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bajic D, Berde CB, Commons KG. Periaqueductal gray neuroplasticity following chronic morphine varies with age: role of oxidative stress. Neuroscience 2012; 226: 165–77. 10.1016/j.neuroscience.2012.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muñoz P. Iron-mediated redox modulation in neural plasticity. Commun Integr Biol 2012; 5: 166–8. 10.4161/cib.18710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neul JL, Fang P, Barrish J, Lane J, Caeg EB, Smith EO, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008;70: 1313–1321. 10.1212/01.wnl.0000291011.54508.aa [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caselli R, Speciale C, Pescucci C, Uliana V, Sampieri K, Bruttini M, et al. Retinoblastoma and mental retardation microdeletion syndrome: clinical characterization and molecular dissection using array CGH. J Hum Genet 2007; 52: 535–542. [DOI] [PubMed] [Google Scholar]
- 26.De Felice C, Ciccoli L, Leoncini S, Signorini C, Rossi M, Vannuccini L, et al. Systemic oxidative stress in classic Rett syndrome. Free Radic Biol Med 2009; 47: 440–448. 10.1016/j.freeradbiomed.2009.05.016 [DOI] [PubMed] [Google Scholar]
- 27.Signorini C, De Felice C, Leoncini S, Giardini A, D'Esposito M, Filosa S, et al. F4-neuroprostanes mediate neurological severity in Rett syndrome. Clin Chim Acta 2011; 412: 1399–1406. 10.1016/j.cca.2011.04.016 [DOI] [PubMed] [Google Scholar]
- 28.De Felice C, Signorini C, Durand T, Oger C, Guy A, Bultel-Poncé V, et al. F2-dihomo-isoprostanes as potential early biomarkers of lipid oxidative damage in Rett syndrome. J Lipid Res 2011; 52: 2287–2297. 10.1194/jlr.P017798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allain CC, Poon LS, Chan CS, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clin Chem. 1974;20: 470–475. [PubMed] [Google Scholar]
- 30.Harris N, Galpchian V, Rifai N. Three routine methods for measuring high-density lipoprotein cholesterol compared with the Reference method. Clin Chem 1996; 42: 738–743. [PubMed] [Google Scholar]
- 31.Tietz NW. Clinical Guide to Laboratory Tests, 3 rd ed. WB Saunders Company, Philadelphia; 1995. pp. 610–611. [Google Scholar]
- 32.Wagner C, Dati F. Fibrinogen, in: Thomas L. ed., Clinical Laboratory Diagnostics, THBook Verlagsgeselisshaft, Frankfurt, 1998. pp. 609–612. [Google Scholar]
- 33.Piva E, Pajola R, Temporin V, Plebani M. A new turbidimetric standard to improve the qualityassurance of the erythrocyte sedimentation rate measurement. Clin Biochem 2007; 40: 491–495. [DOI] [PubMed] [Google Scholar]
- 34.Hill VL, Simpson VZ, Higgins JM, Hu Z, Stevens RA, Metcalf JA, et al. Evaluation of the Performance of the Sysmex XT-2000i Hematology Analyzer With Whole Bloods Stored at Room Temperature. Lab Med 2009; 40: 709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine, 5 th ed. Oxford University Press; 2015. [Google Scholar]
- 36.Pozzo-Miller L, Pati S, Percy AK. Rett Syndrome: Reaching for Clinical Trials. Neurotherapeutics 2015; 12: 631–640. 10.1007/s13311-015-0353-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samaco RC, Neul JL. Complexities of Rett syndrome and MeCP2. J Neurosci 2011; 31: 7951–7959. 10.1523/JNEUROSCI.0169-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gold WA, Williamson SL, Kaur S, Hargreaves IP, Land JM, Pelka GJ, et al. Mitochondrial dysfunction in the skeletal muscle of a mouse model of Rett syndrome (RTT): implications for the disease phenotype. Mitochondrion 2014; 15:10–7. 10.1016/j.mito.2014.02.012 [DOI] [PubMed] [Google Scholar]
- 39.Pecorelli A, Leoni G, Cervellati F, Canali R, Signorini C, Leoncini S, et al. Genes related to mitochondrial functions, protein degradation, and chromatin folding are differentially expressed in lymphomonocytes of Rett syndrome patients. Mediators Inflamm 2013; 2013:137629 Available: http://www.hindawi.com/journals/mi/2013/137629/10.1155/2013/137629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kriaucionis S, Paterson A, Curtis J, Guy J, Macleod N, Bird A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol 2006; 26: 5033–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cardaioli E, Dotti MT, Hayek G, Zappella M, Federico A. Studies on mitochondrial pathogenesis of Rett syndrome: ultrastructural data from skin and muscle biopsies and mutational analysis at mtDNA nucleotides 10463 and 2835. J Submicrosc Cytol Pathol 1999; 31: 301–304. [PubMed] [Google Scholar]
- 42.Signorini C, Leoncini S, De Felice C, Pecorelli A, Meloni I, Ariani F, et al. Redox imbalance and morphological changes in skin fibroblasts in typical Rett syndrome. Oxid Med Cell Longev 2014: 2014:195935 Available: http://www.hindawi.com/journals/omcl/2014/195935/10.1155/2014/195935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Musiek ES, Cha JK, Yin H, Zackert WE, Terry ES, Porter NA, et al. Quantification of F-ring isoprostane-like compounds (F4-neuroprostanes) derived from docosahexaenoic acid in vivo in humans by a stable isotope dilution mass spectrometric assay. J Chrom B Analyt Technol Biomed Life Sci 2004; 799: 95–102. [DOI] [PubMed] [Google Scholar]
- 44.Kuo HC, Yen HC, Huang CC, Hsu WC, Wei HJ, Lin CL. Cerebrospinal fluid biomarkers for neuropsychological symptoms in early stage of late-onset Alzheimer's disease. Int J Neurosci. 2015; 125: 747–754. 10.3109/00207454.2014.971787 [DOI] [PubMed] [Google Scholar]
- 45.VanRollins M, Woltjer RL, Yin H, Morrow JD, Montine TJ. F2-dihomo-isoprostanes arise from free radical attack on adrenic acid. J Lipid Res 2008; 49: 995–1005. 10.1194/jlr.M700503-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El Chehadeh S, Faivre L, Mosca-Boidron AL, Malan V, Amiel J, Nizon M, et al. Large national series of patients with Xq28 duplication involving MECP2: Delineation of brain MRI abnormalities in 30 affected patients. Am J Med Genet Part A 2015; 9999A: 1–14. [DOI] [PubMed] [Google Scholar]
- 47.Urdinguio RG, Fernandez AF, Lopez-Nieva P, Rossi S, Huertas D, Kulis M, et al. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 2010; 5: 656–663. 10.4161/epi.5.7.13055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kishi N, MacDonald JL, Ye J, Molyneaux BJ, Azim E, Macklis JD. Reduction of aberrant NF-κB signalling ameliorates Rett syndrome phenotypes in Mecp2-null mice. Nat Commun. 2016; 7:10520 Available: http://www.nature.com/ncomms/2016/160129/ncomms10520/pdf/ncomms10520.pdf10.1038/ncomms10520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bauer M, Kölsch U, Krüger R, Unterwalder N, Hameister K, Kaiser FM, et al. Infectious and immunologic phenotype of MECP2 duplication syndrome. J Clin Immunol. 2015; 35: 168–181. 10.1007/s10875-015-0129-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.